Abstract
Ester-based lubricants have been widely used owing to their excellent overall performance. In this study, the quantitative structure–property relationship (QSPR) approach was combined with molecular descriptors, a genetic algorithm (GA), and an artificial neural network (ANN) to systematically predict the key properties—kinematic viscosity at 40 °C and 100 °C, viscosity index, pour point, and flash point—of 64 diester-based lubricants. Quantum chemical calculations were first performed to obtain the equilibrium geometries and electronic information of the molecules. Geometry optimizations and frequency analyses were carried out using the Gaussian 16 software at the B3LYP/6-31G (d, p) level, providing a reliable foundation for molecular descriptor computation. Subsequently, topological, geometrical, and electronic descriptors were calculated using the RDKit toolkit, and the optimal feature subsets were selected by GA and used as ANN inputs for property prediction. The results showed that the ANN models exhibited good performance in predicting viscosity and flash point, with R2 values of 0.9455 and 0.8835, respectively, indicating that the ANN effectively captured the nonlinear relationships between molecular structure and physicochemical properties. In contrast, the prediction accuracy for pour point was relatively lower (R2 = 0.6155), suggesting that it is influenced by complex molecular packing and crystallization behaviors at low temperatures. Overall, the study demonstrates the feasibility of integrating quantum chemical calculations with the QSPR–ANN framework for lubricant property prediction, providing a theoretical basis and data-driven tool for molecular design and performance optimization of ester-based lubricants.
1. Introduction
As an important component of synthetic lubricants, ester-based lubricants occupy an irreplaceable position in modern lubrication technology. The polar carbonyl and ether groups in ester molecules can form a stable adsorption film on metal surfaces, thereby significantly improving lubricity and anti-wear performance—advantages that are not possessed by mineral or hydrocarbon-based synthetic oils [1,2,3]. In addition, some ester-based lubricants exhibit good biodegradability and low toxicity, making them widely recognized as ideal candidate base oils for achieving green lubrication and sustainable development [4]. Moreover, ester-based lubricants generally possess a high viscosity index, low volatility, and excellent low-temperature fluidity, enabling them to maintain stability across a wide temperature range. Consequently, they are widely used in aerospace applications and high-performance mechanical equipment [5].
In the characterization of lubricant performance, viscosity is the most fundamental and important physicochemical parameter, reflecting the fluidity of the oil and the load-bearing capacity of the lubricant film [6]. The viscosity index measures the degree to which a lubricant’s viscosity changes with temperature and serves as an important parameter for evaluating its ability to perform under a wide range of temperature conditions [7,8]. The pour point reflects the fluidity of a lubricant at low temperatures and determines its low-temperature start-up performance, while the flash point indicates the volatility and safety of the oil—a higher flash point generally implies better thermal stability [9]. These performance parameters constitute the core components of the lubricant evaluation system.
However, the performance of lubricants depends not only on experimental measurements but is also closely related to their molecular structures. QSPR approach provides both a theoretical and technical framework for investigating this correlation. QSPR starts from molecular structures and establishes quantitative models linking structural features of compounds to their physicochemical properties through mathematical and statistical methods. Its core concept is to use molecular descriptors to numerically represent structural information, thereby revealing the intrinsic relationship between molecular structural characteristics and macroscopic properties [10,11]. Among these, quantum chemical calculations provide a reliable theoretical foundation for studying the structures and properties of lubricant molecules. Using density functional theory (DFT), geometric structures, electronic distributions, and energy parameters can be obtained. These quantitative data help to elucidate the microscopic relationships between molecular structure and macroscopic performance and serve as a valuable basis for molecular descriptor calculation and model construction. Nasab et al. predicted the relationships between molecular structures and the viscosity and pour point of 41 ester-based lubricants [12].Zhou et al. also employed structure–property correlation predictions in the design of high-flash-point diester molecules [13]. In addition, numerous studies worldwide have investigated the QSPR of lubricants [14,15,16,17].
Molecular descriptors play a crucial role in this process. They transform complex molecular structures, electronic distributions, and geometrical configurations into manageable numerical parameters, enabling researchers to use mathematical models to predict and optimize lubricant performance [18]. Therefore, molecular descriptors serve not only as a vital link between molecular structure and performance but also as the foundation for conducting QSPR studies and advancing the molecular design and performance improvement of lubricants.
Therefore, the objective of this study is to develop a QSPR predictive model for five key properties of ester-based lubricants, including kinematic viscosity at 40 °C and 100 °C, viscosity index, pour point, and flash point, as illustrated in Figure 1. Molecular descriptors derived from the molecular structure were used as the input variables (X) of the neural network. The hidden layers (h) learn and approximate the nonlinear mapping between structural descriptors and physicochemical behavior, while the output layer (o) produces the predicted lubricant properties. Without the need for extensive experimental work, this model aims to accurately predict the essential performance parameters of diester lubricants. Such an approach not only facilitates a deeper understanding of the quantitative relationships between molecular structure and physicochemical properties but also provides a theoretical basis and efficient tool for the molecular design and optimization of lubricants. Therefore, the method has considerable practical value for accelerating the development of high-performance and environmentally friendly lubricants.
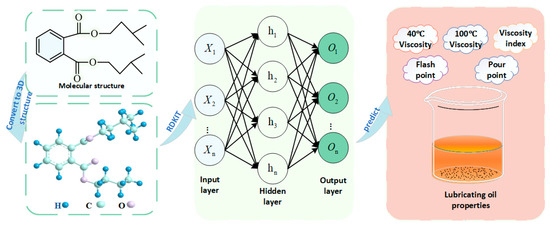
Figure 1.
Workflow of the present study.
2. Materials and Methods
2.1. Dataset
The dataset used in this study consists of 64 diester lubricant molecules collected from the literature [13], their SMILES notations, along with the corresponding kinematic viscosity at 40 °C and 100 °C, viscosity index, flash point, and pour point, are listed in Table 1.

Table 1.
SMILES notations and physicochemical properties of 64 diester molecules.
2.2. Quantum Chemistry Software
Obtaining accurate molecular structure and energy information is a prerequisite for establishing subsequent models in the study of the relationship between lubricant molecular structure and performance. This article uses Gaussian 16 software [19] to first optimize the structure and analyze the vibration frequency of Diisopentyl phthalate (DIPP) molecules using two different methods, B3LYP [20,21] and M062x [22], under three different basis sets: def2svp, 6-31G (d, p), and def2-TZVP. By systematically analyzing and comparing the computational performance of different methods and base groups, determine the optimal computational solution. The B3LYP method, as a widely used hybrid functional, combines Hartree–Fock exchange energy (20%), Becke three-parameter exchange function (B3), and nonlocal correlation function (LYP), demonstrating good applicability in balancing geometric configurations and vibration frequency calculations. Its ability to describe weak interactions and hydrogen bonds is weak, but it is sufficient to meet the simulation needs of the main chain structure of ester compounds in this study. The M062x method belongs to the second generation of hybrid functionals, which enhances the descriptive ability of electron correlation by improving the exchange correlation functionals, and is particularly suitable for accurate calculations of systems containing π bonds. However, this method requires high computational resources and may lead to superlinear scaling behavior. The selection of the three basic groups follows the principle of “minimum necessity”. Def2svp is the minimum splitting valence bond group with high computational efficiency, but it only includes the polarization function (p-orbital), resulting in significant errors in describing bond length and angle. 6-31G (d, p) is a medium-sized basis set that includes the d-orbital polarization function and p-orbital dispersion function, exhibiting balance in equilibrium geometry optimization. Def2-TZVP is a Gaussian orbital basis set, consisting of double zeta series and polarization function (d orbital), with the highest computational accuracy but significantly increased time consumption. Table 2 summarizes the calculation results of key parameters of DIPP molecules. The predicted error of bond length/bond angle calculated by B3LYP/6-31G (d, p) is less than 0.5%, which meets the requirements of molecular dynamics simulation input, and the calculation time for a single configuration is short, supporting batch processing. Based on comprehensive accuracy (consistency with literature values), computational cost (CPU time), and model construction requirements, B3LYP/6-31G (d, p) was ultimately selected. The optimized molecular structure of DIPP with B3LYP/6-31G (d, p) is illustrated in Figure 2.
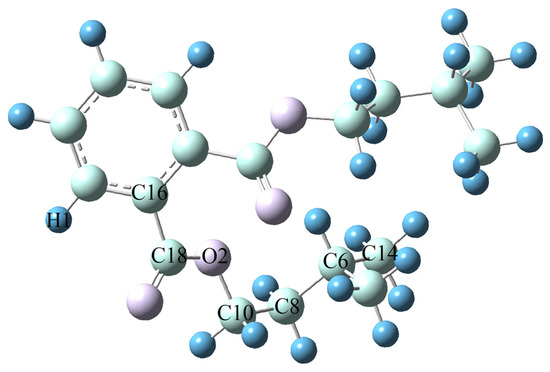
Figure 2.
Optimized Molecular Structure of DIPP with B3LYP/6-31G (d, p).
This step provides reliable geometric structures and related electronic information, serving as the foundation for subsequent molecular descriptor calculations. The optimized output files were converted into SDF format using OpenBabel [23] to ensure compatibility with the molecular descriptor calculation software.
Table 2.
Computational Results for the DIPP Molecule.
Table 2.
Computational Results for the DIPP Molecule.
| Item | B3LYP | M062x | Exp [24] | |||||
|---|---|---|---|---|---|---|---|---|
| def2svp | 6-31G (d, p) | def2-TZVP | def2svp | 6-31G (d, p) | def2-TZVP | |||
| r(C-C)/Å | C14~C6 | 1.531 | 1.532 | 1.528 | 1.527 | 1.528 | 1.525 | 1.529 |
| C16~C18 | 1.499 | 1.496 | 1.494 | 1.500 | 1.498 | 1.496 | 1.457 | |
| C8~C10 | 1.516 | 1.518 | 1.513 | 1.513 | 1.516 | 1.512 | 1.523 | |
| ∠(C-O-C)/o | 118.8 | 116.0 | 111.2 | 116.3 | 115.6 | 116.1 | 116.8 | |
| μ (Debye) | 3.658 | 3.730 | 3.915 | 3.584 | 3.656 | 3.854 | ||
| CPU time (t/h) | 2 h 41 min | 2 h 43 min | 22 h 26 min | 3 h 48 min | 3 h 41 min | 28 h 17 min | ||
2.3. Molecular Descriptors
Molecular descriptors are key parameters that characterize the relationship between molecular structural features and physicochemical properties, serving as the core input variables for constructing QSPR models. They transform complex chemical features—such as molecular structure, electronic distribution, topology, and geometry—into quantifiable mathematical expressions, thereby mapping molecular structural information into numerical feature space. According to their source of information and computational approach, molecular descriptors are generally classified into three categories: one-dimensional (1D), two-dimensional (2D), and three-dimensional (3D) descriptors. The 1D descriptors represent basic molecular composition information, including molecular weight, atom counts, and the numbers of hydrogen bond donors and acceptors. The 2D descriptors reflect molecular topological characteristics, such as connectivity indices, topological polar surface area (TPSA), and LogP values. The 3D descriptors are closely related to molecular geometry and electronic structure, including molecular volume, surface area, orbital energies (HOMO/LUMO), and charge distribution, and are typically calculated based on optimized three-dimensional structures.
In this study, the RDKit software (version:2025.03.6) [24] was used to process the structures of diester molecules and systematically calculate topological, geometrical, electronic, and hybrid types of molecular descriptors.
2.4. Genetic Algorithm
Because the original set of molecular descriptors is large, directly using them for modeling may lead to the curse of dimensionality and overfitting problems. Therefore, effective feature selection is necessary. In this study, the GA was employed to select the optimal descriptors corresponding to different target properties, including kinematic viscosity at 40 °C and 100 °C, viscosity index, pour point, and flash point. GA simulates the processes of natural selection and genetic variation to perform a global search for the optimal subset of features, thereby effectively improving the predictive accuracy and interpretability of the model [12,25,26]. This method avoids local optima while maintaining computational efficiency, making it suitable for studying the structure–property relationships of complex lubricant molecules.
2.5. Predictive Model
To establish the nonlinear mapping relationship between the molecular structure and physicochemical properties of lubricants, a machine learning approach was employed to develop the predictive model, with particular emphasis on the ANN. For each target property, the optimal molecular descriptors selected by the GA were used as input variables, while the corresponding physicochemical parameters served as output variables. The ANN, implemented through a multilayer perceptron (MLP) architecture and trained using the backpropagation algorithm, effectively captures the complex nonlinear relationships between molecular structures and properties. The robustness and generalization ability of the model were evaluated using cross-validation [27]. This modeling strategy provides a more reliable and interpretable predictive tool for investigating the structure–property relationships of lubricant molecules.
3. Results
This section presents the analysis and discussion of the predictive model results developed using the quantitative QSPR and ANN approaches. First, the predictive ability and generalization performance of the model for key properties—viscosity, viscosity index, pour point, and flash point—were evaluated by comparing the experimental and predicted values.
3.1. Prediction Workflow
The smiles representations of the diester molecules were first converted into preliminary 3D structures using the MMFF force field in RDKit. After geometry optimization to obtain equilibrium conformations, RDKit was used to calculate a total of 217 molecular descriptors, including both 2D and 3D structural features. The GA was then applied for feature screening and selection, resulting in 43, 44, 42, 43, and 59 relevant descriptors for viscosity at 40 °C, viscosity at 100 °C, viscosity index, flash point, and pour point, respectively, as summarized in Table 3. A five-fold cross-validation strategy was adopted for model training. The ANN consisted of three hidden layers, each containing 128 neurons and uses the Adam optimizer with a learning rate of 0.0005 The ReLU activation function was used, and dropout regularization was applied to prevent overfitting.
Table 3.
Results of GA-Based Descriptor Selection and Optimization.
In the regression model, the mean absolute error (MAE) and coefficient of determination (R2) were selected as evaluation metrics. The main reason for this choice is that these indicators quantify model performance from different perspectives and collectively capture key aspects such as error magnitude, error distribution, and explanatory power.
N represents the number of samples; yi denotes the true value of the ith sample, which is the experimentally observed result;
represents the predicted value of the ith sample obtained from the regression model; and
denotes the mean of all true values.
3.2. Viscosity Prediction
To evaluate the effectiveness of the developed model in predicting lubricant viscosity, an ANN-based approach was applied to model and predict the kinematic viscosity at 40 °C and 100 °C, as well as the viscosity index. The model inputs were the optimal molecular descriptors selected by the GA, and the outputs were the corresponding viscosity and viscosity index values. The prediction results showed that the ANN model achieved high fitting accuracy, with R2 values of 0.8823, 0.9455, and 0.8425 for viscosity at 40 °C, viscosity at 100 °C, and viscosity index, respectively. The summarized R2 are shown in Figure 3f. The MAE values were controlled within 15, indicating good predictive performance and generalization capability of the model. Figure 3a–c show the regression relationships between the experimental and predicted values for the respective properties, where most data points are distributed close to the diagonal line, demonstrating that the ANN predictions for viscosity are highly consistent with the experimental results.
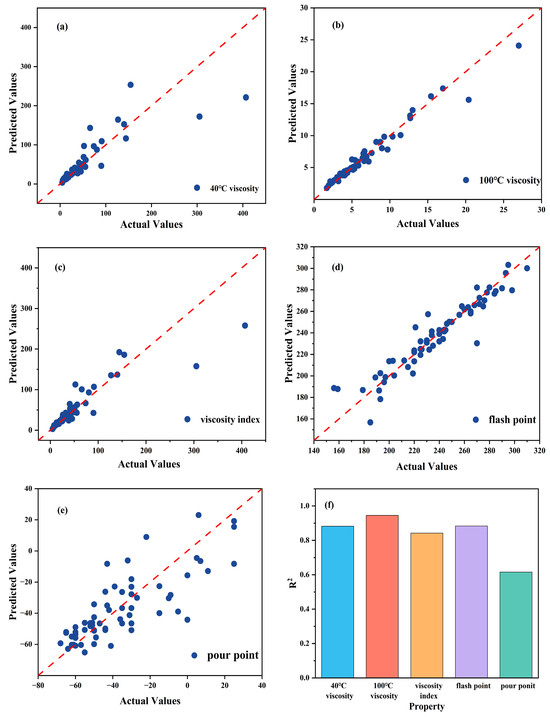
Figure 3.
(a), (b), (c), (d) and (e) show the scatter regression plots for 40 °C viscosity, 100 °C viscosity, viscosity index, flash point and pour point, respectively, while (f) presents the correlation coefficient comparison.
3.3. Pour Point and Flash Point Prediction
Based on the ANN model, the pour point and flash point of diester lubricants were modeled and predicted. The results show that the model performed well in predicting the flash point, achieving an R2 value of 0.8835 with a low MAE, indicating that the ANN effectively captured the relationship between molecular structure and flash point. The residual analysis further confirmed the model’s robustness, as most prediction errors were symmetrically distributed around zero, suggesting the absence of systematic bias. In contrast, the prediction accuracy for the pour point was relatively lower, with an R2 of only 0.6155 and a more dispersed error distribution. This discrepancy reflects the higher sensitivity of pour point to subtle intermolecular interactions that are not easily represented by static molecular descriptors. The comparison between experimental and predicted values is shown in Figure 3d,e, where the flash point predictions exhibit good overall agreement with the experimental data, while some noticeable deviations are observed in the pour point predictions, particularly for molecules with highly branched structures or flexible chains.
Further feature analysis revealed that the superior performance in flash point prediction primarily arises from its strong correlation with descriptors related to molecular weight, molecular polarity, and boiling point. These structural factors exhibit well-defined relationships with molecular thermal stability and volatility, leading to more consistent trends across the dataset. In contrast, the pour point is jointly influenced by more complex factors such as molecular packing, degree of branching, conformational freedom, and intermolecular hydrogen bonding, which are challenging to quantify using conventional 2D or 3D descriptors. Future improvements may involve incorporating molecular dynamics–derived descriptors or ensemble learning models to better capture these subtle structural effects and enhance the prediction of low-temperature properties.
4. Discussion
In this study, a QSPR–ANN model was constructed to predict key physicochemical properties of diester-based lubricants, including viscosity, viscosity index, flash point, and pour point. The results demonstrated that the ANN exhibited a remarkable capability to capture the nonlinear correlations between molecular structures and macroscopic properties. In particular, the prediction accuracy for viscosity and flash point was high (R2 > 0.88), indicating that the GA–ANN framework effectively extracted the key molecular features governing rheological behavior and thermal stability. This superior performance can be attributed to two main factors: (i) the GA-based feature selection significantly reduced descriptor redundancy while retaining variables strongly correlated with target properties, and (ii) the nonlinear activation functions and multilayer neuron architecture of the ANN enabled the model to learn complex, high-dimensional structure–property mapping relationships.
Compared with traditional linear QSPR models, the ANN model shows a stronger adaptability in handling nonlinear and multivariate correlations. Although linear models offer more direct interpretability, they often fail when parameter coupling or multicollinearity among descriptors becomes significant. The ANN, on the other hand, employs multilayer perception and backpropagation algorithms to automatically adjust weights and minimize fitting errors, while the use of dropout regularization helps to prevent overfitting, thereby improving model generalization. In addition, the global search capability of the GA overcomes the limitations of conventional feature selection methods—such as stepwise regression or principal component analysis—which tend to fall into local optima, ensuring that the selected descriptors represent global relevance to the target properties.
Nevertheless, several limitations remain in this study. First, the dataset size was relatively small, consisting of only 64 diester molecules, which may not fully represent the structural diversity of ester-based lubricants, thus affecting model generalization. Second, the chemical interpretability of some molecular descriptors has not been fully clarified, particularly those associated with molecular conformations and intermolecular interactions. Third, the relatively low accuracy in pour point prediction suggests that low-temperature flow behavior is influenced by complex molecular packing and crystallization processes, which cannot be fully captured by static molecular descriptors.
Future work can focus on several directions. Expanding the dataset to include more representative diesters and polyesters would improve the model’s robustness. Integrating molecular dynamics simulations and quantum chemical parameters could introduce dynamic descriptors that better reflect molecular mobility and energy distribution. Furthermore, the application of hybrid or ensemble learning models—such as Random Forest, XGBoost, or LSTM networks—may enhance model stability and interpretability. Sensitivity and contribution analyses could also be employed to identify key structural features affecting specific lubricant properties, providing a quantitative theoretical foundation for lubricant molecular design.
In summary, the GA–ANN model developed in this study offers an efficient and reliable approach for predicting lubricant properties. The framework demonstrates excellent scalability and provides a data-driven pathway for the molecular design and performance optimization of environmentally friendly lubricants.
5. Conclusions
In this study, a predictive model combining molecular descriptors, GA-based feature selection, and ANN was developed based on the QSPR approach to systematically investigate the viscosity, viscosity index, pour point, and flash point of diester lubricants. The model exhibited high accuracy in predicting viscosity and flash point, indicating that the ANN effectively captured the nonlinear relationships between molecular structure and physicochemical properties. The prediction of viscosity at 100 °C achieved an R2 of 0.9322, while the flash point prediction reached an R2 of 0.8835. However, the prediction accuracy for the pour point was relatively lower, suggesting that it is influenced by complex factors such as molecular packing and low-temperature crystallization, which are not fully captured by single-descriptor inputs or the ANN fitting capability.
The results of this study demonstrate that the integration of QSPR and ANN provides an effective strategy for lubricant property prediction and molecular design. Although diesters were selected as the main molecular class, the descriptors and machine-learning framework employed here are broadly applicable and can be extended to other ester-based lubricants, including polyol esters, complex esters, and bio-derived esters. Furthermore, the ability to predict multiple key performance indicators without experimental measurements provides practical guidance for base-oil selection, structural modification, and formulation development. This highlights the direct relevance of the model to real industrial applications. Future work may incorporate more complex molecular conformational features and hybrid or ensemble learning models to further enhance the predictive performance for lubricant properties.
Author Contributions
Conceptualization, writing—review, formal analysis, and editing, H.W. (Hanlu Wang); methodology and software, investigation, resources, data curation, writing—original draft preparation, Y.T.; validation, funding acquisition, H.W. (Hui Wang); project administration, P.P.; resources, Y.Z.; visualization, supervision, project administration, X.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Guangdong Provincial Key Laboratory of Advanced Green Lubricating Materials, grant number 2023B1212020002.
Data Availability Statement
The original contributions presented in this study are included in the article. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| PP | Pour Point |
| FP | Flash Point |
| VI | Viscosity Index |
| QSPR | Quantitative Structure–Property Relationship |
| ANN | Artificial Neural Network |
| DIPP | Diisopentyl Phthalate |
| GA | Genetic Algorithm |
| cSt | Centistokes |
References
- Flexa Ribeiro Filho, P.R.C.; dos Santos e Santos, L. The Influence of Unsaturation Modifications on the Tribological Characteristics of Bio-Based Lubricants Obtained from Vegetable Oils: A Review. J. Braz. Soc. Mech. Sci. Eng. 2025, 47, 216. [Google Scholar] [CrossRef]
- Shan, J.Y.; Song, Y.H.; Wen, P.; Dong, R.; Fan, M.J. Molecular Structure Insight into the Lubricating Performance of Heterocyclic Ester Oil and Their Film-Forming Interaction. Tribol. Int. 2026, 214, 111236. [Google Scholar] [CrossRef]
- Ferreira, E.N.; Arruda, T.B.M.G.; Rodrigues, F.E.A.; Moreira, D.R.; Chaves, P.O.B.; da Silva Rocha, W.; Silva, L.M.R.d.; Petzhold, C.L.; Ricardo, N.M.P.S. Pequi Oil Esters as an Alternative to Environmentally Friendly Lubricant for Industrial Purposes. ACS Sustain. Chem. Eng. 2022, 10, 1093–1102. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, X.; Ma, L.; Fan, M. Facilitating Investigations of Sustainable Ester Oils: Lubricating Performance, Phytotoxicity Indicators, and Their Structure–Activity Relationship. ACS Sustain. Chem. Eng. 2024, 12, 2790–2801. [Google Scholar] [CrossRef]
- Chen, T.; Yang, S.Z.; Ma, J.; Gao, H.S.; Xu, X.; Xie, F.; Cao, J.P.; Hu, J.Q. Real-Time Oxidation and Coking Behavior of Ester Aviation Lubricating Oil in Aircraft Engines. Tribol. Int. 2024, 192, 109240. [Google Scholar] [CrossRef]
- Nasser, R.M.; Sedeq, W.E.; Kolaiby, M.M. Comparative Analysis of Oleate Esters as Ester-Based Synthetic Lubricating Oils: A Physicochemical, Quantum, and Functional Study. Pet. Sci. Technol. 2025, 1–16. [Google Scholar] [CrossRef]
- Wang, Y.; Qiu, Q.; Zhang, P.; Gao, X.; Zhang, Z.; Huang, P. Correlation between Lubricating Oil Characteristic Parameters and Friction Characteristics. Coatings 2023, 13, 881. [Google Scholar] [CrossRef]
- Emel’ianov, V.V.; Krasnykh, E.L.; Sokolov, A.B. Synthetic Oils Based on Pentaerythritol Esters. Kinematic Viscosity. Fluid Phase Equilib. 2024, 581, 114074. [Google Scholar] [CrossRef]
- Zainal, N.A.; Zulkifli, N.W.M.; Gulzar, M.; Masjuki, H.H. A Review on the Chemistry, Production, and Technological Potential of Bio-Based Lubricants. Renew. Sustain. Energy Rev. 2018, 82, 80–102. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Yi, C.; Zhang, R.; Yang, S.; Liu, T.; Jia, D.; Yang, Q.; Peng, S. Evaluation of Antioxidant Properties and Molecular Design of Lubricant Antioxidants Based on Qspr Model. Lubricants 2024, 12, 3. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Yang, S.; Yi, C.; Liu, T.; Zhang, R.; Jia, D.; Peng, S.; Yang, Q. Prediction of Lubrication Performances of Vegetable Oils by Genetic Functional Approximation Algorithm. Lubricants 2024, 12, 226. [Google Scholar] [CrossRef]
- Nasab, S.G.; Semnani, A.; Marini, F.; Biancolillo, A. Prediction of Viscosity Index and Pour Point in Ester Lubricants Using Quantitative Structure-Property Relationship (Qspr). Chemom. Intell. Lab. Syst. 2018, 183, 59–78. [Google Scholar] [CrossRef]
- Zhou, R.; Ma, R.; Bao, L.; Cai, M.; Zhou, F.; Li, W.; Wang, X. “Lubrication Brain”―A Machine Learning Framework of Lubrication Oil Molecule Design. Tribol. Int. 2023, 183, 108381. [Google Scholar] [CrossRef]
- Liu, J.; Yi, C.; Zhang, Y.; Yang, S.; Liu, T.; Zhang, R.; Jia, D.; Peng, S.; Yang, Q. Structure-Activity Relationship Study of Anti-Wear Additives in Rapeseed Oil Based on Machine Learning and Logistic Regression. RSC Adv. 2024, 14, 8464–8480. [Google Scholar] [CrossRef]
- Jia, D.; Li, J.; Zhan, S.; Jin, Y.; Cheng, B.; Tu, J.; Li, Y.; Duan, H. Quantum Mechanics/Molecular Mechanics Studies on the Intrinsic Properties of Typical Ester Oil Molecules. Mater. Res. Express 2022, 9, 045102. [Google Scholar] [CrossRef]
- Wang, Y.; Song, Y.; Wang, H.; Ma, L.; Fan, M. Structure-Activity Relationship Study on the Phytotoxicity of Polyether Lubricants: Experimental Investigation and Theoretical Prediction Based on Machine Learning Models. J. Mol. Liq. 2025, 428, 127527. [Google Scholar] [CrossRef]
- Wang, H.; Wang, Y.; Wen, P.; Ma, L.; Fan, M.; Dong, R.; Zhang, C. Low-Viscosity Oligoether Esters (Oees) as High-Efficiency Lubricating Oils: Insight on Their Structure–Lubricity Relationship. Friction 2024, 12, 1133–1153. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, C.; Yu, X.; Li, Y. Evaluating Wear Volume of Oligoether Esters with an Interpretable Machine Learning Approach. Tribol. Lett. 2023, 71, 43. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. Iii. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef]
- Rdkit: Open-Source Cheminformatics. Available online: https://www.rdkit.org (accessed on 15 October 2025).
- Yu, T.; Yin, P.; Zhang, W.; Song, Y.; Zhang, X. A Compounding-Model Comprising Back Propagation Neural Network and Genetic Algorithm for Performance Prediction of Bio-Based Lubricant Blending with Functional Additives. Ind. Lubr. Tribol. 2020, 73, 246–252. [Google Scholar] [CrossRef]
- Dhanarajan, G.; Rangarajan, V.; Bandi, C.; Dixit, A.; Das, S.; Ale, K.; Sen, R. Biosurfactant-Biopolymer Driven Microbial Enhanced Oil Recovery (Meor) and Its Optimization by an Ann-Ga Hybrid Technique. J. Biotechnol. 2017, 256, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Zaidan, M.A.; Canova, F.F.; Laurson, L.; Foster, A.S. Mixture of Clustered Bayesian Neural Networks for Modeling Friction Processes at the Nanoscale. J. Chem. Theory Comput. 2016, 13, 3–8. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).