Deciphering the Structural Basis of High Thermostability of Dehalogenase from Psychrophilic Bacterium Marinobacter sp. ELB17

 ,
,  ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Gene Synthesis, Expression, and Purification
2.2. Analysis of Secondary Structure and Thermostability
2.3. Native Polyacrylamide Gel Electrophoresis
2.4. Specific Activity Assay
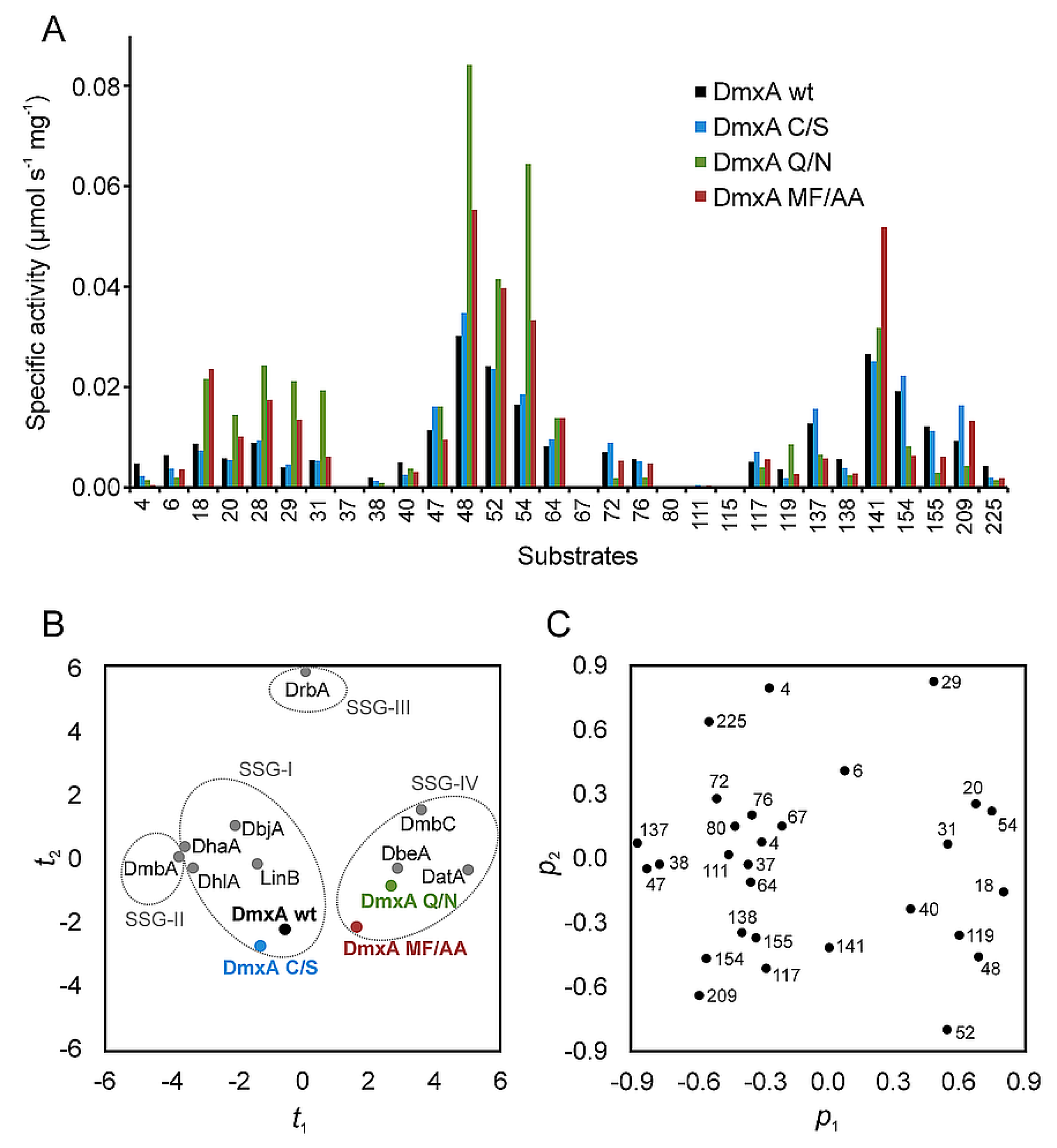
2.5. Principal Component Analysis (PCA) Analysis
2.6. Freeze–Thaw Stability Testing
2.7. Steady-State Kinetics
2.8. Measurement of Enantioselectivity
2.9. Enzyme Crystallization
2.10. Data Collection and Processing
2.11. Structure Determination and Refinement
2.12. Molecular Dynamics
2.13. Tunnel Analysis
2.14. Construction of Mutants
2.15. Site-Directed Mutagenesis
3. Results
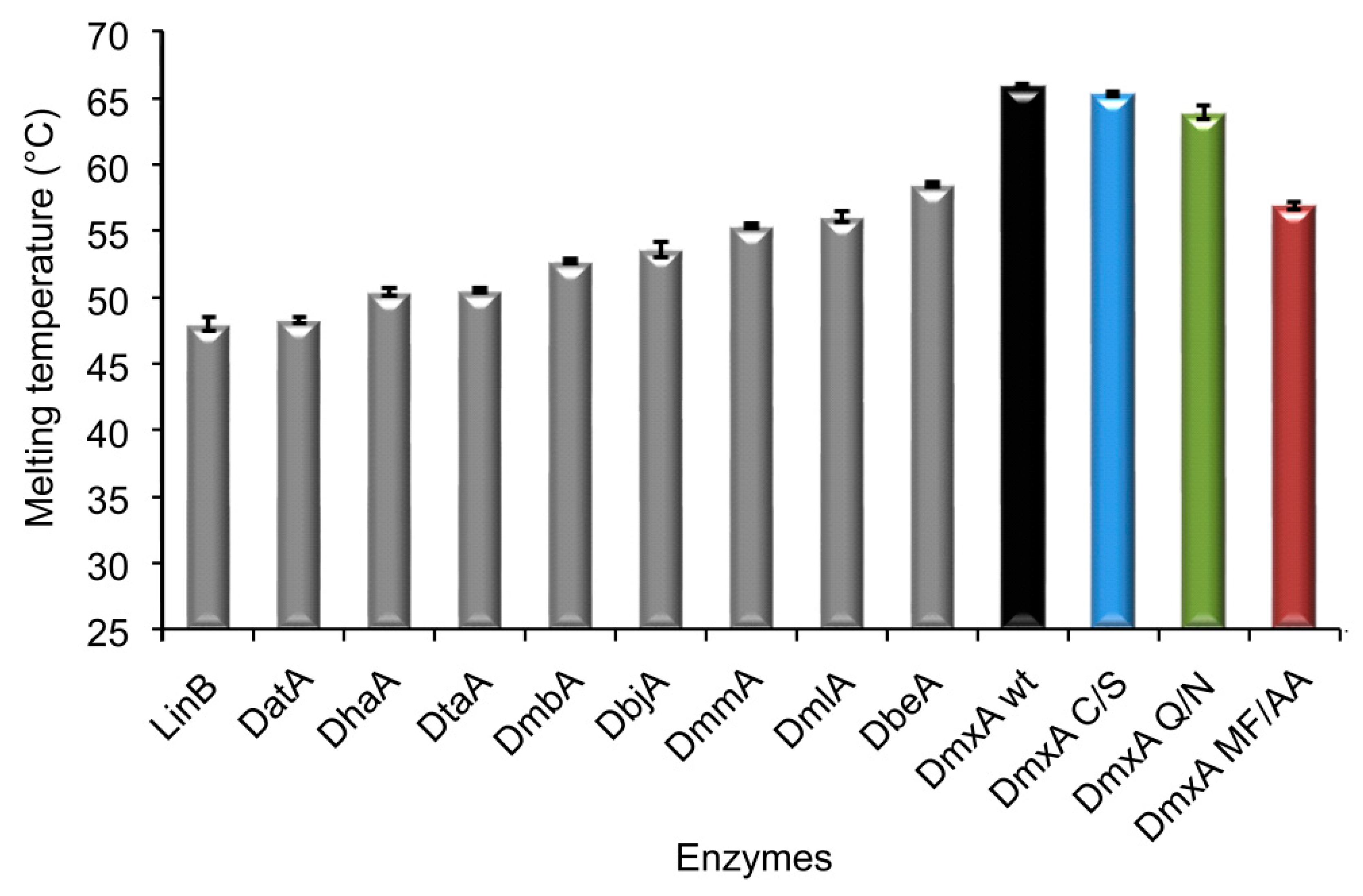
3.1. Expression and Biochemical Characterization of DmxA
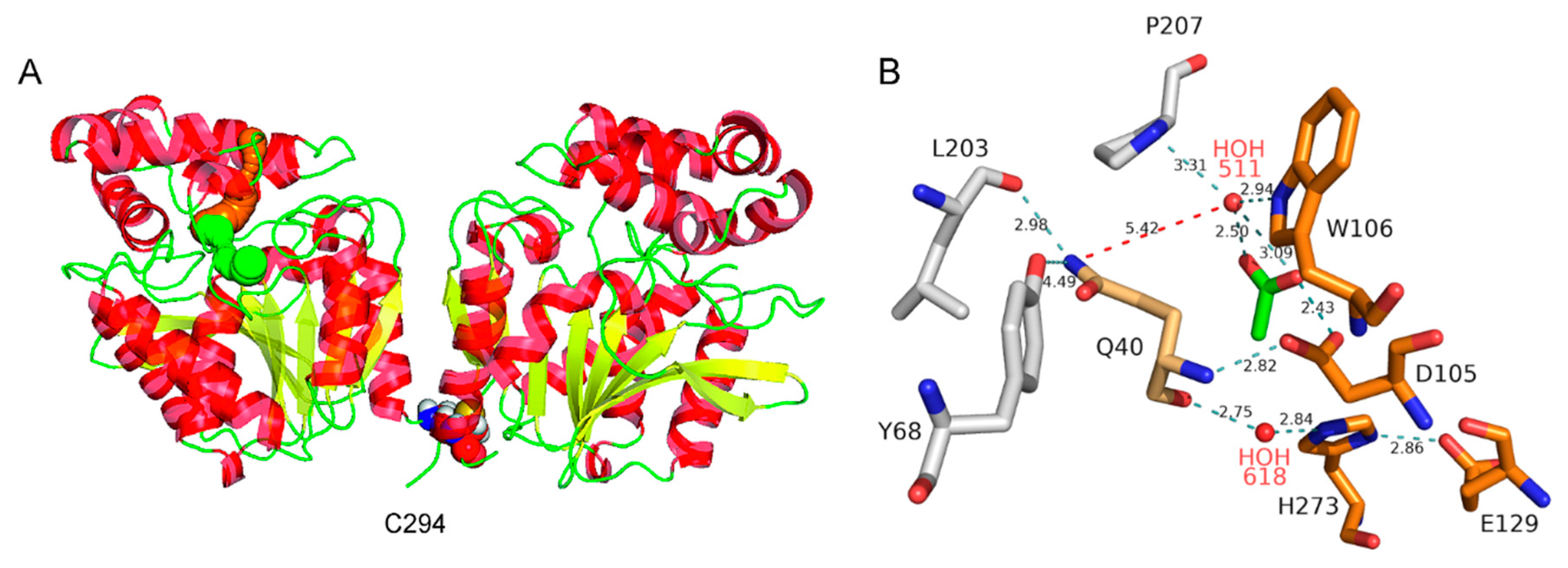
3.2. Structural Characterization of DmxA
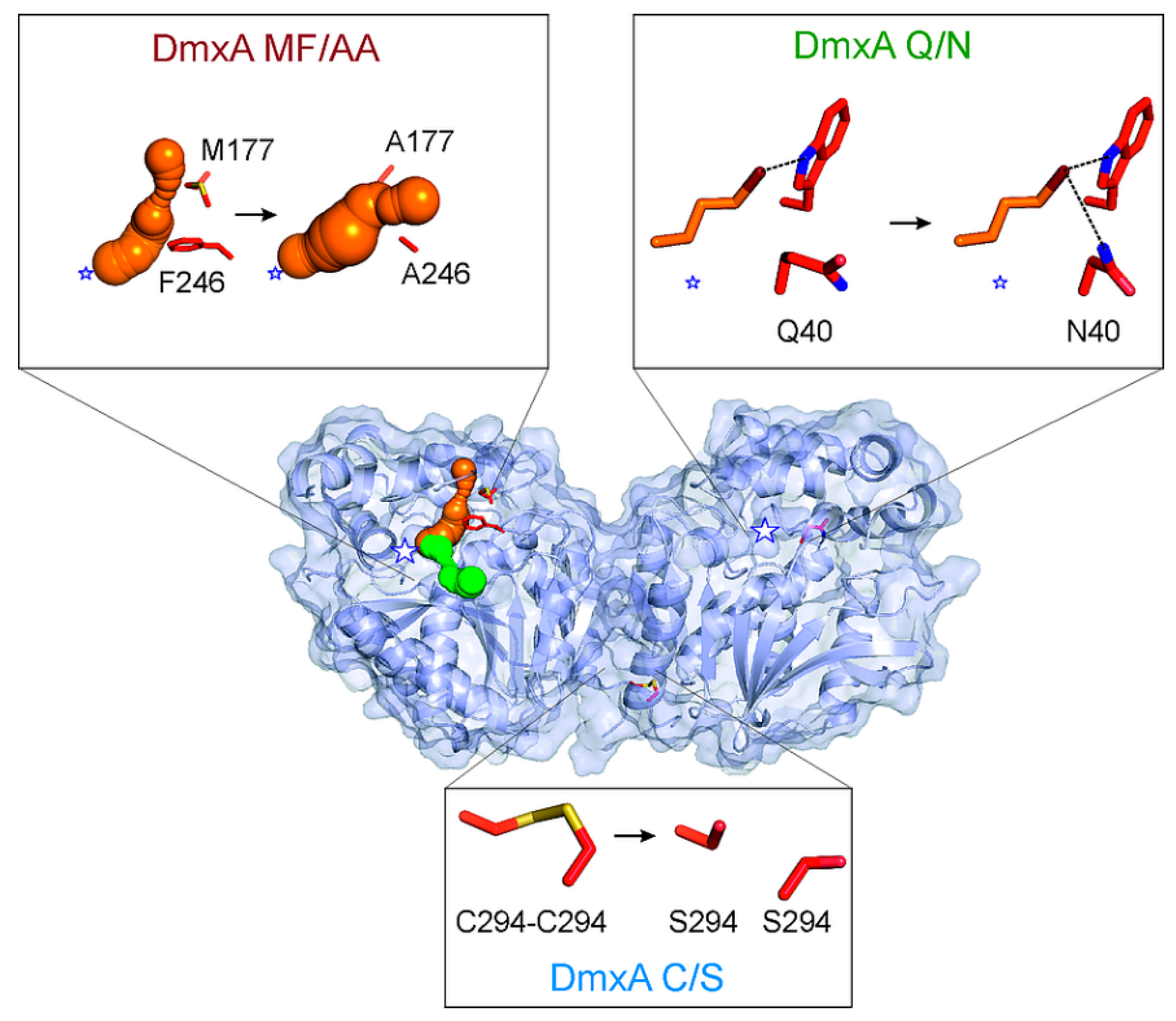
3.3. Construction of Variant DmxA C/S with Eliminated Cysteine Bridge
3.4. Analysis of Tunnel Network and Q40 in DmxA Wild-Type
3.5. Construction of Variant DmxA Q/N with Substituted Halide-Stabilizing Residue
3.6. Mutagenesis of Tunnel Bottlenecks and Construction of DmxA MF/AA Variant
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Janssen, D.B. Evolving haloalkane dehalogenases. Curr. Opin. Chem. Biol. 2004, 8, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Damborsky, J.; Chaloupkova, R.; Pavlova, M.; Chovancova, E.; Brezovsky, J. Structure–Function Relationships and Engineering of Haloalkane Dehalogenases. In Handbook of Hydrocarbon and Lipid Microbiology; Timmins, K.N., Ed.; Springer: New York, NY, USA, 2010; pp. 1081–1098. [Google Scholar]
- Ollis, D.L.; Cheah, E.; Cygler, M.; Dijkstra, B.; Frolow, F.; Franken, S.M.; Harel, M.; Remington, S.J.; Silman, I.; Schrag, J.; et al. Verschueren KHG & Goldman A the Alpha/Beta-Hydrolase Fold. Protein Eng. 1992, 5, 197–211. [Google Scholar] [PubMed]
- Vévodová, J.; Kutá-Smatanová, I.; Svensson, L.A.; Marek, J.; Nagata, Y.; Newman, J.; Takagi, M.; Damborsky, J. Crystal structure of the haloalkane dehalogenase from Sphingomonas paucimobilis UT26. Biochemistry 2000, 39, 14082–14086. [Google Scholar]
- Kmunicek, J.; Hynková, K.; Jedlicka, T.; Nagata, Y.; Negri, A.; Gago, F.; Wade, R.C.; Damborsky, J. Quantitative Analysis of Substrate Specificity of Haloalkane Dehalogenase LinB from Sphingomonas paucimobilis UT26. Biochemistry 2005, 44, 3390–3401. [Google Scholar] [CrossRef] [PubMed]
- Otyepka, M.; Damborsky, J. Functionally relevant motions of haloalkane dehalogenases occur in the specificity-modulating cap domains. Protein Sci. 2002, 11, 1206–1217. [Google Scholar] [CrossRef][Green Version]
- Sýkora, J.; Brezovský, J.; Koudelakova, T.; Lahoda, M.; Fortova, A.; Chernovets, T.; Chaloupkova, R.; Stepankova, V.; Prokop, Z.; Smatanová, I.K.; et al. Dynamics and hydration explain failed functional transformation in dehalogenase design. Nat. Methods 2014, 10, 428–430. [Google Scholar] [CrossRef]
- Chovancová, E.; Kosinski, J.; Bujnicki, J.M.; Damborský, J. Phylogenetic analysis of haloalkane dehalogenases. Proteins Struct. Funct. Bioinform. 2007, 67, 305–316. [Google Scholar] [CrossRef]
- Hasan, K.; Fortova, A.; Koudelakova, T.; Chaloupkova, R.; Ishitsuka, M.; Nagata, Y.; Damborsky, J.; Prokop, Z. Biochemical Characteristics of the Novel Haloalkane Dehalogenase DatA, Isolated from the Plant Pathogen Agrobacterium tumefaciens C58. Appl. Environ. Microb. 2011, 77, 1881–1884. [Google Scholar] [CrossRef]
- Koudelakova, T.; Chovancová, E.; Brezovský, J.; Monincová, M.; Fortova, A.; Jarkovsky, J.; Damborsky, J. Substrate specificity of haloalkane dehalogenases. Biochem. J. 2011, 435, 345–354. [Google Scholar] [CrossRef]
- Koudelakova, T.; Bidmanova, S.; Dvorak, P.; Pavelka, A.; Chaloupkova, R.; Prokop, Z.; Damborsky, J. Haloalkane dehalogenases: Biotechnological applications. Biotechnol. J. 2013, 8, 32–45. [Google Scholar] [CrossRef]
- Nagata, Y.; Ohtsubo, Y.; Tsuda, M. Properties and biotechnological applications of natural and engineered haloalkane dehalogenases. Appl. Microbiol. Biotechnol. 2015, 99, 9865–9881. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.; Steiner, W. The biocatalytic potential of extremophiles and extremozymes. Food Technol. Biotech. 2004, 42, 223–235. [Google Scholar]
- Hough, D.W.; Danson, M.J. Extremozymes. Curr. Opin. Chem. Biol. 1999, 3, 39–46. [Google Scholar] [CrossRef]
- Pace, C.N.; Fu, H.; Fryar, K.L.; Landua, J.; Trevino, S.R.; Shirley, B.A.; Hendricks, M.M.; Iimura, S.; Gajiwala, K.; Scholtz, J.M.; et al. Contribution of hydrophobic interactions to protein stability. J. Mol. Boil. 2011, 408, 514–528. [Google Scholar] [CrossRef] [PubMed]
- Feller, G. Protein stability and enzyme activity at extreme biological temperatures. J. Phys. Condens. Matter 2010, 22, 323101. [Google Scholar] [CrossRef]
- Chiuri, R.; Maiorano, G.; Rizzello, A.; Del Mercato, L.L.; Cingolani, R.; Rinaldi, R.; Maffia, M.; Pompa, P. Exploring Local Flexibility/Rigidity in Psychrophilic and Mesophilic Carbonic Anhydrases. Biophys. J. 2009, 96, 1586–1596. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nussinov, R. How do thermophilic proteins deal with heat? Cell Mol. Life Sci. 2001, 58, 1216–1233. [Google Scholar] [CrossRef]
- Lam, S.Y.; Yeung, R.C.Y.; Yu, T.-H.; Sze, K.-H.; Wong, K.-B. A rigidifying salt-bridge favors the activity of thermophilic enzyme at high temperatures at the expense of low-temperature activity. PLoS Biol. 2011, 9, e1001027. [Google Scholar] [CrossRef]
- Ding, Y.R.; Cai, Y.J.; Han, Y.G.; Zhao, B.Q. Comparison of the structural basis for thermal stability between archaeal and bacterial proteins. Extremophiles 2012, 16, 67–78. [Google Scholar] [CrossRef]
- Yamanaka, Y.; Kazuoka, T.; Yoshida, M.; Yamanaka, K.; Oikawa, T.; Soda, K. Thermostable aldehyde dehydrogenase from psychrophile, Cytophaga sp. KUC-1: Enzymological characteristics and functional properties. Biochem. Biophys. Res. Commun. 2002, 298, 632–637. [Google Scholar] [CrossRef]
- Kazuoka, T.; Masuda, Y.; Oikawa, T.; Soda, K. Thermostable aspartase from a marine psychrophile, Cytophaga sp. KUC-1: Molecular characterization and primary structure. J. Biochem. 2003, 133, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Kazuoka, T.; Oikawa, T.; Muraoka, I.; Kuroda, S.; Soda, K. A cold-active and thermostable alcohol dehydrogenase of a psychrotorelant from Antarctic seawater, Flavobacterium frigidimaris KUC-1. Extremophiles 2007, 11, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.; Duan, X.Y.; Lu, X.M.; Gao, P.J. A novel thermophilic endoglucanase from a mesophilic fungus Fusarium oxysporum. Chin. Sci. Bull. 2006, 51, 191–197. [Google Scholar] [CrossRef]
- Fedøy, A.-E.; Yang, N.; Martinez, A.; Leiros, H.-K.S.; Steen, I.H. Structural and Functional Properties of Isocitrate Dehydrogenase from the Psychrophilic Bacterium Desulfotalea psychrophila Reveal a Cold-active Enzyme with an Unusual High Thermal Stability. J. Mol. Boil. 2007, 372, 130–149. [Google Scholar] [CrossRef]
- Novak, H.R.; Sayer, C.; Panning, J.; Littlechild, J.A. Characterisation of an l-Haloacid Dehalogenase from the Marine Psychrophile Psychromonas ingrahamii with Potential Industrial Application. Mar. Biotechnol. 2013, 15, 695–705. [Google Scholar] [CrossRef]
- Lennox, E.S. Transduction of Linked Genetic Characters of the Host by Bacteriophage-Pl. Virology 1955, 1, 190–206. [Google Scholar] [CrossRef]
- Bradford, M.M. Rapid and Sensitive Method for Quantitation of Microgram Quantities of Protein Utilizing Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Iwasaki, I.; Utsumi, S.; Ozawa, T. New Colorimetric Determination of Chloride using Mercuric Thiocyanate and Ferric Ion. Bull. Chem. Soc. Jpn. 1952, 25, 226. [Google Scholar] [CrossRef]
- Tratsiak, K.; Degtjarik, O.; Drienovska, I.; Chrast, L.; Řezáčová, P.; Kuty, M.; Chaloupkova, R.; Damborsky, J.; Smatanova, I.K. Crystallographic analysis of new psychrophilic haloalkane dehalogenases: DpcA from Psychrobacter cryohalolentis K5 and DmxA from Marinobacter sp. ELB17. Acta Crystallogr. Sect. F Struct. Boil. Cryst. Commun. 2013, 69, 683–688. [Google Scholar] [CrossRef]
- De Sanctis, D.; Beteva, A.; Caserotto, H.; Dobias, F.; Gabadinho, J.; Giraud, T.; Gobbo, A.; Guijarro, M.; Lentini, M.; Lavault, B.; et al. ID29: A high-intensity highly automated ESRF beamline for macromolecular crystallography experiments exploiting anomalous scattering. J. Synchrotron Radiat. 2012, 19, 455–461. [Google Scholar] [CrossRef]
- Kabsch, W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993, 26, 795–800. [Google Scholar] [CrossRef]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Boil. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Xds. Acta Crystallogr. D 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Stepankova, V.; Khabiri, M.; Brezovský, J.; Pavelka, A.; Sýkora, J.; Amaro, M.; Minofar, B.; Prokop, Z.; Hof, M.; Ettrich, R.; et al. Expansion of Access Tunnels and Active-Site Cavities Influence Activity of Haloalkane Dehalogenases in Organic Cosolvents. ChemBioChem 2013, 14, 890–897. [Google Scholar] [CrossRef]
- Painter, J.; Merritt, E.A. Optimal description of a protein structure in terms of multiple groups undergoing TLS motion. Acta Crystallogr. D 2006, 62, 439–450. [Google Scholar] [CrossRef]
- Painter, J.; Merritt, E.A. TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 2006, 39, 109–111. [Google Scholar] [CrossRef]
- Winn, M.; Isupov, M.; Murshudov, G. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. Sect. D Boil. Crystallogr. 2001, 57, 122–133. [Google Scholar] [CrossRef]
- Winn, M.D.; Murshudov, G.N.; Papiz, M.Z. Macromolecular TLS Refinement in REFMAC at Moderate Resolutions. Methods Enzymol. 2003, 374, 300–321. [Google Scholar]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of Macromolecular Structures by the Maximum-Likelihood Method. Acta Crystallogr. Sect. D Boil. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Joosten, R.P.; Long, F.; Murshudov, G.N.; Perrakis, A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 2014, 1, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Vaguine, A.A.; Richelle, J.; Wodak, S.J. SFCHECK: A unified set of procedures for evaluating the quality of macromolecular structure-factor data and their agreement with the atomic model. Acta Crystallogr. Sect. D Boil. Crystallogr. 1999, 55, 191–205. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide Protein Data Bank. Nat. Struct. Mol. Boil. 2003, 10, 980. [Google Scholar] [CrossRef]
- Kabsch, W. A solution for the best rotation to relate two sets of vectors. Acta Crystallogr. Sect. A Cryst. Phys. Diffr. Theor. Gen. Crystallogr. 1976, 32, 922–923. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Gordon, J.C.; Myers, J.B.; Folta, T.; Shoja, V.; Heath, L.S.; Onufriev, A. H++: A server for estimating pK(a)s and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–W371. [Google Scholar] [CrossRef]
- Chandrasekhar, J.; Impey, R.W.; Jorgensen, W.L.; Madura, J.D.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar]
- Doerr, S.; Harvey, M.J.; Noé, F.; De Fabritiis, G. HTMD: High-Trhoughput Molecular Dynamics for Molecular Discovery. J. Chem. Theory Comput. 2016, 12, 1845–1852. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Le Grand, S.; Gotz, A.W.; Walker, R.C. SPFP: Speed without compromise—A mixed precision model for GPU accelerated molecular dynamics simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Gotz, A.W.; Williamson, M.J.; Xu, D.; Poole, D.; Le Grand, S.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 1. Generalized Born. J. Chem. Theory Comput. 2012, 8, 1542–1555. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Molecular Dynamics Simulations of the Dynamic and Energetic Properties of Alkali and Halide Ions Using Water-Model-Specific Ion Parameters. J. Phys. Chem. B 2009, 113, 13279–13290. [Google Scholar] [CrossRef]
- Harvey, M.J.; De Fabritiis, G. An implementation of the smooth particle mesh Ewald method on GPU hardware. J. Chem. Theory Comput. 2009, 5, 2371–2377. [Google Scholar] [CrossRef]
- Harvey, M.J.; Giupponi, G.; De Fabritiis, G.; Harvey, M. ACEMD: Accelerating Biomolecular Dynamics in the Microsecond Time Scale. J. Chem. Theory Comput. 2009, 5, 1632–1639. [Google Scholar] [CrossRef]
- Feenstra, K.A.; Hess, B.; Berendsen, H.J.C. Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 1999, 20, 786–798. [Google Scholar] [CrossRef]
- Hopkins, C.W.; Le Grand, S.; Walker, R.C.; Roitberg, A.E. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 2015, 11, 1864–1874. [Google Scholar] [CrossRef] [PubMed]
- The PyMOL Molecular Graphic System; Version 1.2r2; Schroedinger LLC: New York, NY, USA, 2009.
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. Model. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Chovancova, E.; Pavelka, A.; Benes, P.; Strnad, O.; Brezovsky, J.; Kozlíková, B.; Gora, A.; Sustr, V.; Klvana, M.; Medek, P.; et al. CAVER 3.0: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures. PLoS Comput. Boil. 2012, 8, e1002708. [Google Scholar] [CrossRef]
- Kellogg, E.H.; Leaver-Fay, A.; Baker, D. Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins 2011, 79, 830–838. [Google Scholar] [CrossRef]
- Sanchis, J.; Fernández, L.; Carballeira, J.D.; Drone, J.; Gumulya, Y.; Höbenreich, H.; Kahakeaw, D.; Kille, S.; Lohmer, R.; Peyralans, J.J.-P.; et al. Improved PCR method for the creation of saturation mutagenesis libraries in directed evolution: Application to difficult-to-amplify templates. Appl. Microbiol. Biotechnol. 2008, 81, 387–397. [Google Scholar] [CrossRef]
- Ward, B.B.; Priscu, J.C. Detection and characterization of denitrifying bacteria from a permanently ice-covered Antarctic lake. Hydrobiologia 1997, 347, 57–68. [Google Scholar] [CrossRef]
- Fasman, G.D. Circular Dichroism and the Conformational Analysis of Biomolecules; Plenum Press: New York, NY, USA, 1996. [Google Scholar]
- Prokop, Z.; Sato, Y.; Brezovský, J.; Mozga, T.; Chaloupkova, R.; Koudelakova, T.; Jeřabek, P.; Stepankova, V.; Natsume, R.; Van Leeuwen, J.G.E.; et al. Enantioselectivity of Haloalkane Dehalogenases and its Modulation by Surface Loop Engineering. Angew. Chem. 2010, 122, 6247–6251. [Google Scholar] [CrossRef]
- Holm, L.; Rosenstrom, P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010, 38, 545–549. [Google Scholar] [CrossRef]
- Brünger, A.T. Free R value: A novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [Google Scholar] [CrossRef]
- Klvana, M.; Pavlová, M.; Koudelakova, T.; Chaloupkova, R.; Dvorak, P.; Prokop, Z.; Stsiapanava, A.; Kuty, M.; Kuta-Smatanova, I.; Dohnalek, J.; et al. Pathways and Mechanisms for Product Release in the Engineered Haloalkane Dehalogenases Explored Using Classical and Random Acceleration Molecular Dynamics Simulations. J. Mol. Boil. 2009, 392, 1339–1356. [Google Scholar] [CrossRef] [PubMed]
- Chaloupkova, R.; Prokop, Z.; Sato, Y.; Nagata, Y.; Damborsky, J. Stereoselectivity and conformational stability of haloalkane dehalogenase DbjA from Bradyrhizobium japonicum USDA110: The effect of pH and temperature. FEBS J. 2011, 278, 2728–2738. [Google Scholar] [CrossRef] [PubMed]
- Vanacek, P.; Sebestova, E.; Babkova, P.; Bidmanova, S.; Daniel, L.; Dvořák, P.; Stepankova, V.; Chaloupkova, R.; Brezovsky, J.; Prokop, Z.; et al. Exploration of Enzyme Diversity by Integrating Bioinformatics with Expression Analysis and Biochemical Characterization. ACS Catal. 2018, 8, 2402–2412. [Google Scholar] [CrossRef]
- Oikawa, T.; Kazuoka, T.; Soda, K. Paradoxical thermostable enzymes from psychrophile: Molecular characterization and potentiality for biotechnological application. J. Mol. Catal. B: Enzym. 2003, 23, 65–70. [Google Scholar] [CrossRef]
- Chaloupkova, R.; Prudnikova, T.; Řezáčová, P.; Prokop, Z.; Koudelakova, T.; Daniel, L.; Brezovský, J.; Ikeda-Ohtsubo, W.; Sato, Y.; Kuty, M.; et al. Structural and functional analysis of a novel haloalkane dehalogenase with two halide-binding sites. Acta Crystallogr. Sect. D Boil. Crystallogr. 2014, 70, 1884–1897. [Google Scholar] [CrossRef] [PubMed]
- Hasan, K.; Gora, A.; Brezovský, J.; Chaloupkova, R.; Moskalíková, H.; Fortova, A.; Nagata, Y.; Damborsky, J.; Prokop, Z. The effect of a unique halide-stabilizing residue on the catalytic properties of haloalkane dehalogenase DatA fromAgrobacterium tumefaciensC58. FEBS J. 2013, 280, 3149–3159. [Google Scholar] [CrossRef]
- Brezovsky, J.; Babkova, P.; Degtjarik, O.; Fortova, A.; Gora, A.; Iermak, I.; Rezacova, P.; Dvorak, P.; Smatanova, I.K.; Prokop, Z.; et al. Engineering a de Novo Transport Tunnel. ACS Catal. 2016, 6, 7597–7610. [Google Scholar] [CrossRef]
- Koudelakova, T.; Chaloupkova, R.; Brezovský, J.; Prokop, Z.; Sebestova, E.; Hesseler, M.; Khabiri, M.; Plevaka, M.; Kulik, D.; Smatanova, I.K.; et al. Engineering Enzyme Stability and Resistance to an Organic Cosolvent by Modification of Residues in the Access Tunnel. Angew. Chem. Int. Ed. 2013, 52, 1959–1963. [Google Scholar] [CrossRef]
- Lišková, V.; Bednar, D.; Prudnikova, T.; Řezáčová, P.; Koudelakova, T.; Sebestova, E.; Smatanová, I.K.; Brezovský, J.; Chaloupkova, R.; Damborsky, J. Balancing the Stability-Activity Trade-Off by Fine-Tuning Dehalogenase Access Tunnels. ChemCatChem 2015, 7, 648–659. [Google Scholar] [CrossRef]
- Xie, Y.; An, J.; Yang, G.; Wu, G.; Zhang, Y.; Cui, L.; Feng, Y. Enhanced Enzyme Kinetic Stability by Increasing Rigidity within the Active Site. J. Boil. Chem. 2014, 289, 7994–8006. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection Statistics | |
|---|---|
| X-ray Source | ESRF Grenoble, ID29 |
| Wavelength (Å) | 0.972 |
| Resolution range (Å) | 100.0–1.45 (1.49–1.45) |
| Space group | P212121 |
| Unit-cell parameters (Å; °) | a = 43.37, b = 78.34, c = 150.5; α = γ = β = 90.0 |
| Total no. of measured intensities | 484,657 (37,044) |
| Number of unique reflections | 39,029 (5978) |
| Redundancy | 5.28 (5.52) |
| Average I/σ(I) | 8.02 (2.1) |
| Completeness (%) | 99.7 (99.9) |
| Rmeas a (%) | 9.1 (71.9) |
| Rmerge b (%) | 11.2 (62.1) |
| Wilson B (Å2) | 21.048 |
| Refinement | |
| Resolution range(Å) | 75.26–1.45 (1.48–1.45) |
| No. of reflections in working set | 86,980 (6373) |
| No. of reflections in test set | 4589 (329) |
| R value (%) c | 17.32 (28) |
| Rfree value (%) d | 21.38 (30.5) |
| RMSD, bond lengths (Å) | 0.0188 |
| RMSD, angles (°) | 1.9274 |
| No. of atoms in AU | 5475 |
| No. of water molecules in AU | 599 |
| No. of acetate ions in AU | 3 |
| Mean B value (Å2) | 18.62 |
| Ramachandran plot statistics: | |
| Residues in preferred regions (%) | 91.5 |
| Residues in allowed regions (%) | 3.76 |
| Residues outliers (%) | 1.08 |
| PDB code | 5MXP |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrast, L.; Tratsiak, K.; Planas-Iglesias, J.; Daniel, L.; Prudnikova, T.; Brezovsky, J.; Bednar, D.; Kuta Smatanova, I.; Chaloupkova, R.; Damborsky, J. Deciphering the Structural Basis of High Thermostability of Dehalogenase from Psychrophilic Bacterium Marinobacter sp. ELB17. Microorganisms 2019, 7, 498. https://doi.org/10.3390/microorganisms7110498
Chrast L, Tratsiak K, Planas-Iglesias J, Daniel L, Prudnikova T, Brezovsky J, Bednar D, Kuta Smatanova I, Chaloupkova R, Damborsky J. Deciphering the Structural Basis of High Thermostability of Dehalogenase from Psychrophilic Bacterium Marinobacter sp. ELB17. Microorganisms. 2019; 7(11):498. https://doi.org/10.3390/microorganisms7110498
Chicago/Turabian StyleChrast, Lukas, Katsiaryna Tratsiak, Joan Planas-Iglesias, Lukas Daniel, Tatyana Prudnikova, Jan Brezovsky, David Bednar, Ivana Kuta Smatanova, Radka Chaloupkova, and Jiri Damborsky. 2019. "Deciphering the Structural Basis of High Thermostability of Dehalogenase from Psychrophilic Bacterium Marinobacter sp. ELB17" Microorganisms 7, no. 11: 498. https://doi.org/10.3390/microorganisms7110498
APA StyleChrast, L., Tratsiak, K., Planas-Iglesias, J., Daniel, L., Prudnikova, T., Brezovsky, J., Bednar, D., Kuta Smatanova, I., Chaloupkova, R., & Damborsky, J. (2019). Deciphering the Structural Basis of High Thermostability of Dehalogenase from Psychrophilic Bacterium Marinobacter sp. ELB17. Microorganisms, 7(11), 498. https://doi.org/10.3390/microorganisms7110498