
Therapeutic Potential of Emerging NAD+-Increasing Strategies for Cardiovascular Diseases

 , , , , ,
, , , , , 
 ,
,  , and
, and 
Abstract
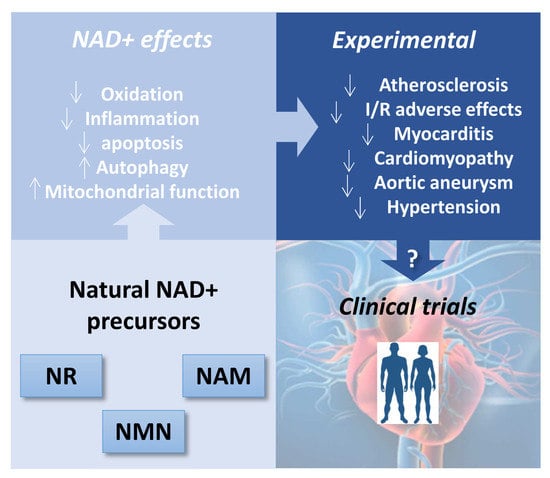
:
1. Introduction
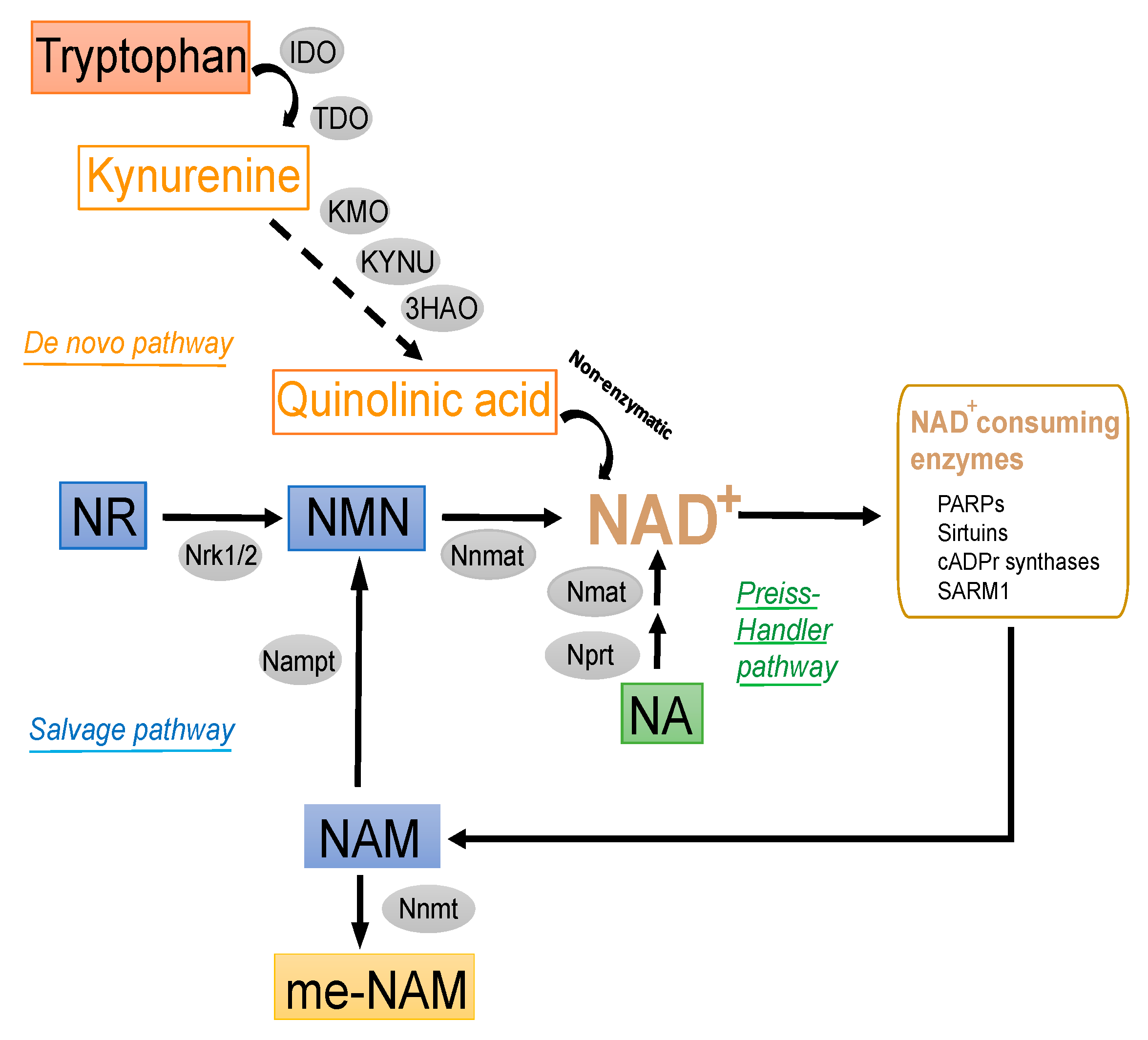
2. Preferential Physiological Sources of NAD+
2.1. NAD+ Sources
2.1.1. De Novo Synthesis Pathway
2.1.2. Salvage Pathway
2.1.3. Other Modulators of Intracellular NAD+ Pools
2.2. Administration and Bioavailability of NAD+ Precursors
2.3. NAD+-Consuming Enzymes as Determinants of the NAD+ Content
3. In Vitro Cardioprotective Effects of NAD+ Sources
3.1. Ex Vivo Properties of NAD+ Precursors Relevant to Cardiovascular Diseases
3.2. Cellular Effects of NAD+-Increasing Strategies Relevant to Cardiovascular Diseases
3.2.1. Oxidation
3.2.2. Inflammation
3.2.3. Apoptosis
3.2.4. Mitochondrial Stress
3.2.5. Autophagy
3.2.6. Fibrosis
4. In Vivo Cardiovascular Effects of NAD+-Boosting Strategies: Lessons from Animal Models
4.1. Atherosclerosis
4.2. Ischemic-Infarcted Myocardium or Ischemia-Reperfusion
4.3. Cardiomyopathy and Heart Failure (HF)
4.4. Cardiotoxicity
4.5. Aortic Aneurysm: Vascular Disease
4.6. Hypertension

{kind=link}
{kind=link}
{kind=link}
| Form of Suppl. B3 | Animal Model | Dose and Route of Administration | Duration | Outcome | Reference |
|---|---|---|---|---|---|
| Atherosclerosis | |||||
| NA | Ldlr−/− mice | Dietary supplementation of NA (0.3% w/w) in a high-fat diet containing 21% butter fat and 1.5% cholesterol | 10 weeks | Reduced aortic atherosclerotic plaque area | [137] |
| NA | Apoe−/− mice | Dietary supplementation NA (0.5% w/w) in a regular chow diet | 27 weeks | Reduced atherosclerotic lesions within the innominate artery | [138] |
| NA | Apoe−/− mice and Ldlr−/− mice | Dietary supplementation of NA (3% w/w) in a high-fat diet containing 21% of fat and 0.2% cholesterol | 8 weeks | Reduced aortic cholesterol and whole atherosclerotic plaque area | [139] |
| NA | Apoe−/− mice | Dietary supplementation NA (0.5% w/w) in a regular chow diet containing 0.2% cholesterol | 14 weeks | No changes in aortic root atherosclerotic area | [140] |
| NA | Apoe*3Leiden transgenic mice expressing human CETP | Dietary supplementation NA (0.1% w/w) in high fat diet containing 15% cacao butter and 0.1% cholesterol | 18 weeks | Reduced aortic root atherosclerotic area | [141] |
| NA and pentaerythritoltetran NA | Rabbit | Dietary supplementation of NA or pentaerythritoltetran NA (0.5% w/w) in a cholesterol-containing (1%) diet | 71 days | Only pentaerythritoltetran NA reduced the lipid infiltrated area of the aorta | [132] |
| NA and pentaerythritoltetran NA | Rabbit | Dietary supplementation of NA or pentaerythritoltetran NA (0.5% w/w) in a cholesterol- and coconut oil- containing (1% and 3%, respectively) diet | 81 days | Both drugs reduced the lipid infiltrated area of the aorta | [132] |
| Pentaerythritoltetran NA | Rabbit | Dietary supplementation of pentaerythritoltetran NA (0.5% w/w) in a coconut oil- containing (8% and 15%) diet | 120 and 160 days | Reduced lipid infiltration in the aorta | [131] |
| Pentaerythritoltetran NA | Rabbit | Dietary supplementation of pentaerythritoltetran NA (0.75% w/w) in a coconut oil- containing (3%) diet. The concentration of cholesterol in the diet was adjusted for each rabbit andt the animals attained a mean plasma cholesterol level of about 6 mg/mL | 160 days | Reduced aortic cholesterol | [130] |
| NA and Pentaerythritoltetran NA | Mini-pigs | Dietary supplementation of NA or pentaerythritoltetran NA (0.25–0.75% w/w) in egg yolk- and cholesterol- (11 and 0.5–0.75%, respectively) containing diet | 12–19 months | Reduced lipid infiltration in the aorta | [134] |
| Me-NAM and NA | Double Ldlr−/−/Apoe−/− mice | Administration of me-NAM or NA (0.1 g/kg/day) in the drinking water together a regular chow diet | 4 weeks | Both drugs reduced aortic root atherosclerotic area | [29] |
| NAM | Apoe−/− mice | Administration of NAM (0.25 and 1% w/v equivalent to 0.5 and 1.9 g/kg/day, respectively) in the drinking water together a high-fat diet containing 21% of fat and 0.2% cholesterol | 4 weeks | Reduced aortic root atherosclerotic area | [25] |
| me-NAM | Apoe−/− mice | Dietary supplementation of me-NAM (0.0057 and 0.017% w/w) in a high-fat diet containing 21% of fat and 0.2% cholesterol | 8 weeks | Reduced aortic root atherosclerotic area | [30] |
| Cardiotoxicity | |||||
| NAM | Sprague Dawley male rats treated with DOX (5 mg/kg, i.p.) once/week for four consecutive weeks | Oral dose of NAM (600 mg/kg by oral gavage). | 28 consecutive days | Amelioration of cardiotoxic serum cardiotoxicity indices, conduction and histopathological abnormalities | [210] |
| NR | Male mice (aged 2 months) were injected with a single dose of DOX (20 mg/kg, i.p.) | 0, 100, 300, or 500 mg/kg (i.p.) given 30 min prior DOX injection | 5 days | Reduced cardiac injury and myocardial dysfunction | [120] |
| Myocardial ischemia/reperfusion injury | |||||
| NAD+ | Male Wistar rats | 10–20 mg/kg intravenous (i.v) (approximately 85% reduction of the infarct at the dosage of 20 mg/kg) | A single dose immendiately before ischemia The rats were sacrificed 6 and 24 h after reperfusion. | Reduced the infarct size after ischemia/reperfusion Attenuated apoptotic damage and enhancing the antioxidant capacity | [159] |
| NAD+ | C57BL/6 wild-type mice | i.p. administration of 0.2 g/kg NAD | 1 dose before myocardial injury | Reduced myocardial infarct size after ischemia/reperfusion | [161] |
| NAD+ | Bama miniature pigs (a swine model of ischemia/reperfusion injury) | 20 mg/kg NAD+ or saline, i.v. | Before reperfusion | Dysinflammation, less cardiac fibrosis, and better ventricular compliance; reduced myocardial necrosis, and promoted cardiac function recovery | [162] |
| NAD+ | Specific-pathogen-free male Sprague-Dawley rats | 10 mg/kg i.p. | 14 days | Attenuation the depression of cardiac function in the isolated rat hearts after ischemia-reperfusion | [163] |
| NMN | C57BL/6 wild-type mice | administration of 0.5 g/kg i.p. | 30 min before ischemia and repetitive administration just before and during reperfusion | Reduced the infarct size after ischemia/reperfusion | [164] |
| NR | Male Wistar rats | i.v. infusion of 50 mg/kg NR | NR infusion for 5 min before and 15 min after the beginning of reperfusion | Restoration of small intestine microcirculation after mesenteric ischaemia/reperfusion; improved small intestine mucosa damage. | [167] |
| Cardiomyopathy | |||||
| NR soft pellets | Srfh−/− and Sf/Sf control littermates | Chow diet or NR-supplemented with 400 mg/Kg of body weight/day | 50 days | Protection against cardiac dysfunction (measure LVEF and FS, dilatation and thinning of the LV wall) | [171] |
| NR soft pellets or in water | LmnaH222P/H222P and wild-type mice | Chow diet or NR-supplemented with 400 mg/kg of body weight/day | 16 days | Increased NAD+ content in liver and heart and partially restore the left ventricular function and increase survival | [177] |
| NR soft pellets or in water | LmnaH222P/H222P mice and wild-type mice | For post-symptomatic treatment, LmnaH222P/H222P mice received 400 mg/kg per day orally by gavage | 9 weeks | Stable left ventricular dimensions and fractional shortening | [177] |
| NAM | LmnaH222P/H222P and wild-type mice | Received 500 mg/kg i.p. | every other day during 9 weeks | Treatment with NAM is not efficient to restore the cardiac NAD+ content and cardiac function | [177] |
| NR | C57BL/6 CD or HFD+L-NAME (HFpEF mice model) | 400 mg/kg body weight/d | 5 day per week for 4 weeks | Improved mitochondrial function, ameliorates cardiac hypertrophy, attenuates diastolic dysfunction, improves exercise capacity, and reduces lung capacity | [179] |
| NR medium and high dose | MutUNG1 mice and control littermates | CD, NR-supplemented with 400 mg/kg (medium dose) or 1,000 mg/kg (high dose) | 2 weeks | NR high dose: inhibits SIRT3 activity due to an enhance levels of NAM and promotes mitochondrial dysfunction. Reduction of NAD+ levels in cardiac tissue and loss of mitochondrial deacetylation | [182] |
| NMN | Myh6-Cre:Klf4 fl/fl mice, designated CM-K4KO (C57BL/6J background) | 500 mg/kg/day i.p. | 5 days | NMN protected the mutant mice from pressure overload-induced HF | [194] |
| NR | Mouse model of cardiac hypertrophy established using Transverse aortic constriction surgery (C57BL/6J background) | 400 mg/kg/day (daily oral gavage) | 8 weeks | NR alleviated from cardiac hypertrophy and dysfunction | [202] |
| Aortic Aneurysm | |||||
| NR | Marfan Syndrome mouse model Fbn1c1039g/+ | i.p. injections 1,000 mg/kg every second day | 28 days | Elevation of TFAM levels, improvement of mitochondrial metabolism, and normalization of aortic function and diameter | [221] |
| NA NAM | Apoe−/− mice infused with Ang II and C57BL/6J mouse CaCl2 induced AAA | Drinking water supplemented with NA 0.3% w/v, NAM 0.1% or 0.4% w/v | 2 days prior AAA induction to the end of the study | Protection against AAA formation mediated through NAD+ repletion that lead to SIRT1 activation | [24] |
| Me-NAM | Apoe−/− mice | Dietary supplementation of me-NAM (0.0057 and 0.017% w/w) in a high-fat diet containing 21% of fat and 0.2% cholesterol | 8 weeks | Reduced aortic root atherosclerotic area | [30] |
| Hypertension | |||||
| NA | Male Sprague-Dawley rats | NA dissolved in the drinking water (50 mg/kg/day) | 12 weeks | Ameliorated hypertension and partial reversal of upregulation of oxidative, inflammatory, and profibrotic mediators in the remnant kidney | [249] |
| NAD+ | Sirt1 transgenic and C57BL/6J mice | Incubation of NAD+ in whole aorta homogenates to measure SIRT1 deacetylase activity | 80 min | Improvement of vascular remodeling in the aortas of transgenic mice resulting in a decreased SBP | [244] |
| NADH | Spontaneously hypertensive male rats | 5 mg administered in single tablets daily | 60 days | Lower SPB in treated rats vs. non-treated ones | [248] |
| β-NAD+ and NAM | Sea urchin egg | Incubation in the medium, Various concentrations of β-NAD+ (0–4 mM) and nicotinamide (0–5 mM) | 90 min | vasorelaxation by inhibition of ADP ribosyl cyclase | [245] |
| β-NAD+ β-NADH NGD+ NGD and NAM | Male New Zeland white rabbit | Incubation in smooth muscle homogenates of pulmonary arteries β-NAD+ (2.5 mM) β-NADH (25 mM) NGD+ (250 µM) NGD+ (250 µM) + NAM (10 mM) | 60 min | Vasorelaxation by inhibition of ADP ribosyl cyclase | [247] |
| NAM | Sprague-Dawley rats, wild-type C57BL/6 mice, and CD38/mice on a C57BL6 background | Injection of 6 mg/kg/min in the renal artery | 3 min before and 5 min following endothelin-1 or sarafotoxin-6c injection | Vasorelaxation by inhibition of ADP ribosyl cyclase | [246] |
| NAM | Dahl salt-sensitive rats | 40 mM oral supplementation in the drinking water | 5 weeks | improved diastolic dysfunction | [31] |
| NAM | RUPP mice | Daily oral gavage (500 mg/kg/day) | In between 14.5 and 18.5 days postcoitus | Improving and preventing hypertension, fetal growth restriction, and premature birth | [250] |
| NAM | 1-Non-pregnant female C57BL/6J 2-Pregnant female ICR 3-Asb4−/− females | Daily oral gavage (500 mg/kg/day) | In between 12.5 and 18.5 days postcoitus | Improving and preventing hypertension, proteinuria, miscarriage, and premature birth in preeclampsia | [251] |
| NAM | 1-C57BL/6J male mice 2-Renin-transgenic male mice 3-eNOS-null female and male mice | NAM dissolved in the drinking water (500 mg/kg/day) L-NAME: 50 mg/kg/day in drinking water L-NAME + NAM same dose as above in drinking water | 60 days | Normalized blood pressure in mice with impaired eNOS function via suppressing inflammation | [252] |
| NMN | C57BL/6J mice | i.p. injection of NMN (500 mg/kg) | twice a day during 7 consecutive days | Preservation of myocardial NAD+ levels and functional compensation against pressure overload | [253] |
5. NAD+-Increasing Strategies in Human Heart Health: From Bench to Bed
5.1. NA
5.2. NAM
5.3. NR
5.4. NMN
| Interventions | Aimed Cardiovascular-Related Outcomes | Study Start Date | Finish Date | Time Frame | Dosage | NCT |
|---|---|---|---|---|---|---|
| NA | Change in the mean severity of proximal stenosis | Jan 1984 | Aug 1989 | 2.5 years | NA was started at 125 mg twice a day and gradually increased to 500 mg four times a day (with meals and at bedtime) at one month and 1 g four times a day at two months. If the LDL cholesterol level did not fall below 3.1 mmol per liter (120 mg per deciliter) after three months, the dose of niacin was increased to 1.5 g (three tablets) four times a day, but no further | NCT00000512 |
| NA | Change in minimal diameter of coronary artery lesions | Dec 1986 | Nov 1992 | Not available | Not available | NCT00000461 |
| NA | Change in proximal obstructive disease | Sep 1994 | Aug 1999 | 2.5 years | Not available | NCT00000553 |
| NA | Change in plaque morphology | Jan 2000 | Sep 2005 | 12 months | NA 20 mg daily | NCT00307307 |
| NA | Changes in carotid plaque composition | Jun 2001 | Apr 2019 | 40 months | NA 2000 mg daily | NCT00715273 |
| NA | Inflammation and clot formation and blood vessel health | Jun 2002 | Nov 2005 | 16 weeks | NA 1500 mg daily | NCT00590629 |
| NA | Endothelial function | Jun 2003 | Jun 2005 | 16 weeks | NA 1.5 g daily | NCT01921010 |
| NA | Changes in aortic and carotid plaque architecture and composition | Sep 2003 | Dec 2008 | 18 months | NA extended release 0.5 to 3.0 g daily | NCT00127218 |
| NA | Change in superficial femoral artery wall volume | Apr 2004 | Dec 2021 | 24 months | NA 1500 mg daily | NCT00687076 |
| NA | Brachial artery flow mediated dilation | Sep 2005 | Aug 2008 | 3 months | NA extended release 1500 mg daily | NCT00150722 |
| NA | Relative effect on flow-mediated dilatation of radial artery | Mar 2006 | Jun 2009 | 6 months | NA extended-release | NCT00298909 |
| NA | Endothelial function by high resolution echography in response to nitric agent | Jun 2007 | May 2009 | 3 months | Not available | NCT00855257 |
| NA | Mean plaque lipid composition in carotid arteries | Mar 2008 | Feb 2015 | 24 months | NA extended-release 1500 mg or 2000 mg daily | NCT01178320 |
| NA | Composite score of plaque inflammation/stability, plaque instability protein composite score, total cholesterol, and free cholesterol | Apr 2009 | Oct 2010 | 12 weeks | NA extended-release tablet 2 g daily | NCT00804843 |
| NA | Endothelial dependant dilatation of the arterial wall | Sep 2010 | Oct 2011 | 12 weeks | NA 2000 mg/40 mg | NCT01126073 |
| NA | Change in percent atheroma volume by intravascular ultrasonography | Oct 2010 | Nov 2015 | 12 months | NA extended release 1500–2000 mg daily | NCT01221402 |
| NA | Change from baseline in arterial fluorodeoxyglucose uptake | Mar 2012 | Jan 2013 | 12 weeks | NA titrated to 6000 mg daily | NCT02003638 |
| NA | Changes in protein or lipid composition of any lipoprotein fraction and changes in vascular compliance | Mar 2015 | Jun 2019 | 14 weeks | NA extended release 2000 mg/day | NCT02322203 |
| NAM | Number of participants with adverse events (early onset Preeclampsia) | Aug 2014 | Nov 2018 | 48 h | Either 500 mg or 1000 mg by mouth each morning until delivery or 14 days, whichever occurs first | NCT02213094 |
| NAM | NAD+ augmentation in cardiac surgery associated myocardial injury trial. Troponin T (area under the curve) | Feb 2021 | Sep 2021 | From baseline to three days after surgery | 3 g on the day of surgery and post-surgical days one and two | NCT04750616 |
| NR | Mean IL-1beta release From peripheral blood mononuclear cells during refeeding after 24 h fast | Jun 2016 | Aug 2018 | 4 weeks | Either NR at 1000 mg/day or placebo for one week, followed by a washout period of 2–3 weeks, then a crossover to placebo or NR at 1000 mg/day for one additional week. The end point was analyzed at end of each treatment. | NCT02812238 |
| NR | Bioavailability (pharmacokinetics), safety (blood pressure, pulse, etc.) and impact on mitochondrial disease symptoms * | Dec 2017 | Dec 2019 | 24 h (bioavailability and safety) and 4 weeks (mitochondrial characteristics) | not defined (open-label experimental medicine study; all subjects will receive the same dosage of the supplement) | NCT03432871 |
| NR | Number of participants without heart failure linked inflammation in patients with stable, systolic heart failure | Jun 2018 | Oct 2020 | 12 weeks | Starting at 500 mg daily (250 mg BID) be increased at two weekly intervals by 250 mg/dose (BID) (500 mg/day) to a final dose of 1000 mg PO BID (2000 mg/day) | NCT03565328 |
| NR | Incidence of treatment-emergent adverse events (safety and tolerability), whereby the main aim was to assess the preoperative effect of NR supplementation in patients undergoing elective left ventricular assist device (LVAD) implantation | Nov 2018 | Sep 2020 | Up to 14 days | Dose Escalation: Day 1: 250 mg (1 capsule) twice daily (total daily intake = 500 mg) Day 2: 500 mg (2 capsules) twice daily Day 3: 1000 mg (4 capsules) twice daily (total daily intake = 2000 mg) Dose Maintenance: Day 4: 1000 mg (4 capsules) twice daily Day 5–14 as applicable through Day Before Surgery: 1000 mg (4 capsules) twice daily(total daily intake = 1000 mg) | NCT03727646 |
| NR | Exploratory endpoint: effect of NR on left ventricular diastolic and systolic function | May 2019 | Jun 2019 | 12 weeks | The initial dose will be 1 capsule twice daily, followed by weekly up-titration by 1 capsule/dose to a final dose of 4 capsules (1000 mg) twice daily at the end of week 4; participants will be continued on the final dose up to the final follow up visit (week 12) | NCT03423342 |
| NR | Carotid-femoral pulse wave velocity (primary) and systolic and diastolic blood pressure (secondary) | Nov 2019 | Sep 2014 | 3 months | 500 mg by mouth twice | NCT04040959 |
| NR | Systolic blood pressure | May 2019 | Dec 2023 | 3 months | 500 mg of the vitamin B3-precursor, nicotinamide riboside (NIAGEN) twice per day (1000 mg per day total) | NCT03821623 |
| NR | Between-group comparisons of myocardial NAD(H) levels, myocardial mitochondrial morphology, myocardial mitochondrial respiratory function, myocardial protein acetylation, myocardial gene expression by RNA-seq and the myocardial epigenome by ATAC-seq, inflammatory markers in myocardium | Sep 2020 | Aug 2024 | Up to 14 days | Dose Escalation: Day 1: 250 mg (1 capsule) twice daily (total daily intake = 500 mg) Day 2: 500 mg (2 capsules) twice daily Day 3: 1000 mg (4 capsules) twice daily (total daily intake = 2000 mg) Dose Maintenance: Day 4: 1000 mg (4 capsules) twice daily Day 5–14 as applicable through Day Before Surgery: 1000 mg (4 capsules) twice daily(total daily intake = 1000 mg) | NCT04528004 |
| NR | Change in Systolic blood pressure (primary), and change in arterial stiffness (secondary) | Jul 2020 | May 2022 | 6 week | 1000 mg/day | NCT04112043 |
| NMN | Effect of NMN on flow mediated dilation and brachial-ankle pulse wave velocity | Jun 2021 | Jul 2022 | 2 months | NMN10,000 WRIGHT LIFE® + lifestyle modification | NCT04903210 |
| NR | Vasodilatory Reserve (Percent change in systemic vascular resistance at baseline vs. exhaustion) and Kansas City Cardiomyopathy Questionnaire Overall Summary Score (Assess the impact of our interventions on quality of life) | Oct 2021 | Sep 2026 | 6 days | Potassium Nitrate (KNO3) 6 mmol three times daily + Propionyl-L-Carnitine (PLC) 1000 mg twice daily + NR 300 mg three times daily | NCT04913805 |
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fajemiroye, J.O.; da Cunha, L.C.; Saavedra-Rodriguez, R.; Rodrigues, K.L.; Naves, L.M.; Mourao, A.A.; da Silva, E.F.; Williams, N.E.E.; Martins, J.L.R.; Sousa, R.B.; et al. Aging-Induced Biological Changes and Cardiovascular Diseases. Biomed. Res. Int. 2018, 2018, 7156435. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prolla, T.A.; Denu, J.M. NAD+ deficiency in age-related mitochondrial dysfunction. Cell Metab. 2014, 19, 178–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, M.B.; Sinclair, D.A. Why NAD(+) Declines during Aging: It’s Destroyed. Cell Metab. 2016, 23, 965–966. [Google Scholar] [CrossRef] [Green Version]
- Okabe, K.; Yaku, K.; Tobe, K.; Nakagawa, T. Implications of altered NAD metabolism in metabolic disorders. J. Biomed. Sci. 2019, 26, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Zuo, W.; Liu, Y.; Wu, K.; Liu, Q. NAD(+) and cardiovascular diseases. Clin. Chim. Acta 2021, 515, 104–110. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Sun, A.; Je, G. The effects of nicotinamide adenine dinucleotide in cardiovascular diseases: Molecular mechanisms, roles and therapeutic potential. Genes Dis. 2021, in press. [Google Scholar] [CrossRef]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef] [Green Version]
- de Picciotto, N.E.; Gano, L.B.; Johnson, L.C.; Martens, C.R.; Sindler, A.L.; Mills, K.F.; Imai, S.; Seals, D.R. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell 2016, 15, 522–530. [Google Scholar] [CrossRef]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [Green Version]
- Mills, K.F.; Yoshida, S.; Stein, L.R.; Grozio, A.; Kubota, S.; Sasaki, Y.; Redpath, P.; Migaud, M.E.; Apte, R.S.; Uchida, K.; et al. Long-Term Administration of Nicotinamide Mononucleotide Mitigates Age-Associated Physiological Decline in Mice. Cell Metab. 2016, 24, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Mendez-Lara, K.A.; Rodriguez-Millan, E.; Sebastian, D.; Blanco-Soto, R.; Camacho, M.; Nan, M.N.; Diarte-Anazco, E.M.G.; Mato, E.; Lope-Piedrafita, S.; Roglans, N.; et al. Nicotinamide Protects Against Diet-Induced Body Weight Gain, Increases Energy Expenditure, and Induces White Adipose Tissue Beiging. Mol. Nutr. Food Res. 2021, 65, e2100111. [Google Scholar] [CrossRef]
- Mitchell, S.J.; Bernier, M.; Aon, M.A.; Cortassa, S.; Kim, E.Y.; Fang, E.F.; Palacios, H.H.; Ali, A.; Navas-Enamorado, I.; Di Francesco, A.; et al. Nicotinamide Improves Aspects of Healthspan, but Not Lifespan, in Mice. Cell Metab. 2018, 27, 667–676.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [CrossRef] [Green Version]
- Masana, L.; Cabre, A.; Heras, M.; Amigo, N.; Correig, X.; Martinez-Hervas, S.; Real, J.T.; Ascaso, J.F.; Quesada, H.; Julve, J.; et al. Remarkable quantitative and qualitative differences in HDL after niacin or fenofibrate therapy in type 2 diabetic patients. Atherosclerosis 2015, 238, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Lukasova, M.; Hanson, J.; Tunaru, S.; Offermanns, S. Nicotinic acid (niacin): New lipid-independent mechanisms of action and therapeutic potentials. Trends Pharmacol. Sci. 2011, 32, 700–707. [Google Scholar] [CrossRef]
- Romani, M.; Hofer, D.C.; Katsyuba, E.; Auwerx, J. Niacin: An old lipid drug in a new NAD(+) dress. J. Lipid Res. 2019, 60, 741–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, T.J.; Boden, W.E.; Desvigne-Nickens, P.; Fleg, J.L.; Kashyap, M.L.; McBride, R.; Probstfield, J.L. Safety profile of extended-release niacin in the AIM-HIGH trial. N. Engl. J. Med. 2014, 371, 288–290. [Google Scholar] [CrossRef] [Green Version]
- Investigators, A.-H.; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [Green Version]
- HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomi.ized placebo-controlled trial in 25,673 high-risk patients of ER niacin/laropiprant: Trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur. Heart J. 2013, 34, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Li, J.; Shen, Y.; Liu, G.; Zuo, S.; Tao, B.; Ji, Y.; Lu, A.; Lazarus, M.; Breyer, R.M.; et al. Niacin Promotes Cardiac Healing after Myocardial Infarction through Activation of the Myeloid Prostaglandin D2 Receptor Subtype 1. J. Pharmacol. Exp. Ther. 2017, 360, 435–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horimatsu, T.; Blomkalns, A.L.; Ogbi, M.; Moses, M.; Kim, D.; Patel, S.; Gilreath, N.; Reid, L.; Benson, T.W.; Pye, J.; et al. Niacin protects against abdominal aortic aneurysm formation via GPR109A independent mechanisms: Role of NAD+/nicotinamide. Cardiovasc. Res. 2020, 116, 2226–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
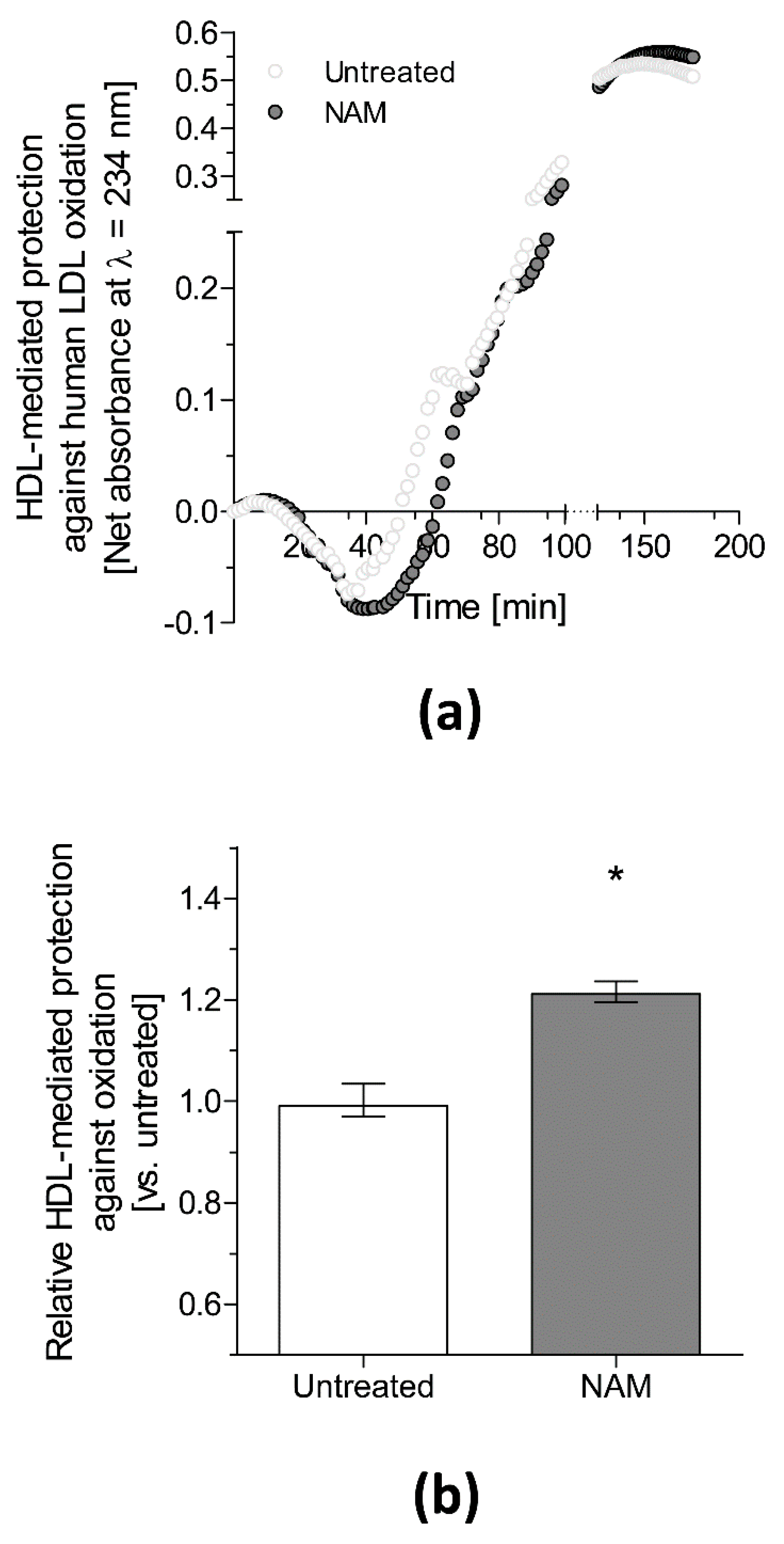
- Mendez-Lara, K.A.; Letelier, N.; Farre, N.; Diarte-Anazco, E.M.G.; Nieto-Nicolau, N.; Rodriguez-Millan, E.; Santos, D.; Pallares, V.; Escola-Gil, J.C.; Vazquez Del Olmo, T.; et al. Nicotinamide Prevents Apolipoprotein B-Containing Lipoprotein Oxidation, Inflammation and Atherosclerosis in Apolipoprotein E-Deficient Mice. Antioxidants 2020, 9, 1162. [Google Scholar] [CrossRef]
- Elliott, R.B.; Pilcher, C.C.; Stewart, A.; Fergusson, D.; McGregor, M.A. The use of nicotinamide in the prevention of type 1 diabetes. Ann. N. Y. Acad. Sci. 1993, 696, 333–341. [Google Scholar] [CrossRef]
- Gale, E.A.; Bingley, P.J.; Emmett, C.L.; Collier, T. European Nicotinamide Diabetes Intervention Trial (ENDIT): A randomised controlled trial of intervention before the onset of type 1 diabetes. Lancet 2004, 363, 925–931. [Google Scholar] [CrossRef]
- Hawrylyshyn, K.M. Nicotinamide Riboside Delivery Generates NAD+ Reserves to Protect Vascular Cells against Oxidative Damage; The University of Western Ontario: London, ON, USA, 2015. [Google Scholar]
- Mateuszuk, L.; Jasztal, A.; Maslak, E.; Gasior-Glogowska, M.; Baranska, M.; Sitek, B.; Kostogrys, R.; Zakrzewska, A.; Kij, A.; Walczak, M.; et al. Antiatherosclerotic Effects of 1-Methylnicotinamide in Apolipoprotein E/Low-Density Lipoprotein Receptor-Deficient Mice: A Comparison with Nicotinic Acid. J. Pharmacol. Exp. Ther. 2016, 356, 514–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, N.; Wang, M.; Song, J.; Liu, Y.; Chen, H.; Mu, D.; Xia, M. N-methylnicotinamide protects against endothelial dysfunction and attenuates atherogenesis in apolipoprotein E-deficient mice. Mol. Nutr. Food Res. 2016, 60, 1625–1636. [Google Scholar] [CrossRef]
- Abdellatif, M.; Trummer-Herbst, V.; Koser, F.; Durand, S.; Adao, R.; Vasques-Novoa, F.; Freundt, J.K.; Voglhuber, J.; Pricolo, M.R.; Kasa, M.; et al. Nicotinamide for the treatment of heart failure with preserved ejection fraction. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [Green Version]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magni, G.; Amici, A.; Emanuelli, M.; Orsomando, G.; Raffaelli, N.; Ruggieri, S. Enzymology of NAD+ homeostasis in man. Cell. Mol. Life Sci. 2004, 61, 19–34. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [Green Version]
- Chi, Y.; Sauve, A.A. Nicotinamide riboside, a trace nutrient in foods, is a vitamin B3 with effects on energy metabolism and neuroprotection. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, R.; Forteza, M.J.; Ketelhuth, D.F.J. The interplay between cytokines and the Kynurenine pathway in inflammation and atherosclerosis. Cytokine 2019, 122, 154148. [Google Scholar] [CrossRef]
- Bender, D.A. Biochemistry of tryptophan in health and disease. Mol. Aspects Med. 1983, 6, 101–197. [Google Scholar] [CrossRef]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef] [Green Version]
- Sauve, A.A. NAD+ and vitamin B3: From metabolism to therapies. J. Pharmacol. Exp. Ther. 2008, 324, 883–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polyzos, K.A.; Ketelhuth, D.F. The role of the kynurenine pathway of tryptophan metabolism in cardiovascular disease. An emerging field. Hamostaseologie 2015, 35, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de novo NAD(+) synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef]
- Kraus, D.; Yang, Q.; Kong, D.; Banks, A.S.; Zhang, L.; Rodgers, J.T.; Pirinen, E.; Pulinilkunnil, T.C.; Gong, F.; Wang, Y.C.; et al. Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 2014, 508, 258–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felici, R.; Lapucci, A.; Ramazzotti, M.; Chiarugi, A. Insight into molecular and functional properties of NMNAT3 reveals new hints of NAD homeostasis within human mitochondria. PLoS ONE 2013, 8, e76938. [Google Scholar] [CrossRef]
- Weinshilboum, R. Methyltransferase pharmacogenetics. Pharmacol. Ther. 1989, 43, 77–90. [Google Scholar] [CrossRef]
- Bartus, M.; Lomnicka, M.; Kostogrys, R.B.; Kazmierczak, P.; Watala, C.; Slominska, E.M.; Smolenski, R.T.; Pisulewski, P.M.; Adamus, J.; Gebicki, J.; et al. 1-Methylnicotinamide (MNA) prevents endothelial dysfunction in hypertriglyceridemic and diabetic rats. Pharmacol. Rep. 2008, 60, 127–138. [Google Scholar]
- Chlopicki, S.; Swies, J.; Mogielnicki, A.; Buczko, W.; Bartus, M.; Lomnicka, M.; Adamus, J.; Gebicki, J. 1-Methylnicotinamide (MNA), a primary metabolite of nicotinamide, exerts anti-thrombotic activity mediated by a cyclooxygenase-2/prostacyclin pathway. Br. J. Pharmacol. 2007, 152, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Mateuszuk, L.; Khomich, T.I.; Slominska, E.; Gajda, M.; Wojcik, L.; Lomnicka, M.; Gwozdz, P.; Chlopicki, S. Activation of nicotinamide N-methyltrasferase and increased formation of 1-methylnicotinamide (MNA) in atherosclerosis. Pharmacol. Rep. 2009, 61, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Sternak, M.; Khomich, T.I.; Jakubowski, A.; Szafarz, M.; Szczepanski, W.; Bialas, M.; Stojak, M.; Szymura-Oleksiak, J.; Chlopicki, S. Nicotinamide N-methyltransferase (NNMT) and 1-methylnicotinamide (MNA) in experimental hepatitis induced by concanavalin A in the mouse. Pharmacol. Rep. 2010, 62, 483–493. [Google Scholar] [CrossRef]
- Watala, C.; Kazmierczak, P.; Dobaczewski, M.; Przygodzki, T.; Bartus, M.; Lomnicka, M.; Slominska, E.M.; Durackova, Z.; Chlopicki, S. Anti-diabetic effects of 1-methylnicotinamide (MNA) in streptozocin-induced diabetes in rats. Pharmacol. Rep. 2009, 61, 86–98. [Google Scholar] [CrossRef]
- Fernstrom, J.D. A Perspective on the Safety of Supplemental Tryptophan Based on Its Metabolic Fates. J. Nutr. 2016, 146, 2601S–2608S. [Google Scholar] [CrossRef] [Green Version]
- EFSA. Scientific Opinion on the safety and efficacy of niacin (nicotinamide) as a feed additivefor all animal species based on a dossier submitted by EUROPE-ASIA Import Export GmbH. EFSA J. 2012, 10, 2789. [Google Scholar]
- McDowell, L.R. Vitamins in Animal and Human Nutrition, 2nd ed.; Wiley-Blackwell: Oxford, UK, 2000; p. 816. [Google Scholar]
- Trammell, S.A.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef]
- Belenky, P.A.; Moga, T.G.; Brenner, C. Saccharomyces cerevisiae YOR071C encodes the high affinity nicotinamide riboside transporter Nrt1. J. Biol. Chem. 2008, 283, 8075–8079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grozio, A.; Sociali, G.; Sturla, L.; Caffa, I.; Soncini, D.; Salis, A.; Raffaelli, N.; De Flora, A.; Nencioni, A.; Bruzzone, S. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J. Biol. Chem. 2013, 288, 25938–25949. [Google Scholar] [CrossRef] [Green Version]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E.; et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, S.; Guida, L.; Zocchi, E.; Franco, L.; De Flora, A. Connexin 43 hemi channels mediate Ca2+-regulated transmembrane NAD+ fluxes in intact cells. FASEB J. 2001, 15, 10–12. [Google Scholar] [CrossRef]
- Ferrero, E.; Lo Buono, N.; Horenstein, A.L.; Funaro, A.; Malavasi, F. The ADP-ribosyl cyclases--the current evolutionary state of the ARCs. Front. Biosci. (Landmark Ed.) 2014, 19, 986–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwer, B.; Verdin, E. Conserved metabolic regulatory functions of sirtuins. Cell Metab. 2008, 7, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Belenky, P.; Bogan, K.L.; Brenner, C. NAD+ metabolism in health and disease. Trends Biochem. Sci. 2007, 32, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Canto, C.; Sauve, A.A.; Bai, P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Aspects Med. 2013, 34, 1168–1201. [Google Scholar] [CrossRef] [Green Version]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.C. Physiological functions of cyclic ADP-ribose and NAADP as calcium messengers. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 317–345. [Google Scholar] [CrossRef]
- Zocchi, E.; Daga, A.; Usai, C.; Franco, L.; Guida, L.; Bruzzone, S.; Costa, A.; Marchetti, C.; De Flora, A. Expression of CD38 increases intracellular calcium concentration and reduces doubling time in HeLa and 3T3 cells. J. Biol. Chem. 1998, 273, 8017–8024. [Google Scholar] [CrossRef] [Green Version]
- Ortolan, E.; Vacca, P.; Capobianco, A.; Armando, E.; Crivellin, F.; Horenstein, A.; Malavasi, F. CD157, the Janus of CD38 but with a unique personality. Cell Biochem. Funct. 2002, 20, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J. Biol. Chem. 2012, 287, 31633–31640. [Google Scholar] [CrossRef] [Green Version]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Bayo Jimenez, M.T.; Vujacic-Mirski, K.; Helmstadter, J.; Kroller-Schon, S.; Munzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxid. Med. Cell. Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betteridge, D.J. What is oxidative stress? Metabolism 2000, 49, 3–8. [Google Scholar] [CrossRef]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Ardanaz, N.; Yang, X.P.; Cifuentes, M.E.; Haurani, M.J.; Jackson, K.W.; Liao, T.D.; Carretero, O.A.; Pagano, P.J. Lack of glutathione peroxidase 1 accelerates cardiac-specific hypertrophy and dysfunction in angiotensin II hypertension. Hypertension 2010, 55, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Chua, C.C.; Gao, J.; Chua, K.W.; Ho, Y.S.; Hamdy, R.C.; Chua, B.H. Prevention of ischemia/reperfusion-induced cardiac apoptosis and injury by melatonin is independent of glutathione peroxdiase 1. J. Pineal Res. 2009, 46, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Zhang, H.; Qiao, Z.; Wang, Y.; Zhang, P.; Yang, D. Loss of thioredoxin 2 alters mitochondrial respiratory function and induces cardiomyocyte hypertrophy. Exp. Cell Res. 2018, 372, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Zhou, H.J.; Zhang, H.; Huang, Y.; Hinojosa-Kirschenbaum, F.; Fan, P.; Yao, L.; Belardinelli, L.; Tellides, G.; Giordano, F.J.; et al. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation 2015, 131, 1082–1097. [Google Scholar] [CrossRef] [Green Version]
- Ogata, S.; Takeuchi, M.; Teradaira, S.; Yamamoto, N.; Iwata, K.; Okumura, K.; Taguchi, H. Radical scavenging activities of niacin-related compounds. Biosci. Biotechnol. Biochem. 2002, 66, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Kamat, J.P.; Devasagayam, T.P. Nicotinamide (vitamin B3) as an effective antioxidant against oxidative damage in rat brain mitochondria. Redox. Rep. 1999, 4, 179–184. [Google Scholar] [CrossRef] [Green Version]
- Osiecki, M.; Ghanavi, P.; Atkinson, K.; Nielsen, L.K.; Doran, M.R. The ascorbic acid paradox. Biochem. Biophys. Res. Commun. 2010, 400, 466–470. [Google Scholar] [CrossRef] [PubMed]
- Palozza, P.; Calviello, G.; Serini, S.; Maggiano, N.; Lanza, P.; Ranelletti, F.O.; Bartoli, G.M. beta-carotene at high concentrations induces apoptosis by enhancing oxy-radical production in human adenocarcinoma cells. Free Radic. Biol. Med. 2001, 30, 1000–1007. [Google Scholar] [CrossRef]
- Bergstrom, T.; Bergman, J.; Moller, L. Vitamin A and C compounds permitted in supplements differ in their abilities to affect cell viability, DNA and the DNA nucleoside deoxyguanosine. Mutagenesis 2011, 26, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, T.; Ersson, C.; Bergman, J.; Moller, L. Vitamins at physiological levels cause oxidation to the DNA nucleoside deoxyguanosine and to DNA--alone or in synergism with metals. Mutagenesis 2012, 27, 511–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escola-Gil, J.C.; Rotllan, N.; Julve, J.; Blanco-Vaca, F. In vivo macrophage-specific RCT and antioxidant and antiinflammatory HDL activity measurements: New tools for predicting HDL atheroprotection. Atherosclerosis 2009, 206, 321–327. [Google Scholar] [CrossRef]
- Mendez-Lara, K.A.; Santos, D.; Farre, N.; Ruiz-Nogales, S.; Leanez, S.; Sanchez-Quesada, J.L.; Zapico, E.; Lerma, E.; Escola-Gil, J.C.; Blanco-Vaca, F.; et al. Administration of CORM-2 inhibits diabetic neuropathy but does not reduce dyslipidemia in diabetic mice. PLoS ONE 2018, 13, e0204841. [Google Scholar] [CrossRef] [Green Version]
- Elliott, R.B.; Chase, H.P. Prevention or delay of type 1 (insulin-dependent) diabetes mellitus in children using nicotinamide. Diabetologia 1991, 34, 362–365. [Google Scholar] [CrossRef] [Green Version]
- Hong, G.; Zheng, D.; Zhang, L.; Ni, R.; Wang, G.; Fan, G.C.; Lu, Z.; Peng, T. Administration of nicotinamide riboside prevents oxidative stress and organ injury in sepsis. Free Radic. Biol. Med. 2018, 123, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Mateuszuk, L.; Campagna, R.; Kutryb-Zajac, B.; Kus, K.; Slominska, E.M.; Smolenski, R.T.; Chlopicki, S. Reversal of endothelial dysfunction by nicotinamide mononucleotide via extracellular conversion to nicotinamide riboside. Biochem. Pharmacol. 2020, 178, 114019. [Google Scholar] [CrossRef]
- Gomes, A.P.; Price, N.L.; Ling, A.J.; Moslehi, J.J.; Montgomery, M.K.; Rajman, L.; White, J.P.; Teodoro, J.S.; Wrann, C.D.; Hubbard, B.P.; et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 2013, 155, 1624–1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiss, T.; Giles, C.B.; Tarantini, S.; Yabluchanskiy, A.; Balasubramanian, P.; Gautam, T.; Csipo, T.; Nyul-Toth, A.; Lipecz, A.; Szabo, C.; et al. Nicotinamide mononucleotide (NMN) supplementation promotes anti-aging miRNA expression profile in the aorta of aged mice, predicting epigenetic rejuvenation and anti-atherogenic effects. Geroscience 2019, 41, 419–439. [Google Scholar] [CrossRef]
- Kang, H.; Park, Y.K.; Lee, J.Y. Nicotinamide riboside, an NAD(+) precursor, attenuates inflammation and oxidative stress by activating sirtuin 1 in alcohol-stimulated macrophages. Lab. Invest. 2021, 101, 1225–1237. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.M.; Castoldi, A.; Sanin, D.E.; Flachsmann, L.J.; Field, C.S.; Puleston, D.J.; Kyle, R.L.; Patterson, A.E.; Hassler, F.; Buescher, J.M.; et al. Inflammatory macrophage dependence on NAD(+) salvage is a consequence of reactive oxygen species-mediated DNA damage. Nat. Immunol. 2019, 20, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Langston, P.K.; Shibata, M.; Horng, T. Metabolism Supports Macrophage Activation. Front. Immunol. 2017, 8, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiss, T.; Nyul-Toth, A.; Balasubramanian, P.; Tarantini, S.; Ahire, C.; Yabluchanskiy, A.; Csipo, T.; Farkas, E.; Wren, J.D.; Garman, L.; et al. Nicotinamide mononucleotide (NMN) supplementation promotes neurovascular rejuvenation in aged mice: Transcriptional footprint of SIRT1 activation, mitochondrial protection, anti-inflammatory, and anti-apoptotic effects. Geroscience 2020, 42, 527–546. [Google Scholar] [CrossRef] [PubMed]
- Weiss, R.; Schilling, E.; Grahnert, A.; Kolling, V.; Dorow, J.; Ceglarek, U.; Sack, U.; Hauschildt, S. Nicotinamide: A vitamin able to shift macrophage differentiation toward macrophages with restricted inflammatory features. Innate Immun. 2015, 21, 813–826. [Google Scholar] [CrossRef]
- Hiromatsu, Y.; Sato, M.; Tanaka, K.; Ishisaka, N.; Kamachi, J.; Nonaka, K. Inhibitory effects of nicotinamide on intercellular adhesion molecule-1 expression on cultured human thyroid cells. Immunology 1993, 80, 330–332. [Google Scholar]
- Hiromatsu, Y.; Sato, M.; Yamada, K.; Nonaka, K. Inhibitory effects of nicotinamide on recombinant human interferon-gamma-induced intercellular adhesion molecule-1 (ICAM-1) and HLA-DR antigen expression on cultured human endothelial cells. Immunol. Lett. 1992, 31, 35–39. [Google Scholar] [CrossRef]
- Biedron, R.; Ciszek, M.; Tokarczyk, M.; Bobek, M.; Kurnyta, M.; Slominska, E.M.; Smolenski, R.T.; Marcinkiewicz, J. 1-Methylnicotinamide and nicotinamide: Two related anti-inflammatory agents that differentially affect the functions of activated macrophages. Arch. Immunol. Ther. Exp. (Warsz) 2008, 56, 127–134. [Google Scholar] [CrossRef] [Green Version]
- Fukuzawa, M.; Satoh, J.; Muto, G.; Muto, Y.; Nishimura, S.; Miyaguchi, S.; Qiang, X.L.; Toyota, T. Inhibitory effect of nicotinamide on in vitro and in vivo production of tumor necrosis factor-alpha. Immunol. Lett. 1997, 59, 7–11. [Google Scholar] [CrossRef]
- Ungerstedt, J.S.; Heimersson, K.; Soderstrom, T.; Hansson, M. Nicotinamide inhibits endotoxin-induced monocyte tissue factor expression. J. Thromb. Haemost. 2003, 1, 2554–2560. [Google Scholar] [CrossRef] [Green Version]
- Ungerstedt, J.S.; Blomback, M.; Soderstrom, T. Nicotinamide is a potent inhibitor of proinflammatory cytokines. Clin. Exp. Immunol. 2003, 131, 48–52. [Google Scholar] [CrossRef]
- Grange, P.A.; Raingeaud, J.; Calvez, V.; Dupin, N. Nicotinamide inhibits Propionibacterium acnes-induced IL-8 production in keratinocytes through the NF-kappaB and MAPK pathways. J. Dermatol. Sci. 2009, 56, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M.; Permezel, M. The anti-inflammatory and antioxidative effects of nicotinamide, a vitamin B(3) derivative, are elicited by FoxO3 in human gestational tissues: Implications for preterm birth. J. Nutr. Biochem. 2011, 22, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Sack, M.N. Mitochondrial fidelity and metabolic agility control immune cell fate and function. J. Clin. Invest. 2018, 128, 3651–3661. [Google Scholar] [CrossRef]
- Kirkman, D.L.; Robinson, A.T.; Rossman, M.J.; Seals, D.R.; Edwards, D.G. Mitochondrial contributions to vascular endothelial dysfunction, arterial stiffness, and cardiovascular diseases. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H2080–H2100. [Google Scholar] [CrossRef]
- Zhou, B.; Wang, D.D.; Qiu, Y.; Airhart, S.; Liu, Y.; Stempien-Otero, A.; O’Brien, K.D.; Tian, R. Boosting NAD level suppresses inflammatory activation of PBMCs in heart failure. J. Clin. Invest. 2020, 130, 6054–6063. [Google Scholar] [CrossRef] [PubMed]
- Lenaz, G. Role of mitochondria in oxidative stress and ageing. Biochim. Biophys. Acta 1998, 1366, 53–67. [Google Scholar] [CrossRef] [Green Version]
- Wang, X. The expanding role of mitochondria in apoptosis. Genes Dev. 2001, 15, 2922–2933. [Google Scholar] [PubMed]
- Lai, Y.F.; Wang, L.; Liu, W.Y. Nicotinamide pretreatment alleviates mitochondrial stress and protects hypoxic myocardial cells via AMPK pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1797–1806. [Google Scholar] [PubMed]
- Tong, D.L.; Zhang, D.X.; Xiang, F.; Teng, M.; Jiang, X.P.; Hou, J.M.; Zhang, Q.; Huang, Y.S. Nicotinamide pretreatment protects cardiomyocytes against hypoxia-induced cell death by improving mitochondrial stress. Pharmacology 2012, 90, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhu, L.; Ruan, Z.B.; Wang, M.X.; Ren, Y.; Lu, W. Nicotinamide protects chronic hypoxic myocardial cells through regulating mTOR pathway and inducing autophagy. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5503–5511. [Google Scholar]
- Unno, K.; Isobe, S.; Izawa, H.; Cheng, X.W.; Kobayashi, M.; Hirashiki, A.; Yamada, T.; Harada, K.; Ohshima, S.; Noda, A.; et al. Relation of functional and morphological changes in mitochondria to myocardial contractile and relaxation reserves in asymptomatic to mildly symptomatic patients with hypertrophic cardiomyopathy. Eur. Heart J. 2009, 30, 1853–1862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Pulakat, L.; Whaley-Connell, A.; Sowers, J.R. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med. 2010, 88, 993–1001. [Google Scholar] [CrossRef] [Green Version]
- Karamanlidis, G.; Lee, C.F.; Garcia-Menendez, L.; Kolwicz, S.C., Jr.; Suthammarak, W.; Gong, G.; Sedensky, M.M.; Morgan, P.G.; Wang, W.; Tian, R. Mitochondrial complex I deficiency increases protein acetylation and accelerates heart failure. Cell Metab. 2013, 18, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Smyrnias, I.; Gray, S.P.; Okonko, D.O.; Sawyer, G.; Zoccarato, A.; Catibog, N.; Lopez, B.; Gonzalez, A.; Ravassa, S.; Diez, J.; et al. Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J. Am. Coll. Cardiol. 2019, 73, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Chong, Z.Z.; Lin, S.H.; Maiese, K. The NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J. Cereb. Blood Flow Metab. 2004, 24, 728–743. [Google Scholar] [CrossRef] [Green Version]
- Maiese, K. New Insights for nicotinamide: Metabolic disease, autophagy, and mTOR. Front. Biosci. 2020, 25, 1925–1973. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Kroemer, G. Mechanisms of apoptotic phosphatidylserine exposure. Cell Res. 2013, 23, 1247–1248. [Google Scholar] [CrossRef] [Green Version]
- Pajuelo, D.; Gonzalez-Juarbe, N.; Tak, U.; Sun, J.; Orihuela, C.J.; Niederweis, M. NAD(+) Depletion Triggers Macrophage Necroptosis, a Cell Death Pathway Exploited by Mycobacterium tuberculosis. Cell Rep. 2018, 24, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, C.L.; Payne, C.M.; Bernstein, H.; Bernstein, C.; Roe, D. The NAD+ precursors, nicotinic acid and nicotinamide protect cells against apoptosis induced by a multiple stress inducer, deoxycholate. Cell Death Differ. 2000, 7, 314–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Zhang, Y.; Zheng, M.; Cao, T.; Wang, G.; Zhang, L.; Ni, R.; Brockman, J.; Zhong, H.; Fan, G.C.; et al. Nicotinamide riboside promotes autolysosome clearance in preventing doxorubicin-induced cardiotoxicity. Clin. Sci. 2019, 133, 1505–1521. [Google Scholar] [CrossRef]
- Pham, T.X.; Bae, M.; Kim, M.B.; Lee, Y.; Hu, S.; Kang, H.; Park, Y.K.; Lee, J.Y. Nicotinamide riboside, an NAD+ precursor, attenuates the development of liver fibrosis in a diet-induced mouse model of liver fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2451–2463. [Google Scholar] [CrossRef] [PubMed]
- Traister, A.; Breitman, I.; Bar-Lev, E.; Zvibel, I.; Harel, A.; Halpern, Z.; Oren, R. Nicotinamide induces apoptosis and reduces collagen I and pro-inflammatory cytokines expression in rat hepatic stellate cells. Scand. J. Gastroenterol. 2005, 40, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Lee, K.B.; Park, S.Y.; Jang, J.J. Nicotinamide inhibits hepatic fibrosis by suppressing DNA synthesis and enhancing apoptosis of hepatic stellate cells. Virchows Arch. 2011, 458, 689–696. [Google Scholar] [CrossRef] [PubMed]
- Arauz, J.; Rivera-Espinoza, Y.; Shibayama, M.; Favari, L.; Flores-Beltran, R.E.; Muriel, P. Nicotinic acid prevents experimental liver fibrosis by attenuating the prooxidant process. Int. Immunopharmacol. 2015, 28, 244–251. [Google Scholar] [CrossRef]
- Wolf, D.; Ley, K. Immunity and Inflammation in Atherosclerosis. Circ. Res. 2019, 124, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Mallat, Z. Macrophages. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2509–2519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feil, S.; Fehrenbacher, B.; Lukowski, R.; Essmann, F.; Schulze-Osthoff, K.; Schaller, M.; Feil, R. Transdifferentiation of vascular smooth muscle cells to macrophage-like cells during atherogenesis. Circ. Res. 2014, 115, 662–667. [Google Scholar] [CrossRef]
- Sorci-Thomas, M.G.; Thomas, M.J. Microdomains, Inflammation, and Atherosclerosis. Circ. Res. 2016, 118, 679–691. [Google Scholar] [CrossRef] [Green Version]
- Williams, K.J.; Tabas, I. The response-to-retention hypothesis of early atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 551–561. [Google Scholar] [CrossRef] [Green Version]
- Brattsand, R.; Petersen, H.; Lundholm, L. Action of niceritrol (pentaerythritoltetranicotinate) on lipid accumulation in aortas of cholesterol-fed rabbits independent of contemporary reduction of serum lipids. Atherosclerosis 1974, 20, 469–479. [Google Scholar] [CrossRef]
- Brattsand, R. The effect of niceritrol (pentaerythritoltetranicotinate) and clofibrate upon hyperlipemia and atherosclerosis induced in rabbits by cholesterol-free semisynthetic diets. Atherosclerosis 1974, 20, 453–467. [Google Scholar] [CrossRef]
- Brattsand, R.; Lundholm, L. The effect of nicotinic acid and pentaerythritolteranicotinate upon experimental atherosclerosis in the rabbit. Atherosclerosis 1971, 14, 91–105. [Google Scholar] [CrossRef]
- Brattsand, R. Actions of vitamins A and E and some nicotinic acid derivatives on plasma lipids and on lipid infiltration of aorta in cholesterol-fed rabbits. Atherosclerosis 1975, 22, 47–61. [Google Scholar] [CrossRef]
- Lundholm, L.; Jacobsson, L.; Brattsand, R.; Magnusson, O. Influence of nicotinic acid, niceritrol and beta-pyridylcarbinol on experimental hyperlipidemia and atherosclerosis in mini-pigs. Atherosclerosis 1978, 29, 217–239. [Google Scholar] [CrossRef]
- Ost, C.R.; Stenson, S. Regression of peripheral atherosclerosis during therapy with high doses of nicotinic acid. Scand. J. Clin. Lab. Invest. Suppl. 1967, 99, 241–245. [Google Scholar]
- Lee, J.M.; Robson, M.D.; Yu, L.M.; Shirodaria, C.C.; Cunnington, C.; Kylintireas, I.; Digby, J.E.; Bannister, T.; Handa, A.; Wiesmann, F.; et al. Effects of high-dose modified-release nicotinic acid on atherosclerosis and vascular function: A randomized, placebo-controlled, magnetic resonance imaging study. J. Am. Coll. Cardiol. 2009, 54, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Lukasova, M.; Malaval, C.; Gille, A.; Kero, J.; Offermanns, S. Nicotinic acid inhibits progression of atherosclerosis in mice through its receptor GPR109A expressed by immune cells. J. Clin. Invest. 2011, 121, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Holzhauser, E.; Albrecht, C.; Zhou, Q.; Buttler, A.; Preusch, M.R.; Blessing, E.; Katus, H.A.; Bea, F. Nicotinic acid has anti-atherogenic and anti-inflammatory properties on advanced atherosclerotic lesions independent of its lipid-modifying capabilities. J. Cardiovasc. Pharmacol. 2011, 57, 447–454. [Google Scholar] [CrossRef]
- Strack, A.M.; Carballo-Jane, E.; Wang, S.P.; Xue, J.; Ping, X.; McNamara, L.A.; Thankappan, A.; Price, O.; Wolff, M.; Wu, T.J.; et al. Nicotinic acid and DP1 blockade: Studies in mouse models of atherosclerosis. J. Lipid Res. 2013, 54, 177–188. [Google Scholar] [CrossRef] [Green Version]
- Declercq, V.; Yeganeh, B.; Moshtaghi-Kashanian, G.R.; Khademi, H.; Bahadori, B.; Moghadasian, M.H. Paradoxical effects of fenofibrate and nicotinic acid in apo E-deficient mice. J. Cardiovasc. Pharmacol. 2005, 46, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Kuhnast, S.; Louwe, M.C.; Heemskerk, M.M.; Pieterman, E.J.; van Klinken, J.B.; van den Berg, S.A.; Smit, J.W.; Havekes, L.M.; Rensen, P.C.; van der Hoorn, J.W.; et al. Niacin Reduces Atherosclerosis Development in APOE*3Leiden.CETP Mice Mainly by Reducing NonHDL-Cholesterol. PLoS ONE 2013, 8, e66467. [Google Scholar] [CrossRef] [Green Version]
- Kane, A.E.; Sinclair, D.A. Sirtuins and NAD(+) in the Development and Treatment of Metabolic and Cardiovascular Diseases. Circ. Res. 2018, 123, 868–885. [Google Scholar] [CrossRef]
- Li, X.; Zhang, S.; Blander, G.; Tse, J.G.; Krieger, M.; Guarente, L. SIRT1 deacetylates and positively regulates the nuclear receptor LXR. Mol. Cell 2007, 28, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Lara, K.A.; Santos, D.; Farre, N.; Nan, M.N.; Pallares, V.; Perez-Perez, A.; Alonso, N.; Escola-Gil, J.C.; Blanco-Vaca, F.; Julve, J. Vitamin B3 impairs reverse cholesterol transport in Apolipoprotein E-deficient mice. Clin. Investig. Arterioscler. 2019, 31, 251–260. [Google Scholar] [PubMed]
- Nencioni, A.; da Silva, R.F.; Fraga-Silva, R.A.; Steffens, S.; Fabre, M.; Bauer, I.; Caffa, I.; Magnone, M.; Sociali, G.; Quercioli, A.; et al. Nicotinamide phosphoribosyltransferase inhibition reduces intraplaque CXCL1 production and associated neutrophil infiltration in atherosclerotic mice. Thromb. Haemost. 2014, 111, 308–322. [Google Scholar]
- Li, S.; Wang, C.; Li, K.; Li, L.; Tian, M.; Xie, J.; Yang, M.; Jia, Y.; He, J.; Gao, L.; et al. NAMPT knockdown attenuates atherosclerosis and promotes reverse cholesterol transport in ApoE KO mice with high-fat-induced insulin resistance. Sci. Rep. 2016, 6, 26746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, Y.Y.; Li, G.Q.; Zhang, W.J.; Hua, X.; Zhou, C.C.; Xu, T.Y.; Li, Z.Y.; Wang, P.; Miao, C.Y. Nicotinamide phosphoribosyltransferase aggravates inflammation and promotes atherosclerosis in ApoE knockout mice. Acta Pharmacol. Sin. 2019, 40, 1184–1192. [Google Scholar] [CrossRef]
- Bermudez, B.; Dahl, T.B.; Medina, I.; Groeneweg, M.; Holm, S.; Montserrat-de la Paz, S.; Rousch, M.; Otten, J.; Herias, V.; Varela, L.M.; et al. Leukocyte Overexpression of Intracellular NAMPT Attenuates Atherosclerosis by Regulating PPARgamma-Dependent Monocyte Differentiation and Function. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1157–1167. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Bai, P.; Little, P.J.; Liu, P. Poly(ADP-ribose) polymerase 1 (PARP1) in atherosclerosis: From molecular mechanisms to therapeutic implications. Med. Res. Rev. 2014, 34, 644–675. [Google Scholar] [CrossRef]
- Oumouna-Benachour, K.; Hans, C.P.; Suzuki, Y.; Naura, A.; Datta, R.; Belmadani, S.; Fallon, K.; Woods, C.; Boulares, A.H. Poly(ADP-ribose) polymerase inhibition reduces atherosclerotic plaque size and promotes factors of plaque stability in apolipoprotein E-deficient mice: Effects on macrophage recruitment, nuclear factor-kappaB nuclear translocation, and foam cell death. Circulation 2007, 115, 2442–2450. [Google Scholar] [CrossRef] [Green Version]
- von Lukowicz, T.; Hassa, P.O.; Lohmann, C.; Boren, J.; Braunersreuther, V.; Mach, F.; Odermatt, B.; Gersbach, M.; Camici, G.G.; Stahli, B.E.; et al. PARP1 is required for adhesion molecule expression in atherogenesis. Cardiovasc. Res. 2008, 78, 158–166. [Google Scholar] [CrossRef]
- Hans, C.P.; Zerfaoui, M.; Naura, A.S.; Catling, A.; Boulares, A.H. Differential effects of PARP inhibition on vascular cell survival and ACAT-1 expression favouring atherosclerotic plaque stability. Cardiovasc. Res. 2008, 78, 429–439. [Google Scholar] [CrossRef]
- Hans, C.P.; Zerfaoui, M.; Naura, A.S.; Troxclair, D.; Strong, J.P.; Matrougui, K.; Boulares, A.H. Thieno[2,3-c]isoquinolin-5-one, a potent poly(ADP-ribose) polymerase inhibitor, promotes atherosclerotic plaque regression in high-fat diet-fed apolipoprotein E-deficient mice: Effects on inflammatory markers and lipid content. J. Pharmacol. Exp. Ther. 2009, 329, 150–158. [Google Scholar] [CrossRef] [Green Version]
- Zha, S.; Wang, F.; Li, Z.; Ma, Z.; Yang, L.; Liu, F. PJ34, a PARP1 inhibitor, promotes endothelial repair in a rabbit model of high fat diet-induced atherosclerosis. Cell Cycle 2019, 18, 2099–2109. [Google Scholar] [CrossRef]
- Lindsey, M.L.; Bolli, R.; Canty, J.M., Jr.; Du, X.J.; Frangogiannis, N.G.; Frantz, S.; Gourdie, R.G.; Holmes, J.W.; Jones, S.P.; Kloner, R.A.; et al. Guidelines for experimental models of myocardial ischemia and infarction. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H812–H838. [Google Scholar] [CrossRef] [PubMed]
- Nunez, R.; Calva, E.; Marsh, M. Nicotinamide adenine dinucleotide degradation in infarcted cardiac muscle. Recent Adv. Stud. Cardiac Struct. Metab. 1975, 10, 241–250. [Google Scholar]
- Nunez, R.; Calva, E.; Marsch, M.; Briones, E.; Lopez-Soriano, F. NAD glycohydrolase activity in hearts with acute experimental infarction. Am. J. Physiol. 1976, 231, 1173–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, H.H.; Schaper, J.; Puschmann, S.; Nienaber, C.; Kreuzer, H.; Schaper, W. Loss of canine myocardial nicotinamide adenine dinucleotides determines the transition from reversible to irreversible ischemic damage of myocardial cells. Basic Res. Cardiol. 1981, 76, 612–621. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, B.; Fu, X.; Guan, S.; Han, W.; Zhang, J.; Gan, Q.; Fang, W.; Ying, W.; Qu, X. Exogenous NAD(+) administration significantly protects against myocardial ischemia/reperfusion injury in rat model. Am. J. Transl. Res. 2016, 8, 3342–3350. [Google Scholar] [PubMed]
- Hsu, C.P.; Oka, S.; Shao, D.; Hariharan, N.; Sadoshima, J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ. Res. 2009, 105, 481–491. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.H.; Liu, X.H.; Hong, X.; Zhao, N.; Xiao, Y.F.; Wang, L.F.; Tang, L.; Jiang, K.; Qian, Y.S.; Deng, K.Y.; et al. CD38 Deficiency Protects the Heart from Ischemia/Reperfusion Injury through Activating SIRT1/FOXOs-Mediated Antioxidative Stress Pathway. Oxid. Med. Cell. Longev. 2016, 2016, 7410257. [Google Scholar] [CrossRef] [Green Version]
- Zhai, X.; Han, W.; Wang, M.; Guan, S.; Qu, X. Exogenous supplemental NAD+ protect myocardium against myocardial ischemic/reperfusion injury in swine model. Am. J. Transl. Res. 2019, 11, 6066–6074. [Google Scholar] [PubMed]
- Liu, L.; Wang, Q.; Zhao, B.; Wu, Q.; Wang, P. Exogenous nicotinamide adenine dinucleotide administration alleviates ischemia/reperfusion-induced oxidative injury in isolated rat hearts via Sirt5-SDH-succinate pathway. Eur. J. Pharmacol. 2019, 858, 172520. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Byun, J.; Zhai, P.; Ikeda, Y.; Oka, S.; Sadoshima, J. Nicotinamide mononucleotide, an intermediate of NAD+ synthesis, protects the heart from ischemia and reperfusion. PLoS ONE 2014, 9, e98972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, L.; Vafaee, M.S.; Badalzadeh, R. Melatonin and Nicotinamide Mononucleotide Attenuate Myocardial Ischemia/Reperfusion Injury via Modulation of Mitochondrial Function and Hemodynamic Parameters in Aged Rats. J. Cardiovasc. Pharmacol. Ther. 2020, 25, 240–250. [Google Scholar] [CrossRef]
- Ahmad, F.; Tomar, D.; Aryal, A.C.S.; Elmoselhi, A.B.; Thomas, M.; Elrod, J.W.; Tilley, D.G.; Force, T. Nicotinamide riboside kinase-2 alleviates ischemia-induced heart failure through P38 signaling. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165609. [Google Scholar] [CrossRef] [PubMed]
- Toropova, Y.G.; Pechnikova, N.A.; Zelinskaya, I.A.; Zhuravsky, S.G.; Kornyushin, O.V.; Gonchar, A.I.; Ivkin, D.Y.; Leonova, Y.V.; Karev, V.E.; Karabak, I.A. Nicotinamide riboside has protective effects in a rat model of mesenteric ischaemia-reperfusion. Int. J. Exp. Pathol. 2018, 99, 304–311. [Google Scholar] [CrossRef]
- Pinto, Y.M.; Elliott, P.M.; Arbustini, E.; Adler, Y.; Anastasakis, A.; Bohm, M.; Duboc, D.; Gimeno, J.; de Groote, P.; Imazio, M.; et al. Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. Eur. Heart J. 2016, 37, 1850–1858. [Google Scholar] [CrossRef] [Green Version]
- Ussher, J.R.; Jaswal, J.S.; Lopaschuk, G.D. Pyridine nucleotide regulation of cardiac intermediary metabolism. Circ. Res. 2012, 111, 628–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.F.; Tian, R. Mitochondrion as a Target for Heart Failure Therapy- Role of Protein Lysine Acetylation. Circ. J. 2015, 79, 1863–1870. [Google Scholar] [CrossRef] [Green Version]
- Diguet, N.; Trammell, S.A.J.; Tannous, C.; Deloux, R.; Piquereau, J.; Mougenot, N.; Gouge, A.; Gressette, M.; Manoury, B.; Blanc, J.; et al. Nicotinamide Riboside Preserves Cardiac Function in a Mouse Model of Dilated Cardiomyopathy. Circulation 2018, 137, 2256–2273. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.F.; Chavez, J.D.; Garcia-Menendez, L.; Choi, Y.; Roe, N.D.; Chiao, Y.A.; Edgar, J.S.; Goo, Y.A.; Goodlett, D.R.; Bruce, J.E.; et al. Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation 2016, 134, 883–894. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.S.; Abraham, D.M.; Hershberger, K.A.; Bhatt, D.P.; Mao, L.; Cui, H.; Liu, J.; Liu, X.; Muehlbauer, M.J.; Grimsrud, P.A.; et al. Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich’s ataxia cardiomyopathy model. JCI Insight 2017, 2, e93885. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.L.; Martin, O.J.; Lai, L.; Riley, N.M.; Richards, A.L.; Vega, R.B.; Leone, T.C.; Pagliarini, D.J.; Muoio, D.M.; Bedi, K.C., Jr.; et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2016, 2, e84897. [Google Scholar] [CrossRef]
- Pillai, V.B.; Sundaresan, N.R.; Kim, G.; Gupta, M.; Rajamohan, S.B.; Pillai, J.B.; Samant, S.; Ravindra, P.V.; Isbatan, A.; Gupta, M.P. Exogenous NAD blocks cardiac hypertrophic response via activation of the SIRT3-LKB1-AMP-activated kinase pathway. J. Biol. Chem. 2010, 285, 3133–3144. [Google Scholar] [CrossRef] [Green Version]
- Fatkin, D.; MacRae, C.; Sasaki, T.; Wolff, M.R.; Porcu, M.; Frenneaux, M.; Atherton, J.; Vidaillet, H.J., Jr.; Spudich, S.; De Girolami, U.; et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N. Engl. J. Med. 1999, 341, 1715–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignier, N.; Chatzifrangkeskou, M.; Morales Rodriguez, B.; Mericskay, M.; Mougenot, N.; Wahbi, K.; Bonne, G.; Muchir, A. Rescue of biosynthesis of nicotinamide adenine dinucleotide protects the heart in cardiomyopathy caused by lamin A/C gene mutation. Hum. Mol. Genet. 2018, 27, 3870–3880. [Google Scholar] [CrossRef] [PubMed]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef]
- Tong, D.; Schiattarella, G.G.; Jiang, N.; Altamirano, F.; Szweda, P.A.; Elnwasany, A.; Lee, D.I.; Yoo, H.; Kass, D.A.; Szweda, L.I.; et al. NAD(+) Repletion Reverses Heart Failure With Preserved Ejection Fraction. Circ. Res. 2021, 128, 1629–1641. [Google Scholar] [CrossRef]
- Tannous, C.; Deloux, R.; Karoui, A.; Mougenot, N.; Burkin, D.; Blanc, J.; Coletti, D.; Lavery, G.; Li, Z.; Mericskay, M. NMRK2 Gene Is Upregulated in Dilated Cardiomyopathy and Required for Cardiac Function and NAD Levels during Aging. Int. J. Mol. Sci. 2021, 22, 3534. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Barrientos, T.; Mao, L.; Rockman, H.A.; Sauve, A.A.; Andrews, N.C. Lethal Cardiomyopathy in Mice Lacking Transferrin Receptor in the Heart. Cell Rep. 2015, 13, 533–545. [Google Scholar] [CrossRef] [Green Version]
- Lauritzen, K.H.; Olsen, M.B.; Ahmed, M.S.; Yang, K.; Rinholm, J.E.; Bergersen, L.H.; Esbensen, Q.Y.; Sverkeli, L.J.; Ziegler, M.; Attramadal, H.; et al. Instability in NAD(+) metabolism leads to impaired cardiac mitochondrial function and communication. Elife 2021, 10, e59828. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.; Oka, S.I.; Imai, N.; Huang, C.Y.; Ralda, G.; Zhai, P.; Ikeda, Y.; Ikeda, S.; Sadoshima, J. Both gain and loss of Nampt function promote pressure overload-induced heart failure. Am. J. Physiol. Heart Circ. Physiol. 2019, 317, H711–H725. [Google Scholar] [CrossRef]
- Wu, K.; Li, B.; Lin, Q.; Xu, W.; Zuo, W.; Li, J.; Liu, N.; Tu, T.; Zhang, B.; Xiao, Y.; et al. Nicotinamide mononucleotide attenuates isoproterenol-induced cardiac fibrosis by regulating oxidative stress and Smad3 acetylation. Life Sci. 2021, 274, 119299. [Google Scholar] [CrossRef]
- Francis, G.S. Diabetic cardiomyopathy: Fact or fiction? Heart 2001, 85, 247–248. [Google Scholar] [CrossRef] [Green Version]
- Picano, E. Diabetic cardiomyopathy. the importance of being earliest. J. Am. Coll. Cardiol. 2003, 42, 454–457. [Google Scholar] [CrossRef] [Green Version]
- Avogaro, A.; Vigili de Kreutzenberg, S.; Negut, C.; Tiengo, A.; Scognamiglio, R. Diabetic cardiomyopathy: A metabolic perspective. Am. J. Cardiol. 2004, 93, 13A–16A. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.; Weidemann, B.J.; Chadda, A.; Yorek, M.S.; Holmes, A.; Coppey, L.J.; Obrosov, A.; Kardon, R.H.; Yorek, M.A.; Brenner, C. Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Sci. Rep. 2016, 6, 26933. [Google Scholar] [CrossRef] [Green Version]
- Ido, Y.; Kilo, C.; Williamson, J.R. Cytosolic NADH/NAD+, free radicals, and vascular dysfunction in early diabetes mellitus. Diabetologia 1997, 40 (Suppl. 2), S115–S117. [Google Scholar] [CrossRef] [Green Version]
- Williamson, J.R.; Chang, K.; Frangos, M.; Hasan, K.S.; Ido, Y.; Kawamura, T.; Nyengaard, J.R.; van den Enden, M.; Kilo, C.; Tilton, R.G. Hyperglycemic pseudohypoxia and diabetic complications. Diabetes 1993, 42, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Jin, Z.; Zheng, H.; Yan, L.J. Sources and implications of NADH/NAD(+) redox imbalance in diabetes and its complications. Diabetes Metab. Syndr. Obes. 2016, 9, 145–153. [Google Scholar]
- Berthiaume, J.M.; Hsiung, C.H.; Austin, A.B.; McBrayer, S.P.; Depuydt, M.M.; Chandler, M.P.; Miyagi, M.; Rosca, M.G. Methylene blue decreases mitochondrial lysine acetylation in the diabetic heart. Mol. Cell. Biochem. 2017, 432, 7–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, E.J.; Berthiaume, J.M.; Kamath, V.; Achike, O.; Buchanan, E.; Montano, M.M.; Chandler, M.P.; Miyagi, M.; Rosca, M.G. Mitochondrial complex I defect and increased fatty acid oxidation enhance protein lysine acetylation in the diabetic heart. Cardiovasc. Res. 2015, 107, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Shen, Y.; Zhou, L.; Sangwung, P.; Fujioka, H.; Zhang, L.; Liao, X. Short-term administration of Nicotinamide Mononucleotide preserves cardiac mitochondrial homeostasis and prevents heart failure. J. Mol. Cell. Cardiol. 2017, 112, 64–73. [Google Scholar] [CrossRef]
- Chiao, Y.A.; Chakraborty, A.D.; Light, C.M.; Tian, R.; Sadoshima, J.; Shi, X.; Gu, H.; Lee, C.F. NAD(+) Redox Imbalance in the Heart Exacerbates Diabetic Cardiomyopathy. Circ. Heart Fail. 2021, 14, e008170. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Haldar, S.M.; Lu, Y.; Jeyaraj, D.; Paruchuri, K.; Nahori, M.; Cui, Y.; Kaestner, K.H.; Jain, M.K. Kruppel-like factor 4 regulates pressure-induced cardiac hypertrophy. J. Mol. Cell. Cardiol. 2010, 49, 334–338. [Google Scholar] [CrossRef] [Green Version]
- Liao, X.; Zhang, R.; Lu, Y.; Prosdocimo, D.A.; Sangwung, P.; Zhang, L.; Zhou, G.; Anand, P.; Lai, L.; Leone, T.C.; et al. Kruppel-like factor 4 is critical for transcriptional control of cardiac mitochondrial homeostasis. J. Clin. Invest. 2015, 125, 3461–3476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, B.B.; Yang, V.W. Mammalian Kruppel-like factors in health and diseases. Physiol. Rev. 2010, 90, 1337–1381. [Google Scholar] [CrossRef] [PubMed]
- Zeitz, M.J.; Smyth, J.W. Translating Translation to Mechanisms of Cardiac Hypertrophy. J. Cardiovasc. Dev. Dis. 2020, 7, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Samak, M.; Fatullayev, J.; Sabashnikov, A.; Zeriouh, M.; Schmack, B.; Farag, M.; Popov, A.F.; Dohmen, P.M.; Choi, Y.H.; Wahlers, T.; et al. Cardiac Hypertrophy: An Introduction to Molecular and Cellular Basis. Med. Sci. Monit. Basic Res. 2016, 22, 75–79. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.; Feng, J.; Lin, X.; Liu, J.; Tang, Y.; Nie, S.; Gong, J.; Wang, L. Nicotinamide Riboside Alleviates Cardiac Dysfunction and Remodeling in Pressure Overload Cardiac Hypertrophy. Oxid. Med. Cell. Longev. 2021, 2021, 5546867. [Google Scholar] [CrossRef]
- Lyon, A.R.; Dent, S.; Stanway, S.; Earl, H.; Brezden-Masley, C.; Cohen-Solal, A.; Tocchetti, C.G.; Moslehi, J.J.; Groarke, J.D.; Bergler-Klein, J.; et al. Baseline cardiovascular risk assessment in cancer patients scheduled to receive cardiotoxic cancer therapies: A position statement and new risk assessment tools from the Cardio-Oncology Study Group of the Heart Failure Association of the European Society of Cardiology in collaboration with the International Cardio-Oncology Society. Eur. J. Heart Fail. 2020, 22, 1945–1960. [Google Scholar]
- Battisti, N.M.L.; Andres, M.S.; Lee, K.A.; Ramalingam, S.; Nash, T.; Mappouridou, S.; Senthivel, N.; Asavisanu, K.; Obeid, M.; Tripodaki, E.S.; et al. Incidence of cardiotoxicity and validation of the Heart Failure Association-International Cardio-Oncology Society risk stratification tool in patients treated with trastuzumab for HER2-positive early breast cancer. Breast Cancer Res. Treat. 2021, 188, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zheng, D.; Wei, M.; Ma, J.; Yu, Y.; Chen, R.; Lacefield, J.C.; Xu, H.; Peng, T. Over-expression of calpastatin aggravates cardiotoxicity induced by doxorubicin. Cardiovasc. Res. 2013, 98, 381–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Wang, Y.; Zheng, D.; Wei, M.; Xu, H.; Peng, T. Rac1 signalling mediates doxorubicin-induced cardiotoxicity through both reactive oxygen species-dependent and -independent pathways. Cardiovasc. Res. 2013, 97, 77–87. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, J.; et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016, 22, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Wang, H.; Han, D.; Xie, E.; Yang, X.; Wei, J.; Gu, S.; Gao, F.; Zhu, N.; Yin, X.; et al. Ferroptosis as a target for protection against cardiomyopathy. Proc. Natl. Acad. Sci. USA 2019, 116, 2672–2680. [Google Scholar] [CrossRef] [Green Version]
- Armenian, S.H.; Lacchetti, C.; Barac, A.; Carver, J.; Constine, L.S.; Denduluri, N.; Dent, S.; Douglas, P.S.; Durand, J.B.; Ewer, M.; et al. Prevention and Monitoring of Cardiac Dysfunction in Survivors of Adult Cancers: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2017, 35, 893–911. [Google Scholar] [CrossRef]
- Awad, H.H.; El-Derany, M.O.; Mantawy, E.M.; Michel, H.E.; El-Naa, M.M.; Salah El-Din, R.A.; El-Brairy, A.I.; El-Demerdash, E. Comparative study on beneficial effects of vitamins B and D in attenuating doxorubicin induced cardiotoxicity in rats: Emphasis on calcium homeostasis. Biomed. Pharmacother. 2021, 140, 111679. [Google Scholar] [CrossRef]
- Kuivaniemi, H.; Platsoucas, C.D.; Tilson, M.D., 3rd. Aortic aneurysms: An immune disease with a strong genetic component. Circulation 2008, 117, 242–252. [Google Scholar] [CrossRef] [Green Version]
- van der Veer, E.; Nong, Z.; O’Neil, C.; Urquhart, B.; Freeman, D.; Pickering, J.G. Pre-B-cell colony-enhancing factor regulates NAD+-dependent protein deacetylase activity and promotes vascular smooth muscle cell maturation. Circ. Res. 2005, 97, 25–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Veer, E.; Ho, C.; O’Neil, C.; Barbosa, N.; Scott, R.; Cregan, S.P.; Pickering, J.G. Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J. Biol. Chem. 2007, 282, 10841–10845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.; van der Veer, E.; Frontini, M.J.; Thibert, V.; O’Neil, C.; Watson, A.; Szasz, P.; Chu, M.W.; Pickering, J.G. Intrinsic directionality of migrating vascular smooth muscle cells is regulated by NAD(+) biosynthesis. J. Cell Sci. 2012, 125, 5770–5780. [Google Scholar] [CrossRef] [Green Version]
- Golledge, J.; Muller, J.; Daugherty, A.; Norman, P. Abdominal aortic aneurysm: Pathogenesis and implications for management. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2605–2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Li, W.L.; Liu, J.M.; Miao, C.Y. NAMPT and NAMPT-controlled NAD Metabolism in Vascular Repair. J. Cardiovasc. Pharmacol. 2016, 67, 474–481. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.; Nong, Z.; Yin, H.; O’Neil, C.; Fox, S.; Balint, B.; Guo, L.; Leo, O.; Chu, M.W.A.; Gros, R.; et al. Nicotinamide Phosphoribosyltransferase in Smooth Muscle Cells Maintains Genome Integrity, Resists Aortic Medial Degeneration, and Is Suppressed in Human Thoracic Aortic Aneurysm Disease. Circ. Res. 2017, 120, 1889–1902. [Google Scholar] [CrossRef] [PubMed]
- McCrudden, C.M.; O’Rourke, M.G.; Cherry, K.E.; Yuen, H.F.; O’Rourke, D.; Babur, M.; Telfer, B.A.; Thomas, H.D.; Keane, P.; Nambirajan, T.; et al. Vasoactivity of rucaparib, a PARP-1 inhibitor, is a complex process that involves myosin light chain kinase, P2 receptors, and PARP itself. PLoS ONE 2015, 10, e0118187. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Xu, T.Y.; Guan, Y.F.; Su, D.F.; Fan, G.R.; Miao, C.Y. Perivascular adipose tissue-derived visfatin is a vascular smooth muscle cell growth factor: Role of nicotinamide mononucleotide. Cardiovasc. Res. 2009, 81, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Howard, D.P.; Banerjee, A.; Fairhead, J.F.; Perkins, J.; Silver, L.E.; Rothwell, P.M. Population-based study of incidence and outcome of acute aortic dissection and premorbid risk factor control: 10-year results from the Oxford Vascular Study. Circulation 2013, 127, 2031–2037. [Google Scholar] [CrossRef] [Green Version]
- Oller, J.; Gabande-Rodriguez, E.; Ruiz-Rodriguez, M.J.; Desdin-Mico, G.; Aranda, J.F.; Rodrigues-Diez, R.; Ballesteros-Martinez, C.; Blanco, E.M.; Roldan-Montero, R.; Acuna, P.; et al. Extracellular Tuning of Mitochondrial Respiration Leads to Aortic Aneurysm. Circulation 2021, 143, 2091–2109. [Google Scholar] [CrossRef] [PubMed]
- Ganji, S.H.; Qin, S.; Zhang, L.; Kamanna, V.S.; Kashyap, M.L. Niacin inhibits vascular oxidative stress, redox-sensitive genes, and monocyte adhesion to human aortic endothelial cells. Atherosclerosis 2009, 202, 68–75. [Google Scholar] [CrossRef]
- Digby, J.E.; Martinez, F.; Jefferson, A.; Ruparelia, N.; Chai, J.; Wamil, M.; Greaves, D.R.; Choudhury, R.P. Anti-inflammatory effects of nicotinic acid in human monocytes are mediated by GPR109A dependent mechanisms. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 669–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzik, B.; Sagan, A.; Ludew, D.; Mrowiecki, W.; Chwala, M.; Bujak-Gizycka, B.; Filip, G.; Grudzien, G.; Kapelak, B.; Zmudka, K.; et al. Mechanisms of oxidative stress in human aortic aneurysms--association with clinical risk factors for atherosclerosis and disease severity. Int. J. Cardiol. 2013, 168, 2389–2396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, T.; Morishita, R. Pharmacological treatment of abdominal aortic aneurysm. Cardiovasc. Res. 2009, 83, 436–443. [Google Scholar] [CrossRef] [Green Version]
- Lavu, S.; Boss, O.; Elliott, P.J.; Lambert, P.D. Sirtuins--novel therapeutic targets to treat age-associated diseases. Nat. Rev. Drug Discov. 2008, 7, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Pillarisetti, S. A review of Sirt1 and Sirt1 modulators in cardiovascular and metabolic diseases. Recent Pat. Cardiovasc. Drug Discov. 2008, 3, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Dimmeler, S. Emerging roles of SIRT1 in vascular endothelial homeostasis. Cell Cycle 2008, 7, 2117–2122. [Google Scholar] [CrossRef]
- Imai, S. A possibility of nutriceuticals as an anti-aging intervention: Activation of sirtuins by promoting mammalian NAD biosynthesis. Pharmacol. Res. 2010, 62, 42–47. [Google Scholar] [CrossRef] [Green Version]
- Satoh, A.; Stein, L.; Imai, S. The role of mammalian sirtuins in the regulation of metabolism, aging, and longevity. Handb. Exp. Pharmacol. 2011, 206, 125–162. [Google Scholar] [PubMed] [Green Version]
- Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Cerutti, R.; Pirinen, E.; Lamperti, C.; Marchet, S.; Sauve, A.A.; Li, W.; Leoni, V.; Schon, E.A.; Dantzer, F.; Auwerx, J.; et al. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 2014, 19, 1042–1049. [Google Scholar] [CrossRef] [Green Version]
- Hughes-Large, J.M.; Pang, D.K.; Robson, D.L.; Chan, P.; Toma, J.; Borradaile, N.M. Niacin receptor activation improves human microvascular endothelial cell angiogenic function during lipotoxicity. Atherosclerosis 2014, 237, 696–704. [Google Scholar] [CrossRef]
- Marcu, R.; Kotha, S.; Zhi, Z.; Qin, W.; Neeley, C.K.; Wang, R.K.; Zheng, Y.; Hawkins, B.J. The mitochondrial permeability transition pore regulates endothelial bioenergetics and angiogenesis. Circ. Res. 2015, 116, 1336–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.Z.; Wang, F.; Gao, P.; Pei, J.F.; Liu, Y.; Xu, T.T.; Tang, X.; Fu, W.Y.; Lu, J.; Yan, Y.F.; et al. Age-Associated Sirtuin 1 Reduction in Vascular Smooth Muscle Links Vascular Senescence and Inflammation to Abdominal Aortic Aneurysm. Circ. Res. 2016, 119, 1076–1088. [Google Scholar] [CrossRef]
- Zhang, Z.; Xu, J.; Liu, Y.; Wang, T.; Pei, J.; Cheng, L.; Hao, D.; Zhao, X.; Chen, H.Z.; Liu, D.P. Mouse macrophage specific knockout of SIRT1 influences macrophage polarization and promotes angiotensin II-induced abdominal aortic aneurysm formation. J. Genet. Genom. 2018, 45, 25–32. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, T.T.; Zhang, R.; Fu, W.Y.; Wang, X.; Wang, F.; Gao, P.; Ding, Y.N.; Xie, Y.; Hao, D.L.; et al. Calorie restriction protects against experimental abdominal aortic aneurysms in mice. J. Exp. Med. 2016, 213, 2473–2488. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Tu, Y.; Gao, Y.; Chen, H.; Liu, J.; Zheng, J. Smooth Muscle Sirtuin 1 Blocks Thoracic Aortic Aneurysm/Dissection Development in Mice. Cardiovasc. Drugs Ther. 2020, 34, 641–650. [Google Scholar] [CrossRef]
- Sanchez-Infantes, D.; Nus, M.; Navas-Madronal, M.; Fite, J.; Perez, B.; Barros-Membrilla, A.J.; Soto, B.; Martinez-Gonzalez, J.; Camacho, M.; Rodriguez, C.; et al. Oxidative Stress and Inflammatory Markers in Abdominal Aortic Aneurysm. Antioxidants 2021, 10, 602. [Google Scholar] [CrossRef]
- Oparil, S.; Acelajado, M.C.; Bakris, G.L.; Berlowitz, D.R.; Cifkova, R.; Dominiczak, A.F.; Grassi, G.; Jordan, J.; Poulter, N.R.; Rodgers, A.; et al. Hypertension. Nat. Rev. Dis. Primers 2018, 4, 18014. [Google Scholar] [CrossRef] [Green Version]
- Kjeldsen, S.E.; Narkiewicz, K.; Burnier, M.; Oparil, S. 2018 Practice guidelines for the management of arterial hypertension of the European Society of Hypertension. Blood Press 2018, 27, 313. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Wang, Z.; Wu, J.; Liu, M.; Li, M.; Sun, Y.; Huang, W.; Li, Y.; Zhang, Y.; Tang, W.; et al. Endothelial SIRT6 Is Vital to Prevent Hypertension and Associated Cardiorenal Injury Through Targeting Nkx3.2-GATA5 Signaling. Circ. Res. 2019, 124, 1448–1461. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Liu, H.; Zhu, S.; Woodson, M.; Liu, R.; Tilton, R.G.; Miller, J.D.; Zhang, W. Sirt6 deficiency results in progression of glomerular injury in the kidney. Aging 2017, 9, 1069–1083. [Google Scholar] [CrossRef] [Green Version]
- Gao, P.; Xu, T.T.; Lu, J.; Li, L.; Xu, J.; Hao, D.L.; Chen, H.Z.; Liu, D.P. Overexpression of SIRT1 in vascular smooth muscle cells attenuates angiotensin II-induced vascular remodeling and hypertension in mice. J. Mol. Med. 2014, 92, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Sethi, J.K.; Empson, R.M.; Galione, A. Nicotinamide inhibits cyclic ADP-ribose-mediated calcium signalling in sea urchin eggs. Biochem. J. 1996, 319 Pt. 2, 613–617. [Google Scholar] [CrossRef] [Green Version]
- Thai, T.L.; Arendshorst, W.J. ADP-ribosyl cyclase and ryanodine receptors mediate endothelin ETA and ETB receptor-induced renal vasoconstriction in vivo. Am. J. Physiol. Renal Physiol. 2008, 295, F360–F368. [Google Scholar] [CrossRef] [Green Version]
- Wilson, H.L.; Dipp, M.; Thomas, J.M.; Lad, C.; Galione, A.; Evans, A.M. Adp-ribosyl cyclase and cyclic ADP-ribose hydrolase act as a redox sensor. a primary role for cyclic ADP-ribose in hypoxic pulmonary vasoconstriction. J. Biol. Chem. 2001, 276, 11180–11188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushehri, N.; Jarrell, S.T.; Lieberman, S.; Mirdamadi-Zonozi, N.; Birkmayer, G.; Preuss, H.G. Oral reduced B-nicotinamide adenine dinucleotide (NADH) affects blood pressure, lipid peroxidation, and lipid profile in hypertensive rats (SHR). Geriatr. Nephrol. Urol. 1998, 8, 95–100. [Google Scholar] [CrossRef]
- Cho, K.H.; Kim, H.J.; Rodriguez-Iturbe, B.; Vaziri, N.D. Niacin ameliorates oxidative stress, inflammation, proteinuria, and hypertension in rats with chronic renal failure. Am. J. Physiol. Renal Physiol. 2009, 297, F106–F113. [Google Scholar] [CrossRef] [Green Version]
- Fushima, T.; Sekimoto, A.; Oe, Y.; Sato, E.; Ito, S.; Sato, H.; Takahashi, N. Nicotinamide ameliorates a preeclampsia-like condition in mice with reduced uterine perfusion pressure. Am. J. Physiol. Renal Physiol. 2017, 312, F366–F372. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Fushima, T.; Oyanagi, G.; Townley-Tilson, H.W.; Sato, E.; Nakada, H.; Oe, Y.; Hagaman, J.R.; Wilder, J.; Li, M.; et al. Nicotinamide benefits both mothers and pups in two contrasting mouse models of preeclampsia. Proc. Natl. Acad. Sci. USA 2016, 113, 13450–13455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huynh, P.K.; Wilder, J.; Hiller, S.; Hagaman, J.; Takahashi, N.; Maeda-Smithies, N.; Li, F. Beneficial effects of nicotinamide on hypertensive mice with impaired endothelial nitric oxide function. J. Exp. Nephrol. 2020, 1, 1–8. [Google Scholar]
- Yano, M.; Akazawa, H.; Oka, T.; Yabumoto, C.; Kudo-Sakamoto, Y.; Kamo, T.; Shimizu, Y.; Yagi, H.; Naito, A.T.; Lee, J.K.; et al. Monocyte-derived extracellular Nampt-dependent biosynthesis of NAD(+) protects the heart against pressure overload. Sci. Rep. 2015, 5, 15857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, R.; Hoffer, A.; Stephen, J.D. Influence of nicotinic acid on serum cholesterol in man. Arch. Biochem. Biophys. 1955, 54, 558–559. [Google Scholar] [CrossRef]
- Investigators, A.-H. The role of niacin in raising high-density lipoprotein cholesterol to reduce cardiovascular events in patients with atherosclerotic cardiovascular disease and optimally treated low-density lipoprotein cholesterol: Baseline characteristics of study participants. The Atherothrombosis Intervention in Metabolic syndrome with low HDL/high triglycerides: Impact on Global Health outcomes (AIM-HIGH) trial. Am. Heart J. 2011, 161, 538–543. [Google Scholar]
- Markel, A. The resurgence of niacin: From nicotinic acid to niaspan/laropiprant. Isr. Med. Assoc. J. 2011, 13, 368–374. [Google Scholar]
- Halder, P.; Bera, D.; Majumder, B.; Venkatraman, D.; Bhandari, M. Dilated cardiomyopathy in a case of pellagra: A extremely rare but treatable entity. Indian J. Med. Res. Pharm. Sci. 2015, 2, 24–27. [Google Scholar]
- Misner, D.L.; Kauss, M.A.; Singh, J.; Uppal, H.; Bruening-Wright, A.; Liederer, B.M.; Lin, T.; McCray, B.; La, N.; Nguyen, T.; et al. Cardiotoxicity Associated with Nicotinamide Phosphoribosyltransferase Inhibitors in Rodents and in Rat and Human-Derived Cells Lines. Cardiovasc. Toxicol. 2017, 17, 307–318. [Google Scholar] [CrossRef]
- Rahman, J.E.; Helou, E.F.; Gelzer-Bell, R.; Thompson, R.E.; Kuo, C.; Rodriguez, E.R.; Hare, J.M.; Baughman, K.L.; Kasper, E.K. Noninvasive diagnosis of biopsy-proven cardiac amyloidosis. J. Am. Coll. Cardiol. 2004, 43, 410–415. [Google Scholar] [CrossRef] [Green Version]
- Frustaci, A.; Verardo, R.; Caldarulo, M.; Acconcia, M.C.; Russo, M.A.; Chimenti, C. Myocarditis in hypertrophic cardiomyopathy patients presenting acute clinical deterioration. Eur. Heart J. 2007, 28, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Rachmilewitz, M.; Braun, K. Electrocardiographic changes in typhoid fever and their reversibility following niacin treatment. Am. Heart J. 1948, 36, 284–294. [Google Scholar] [CrossRef]
- Fine, I.; Brainerd, H.; Sokolow, M. Myocarditis in acute infectious diseases; a clinical and electrocardiographic study. Circulation 1950, 2, 859–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clerkin, K.J.; Fried, J.A.; Raikhelkar, J.; Sayer, G.; Griffin, J.M.; Masoumi, A.; Jain, S.S.; Burkhoff, D.; Kumaraiah, D.; Rabbani, L.; et al. COVID-19 and Cardiovascular Disease. Circulation 2020, 141, 1648–1655. [Google Scholar] [CrossRef] [Green Version]
- Bailey, A.L.; Dmytrenko, O.; Greenberg, L.; Bredemeyer, A.L.; Ma, P.; Liu, J.; Penna, V.; Winkler, E.S.; Sviben, S.; Brooks, E.; et al. SARS-CoV-2 Infects Human Engineered Heart Tissues and Models COVID-19 Myocarditis. JACC Basic Transl. Sci. 2021, 6, 331–345. [Google Scholar] [CrossRef] [PubMed]
- Perez-Bermejo, J.A.; Kang, S.; Rockwood, S.J.; Simoneau, C.R.; Joy, D.A.; Silva, A.C.; Ramadoss, G.N.; Flanigan, W.R.; Fozouni, P.; Li, H.; et al. SARS-CoV-2 infection of human iPSC-derived cardiac cells reflects cytopathic features in hearts of patients with COVID-19. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef]
- Badawy, A.A. Immunotherapy of COVID-19 with poly (ADP-ribose) polymerase inhibitors: Starting with nicotinamide. Biosci. Rep. 2020, 40, BSR20202856. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.; Wentzel, A.R.; Richards, G.A. COVID-19: NAD(+) deficiency may predispose the aged, obese and type2 diabetics to mortality through its effect on SIRT1 activity. Med. Hypotheses 2020, 144, 110044. [Google Scholar] [CrossRef] [PubMed]
- Omran, H.M.; Almaliki, M.S. Influence of NAD+ as an ageing-related immunomodulator on COVID 19 infection: A hypothesis. J. Infect. Public Health 2020, 13, 1196–1201. [Google Scholar] [CrossRef] [PubMed]
- Raines, N.H.; Ganatra, S.; Nissaisorakarn, P.; Pandit, A.; Morales, A.; Asnani, A.; Sadrolashrafi, M.; Maheshwari, R.; Patel, R.; Bang, V.; et al. Niacinamide May Be Associated with Improved Outcomes in COVID-19-Related Acute Kidney Injury: An Observational Study. Kidney360 2021, 2, 33–41. [Google Scholar] [CrossRef]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef]
- Airhart, S.E.; Shireman, L.M.; Risler, L.J.; Anderson, G.D.; Nagana Gowda, G.A.; Raftery, D.; Tian, R.; Shen, D.D.; O’Brien, K.D. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS ONE 2017, 12, e0186459. [Google Scholar] [CrossRef]
- Berthiaume, J.M.; Kurdys, J.G.; Muntean, D.M.; Rosca, M.G. Mitochondrial NAD(+)/NADH Redox State and Diabetic Cardiomyopathy. Antioxid. Redox. Signal. 2019, 30, 375–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, B.; Zhao, Y.; Wang, D.; Zhang, X.; Hao, X.; Hu, M. Nicotinamide mononucleotide supplementation enhances aerobic capacity in amateur runners: A randomized, double-blind study. J. Int. Soc. Sports Nutr. 2021, 18, 54. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.T.; Zheng, L.; Lu, K.; Hou, W.Q. Activation of cardiac Nmnat/NAD+/SIR2 pathways mediates endurance exercise resistance to lipotoxic cardiomyopathy in aging Drosophila. J. Exp. Biol. 2021, 224, jeb242425. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.T.; Zheng, L.; Li, J.X.; Cheng, D.; Liu, Y.; Lu, K.; Hou, W.Q. Endurance exercise resistance to lipotoxic cardiomyopathy is associated with cardiac NAD(+)/dSIR2/PGC-1alpha pathway activation in old Drosophila. Biol. Open 2019, 8, bio044719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershberger, K.A.; Martin, A.S.; Hirschey, M.D. Role of NAD(+) and mitochondrial sirtuins in cardiac and renal diseases. Nat. Rev. Nephrol. 2017, 13, 213–225. [Google Scholar] [CrossRef] [Green Version]

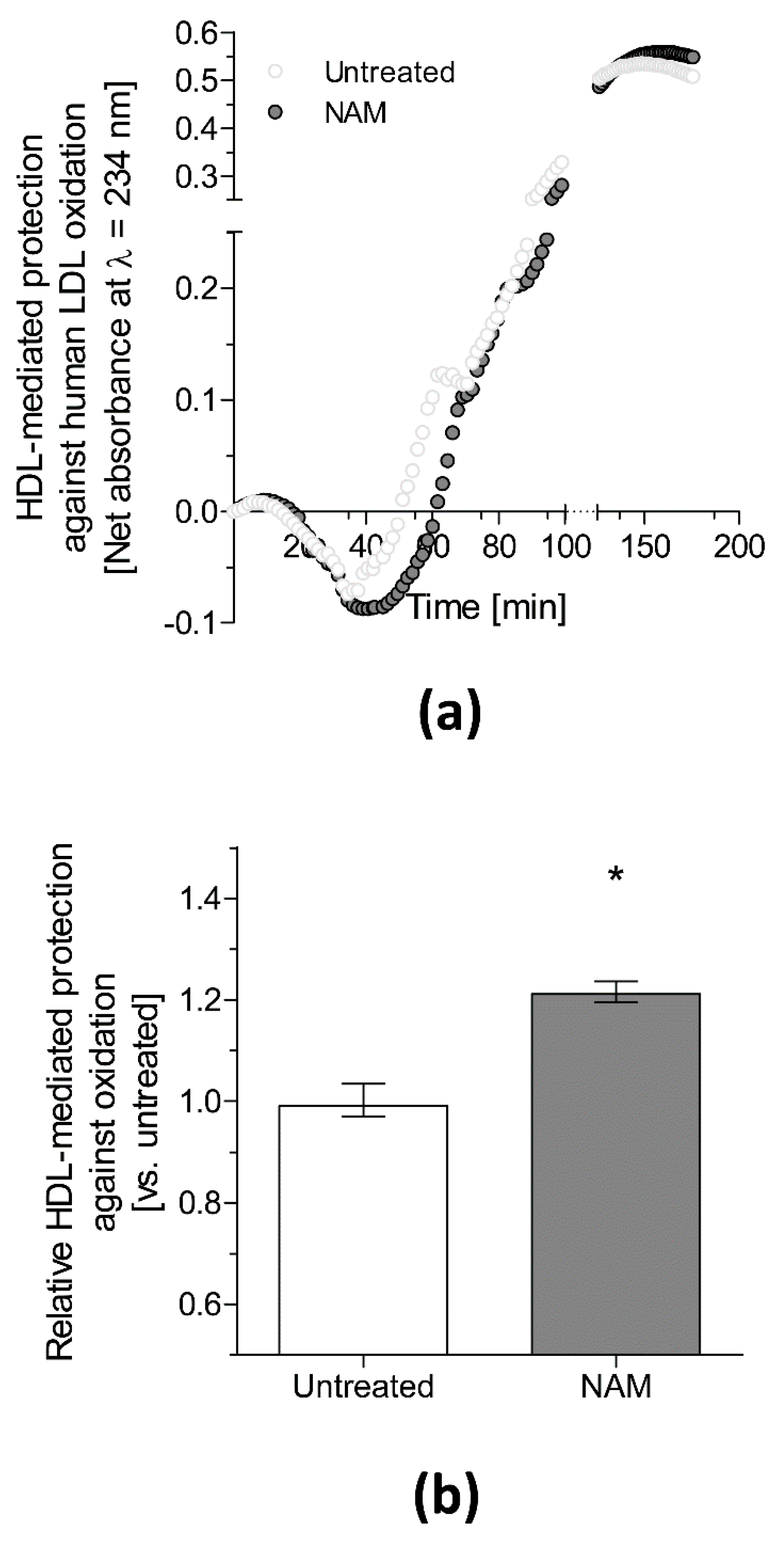
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rotllan, N.; Camacho, M.; Tondo, M.; Diarte-Añazco, E.M.G.; Canyelles, M.; Méndez-Lara, K.A.; Benitez, S.; Alonso, N.; Mauricio, D.; Escolà-Gil, J.C.; et al. Therapeutic Potential of Emerging NAD+-Increasing Strategies for Cardiovascular Diseases. Antioxidants 2021, 10, 1939. https://doi.org/10.3390/antiox10121939
Rotllan N, Camacho M, Tondo M, Diarte-Añazco EMG, Canyelles M, Méndez-Lara KA, Benitez S, Alonso N, Mauricio D, Escolà-Gil JC, et al. Therapeutic Potential of Emerging NAD+-Increasing Strategies for Cardiovascular Diseases. Antioxidants. 2021; 10(12):1939. https://doi.org/10.3390/antiox10121939
Chicago/Turabian StyleRotllan, Noemi, Mercedes Camacho, Mireia Tondo, Elena M. G. Diarte-Añazco, Marina Canyelles, Karen Alejandra Méndez-Lara, Sonia Benitez, Núria Alonso, Didac Mauricio, Joan Carles Escolà-Gil, and et al. 2021. "Therapeutic Potential of Emerging NAD+-Increasing Strategies for Cardiovascular Diseases" Antioxidants 10, no. 12: 1939. https://doi.org/10.3390/antiox10121939
APA StyleRotllan, N., Camacho, M., Tondo, M., Diarte-Añazco, E. M. G., Canyelles, M., Méndez-Lara, K. A., Benitez, S., Alonso, N., Mauricio, D., Escolà-Gil, J. C., Blanco-Vaca, F., & Julve, J. (2021). Therapeutic Potential of Emerging NAD+-Increasing Strategies for Cardiovascular Diseases. Antioxidants, 10(12), 1939. https://doi.org/10.3390/antiox10121939