
Prokaryotic Communities Vary with Cultivation Modes of Shrimp (Litopenaeus vannamei)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Water Sampling
2.2. Size-Fractionated Filtration and Physiochemical Determination
2.3. DNA Extraction
2.4. High-Throughput Sequencing of Microbial 16S rRNA Genes
2.5. Data Processing and Statistical Analyses
3. Results
3.1. Water Parameters of the Aquafarms
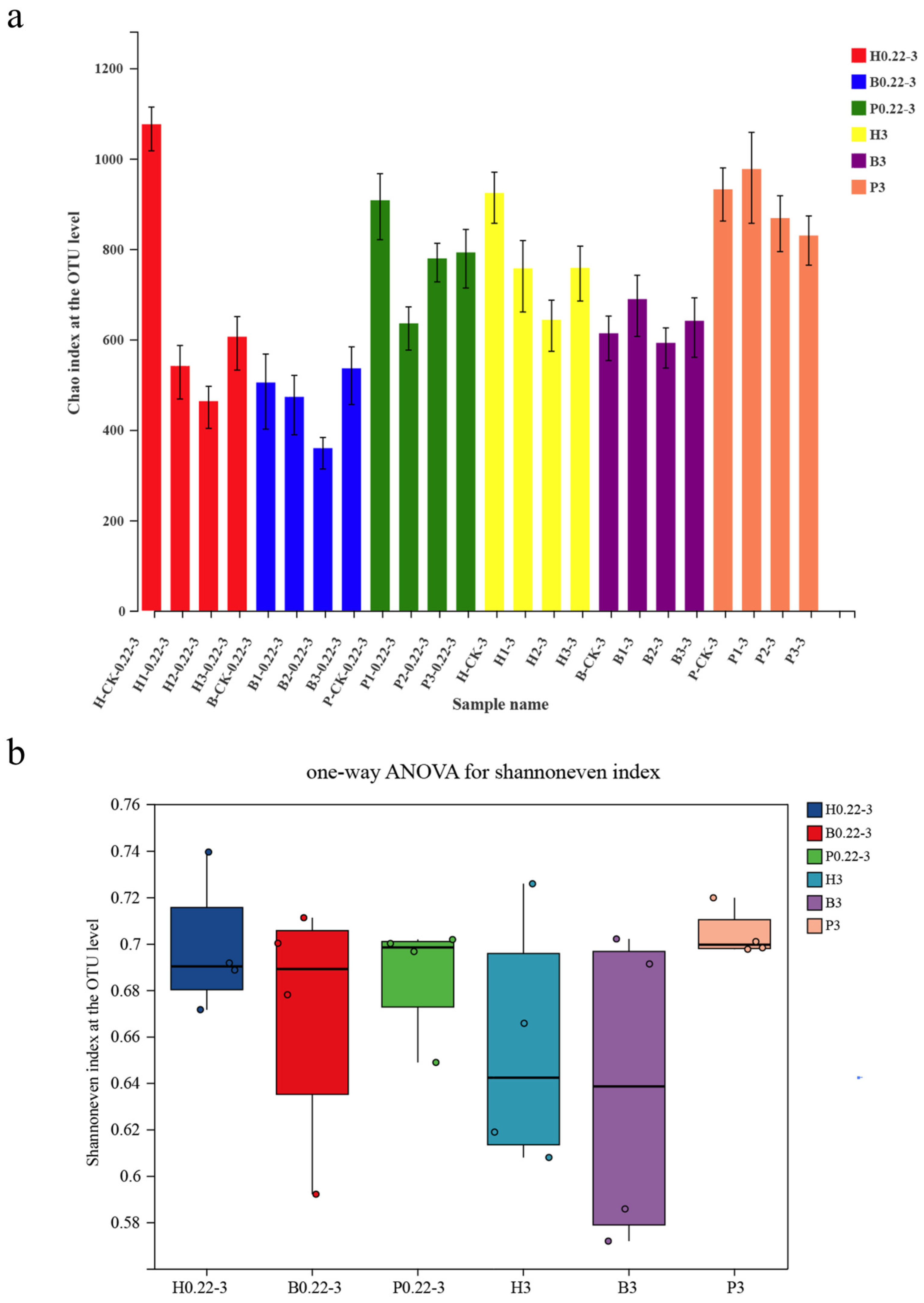
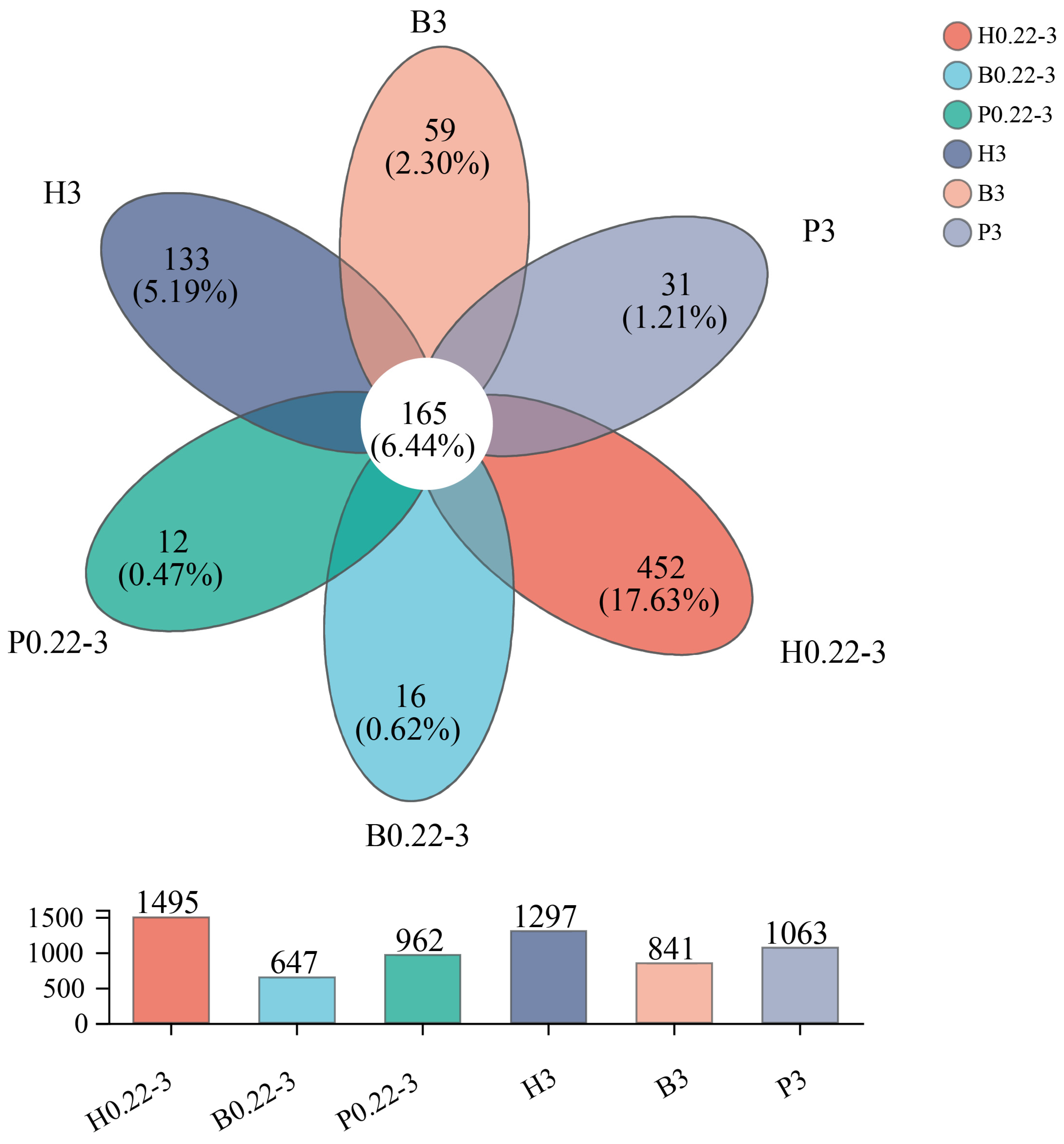
3.2. α-Diversity Index of Microbial Communities and Distribution of Taxonomic Units
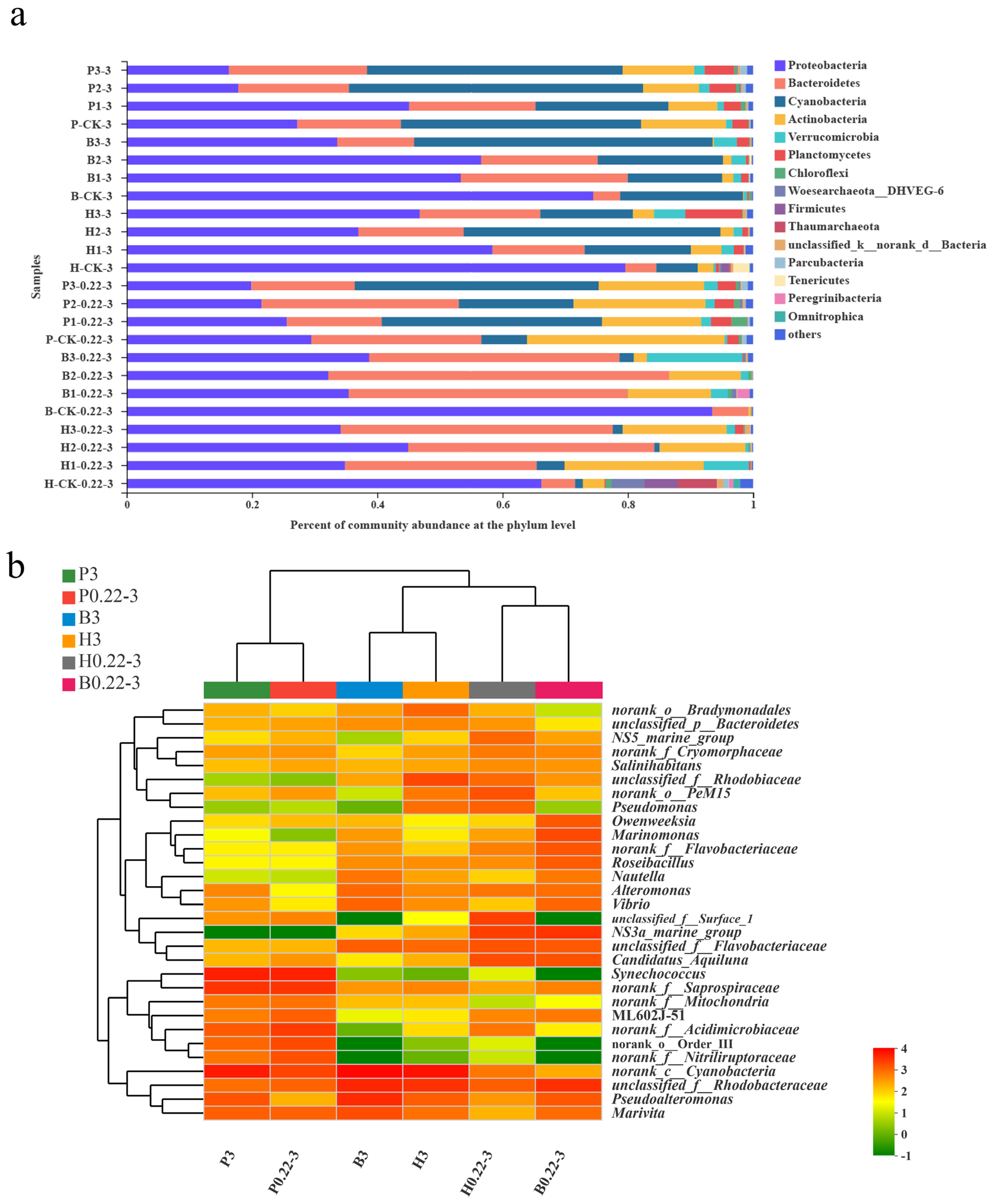
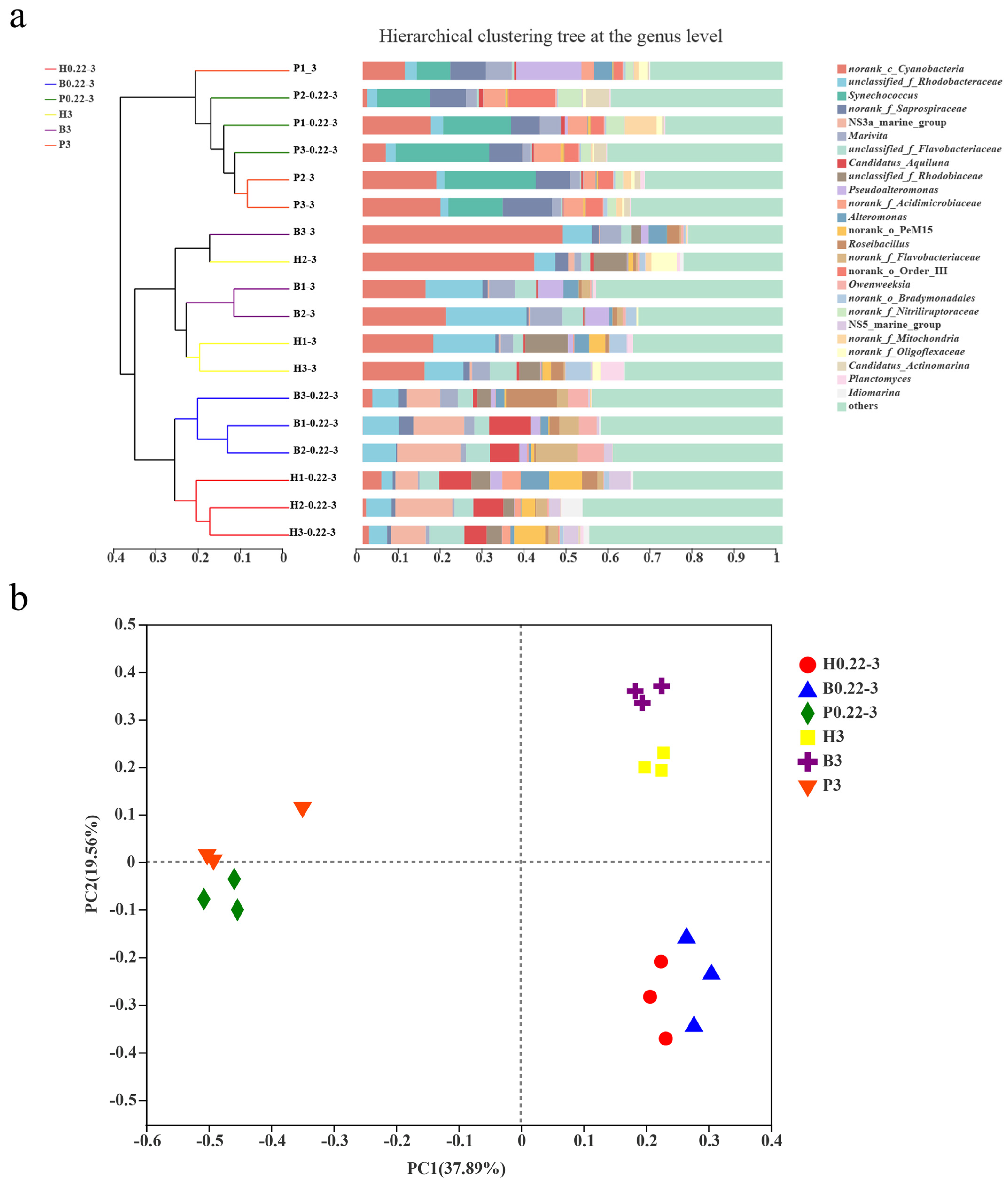
3.3. Microbial Community Compositions
3.4. Differences in Microbial Diversity Between Cultivation Modes
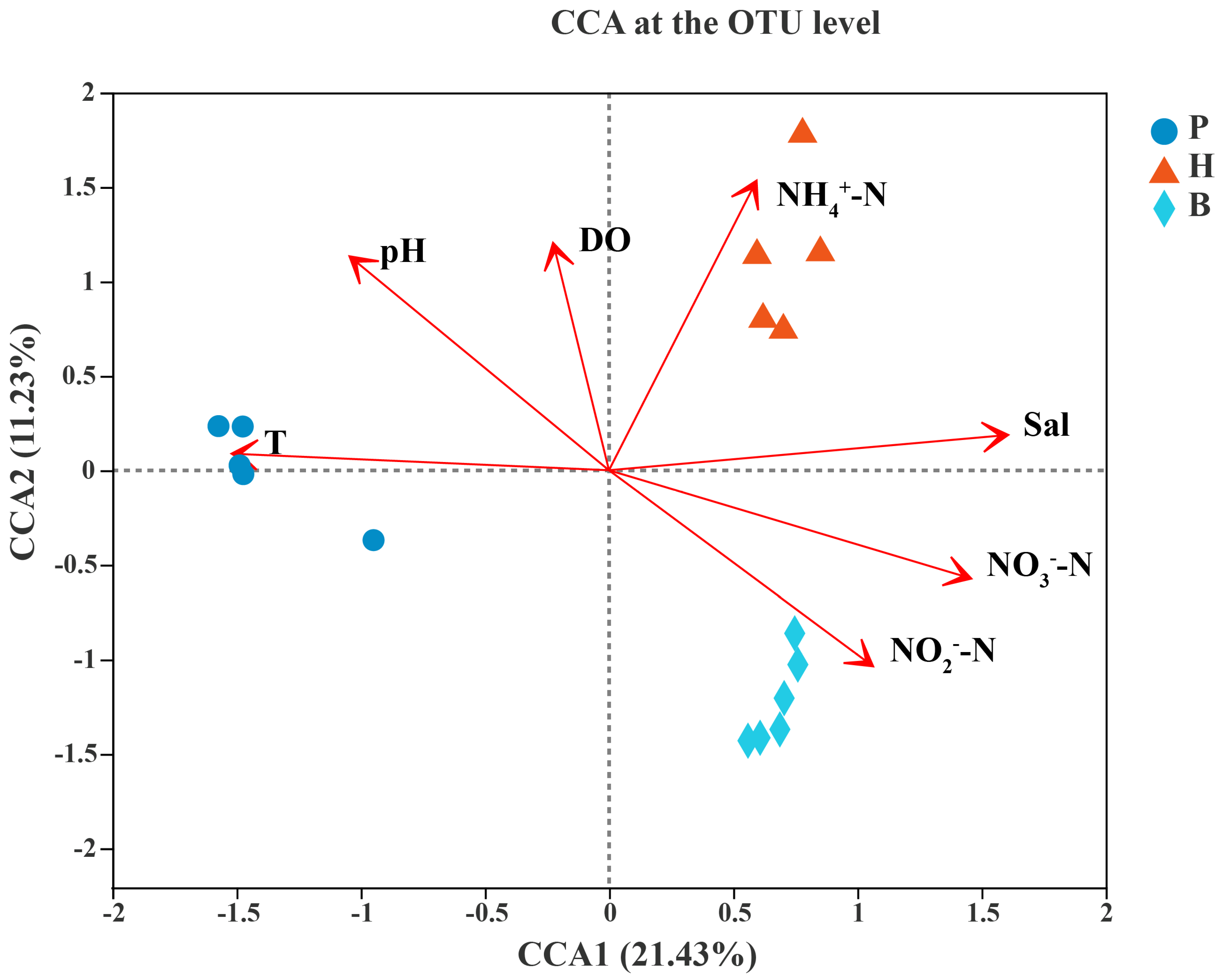
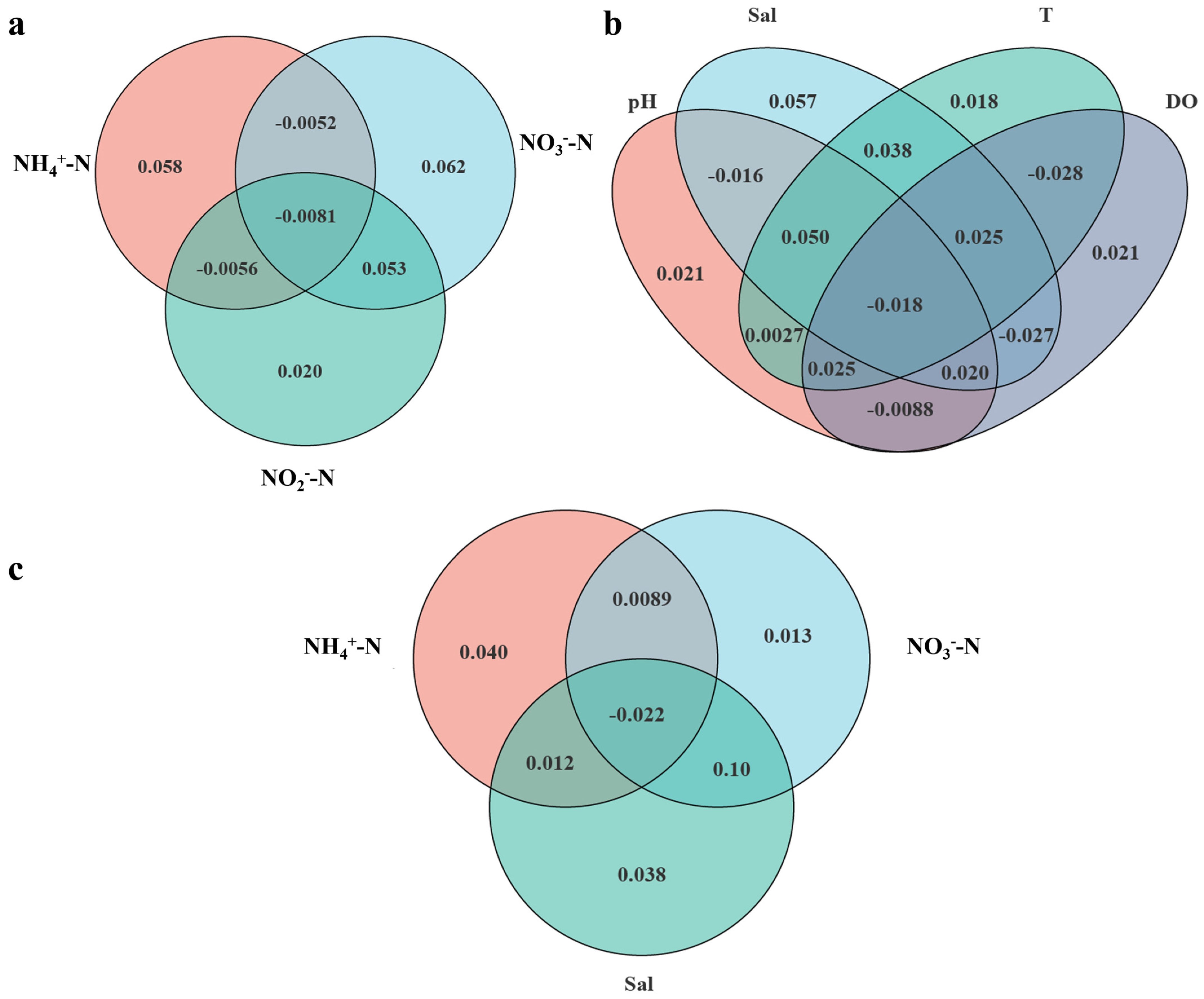
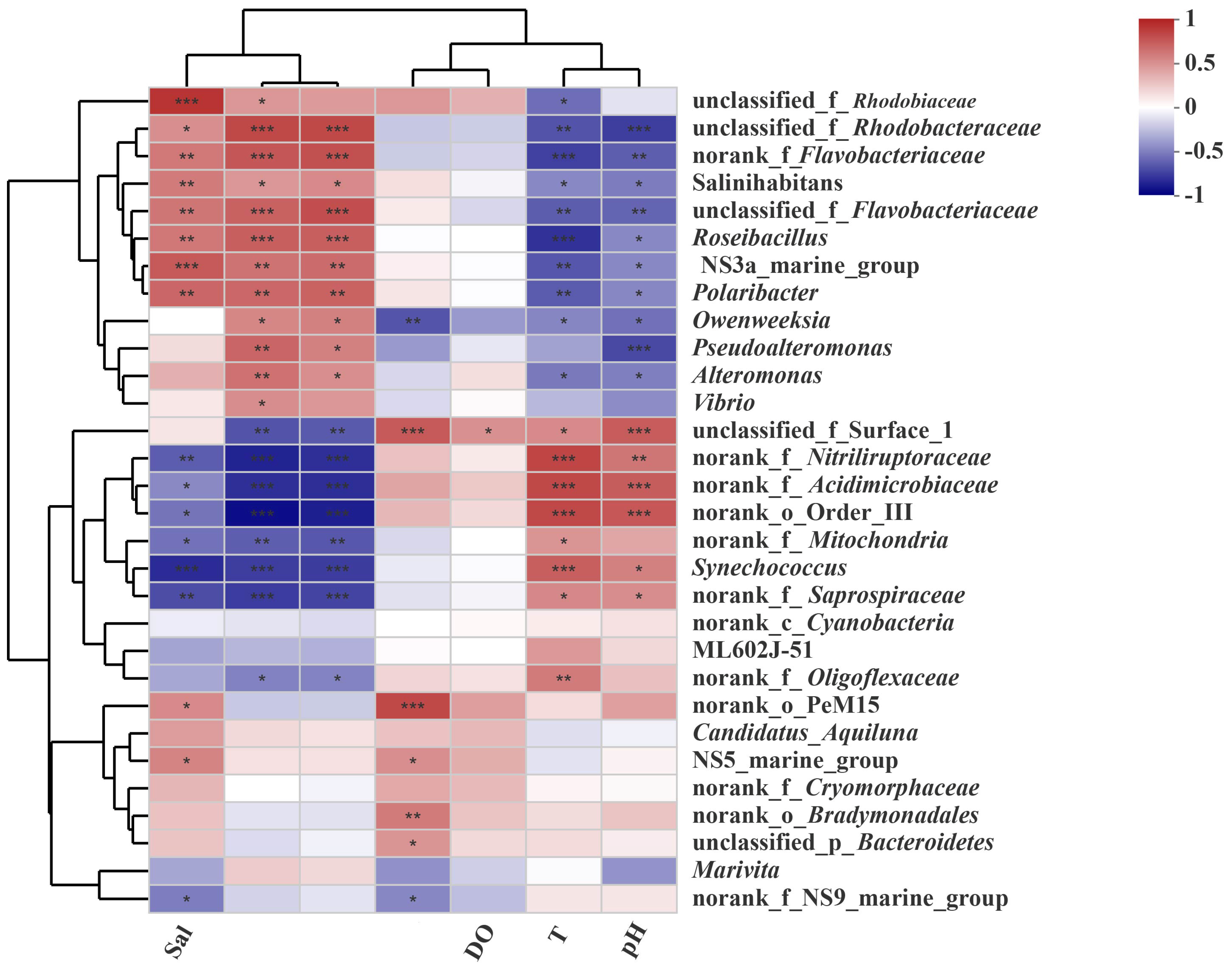
3.5. Relationships Between the Microbial Community and Environmental Factors
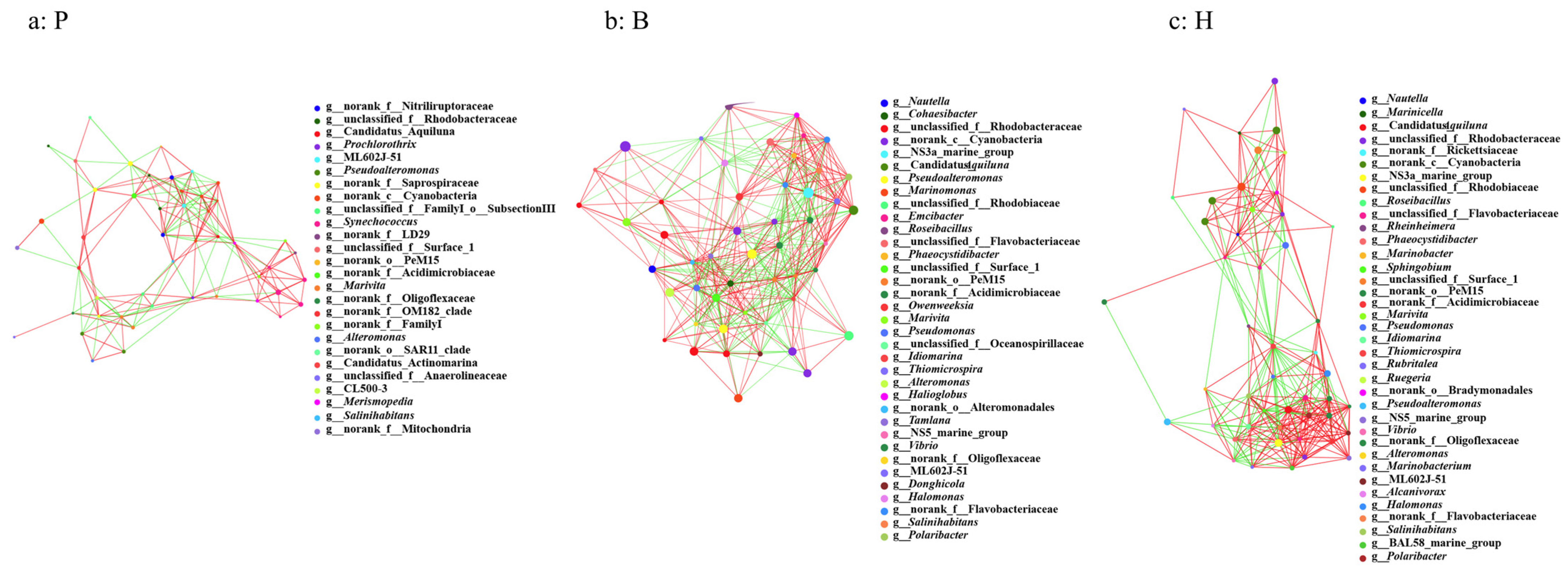
3.6. Network Properties and the Potential Keystone Taxa
4. Discussion
4.1. Aquaculture Mode and Particle Size Are Closely Related to the Microbial Community
4.2. Ammonium, Salinity, and Nitrate Are Three Significant Environmental Factors Influencing the Microbial Community
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Naylor, R.; Goldburg, R.; Primavera, J.; Kautsky, N.; Beveridge, M.; Clay, J.; Folke, C.; Lubchenco, J.; Mooney, H.; Troell, M. Effect of aquaculture on world fish supplies. Nature 2000, 405, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Tacon, A.G. Global trends in aquaculture and compound aquafeed production. World Aquac. 2018, 49, 33–46. [Google Scholar]
- Liao, I.C.; Chien, Y.-H. The pacific white shrimp, Litopenaeus vannamei, in Asia: The world’s most widely cultured alien crustacean. In The Wrong Place-Alien Marine Crustaceans: Distribution, Biology and Impacts; Springer: Dordrecht, The Netherlands, 2011; pp. 489–519. [Google Scholar]
- Chang, Z.Q.; Neori, A.; He, Y.; Li, J.; Qiao, L.; Preston, S.; Liu, P.; Li, J. Development and current state of seawater shrimp farming, with an emphasis on integrated multi-trophic pond aquaculture farms, in China—A review. Rev. Aquacult. 2020, 12, 2544–2558. [Google Scholar] [CrossRef]
- Miao, W.; Yuan, Y. Development trends and future prospects of shrimp culture in China. In Species and System Selection for Sustainable Aquaculture; Leung, P., Lee, C.-S., O’Bryen, P.J., Eds.; Wiley-Blackwell: Valley Center, CA, USA, 2007; pp. 269–282. [Google Scholar]
- Tian, J.; Wu, W.; Li, J.; Wan, X.; Zhao, Z.; Xi, R.; Hu, X.; Pan, M.; Xue, Y.; Yu, W. Development dilemma of Litopenaeus vannamei industry in China, current countermeasures taken and its implications for the world shrimp aquaculture industry. Sraeli J. Aquac. 2024, 76, 106–116. [Google Scholar] [CrossRef]
- Hoppe, H.-G.; Kim, S.-J.; Gocke, K. Microbial decomposition in aquatic environments: Combined process of extracellular enzyme activity and substrate uptake. Appl. Environ. Microbiol. 1988, 54, 784–790. [Google Scholar] [CrossRef]
- Hou, D.; Huang, Z.; Zeng, S.; Liu, J.; Wei, D.; Deng, X.; Weng, S.; He, Z.; He, J. Environmental factors shape water microbial community structure and function in shrimp cultural enclosure ecosystems. Front. Microbiol. 2017, 8, 2359. [Google Scholar] [CrossRef]
- Rajeev, R.; Adithya, K.; Kiran, G.S.; Selvin, J. Healthy microbiome: A key to successful and sustainable shrimp aquaculture. Rev. Aquacult. 2021, 13, 238–258. [Google Scholar] [CrossRef]
- Abraham, T.J.; Ghosh, S.; Nagesh, T.; Sasmal, D. Distribution of bacteria involved in nitrogen and sulphur cycles in shrimp culture systems of West Bengal, India. Aquaculture 2004, 239, 275–288. [Google Scholar] [CrossRef]
- Abraham, T.J.; Ghosh, S.; Sasmal, D. Assessment of nitrogen and sulphur cycle bacteria and shrimp production in ponds treated with biological products. J. Coast. Life Med. 2016, 3, 466–470. [Google Scholar]
- Zhang, M.; Pan, L.; Huang, F.; Gao, S.; Su, C.; Zhang, M.; He, Z. Metagenomic analysis of composition, function and cycling processes of microbial community in water, sediment and effluent of Litopenaeus vannamei farming environments under different culture modes. Aquaculture 2019, 506, 280–293. [Google Scholar] [CrossRef]
- Wei, G.; Shan, D.; Li, G.; Li, X.; Tian, R.; He, J.; Shao, Z. Prokaryotic communities vary with floc size in a biofloc-technology based aquaculture system. Aquaculture 2020, 529, 735632. [Google Scholar] [CrossRef]
- Wade, N.; Bourne, N.; Simon, C.J.A. Influence of marker particle size on nutrient digestibility measurements and particle movement through the digestive system of shrimp. Aquaculture 2018, 491, 273–280. [Google Scholar] [CrossRef]
- Mestre, M.; Ruiz-González, C.; Logares, R.; Duarte, C.M.; Gasol, J.M.; Sala, M.M. Sinking particles promote vertical connectivity in the ocean microbiome. Proc. Natl. Acad. Sci. USA 2018, 115, 6799–6807. [Google Scholar] [CrossRef]
- Walters, W.; Hyde, E.R.; Berg-Lyons, D.; Ackermann, G.; Humphrey, G.; Parada, A.; Gilbert, J.A.; Jansson, J.K.; Caporaso, J.G.; Fuhrman, J.A.; et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems 2016, 1, e00009-15. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity George, M.; Tiedje James, M.; Cole James, R. Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microb. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, 590–596. [Google Scholar] [CrossRef]
- Schloss Patrick, D.; Westcott Sarah, L.; Ryabin, T.; Hall Justine, R.; Hartmann, M.; Hollister Emily, B.; Lesniewski Ryan, A.; Oakley Brian, B.; Parks Donovan, H.; Robinson Courtney, J.; et al. Introducing mothur: Open-Source, platform-Independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microb. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, 256–259. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber RL, V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Deng, Y.; Jiang, Y.-H.; Yang, Y.; He, Z.; Luo, F.; Zhou, J. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 219. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Garcia, A.T.; Olmos, J.S. Quantification by fluorescent in situ hybridization of bacteria associated with Litopenaeus vannamei larvae in Mexican shrimp hatchery. Aquaculture 2007, 262, 211–218. [Google Scholar] [CrossRef]
- Madigan, M.T.; Bender, K.S.; Sattley, W.M. Brock Biology of Microorganisms, 16th ed.; Pearson: London, UK, 2020. [Google Scholar]
- Zeng, Y.; Ma, Y.; Wei, C.; Jiao, N.; Tang, K.; Wu, Z.; Jian, J. Bacterial diversity in various coastal mariculture ponds in Southeast China and in diseased eels as revealed by culture and culture-independent molecular techniques. Aquac. Res. 2010, 41, 172–186. [Google Scholar] [CrossRef]
- Zhang, J.; Jiao, S.; Lu, Y. Biogeographic distribution of bacterial, archaeal and methanogenic communities and their associations with methanogenic capacity in Chinese wetlands. Sci. Total Environ. 2018, 622–623, 664–675. [Google Scholar] [CrossRef]
- Tan, S.; Zhou, J.; Zhu, X.; Yu, S.; Zhan, W.; Wang, B.; Cai, Z. An association network analysis among microeukaryotes and bacterioplankton reveals algal bloom dynamics. J. Phycol. 2015, 51, 120–132. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Krishnan, K.P.; Singh, A.; Thomas, F.A.; Begum, N.; Tiwari, M.; Bhaskar, V.P.; Gopinath, A. Biochemical composition of particles shape particle-attached bacterial community structure in a high Arctic fjord. Ecol. Indic. 2019, 10, 581–592. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiao, W.; Jiao, N. Linking biochemical properties of particles to particle-attached and free-living bacterial community structure along the particle density gradient from freshwater to open ocean. J. Geophys. Res. Biogeosciences 2016, 121, 2261–2274. [Google Scholar] [CrossRef]
- Teeling, H.; Fuchs, B.M.; Becher, D.; Klockow, C.; Gardebrecht, A.; Bennke, C.M.; Kassabgy, M.; Huang, S.; Mann, A.J.; Waldmann, J.; et al. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 2012, 336, 608–611. [Google Scholar] [CrossRef]
- Salazar, G.; Cornejo-Castillo, F.M.; Borrull, E.; Díez-Vives, C.; Lara, E.; Vaqué, D.; Arrieta, J.M.; Duarte, C.M.; Gasol, J.M.; Acinas, S.G. Particle-association lifestyle is a phylogenetically conserved trait in bathypelagic prokaryotes. Mol. Ecol. 2015, 24, 5692–5706. [Google Scholar] [CrossRef]
- Smith, M.W.; Allen, L.Z.; Allen, A.E.; Herfort, L.; Simon, H.M. Contrasting genomic properties of free-living and particle-attached microbial assemblages within a coastal ecosystem. Front. Microbiol. 2013, 4, 120. [Google Scholar] [CrossRef]
- Mestre, M.; Ferrera, I.; Borrull, E.; Ortega-Retuerta, E.; Mbedi, S.; Grossart, H.; Gasol, J.M.; Sala, M.M. Spatial variability of marine bacterial and archaeal communities along the particulate matter continuum. Mol. Ecol. 2017, 26, 6827–6840. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, Y.; Yuan, W.; Ruess, L. The response of microbial communities to environmental factors in bank soil and river sediment: A case study along the mainstream of the Yangtze River. Environ. Res. 2025, 269, 120903. [Google Scholar] [CrossRef]
- Payne, M.S.; Hall, M.R.; Sly, L.; Bourne, D.G. Microbial diversity within early-stage cultured Panulirus ornatus phyllosomas. Appl. Environ. Microbiol. 2007, 73, 1940–1951. [Google Scholar] [CrossRef]
- Priyanka, P.; Chattopadhyay, J.; Srivastava, N. Chapter 6-Response of microbial communities to environmental changes. In Functional Metagenomics; Academic Press: Cambridge, MA, USA, 2024; pp. 143–153. [Google Scholar]
- Niu, S.; Li, C.; Xie, J.; Li, Z.; Zhang, K.; Wang, G.; Yun, X.; Tian, J.; Li, H.; Xie, W.; et al. Influence of aquaculture practices on microbiota composition and pathogen abundance in pond ecosystems in South China. Water Res. X 2025, 27, 100302. [Google Scholar] [CrossRef]
- Yang, J.; Ma, L.; Jiang, H.; Wu, G.; Dong, H. Salinity shapes microbial diversity and community structure in surface sediments of the Qinghai-Tibetan Lakes. Sci. Rep. 2016, 6, 25078. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Wei, G.; Li, J.; Shao, Z. Prokaryotic Communities Vary with Cultivation Modes of Shrimp (Litopenaeus vannamei). Microorganisms 2025, 13, 881. https://doi.org/10.3390/microorganisms13040881
Li G, Wei G, Li J, Shao Z. Prokaryotic Communities Vary with Cultivation Modes of Shrimp (Litopenaeus vannamei). Microorganisms. 2025; 13(4):881. https://doi.org/10.3390/microorganisms13040881
Chicago/Turabian StyleLi, Guizhen, Guangshan Wei, Jianyang Li, and Zongze Shao. 2025. "Prokaryotic Communities Vary with Cultivation Modes of Shrimp (Litopenaeus vannamei)" Microorganisms 13, no. 4: 881. https://doi.org/10.3390/microorganisms13040881
APA StyleLi, G., Wei, G., Li, J., & Shao, Z. (2025). Prokaryotic Communities Vary with Cultivation Modes of Shrimp (Litopenaeus vannamei). Microorganisms, 13(4), 881. https://doi.org/10.3390/microorganisms13040881