

Role of Perturbated Hemostasis in MASLD and Its Correlation with Adipokines

 ,
,  ,
, 

Abstract
1. Introduction
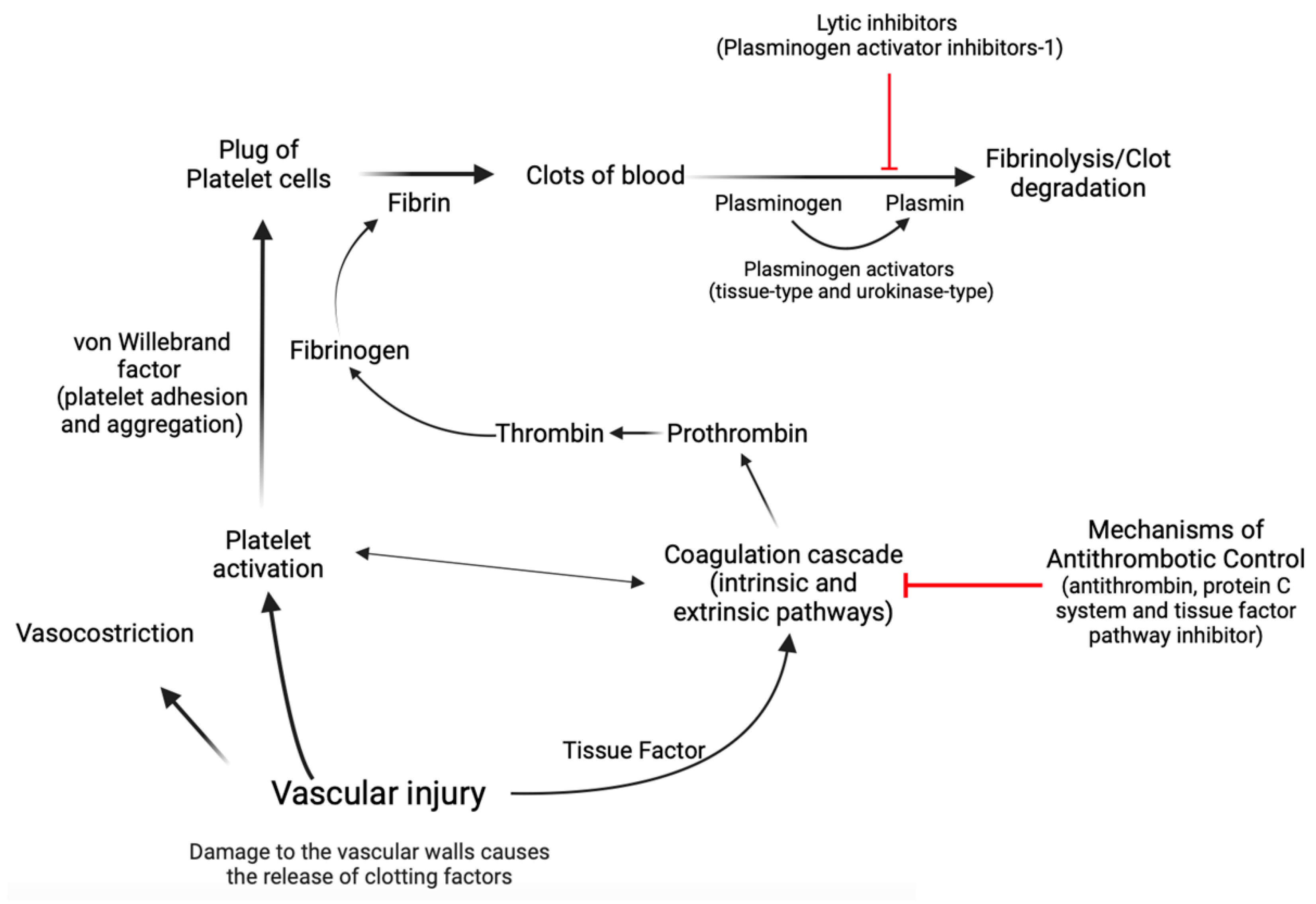
2. Abnormal Hemostasis in Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD)
2.1. Coagulation Dysfunctions
2.2. Dysfunction of Platelets
2.3. Endothelial Dysfunction
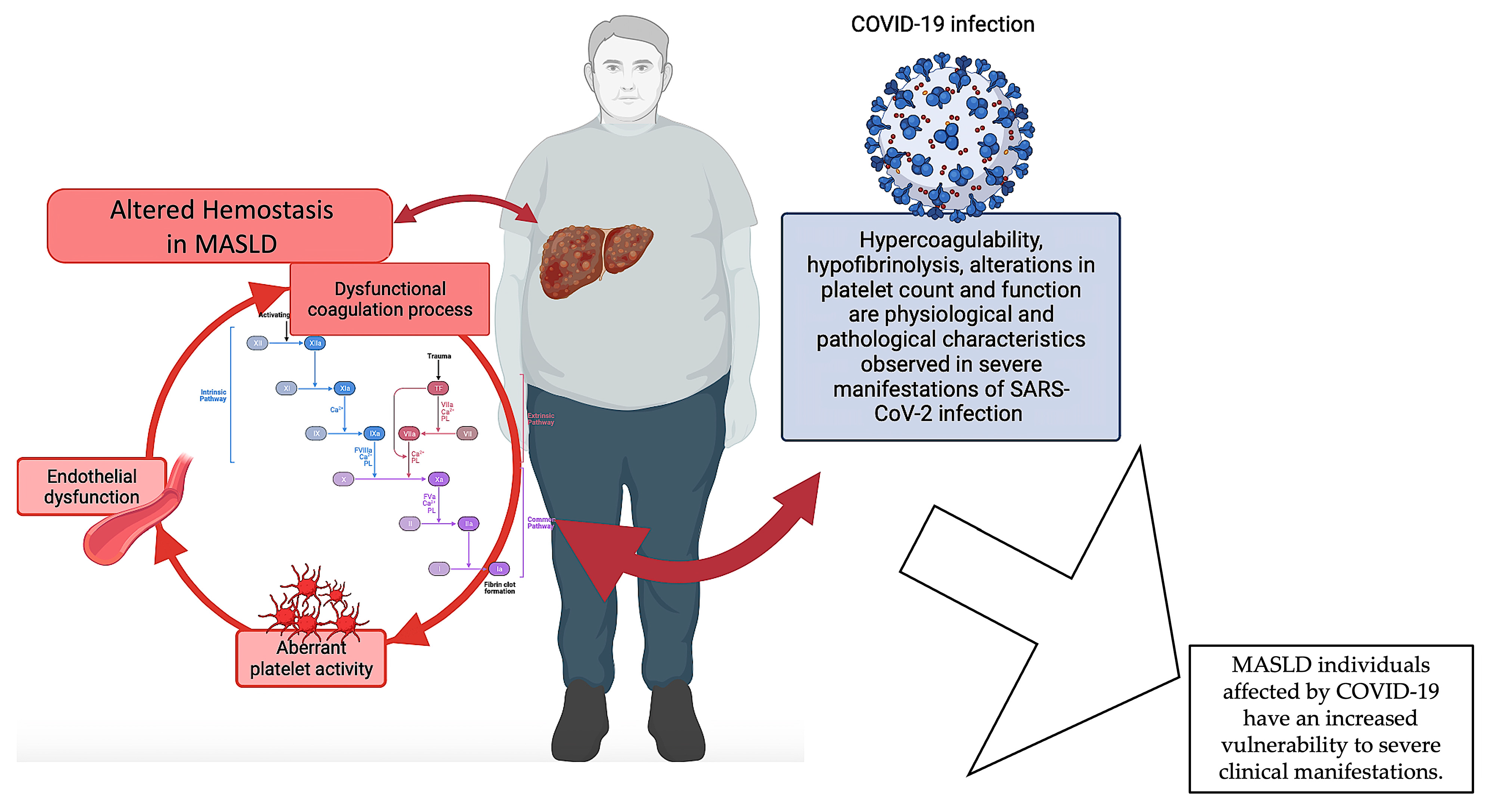
2.4. Hemostasis Dysfunction in MASLD Patients during COVID-19: Recent Learning from the Pandemic
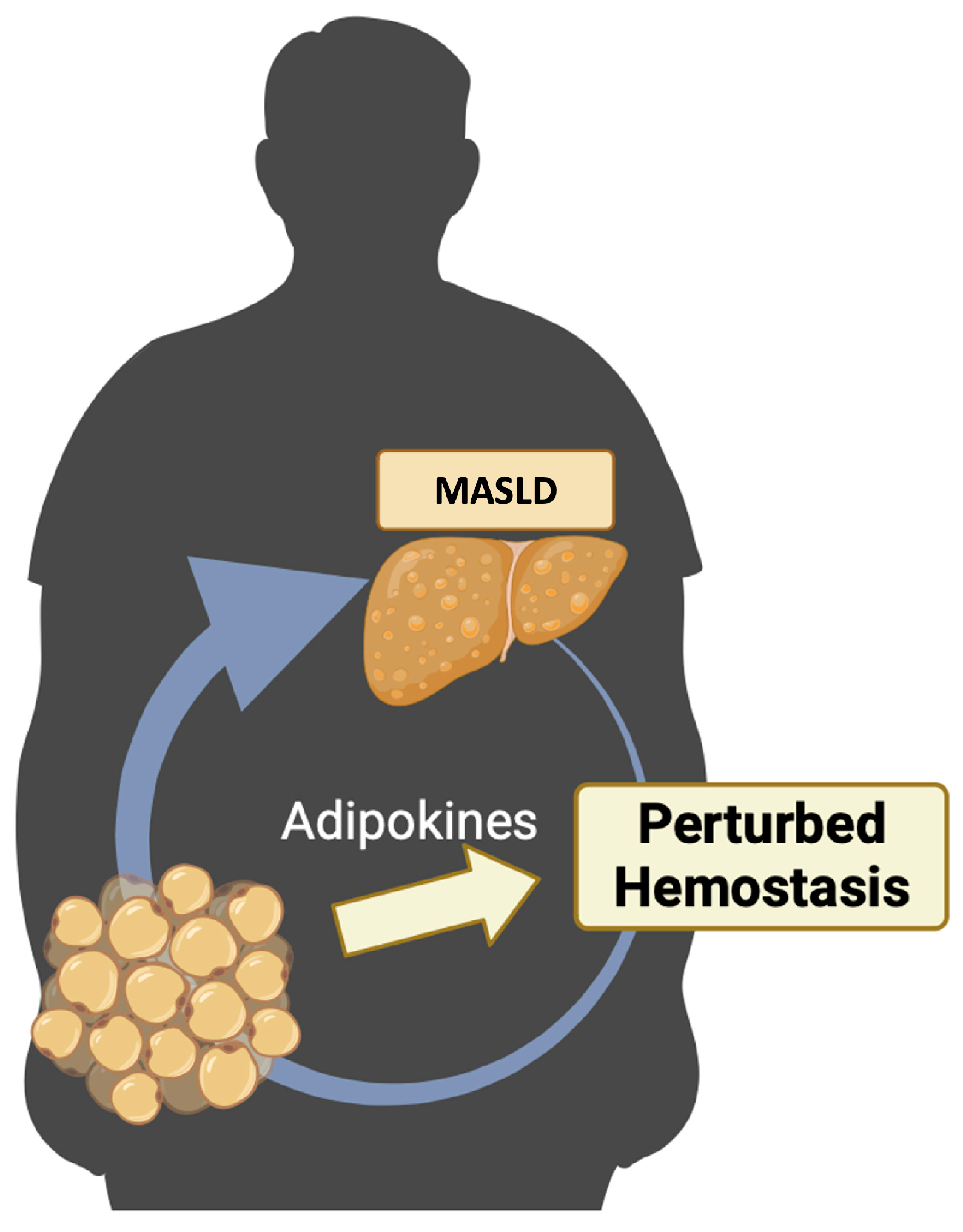
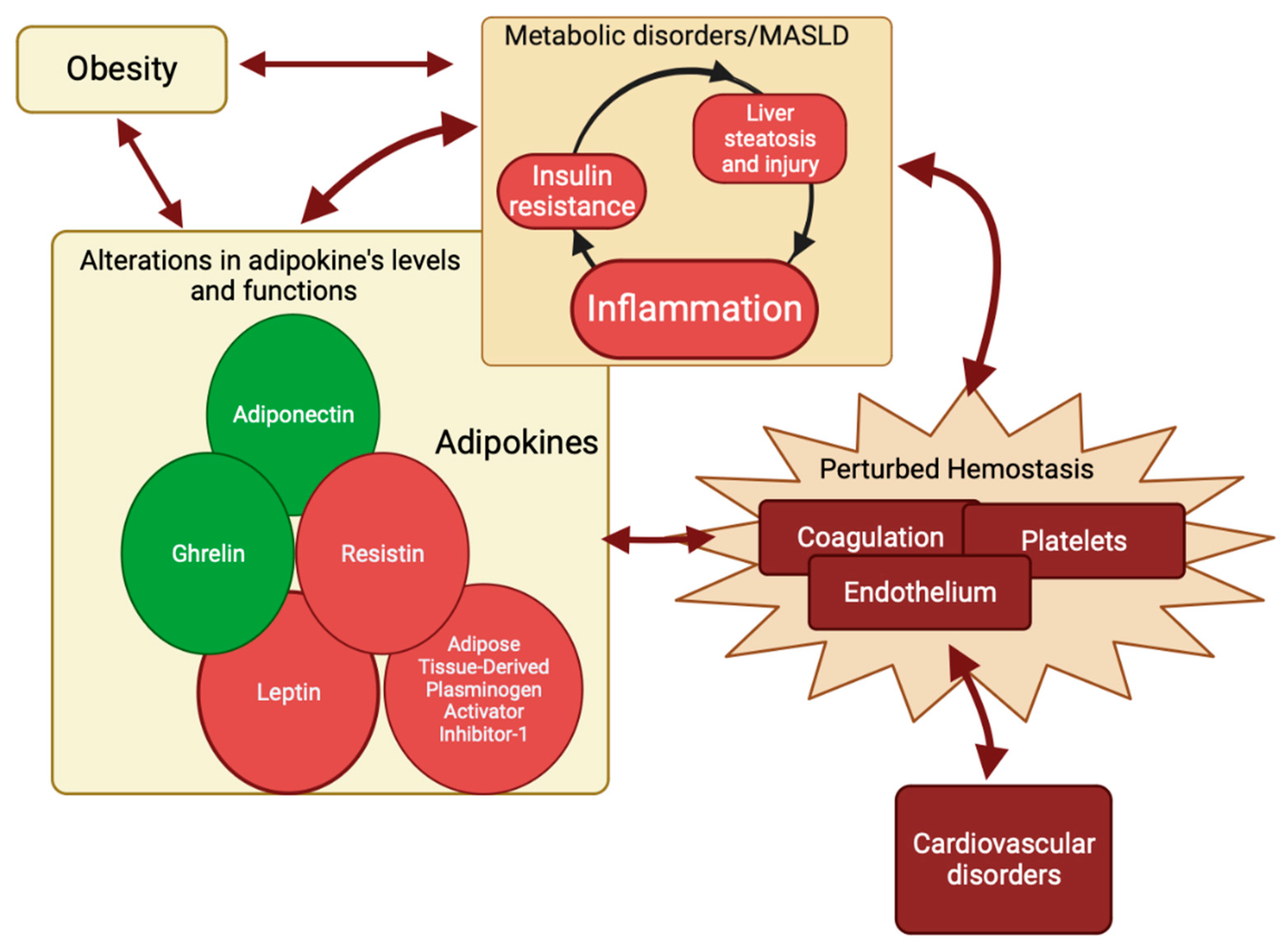
3. Adipokines in Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD) and Their Role in Perturbated Hemostasis
3.1. Adipose Tissue-Derived Plasminogen Activator Inhibitor-1
3.2. Adiponectin
3.3. Leptin
3.4. Resistin
3.5. Ghrelin

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adipokine Levels in Metabolic Syndrome/MASLD | Pathological Outcomes | Ref. |
|---|---|---|
| ↑ Adipose Tissue-Derived Plasminogen Activator Inhibitor-1 |
| [55,57,150,151] |
| ⇓ | ||
| [149,166,167,168] | |
| ↓ Adiponectin |
| [42,55,146,183] |
| [182] | |
| [172,179,180,181] | |
| ⇓ | ||
| [186,187,188,189,192,196] | |
| ↑ Leptin |
| [201,202] |
| [203,204] | |
| [206,207,209] | |
| [208,213,214,215,217,218,219,220,221] | |
| ⇓ | ||
| [105,192,205,208] | |
| ↑ Resistin |
| [237,239,240,245] |
| [241,242,243,252,253] | |
| [239,240,246] | |
| [239,246,247] | |
| ⇓ | ||
| [248,249,250,251] | |
| ↓ Ghrelin |
| [273,277,278,279,280,281,282,283] |
| [282,283,284,285] | |
| [261] | |
| [280,281,282,283,287,288,289,290,295] | |
| ⇓ | ||
| [273,274,275,276] |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A Multisociety Delphi Consensus Statement on New Fatty Liver Disease Nomenclature. Hepatology 2023, 78, 1966–1986. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.L.; Ng, C.H.; Huang, D.Q.; Chan, K.E.; Tan, D.J.; Lim, W.H.; Yang, J.D.; Tan, E.; Muthiah, M.D. Global Incidence and Prevalence of Nonalcoholic Fatty Liver Disease. Clin. Mol. Hepatol. 2023, 29, S32–S42. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Yilmaz, Y.; Yu, M.-L.; Wai-Sun Wong, V.; Fernandez, M.C.; Isakov, V.A.; Duseja, A.K.; Mendez-Sanchez, N.; Eguchi, Y.; Bugianesi, E.; et al. Clinical and Patient-Reported Outcomes From Patients with Nonalcoholic Fatty Liver Disease across the World: Data From the Global Non-Alcoholic Steatohepatitis (NASH)/Non-Alcoholic Fatty Liver Disease (NAFLD) Registry. Clin. Gastroenterol. Hepatol. 2022, 20, 2296–2306.e6. [Google Scholar] [CrossRef] [PubMed]
- Han, S.K.; Baik, S.K.; Kim, M.Y. Non-Alcoholic Fatty Liver Disease: Definition and Subtypes. Clin. Mol. Hepatol. 2023, 29, S5–S16. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Matos, A.F.; Silva Júnior, W.S.; Valerio, C.M. NAFLD as a Continuum: From Obesity to Metabolic Syndrome and Diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef]
- Pouwels, S.; Sakran, N.; Graham, Y.; Leal, A.; Pintar, T.; Yang, W.; Kassir, R.; Singhal, R.; Mahawar, K.; Ramnarain, D. Non-Alcoholic Fatty Liver Disease (NAFLD): A Review of Pathophysiology, Clinical Management and Effects of Weight Loss. BMC Endocr. Disord. 2022, 22, 63. [Google Scholar] [CrossRef]
- Pezzino, S.; Sofia, M.; Faletra, G.; Mazzone, C.; Litrico, G.; La Greca, G.; Latteri, S. Gut-Liver Axis and Non-Alcoholic Fatty Liver Disease: A Vicious Circle of Dysfunctions Orchestrated by the Gut Microbiome. Biology 2022, 11, 1622. [Google Scholar] [CrossRef]
- Pezzino, S.; Sofia, M.; Greco, L.P.; Litrico, G.; Filippello, G.; Sarvà, I.; Greca, G.; Latteri, S. Microbiome Dysbiosis: A Pathological Mechanism at the Intersection of Obesity and Glaucoma. Int. J. Mol. Sci. 2023, 24, 1166. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A Multisystem Disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef]
- Cholongitas, E.; Pavlopoulou, I.; Papatheodoridi, M.; Markakis, G.E.; Bouras, E.; Haidich, A.-B.; Papatheodoridis, G. Epidemiology of Nonalcoholic Fatty Liver Disease in Europe: A Systematic Review and Meta-Analysis. Ann. Gastroenterol. 2021, 34, 404–414. [Google Scholar] [CrossRef]
- Bellentani, S. The Epidemiology of Non-Alcoholic Fatty Liver Disease. Liver Int. 2017, 37 (Suppl. 1), 81–84. [Google Scholar] [CrossRef] [PubMed]
- Marjot, T.; Moolla, A.; Cobbold, J.F.; Hodson, L.; Tomlinson, J.W. Nonalcoholic Fatty Liver Disease in Adults: Current Concepts in Etiology, Outcomes, and Management. Endocr. Rev. 2020, 41, 66–117. [Google Scholar] [CrossRef] [PubMed]
- Divella, R.; Mazzocca, A.; Daniele, A.; Sabbà, C.; Paradiso, A. Obesity, Nonalcoholic Fatty Liver Disease and Adipocytokines Network in Promotion of Cancer. Int. J. Biol. Sci. 2019, 15, 610–616. [Google Scholar] [CrossRef] [PubMed]
- Ogresta, D.; Mrzljak, A.; Cigrovski Berkovic, M.; Bilic-Curcic, I.; Stojsavljevic-Shapeski, S.; Virovic-Jukic, L. Coagulation and Endothelial Dysfunction Associated with NAFLD: Current Status and Therapeutic Implications. J. Clin. Transl. Hepatol. 2022, 10, 339–355. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Byrne, C.D. Diagnosis and Management of Nonalcoholic Fatty Liver Disease and Its Hemostatic/Thrombotic and Vascular Complications. Semin. Thromb. Hemost. 2013, 39, 214–228. [Google Scholar] [CrossRef]
- Tripodi, A.; Lombardi, R.; Primignani, M.; La Mura, V.; Peyvandi, F.; Fracanzani, A.L. Hypercoagulability in Patients with Non-Alcoholic Fatty Liver Disease (NAFLD): Causes and Consequences. Biomedicines 2022, 10, 249. [Google Scholar] [CrossRef]
- Amitrano, L.; Guardascione, M.A.; Brancaccio, V.; Balzano, A. Coagulation Disorders in Liver Disease. Semin. Liver Dis. 2002, 22, 83–96. [Google Scholar] [CrossRef]
- Virović-Jukić, L.; Stojsavljević-Shapeski, S.; Forgač, J.; Kukla, M.; Mikolašević, I. Non-Alcoholic Fatty Liver Disease—A Procoagulant Condition? Croat. Med. J. 2021, 62, 25–33. [Google Scholar] [CrossRef]
- Santos, R.D.; Valenti, L.; Romeo, S. Does Nonalcoholic Fatty Liver Disease Cause Cardiovascular Disease? Current Knowledge and Gaps. Atherosclerosis 2019, 282, 110–120. [Google Scholar] [CrossRef]
- Basili, S.; Raparelli, V.; Violi, F. The Coagulopathy of Chronic Liver Disease: Is There a Causal Relationship with Bleeding? Yes. Eur. J. Intern. Med. 2010, 21, 62–64. [Google Scholar] [CrossRef]
- Li, A.A.; Ahmed, A.; Kim, D. Extrahepatic Manifestations of Nonalcoholic Fatty Liver Disease. Gut Liver 2020, 14, 168–178. [Google Scholar] [CrossRef] [PubMed]
- Azzu, V.; Vacca, M.; Virtue, S.; Allison, M.; Vidal-Puig, A. Adipose Tissue-Liver Cross Talk in the Control of Whole-Body Metabolism: Implications in Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 158, 1899–1912. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Rao, H.; Liu, F.; Wei, L.; Li, H.; Wu, C. Recent Advances in Adipose Tissue Dysfunction and Its Role in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 3300. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Cinti, S. The Endocrine Adipose Organ. Rev. Endocr. Metab. Disord. 2022, 23, 1–4. [Google Scholar] [CrossRef]
- Trayhurn, P.; Beattie, J.H. Physiological Role of Adipose Tissue: White Adipose Tissue as an Endocrine and Secretory Organ. Proc. Nutr. Soc. 2001, 60, 329–339. [Google Scholar] [CrossRef]
- Guerre-Millo, M. Adipose Tissue Hormones. J. Endocrinol. Investig. 2002, 25, 855–861. [Google Scholar] [CrossRef]
- Manne, V.; Handa, P.; Kowdley, K.V. Pathophysiology of Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis. Clin. Liver Dis. 2018, 22, 23–37. [Google Scholar] [CrossRef]
- Milić, S.; Lulić, D.; Štimac, D. Non-Alcoholic Fatty Liver Disease and Obesity: Biochemical, Metabolic and Clinical Presentations. World J. Gastroenterol. 2014, 20, 9330–9337. [Google Scholar] [CrossRef]
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Martín-Rodríguez, A.; Martínez-Guardado, I.; Navarro-Jiménez, E.; Laborde-Cárdenas, C.C.; Tornero-Aguilera, J.F. The Role of Adipokines in Health and Disease. Biomedicines 2023, 11, 1290. [Google Scholar] [CrossRef]
- Blüher, M. Clinical Relevance of Adipokines. Diabetes Metab. J. 2012, 36, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Francisco, V.; Sanz, M.J.; Real, J.T.; Marques, P.; Capuozzo, M.; Ait Eldjoudi, D.; Gualillo, O. Adipokines in Non-Alcoholic Fatty Liver Disease: Are We on the Road toward New Biomarkers and Therapeutic Targets? Biology 2022, 11, 1237. [Google Scholar] [CrossRef] [PubMed]
- Dalbeni, A.; Castelli, M.; Zoncapè, M.; Minuz, P.; Sacerdoti, D. Platelets in Non-Alcoholic Fatty Liver Disease. Front. Pharmacol. 2022, 13, 842636. [Google Scholar] [CrossRef] [PubMed]
- Adolph, T.E.; Grander, C.; Grabherr, F.; Tilg, H. Adipokines and Non-Alcoholic Fatty Liver Disease: Multiple Interactions. Int. J. Mol. Sci. 2017, 18, 1649. [Google Scholar] [CrossRef]
- Tilg, H.; Hotamisligil, G.S. Nonalcoholic Fatty Liver Disease: Cytokine-Adipokine Interplay and Regulation of Insulin Resistance. Gastroenterology 2006, 131, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Insulin Resistance, Inflammation, and Non-Alcoholic Fatty Liver Disease. Trends Endocrinol. Metab. 2008, 19, 371–379. [Google Scholar] [CrossRef]
- Højlund, K. Metabolism and Insulin Signaling in Common Metabolic Disorders and Inherited Insulin Resistance. Dan. Med. J. 2014, 61, B4890. [Google Scholar]
- Fujii, H.; Kawada, N.; Japan Study Group of NAFLD (JSG-NAFLD). The Role of Insulin Resistance and Diabetes in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2020, 21, 3863. [Google Scholar] [CrossRef]
- Armandi, A.; Rosso, C.; Caviglia, G.P.; Bugianesi, E. Insulin Resistance across the Spectrum of Nonalcoholic Fatty Liver Disease. Metabolites 2021, 11, 155. [Google Scholar] [CrossRef]
- Boutari, C.; Tziomalos, K.; Athyros, V.G. The Adipokines in the Pathogenesis and Treatment of Nonalcoholic Fatty Liver Disease. Hippokratia 2016, 20, 259. [Google Scholar]
- Lau, D.C.W.; Dhillon, B.; Yan, H.; Szmitko, P.E.; Verma, S. Adipokines: Molecular Links between Obesity and Atheroslcerosis. Am. J. Physiol.—Heart Circ. Physiol. 2005, 288, H2031–H2041. [Google Scholar] [CrossRef] [PubMed]
- Recinella, L.; Orlando, G.; Ferrante, C.; Chiavaroli, A.; Brunetti, L.; Leone, S. Adipokines: New Potential Therapeutic Target for Obesity and Metabolic, Rheumatic, and Cardiovascular Diseases. Front. Physiol. 2020, 11, 578966. [Google Scholar] [CrossRef] [PubMed]
- Horigome, H.; Katayama, Y.; Yoshinaga, M.; Kato, Y.; Takahashi, H.; Sumazaki, R. Significant Associations Among Hemostatic Parameters, Adipokines, and Components of the Metabolic Syndrome in Japanese Preschool Children. Clin. Appl. Thromb. Hemost. 2012, 18, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Faber, D.R.; De Groot, P.G.; Visseren, F.L.J. Role of Adipose Tissue in Haemostasis, Coagulation and Fibrinolysis. Obes. Rev. 2009, 10, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Kwon, H.; Pessin, J.E. Adipokines Mediate Inflammation and Insulin Resistance. Front. Endocrinol. 2013, 4, 71. [Google Scholar] [CrossRef]
- Antuna-Puente, B.; Feve, B.; Fellahi, S.; Bastard, J.-P. Adipokines: The Missing Link between Insulin Resistance and Obesity. Diabetes Metab. 2008, 34, 2–11. [Google Scholar] [CrossRef]
- Krüger-Genge, A.; Blocki, A.; Franke, R.-P.; Jung, F. Vascular Endothelial Cell Biology: An Update. Int. J. Mol. Sci. 2019, 20, 4411. [Google Scholar] [CrossRef]
- Nasiri-Ansari, N.; Androutsakos, T.; Flessa, C.-M.; Kyrou, I.; Siasos, G.; Randeva, H.S.; Kassi, E.; Papavassiliou, A.G. Endothelial Cell Dysfunction and Nonalcoholic Fatty Liver Disease (NAFLD): A Concise Review. Cells 2022, 11, 2511. [Google Scholar] [CrossRef]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial Cell Control of Thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef]
- LaPelusa, A.; Dave, H.D. Physiology, Hemostasis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Potze, W.; Siddiqui, M.S.; Sanyal, A.J. Vascular Disease in Patients with Nonalcoholic Fatty Liver Disease. Semin. Thromb. Hemost. 2015, 41, 488–493. [Google Scholar] [CrossRef]
- Seré, K.M.; Hackeng, T.M. Basic Mechanisms of Hemostasis. Semin. Vasc. Med. 2003, 3, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Sierra, C.; Moreno, M.; García-Ruiz, J.C. The Physiology of Hemostasis. Blood Coagul. Fibrinolysis 2022, 33, S1–S2. [Google Scholar] [CrossRef] [PubMed]
- Sillen, M.; Declerck, P.J. A Narrative Review on Plasminogen Activator Inhibitor-1 and Its (Patho)Physiological Role: To Target or Not to Target? Int. J. Mol. Sci. 2021, 22, 2721. [Google Scholar] [CrossRef] [PubMed]
- Cesari, M.; Pahor, M.; Incalzi, R.A. Plasminogen Activator Inhibitor-1 (PAI-1): A Key Factor Linking Fibrinolysis and Age-Related Subclinical and Clinical Conditions. Cardiovasc. Ther. 2010, 28, e72–e91. [Google Scholar] [CrossRef] [PubMed]
- Keragala, C.B.; Medcalf, R.L. Plasminogen: An Enigmatic Zymogen. Blood 2021, 137, 2881–2889. [Google Scholar] [CrossRef]
- Levine, J.A.; Oleaga, C.; Eren, M.; Amaral, A.P.; Shang, M.; Lux, E.; Khan, S.S.; Shah, S.J.; Omura, Y.; Pamir, N.; et al. Role of PAI-1 in Hepatic Steatosis and Dyslipidemia. Sci. Rep. 2021, 11, 430. [Google Scholar] [CrossRef]
- Iglesias Morcillo, M.; Freuer, D.; Peters, A.; Heier, M.; Teupser, D.; Meisinger, C.; Linseisen, J. Association between Fatty Liver Index and Blood Coagulation Markers: A Population-Based Study. Lipids Health Dis. 2023, 22, 83. [Google Scholar] [CrossRef]
- Robea, M.A.; Balmus, I.-M.; Girleanu, I.; Huiban, L.; Muzica, C.; Ciobica, A.; Stanciu, C.; Cimpoesu, C.D.; Trifan, A. Coagulation Dysfunctions in Non-Alcoholic Fatty Liver Disease—Oxidative Stress and Inflammation Relevance. Medicina 2023, 59, 1614. [Google Scholar] [CrossRef]
- Peck-Radosavljevic, M. Review Article: Coagulation Disorders in Chronic Liver Disease. Aliment. Pharmacol. Ther. 2007, 26 (Suppl. 1), 21–28. [Google Scholar] [CrossRef]
- Ciavarella, A.; Gnocchi, D.; Custodero, C.; Lenato, G.M.; Fiore, G.; Sabbà, C.; Mazzocca, A. Translational Insight into Prothrombotic State and Hypercoagulation in Nonalcoholic Fatty Liver Disease. Thromb. Res. 2021, 198, 139–150. [Google Scholar] [CrossRef]
- Tripodi, A.; Fracanzani, A.L.; Primignani, M.; Chantarangkul, V.; Clerici, M.; Mannucci, P.M.; Peyvandi, F.; Bertelli, C.; Valenti, L.; Fargion, S. Procoagulant Imbalance in Patients with Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2014, 61, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Tripodi, A.; La Mura, V.; Pelusi, S.; Bianco, C.; Scalambrino, E.; Margarita, S.; Malvestiti, F.; Ronzoni, L.; Clerici, M.; et al. Clinical and Genetic Determinants of the Fatty Liver-Coagulation Balance Interplay in Individuals with Metabolic Dysfunction. JHEP Rep. 2022, 4, 100598. [Google Scholar] [CrossRef] [PubMed]
- Pădureanu, V.; Dop, D.; Drăgoescu, A.N.; Pădureanu, R.; Mușetescu, A.E.; Nedelcu, L. Non-Alcoholic Fatty Liver Disease and Hematologic Manifestations (Review). Exp. Ther. Med. 2021, 22, 1355. [Google Scholar] [CrossRef] [PubMed]
- Rangaswamy, C.; Mailer, R.K.; Englert, H.; Konrath, S.; Renné, T. The Contact System in Liver Injury. Semin. Immunopathol. 2021, 43, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Kopec, A.K.; Joshi, N.; Luyendyk, J.P. Role of Hemostatic Factors in Hepatic Injury and Disease: Animal Models de-Liver. J. Thromb. Haemost. 2016, 14, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Groeneveld, D.; Poole, L.G.; Luyendyk, J.P. Targeting von Willebrand Factor in Liver Diseases: A Novel Therapeutic Strategy? J. Thromb. Haemost. 2021, 19, 1390–1408. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, S.; Carlini, L.E. Coagulation Homeostasis in Liver Disease. Clin. Liver Dis. 2020, 16, 137–141. [Google Scholar] [CrossRef]
- Lv, Y.; Liu, N.; Li, Y.; Wu, J.; Zheng, J.; Li, X.; Zeng, M. Coagulation Dysfunction in Patients with Liver Cirrhosis and Splenomegaly and Its Countermeasures: A Retrospective Study of 1522 Patients. Dis. Markers 2023, 2023, 5560560. [Google Scholar] [CrossRef]
- Pipitone, R.M.; Ciccioli, C.; Infantino, G.; La Mantia, C.; Parisi, S.; Tulone, A.; Pennisi, G.; Grimaudo, S.; Petta, S. MAFLD: A Multisystem Disease. Ther. Adv. Endocrinol. Metab. 2023, 14, 20420188221145549. [Google Scholar] [CrossRef]
- Kaya, E.; Yilmaz, Y. Metabolic-Associated Fatty Liver Disease (MAFLD): A Multi-Systemic Disease Beyond the Liver. J. Clin. Transl. Hepatol. 2022, 10, 329. [Google Scholar] [CrossRef]
- Gidaro, A.; Manetti, R.; Delitala, A.P.; Salvi, E.; Bergamaschini, L.; Vidili, G.; Castelli, R. Prothrombotic and Inflammatory Markers in Elderly Patients with Non-Alcoholic Hepatic Liver Disease before and after Weight Loss: A Pilot Study. J. Clin. Med. 2021, 10, 4906. [Google Scholar] [CrossRef] [PubMed]
- Swan, D.; Lisman, T.; Tripodi, A.; Thachil, J. The Prothrombotic Tendency of Metabolic-Associated Fatty Liver Disease. J. Thromb. Haemost. 2023, 21, 3045–3055. [Google Scholar] [CrossRef] [PubMed]
- Sillen, M.; Declerck, P.J. Targeting PAI-1 in Cardiovascular Disease: Structural Insights Into PAI-1 Functionality and Inhibition. Front. Cardiovasc. Med. 2020, 7, 622473. [Google Scholar] [CrossRef] [PubMed]
- Thuy, S.; Ladurner, R.; Volynets, V.; Wagner, S.; Strahl, S.; Königsrainer, A.; Maier, K.-P.; Bischoff, S.C.; Bergheim, I. Nonalcoholic Fatty Liver Disease in Humans Is Associated with Increased Plasma Endotoxin and Plasminogen Activator Inhibitor 1 Concentrations and with Fructose Intake1. J. Nutr. 2008, 138, 1452–1455. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, S.S.; Siddiqui, K. Plasminogen Activator Inhibitor-1 Mediate Downregulation of Adiponectin in Type 2 Diabetes Patients with Metabolic Syndrome. Cytokine X 2022, 4, 100064. [Google Scholar] [CrossRef] [PubMed]
- Alsharoh, H.; Ismaiel, A.; Leucuta, D.-C.; Popa, S.-L.; Dumitrascu, D.L. Plasminogen Activator Inhibitor-1 Levels in Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. J. Gastrointestin Liver Dis. 2022, 31, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Rivas, G.; Hummer-Bair, B.; Bezinover, D.; Kadry, Z.; Stine, J. Plasminogen Activator Inhibitor Is Significantly Elevated in Liver Transplant Recipients with Decompensated NASH Cirrhosis. BMJ Open Gastroenterol. 2021, 8, e000683. [Google Scholar] [CrossRef] [PubMed]
- Wanless, I.R.; Wong, F.; Blendis, L.M.; Greig, P.; Heathcote, E.J.; Levy, G. Hepatic and Portal Vein Thrombosis in Cirrhosis: Possible Role in Development of Parenchymal Extinction and Portal Hypertension. Hepatology 1995, 21, 1238–1247. [Google Scholar] [CrossRef]
- Li, M.; Wang, H.; Zhang, X.-J.; Cai, J.; Li, H. NAFLD: An Emerging Causal Factor for Cardiovascular Disease. Physiology 2023, 38, 255–265. [Google Scholar] [CrossRef]
- Niederseer, D.; Wernly, B.; Aigner, E.; Stickel, F.; Datz, C. NAFLD and Cardiovascular Diseases: Epidemiological, Mechanistic and Therapeutic Considerations. J. Clin. Med. 2021, 10, 467. [Google Scholar] [CrossRef]
- Lin, H.; Bai, Z.; Guo, X.; Qi, X. Association between Nonalcoholic Fatty Liver Disease and Portal Vein Thrombosis: A Systematic Review and Meta-Analysis. Eur. J. Gastroenterol. Hepatol. 2020, 32, 1405–1406. [Google Scholar] [CrossRef] [PubMed]
- Balta, S. Atherosclerosis and Non-Alcoholic Fatty Liver Disease. Angiology 2022, 73, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Finney, A.C.; Das, S.; Kumar, D.; McKinney, M.P.; Cai, B.; Yurdagul, A.; Rom, O. The Interplay between Nonalcoholic Fatty Liver Disease and Atherosclerotic Cardiovascular Disease. Front. Cardiovasc. Med. 2023, 10, 1116861. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.-Y.; Jung, D.-H.; Shim, J.-Y.; Lee, H.-R. The Association between Non-Alcoholic Hepatic Steatosis and Mean Platelet Volume in an Obese Korean Population. Platelets 2011, 22, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Madan, S.A.; John, F.; Pitchumoni, C.S. Nonalcoholic Fatty Liver Disease and Mean Platelet Volume: A Systemic Review and Meta-Analysis. J. Clin. Gastroenterol. 2016, 50, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Saremi, Z.; Rastgoo, M.; Mohammadifard, M.; Bijari, B.; Akbari, E. Comparison of Platelet Number and Function between Nonalcoholic Fatty Liver Disease and Normal Individuals. J. Res. Med. Sci. 2017, 22, 75. [Google Scholar] [CrossRef]
- Yin, H.; Shi, A.; Wu, J. Platelet-Activating Factor Promotes the Development of Non-Alcoholic Fatty Liver Disease. Diabetes Metab. Syndr. Obes. 2022, 15, 2003–2030. [Google Scholar] [CrossRef]
- Carpino, G.; Del Ben, M.; Pastori, D.; Carnevale, R.; Baratta, F.; Overi, D.; Francis, H.; Cardinale, V.; Onori, P.; Safarikia, S.; et al. Increased Liver Localization of Lipopolysaccharides in Human and Experimental NAFLD. Hepatology 2020, 72, 470–485. [Google Scholar] [CrossRef]
- Yoneda, M.; Fujii, H.; Sumida, Y.; Hyogo, H.; Itoh, Y.; Ono, M.; Eguchi, Y.; Suzuki, Y.; Aoki, N.; Kanemasa, K.; et al. Platelet Count for Predicting Fibrosis in Nonalcoholic Fatty Liver Disease. J. Gastroenterol. 2011, 46, 1300–1306. [Google Scholar] [CrossRef]
- Fujii, H.; Fujii, M.; Iwaki, M.; Hayashi, H.; Toyoda, H.; Oeda, S.; Hyogo, H.; Kawanaka, M.; Morishita, A.; Munekage, K.; et al. Multicenter, Retrospective, Cohort Study Shows Platelet Counts Predict Hepatocellular Carcinoma Development in Patients with Nonalcoholic Fatty Liver Disease. Hepatol. Res. 2023, 53, 391–400. [Google Scholar] [CrossRef]
- Wolberg, A.S. Fibrinogen and Fibrin: Synthesis, Structure, and Function in Health and Disease. J. Thromb. Haemost. 2023, 21, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Brass, L.F. Thrombin and Platelet Activation. Chest 2003, 124, 18S–25S. [Google Scholar] [CrossRef] [PubMed]
- Lisman, T.; Weeterings, C.; de Groot, P.G. Platelet Aggregation: Involvement of Thrombin and Fibrin(Ogen). Front. Biosci. 2005, 10, 2504–2517. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, O.; Feldman, D.M.; Diakow, M.; Sigal, S.H. The Pathophysiology of Thrombocytopenia in Chronic Liver Disease. Hepat. Med. 2016, 8, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Mussbacher, M.; Brunnthaler, L.; Panhuber, A.; Starlinger, P.; Assinger, A. Till Death Do Us Part—The Multifaceted Role of Platelets in Liver Diseases. Int. J. Mol. Sci. 2021, 22, 3113. [Google Scholar] [CrossRef] [PubMed]
- Korbelius, M.; Kuentzel, K.B.; Bradić, I.; Vujić, N.; Kratky, D. Recent Insights into Lysosomal Acid Lipase Deficiency. Trends Mol. Med. 2023, 29, 425–438. [Google Scholar] [CrossRef]
- Ferri, F.; Mischitelli, M.; Tozzi, G.; Messina, E.; Mignini, I.; Mazzuca, S.; Pellone, M.; Parisse, S.; Marrapodi, R.; Visentini, M.; et al. Reduced Lysosomal Acid Lipase Activity in Blood and Platelets Is Associated with Nonalcoholic Fatty Liver Disease. Clin. Transl. Gastroenterol. 2020, 11, e00116. [Google Scholar] [CrossRef]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet GPIbα Is a Mediator and Potential Interventional Target for NASH and Subsequent Liver Cancer. Nat. Med. 2019, 25, 641–655. [Google Scholar] [CrossRef]
- Makhoul, S.; Trabold, K.; Gambaryan, S.; Tenzer, S.; Pillitteri, D.; Walter, U.; Jurk, K. cAMP- and cGMP-Elevating Agents Inhibit GPIbα-Mediated Aggregation but Not GPIbα-Stimulated Syk Activation in Human Platelets. Cell Commun. Signal. 2019, 17, 122. [Google Scholar] [CrossRef]
- Spinosa, M.; Stine, J.G. Nonalcoholic Fatty Liver Disease-Evidence for a Thrombophilic State? Curr. Pharm. Des. 2020, 26, 1036–1044. [Google Scholar] [CrossRef]
- Alberelli, M.A.; Miele, L.; Racco, S.; Biolato, M.; Marrone, G.; Cefalo, C.; Landolfi, R.; Gasbarrini, A.; Candia, E.D.; Grieco, A. Cardiovascular Risk and Inflammation in Nonalcoholic Fatty Liver Disease: The Upregulation of Inflammatory Platelet Transcripts Suggest a Role for Platelets in the “Inflammatory Network” of NAFLD. Dig. Liver Dis. 2015, 47, e222. [Google Scholar] [CrossRef]
- Fontes-Cal, T.C.M.; Mattos, R.T.; Medeiros, N.I.; Pinto, B.F.; Belchior-Bezerra, M.; Roque-Souza, B.; Dutra, W.O.; Ferrari, T.C.A.; Vidigal, P.V.T.; Faria, L.C.; et al. Crosstalk Between Plasma Cytokines, Inflammation, and Liver Damage as a New Strategy to Monitoring NAFLD Progression. Front. Immunol. 2021, 12, 708959. [Google Scholar] [CrossRef] [PubMed]
- Benova, A.; Tencerova, M. Obesity-Induced Changes in Bone Marrow Homeostasis. Front. Endocrinol. 2020, 11, 294. [Google Scholar] [CrossRef]
- Madan, S.A.; John, F.; Pyrsopoulos, N.; Pitchumoni, C.S. Nonalcoholic Fatty Liver Disease and Carotid Artery Atherosclerosis in Children and Adults: A Meta-Analysis. Eur. J. Gastroenterol. Hepatol. 2015, 27, 1237–1248. [Google Scholar] [CrossRef] [PubMed]
- Khoshbaten, M.; Maleki, S.H.; Hadad, S.; Baral, A.; Rocha, A.V.; Poudel, L.; Abdshah, A. Association of Nonalcoholic Fatty Liver Disease and Carotid Media-intima Thickness: A Systematic Review and a Meta-analysis. Health Sci. Rep. 2023, 6, e1554. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-K.; Peng, Z.-G. Targeting Liver Sinusoidal Endothelial Cells: An Attractive Therapeutic Strategy to Control Inflammation in Nonalcoholic Fatty Liver Disease. Front. Pharmacol. 2021, 12, 655557. [Google Scholar] [CrossRef]
- Bourebaba, N.; Marycz, K. Hepatic Stellate Cells Role in the Course of Metabolic Disorders Development—A Molecular Overview. Pharmacol. Res. 2021, 170, 105739. [Google Scholar] [CrossRef]
- Li, J.; Zhao, Y.-R.; Tian, Z. Roles of Hepatic Stellate Cells in Acute Liver Failure: From the Perspective of Inflammation and Fibrosis. World J. Hepatol. 2019, 11, 412–420. [Google Scholar] [CrossRef]
- Cheng, Q.-N.; Yang, X.; Wu, J.-F.; Ai, W.-B.; Ni, Y.-R. Interaction of Non-Parenchymal Hepatocytes in the Process of Hepatic Fibrosis. Mol. Med. Rep. 2021, 23, 364. [Google Scholar] [CrossRef]
- Fargion, S.; Porzio, M.; Fracanzani, A.L. Nonalcoholic Fatty Liver Disease and Vascular Disease: State-of-the-Art. World J. Gastroenterol. 2014, 20, 13306–13324. [Google Scholar] [CrossRef]
- Kasper, P.; Martin, A.; Lang, S.; Kütting, F.; Goeser, T.; Demir, M.; Steffen, H.-M. NAFLD and Cardiovascular Diseases: A Clinical Review. Clin. Res. Cardiol. 2021, 110, 921–937. [Google Scholar] [CrossRef] [PubMed]
- Hamilos, M.; Petousis, S.; Parthenakis, F. Interaction between Platelets and Endothelium: From Pathophysiology to New Therapeutic Options. Cardiovasc. Diagn. Ther. 2018, 8, 568–580. [Google Scholar] [CrossRef] [PubMed]
- Hinsbergh, V.W.M. van Endothelium—Role in Regulation of Coagulation and Inflammation. Semin. Immunopathol. 2012, 34, 93. [Google Scholar] [CrossRef] [PubMed]
- Peche, V.S.; Pietka, T.A.; Jacome-Sosa, M.; Samovski, D.; Palacios, H.; Chatterjee-Basu, G.; Dudley, A.C.; Beatty, W.; Meyer, G.A.; Goldberg, I.J.; et al. Endothelial Cell CD36 Regulates Membrane Ceramide Formation, Exosome Fatty Acid Transfer and Circulating Fatty Acid Levels. Nat. Commun. 2023, 14, 4029. [Google Scholar] [CrossRef] [PubMed]
- Thorp, A.; Stine, J.G. Exercise as Medicine: The Impact of Exercise Training on Nonalcoholic Fatty Liver Disease. Curr. Hepatol. Rep. 2020, 19, 402–411. [Google Scholar] [CrossRef]
- Pugh, C.J.A.; Spring, V.S.; Kemp, G.J.; Richardson, P.; Shojaee-Moradie, F.; Umpleby, A.M.; Green, D.J.; Cable, N.T.; Jones, H.; Cuthbertson, D.J. Exercise Training Reverses Endothelial Dysfunction in Nonalcoholic Fatty Liver Disease. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1298–H1306. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.; Lee, K.L.; Muthiah, M.D.; Wu, K.X.; Chioh, F.W.J.; Tan, K.; Soon, G.S.T.; Shabbir, A.; Loo, W.M.; Low, Z.S.; et al. Endothelial-immune Crosstalk Contributes to Vasculopathy in Nonalcoholic Fatty Liver Disease. EMBO Rep. 2022, 23, e54271. [Google Scholar] [CrossRef]
- Wang, M.; Zhou, B.-G.; Zhang, Y.; Ren, X.-F.; Li, L.; Li, B.; Ai, Y.-W. Association Between Non-Alcoholic Fatty Liver Disease and Risk of Stroke: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 2022, 9, 812030. [Google Scholar] [CrossRef]
- Kjærgaard, K.; Mikkelsen, A.C.D.; Wernberg, C.W.; Grønkjær, L.L.; Eriksen, P.L.; Damholdt, M.F.; Mookerjee, R.P.; Vilstrup, H.; Lauridsen, M.M.; Thomsen, K.L. Cognitive Dysfunction in Non-Alcoholic Fatty Liver Disease—Current Knowledge, Mechanisms and Perspectives. J. Clin. Med. 2021, 10, 673. [Google Scholar] [CrossRef]
- Kelty, T.J.; Dashek, R.J.; Arnold, W.D.; Rector, R.S. Emerging Links between Nonalcoholic Fatty Liver Disease and Neurodegeneration. Semin. Liver Dis. 2023, 43, 77–88. [Google Scholar] [CrossRef]
- Tran, N.; Garcia, T.; Aniqa, M.; Ali, S.; Ally, A.; Nauli, S. Endothelial Nitric Oxide Synthase (eNOS) and the Cardiovascular System: In Physiology and in Disease States. Am. J. Biomed. Sci. Res. 2022, 15, 153–177. [Google Scholar] [PubMed]
- Muniyappa, R.; Quon, M.J. Insulin Action and Insulin Resistance in Vascular Endothelium. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 523–530. [Google Scholar] [CrossRef]
- Takeda, Y.; Matoba, K.; Sekiguchi, K.; Nagai, Y.; Yokota, T.; Utsunomiya, K.; Nishimura, R. Endothelial Dysfunction in Diabetes. Biomedicines 2020, 8, 182. [Google Scholar] [CrossRef] [PubMed]
- Vrsaljko, N.; Samadan, L.; Viskovic, K.; Mehmedović, A.; Budimir, J.; Vince, A.; Papic, N. Association of Nonalcoholic Fatty Liver Disease with COVID-19 Severity and Pulmonary Thrombosis: CovidFAT, a Prospective, Observational Cohort Study. Open Forum Infect. Dis. 2022, 9, ofac073. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Krawczyk, M.; Smyk, W.; Lammert, F.; Di Ciaula, A. COVID-19 and Non-Alcoholic Fatty Liver Disease: Two Intersecting Pandemics. Eur. J. Clin. Investig. 2020, 50, e13338. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Hussain, S.; Antony, B. Non-Alcoholic Fatty Liver Disease and Clinical Outcomes in Patients with COVID-19: A Comprehensive Systematic Review and Meta-Analysis. Diabetes Metab. Syndr. 2021, 15, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Namkoong, H. Susceptibility of the Obese Population to COVID-19. Int. J. Infect. Dis. 2020, 101, 380–381. [Google Scholar] [CrossRef]
- Miranda, C.; Garlatti, E.; Da Porto, A.; Rinaldo, E.; Grazioli, S.; Zanette, G.; Tonizzo, M. Liver Injury in COVID-19 Patients with Non-Alcoholic Fatty Liver Disease: An Update. Arch. Med. Sci. Atheroscler. Dis. 2023, 8, e1–e10. [Google Scholar] [CrossRef]
- Sachdeva, S.; Khandait, H.; Kopel, J.; Aloysius, M.M.; Desai, R.; Goyal, H. NAFLD and COVID-19: A Pooled Analysis. SN Compr. Clin. Med. 2020, 2, 2726–2729. [Google Scholar] [CrossRef]
- Mushtaq, K.; Khan, M.U.; Iqbal, F.; Alsoub, D.H.; Chaudhry, H.S.; Ata, F.; Iqbal, P.; Elfert, K.; Balaraju, G.; Almaslamani, M.; et al. NAFLD Is a Predictor of Liver Injury in COVID-19 Hospitalized Patients but Not of Mortality, Disease Severity on the Presentation or Progression—The Debate Continues. J. Hepatol. 2021, 74, 482–484. [Google Scholar] [CrossRef]
- Jiang, S.-T.; Liu, Y.-G.; Zhang, L.; Sang, X.-T.; Xu, Y.-Y.; Lu, X. Systems Biology Approach Reveals a Common Molecular Basis for COVID-19 and Non-Alcoholic Fatty Liver Disease (NAFLD). Eur. J. Med. Res. 2022, 27, 251. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, M.; Canzano, P.; Becchetti, A.; Tremoli, E.; Camera, M. Alterations in Platelets during SARS-CoV-2 Infection. Platelets 2021, 33, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Bongiovanni, D.; Klug, M.; Lazareva, O.; Weidlich, S.; Biasi, M.; Ursu, S.; Warth, S.; Buske, C.; Lukas, M.; Spinner, C.D.; et al. SARS-CoV-2 Infection Is Associated with a pro-Thrombotic Platelet Phenotype. Cell Death Dis. 2021, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Schrick, D.; Tőkés-Füzesi, M.; Réger, B.; Molnár, T. Plasma Fibrinogen Independently Predicts Hypofibrinolysis in Severe COVID-19. Metabolites 2021, 11, 826. [Google Scholar] [CrossRef] [PubMed]
- Tsutsumi, T.; Saito, M.; Nagai, H.; Yamamoto, S.; Ikeuchi, K.; Lim, L.A.; Adachi, E.; Koga, M.; Okushin, K.; Akai, H.; et al. Association of Coagulopathy with Liver Dysfunction in Patients with COVID-19. Hepatol. Res. 2021, 51, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Abenavoli, L.; Aquila, I.; Sacco, M.A.; Scarlata, G.G.M.; Procopio, A.C.; Boccuto, L.; Scarpellini, E.; Greco, M.; Foti, D.P.; Ricci, P.; et al. Liver Damage and Impaired Coagulation in COVID-19 Patients: A Case Series. Diseases 2023, 11, 141. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Adipokines in Nonalcoholic Fatty Liver Disease. Metabolism 2016, 65, 1062–1079. [Google Scholar] [CrossRef]
- Almeda-Valdés, P.; Cuevas-Ramos, D.; Aguilar-Salinas, C.A. Metabolic Syndrome and Non-Alcoholic Fatty Liver Disease. Ann. Hepatol. 2009, 8 (Suppl. 1), S18–S24. [Google Scholar] [CrossRef]
- Li, M.; Chi, X.; Wang, Y.; Setrerrahmane, S.; Xie, W.; Xu, H. Trends in Insulin Resistance: Insights into Mechanisms and Therapeutic Strategy. Signal Transduct. Target. Ther. 2022, 7, 216. [Google Scholar] [CrossRef]
- Chao, H.-W.; Chao, S.-W.; Lin, H.; Ku, H.-C.; Cheng, C.-F. Homeostasis of Glucose and Lipid in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2019, 20, 298. [Google Scholar] [CrossRef]
- Salavatizadeh, M.; Soltanieh, S.; Ataei Kachouei, A.; Abdollahi Fallahi, Z.; Kord-Varkaneh, H.; Poustchi, H.; Mansour, A.; Khamseh, M.E.; Alaei-Shahmiri, F.; Santos, H.O.; et al. Association between Dietary Glycemic Index and Non-Alcoholic Fatty Liver Disease in Patients with Type 2 Diabetes Mellitus. Front. Endocrinol. 2023, 14, 1228072. [Google Scholar] [CrossRef] [PubMed]
- Kohli, S.; Shahzad, K.; Jouppila, A.; Holthöfer, H.; Isermann, B.; Lassila, R. Thrombosis and Inflammation—A Dynamic Interplay and the Role of Glycosaminoglycans and Activated Protein C. Front. Cardiovasc. Med. 2022, 9, 866751. [Google Scholar] [CrossRef] [PubMed]
- Gehrke, N.; Schattenberg, J.M. Metabolic Inflammation—A Role for Hepatic Inflammatory Pathways as Drivers of Comorbidities in Nonalcoholic Fatty Liver Disease? Gastroenterology 2020, 158, 1929–1947.e6. [Google Scholar] [CrossRef] [PubMed]
- Grander, C.; Grabherr, F.; Moschen, A.R.; Tilg, H. Non-Alcoholic Fatty Liver Disease: Cause or Effect of Metabolic Syndrome. Viszeralmedizin 2016, 32, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Booth, N.A.; Simpson, A.J.; Croll, A.; Bennett, B.; MacGregor, I.R. Plasminogen Activator Inhibitor (PAI-1) in Plasma and Platelets. Br. J. Haematol. 1988, 70, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Zorio, E.; Gilabert-Estellés, J.; España, F.; Ramón, L.A.; Cosín, R.; Estellés, A. Fibrinolysis: The Key to New Pathogenetic Mechanisms. Curr. Med. Chem. 2008, 15, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.; Reynisdottir, S.; Lönnqvist, F.; Stemme, V.; Hamsten, A.; Arner, P. Adipose Tissue Secretion of Plasminogen Activator Inhibitor-1 in Non-Obese and Obese Individuals. Diabetologia 1998, 41, 65–71. [Google Scholar] [CrossRef]
- Kaji, H. Adipose Tissue-Derived Plasminogen Activator Inhibitor-1 Function and Regulation. Compr. Physiol. 2016, 6, 1873–1896. [Google Scholar] [CrossRef]
- Barnard, S.A.; Pieters, M.; De Lange, Z. The Contribution of Different Adipose Tissue Depots to Plasma Plasminogen Activator Inhibitor-1 (PAI-1) Levels. Blood Rev. 2016, 30, 421–429. [Google Scholar] [CrossRef]
- Iwaki, T.; Urano, T.; Umemura, K. PAI-1, Progress in Understanding the Clinical Problem and Its Aetiology. Br. J. Haematol. 2012, 157, 291–298. [Google Scholar] [CrossRef]
- Wang, L.; Chen, L.; Liu, Z.; Liu, Y.; Luo, M.; Chen, N.; Deng, X.; Luo, Y.; He, J.; Zhang, L.; et al. PAI-1 Exacerbates White Adipose Tissue Dysfunction and Metabolic Dysregulation in High Fat Diet-Induced Obesity. Front. Pharmacol. 2018, 9, 1087. [Google Scholar] [CrossRef] [PubMed]
- Rawish, E.; Sauter, M.; Sauter, R.; Nording, H.; Langer, H.F. Complement, Inflammation and Thrombosis. Br. J. Pharmacol. 2021, 178, 2892–2904. [Google Scholar] [CrossRef] [PubMed]
- Kuijpers, M.J.E.; Heemskerk, J.W.M.; Jurk, K. Molecular Mechanisms of Hemostasis, Thrombosis and Thrombo-Inflammation. Int. J. Mol. Sci. 2022, 23, 5825. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, R.; Escaig, R.; Nicolai, L. Hemostasis without Clot Formation: How Platelets Guard the Vasculature in Inflammation, Infection, and Malignancy. Blood 2023, 142, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; McGeough, M.D.; Peña, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 Inflammasome Activation Is Required for Fibrosis Development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Schett, G.; Elewaut, D.; McInnes, I.B.; Dayer, J.-M.; Neurath, M.F. How Cytokine Networks Fuel Inflammation: Toward a Cytokine-Based Disease Taxonomy. Nat. Med. 2013, 19, 822–824. [Google Scholar] [CrossRef]
- Schuliga, M.; Langenbach, S.; Xia, Y.C.; Qin, C.; Mok, J.S.L.; Harris, T.; Mackay, G.A.; Medcalf, R.L.; Stewart, A.G. Plasminogen-Stimulated Inflammatory Cytokine Production by Airway Smooth Muscle Cells Is Regulated by Annexin A2. Am. J. Respir. Cell Mol. Biol. 2013, 49, 751–758. [Google Scholar] [CrossRef]
- Heissig, B.; Salama, Y.; Takahashi, S.; Osada, T.; Hattori, K. The Multifaceted Role of Plasminogen in Inflammation. Cell. Signal. 2020, 75, 109761. [Google Scholar] [CrossRef]
- Kang, S.; Tanaka, T.; Inoue, H.; Ono, C.; Hashimoto, S.; Kioi, Y.; Matsumoto, H.; Matsuura, H.; Matsubara, T.; Shimizu, K.; et al. IL-6 Trans-Signaling Induces Plasminogen Activator Inhibitor-1 from Vascular Endothelial Cells in Cytokine Release Syndrome. Proc. Natl. Acad. Sci. USA 2020, 117, 22351–22356. [Google Scholar] [CrossRef]
- Duan, Y.; Pan, X.; Luo, J.; Xiao, X.; Li, J.; Bestman, P.L.; Luo, M. Association of Inflammatory Cytokines with Non-Alcoholic Fatty Liver Disease. Front. Immunol. 2022, 13, 880298. [Google Scholar] [CrossRef]
- Loskutoff, D.J.; Samad, F. The Adipocyte and Hemostatic Balance in Obesity. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bilgic Gazioglu, S.; Akan, G.; Atalar, F.; Erten, G. PAI-1 and TNF-α Profiles of Adipose Tissue in Obese Cardiovascular Disease Patients. Int. J. Clin. Exp. Pathol. 2015, 8, 15919–15925. [Google Scholar] [PubMed]
- Samad, F.; Pandey, M.; Bell, P.A.; Loskutoff, D.J. Insulin Continues to Induce Plasminogen Activator Inhibitor 1 Gene Expression in Insulin-Resistant Mice and Adipocytes. Mol. Med. 2000, 6, 680–692. [Google Scholar] [CrossRef]
- Bastard, J.P.; Piéroni, L.; Hainque, B. Relationship between Plasma Plasminogen Activator Inhibitor 1 and Insulin Resistance. Diabetes Metab. Res. Rev. 2000, 16, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, D.E. PAI-1 and Atherothrombosis. J. Thromb. Haemost. 2005, 3, 1879–1883. [Google Scholar] [CrossRef] [PubMed]
- Jung, R.G.; Motazedian, P.; Ramirez, F.D.; Simard, T.; Di Santo, P.; Visintini, S.; Faraz, M.A.; Labinaz, A.; Jung, Y.; Hibbert, B. Association between Plasminogen Activator Inhibitor-1 and Cardiovascular Events: A Systematic Review and Meta-Analysis. Thromb. J. 2018, 16, 12. [Google Scholar] [CrossRef]
- Frischmuth, T.; Hindberg, K.; Aukrust, P.; Ueland, T.; Brækkan, S.K.; Hansen, J.; Morelli, V.M. Elevated Plasma Levels of Plasminogen Activator Inhibitor-1 Are Associated with Risk of Future Incident Venous Thromboembolism. J. Thromb. Haemost. 2022, 20, 1618–1626. [Google Scholar] [CrossRef]
- Straub, L.G.; Scherer, P.E. Metabolic Messengers: Adiponectin. Nat. Metab. 2019, 1, 334–339. [Google Scholar] [CrossRef]
- Iwabu, M.; Okada-Iwabu, M.; Yamauchi, T.; Kadowaki, T. Adiponectin/Adiponectin Receptor in Disease and Aging. npj Aging Mech. Dis. 2015, 1, 1–6. [Google Scholar] [CrossRef]
- Filippi, E.; Sentinelli, F.; Trischitta, V.; Romeo, S.; Arca, M.; Leonetti, F.; Di Mario, U.; Baroni, M.G. Association of the Human Adiponectin Gene and Insulin Resistance. Eur. J. Hum. Genet. 2004, 12, 199–205. [Google Scholar] [CrossRef]
- Cho, J.; Koh, Y.; Han, J.; Kim, D.; Kim, T.; Kang, H. Adiponectin Mediates the Additive Effects of Combining Daily Exercise with Caloric Restriction for Treatment of Non-Alcoholic Fatty Liver. Int. J. Obes. 2016, 40, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-S.; Lee, S.-H.; Park, S.G.; Won, B.Y.; Chun, H.; Cho, D.-Y.; Kim, M.-J.; Lee, J.E.; Haam, J.-H.; Han, K. Low Levels of Total and High-Molecular-Weight Adiponectin May Predict Non-Alcoholic Fatty Liver in Korean Adults. Metabolism 2020, 103, 154026. [Google Scholar] [CrossRef]
- Xu, H.; Zhao, Q.; Song, N.; Yan, Z.; Lin, R.; Wu, S.; Jiang, L.; Hong, S.; Xie, J.; Zhou, H.; et al. AdipoR1/AdipoR2 Dual Agonist Recovers Nonalcoholic Steatohepatitis and Related Fibrosis via Endoplasmic Reticulum-Mitochondria Axis. Nat. Commun. 2020, 11, 5807. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.C.; Liang, L.; Hong, F.; Fu, J.F.; Zhao, Z.Y. Serum Adiponectin, Resistin Levels and Non-Alcoholic Fatty Liver Disease in Obese Children. Endocr. J. 2005, 52, 519–524. [Google Scholar] [CrossRef]
- Lebensztejn, D.M.; Wojtkowska, M.; Skiba, E.; Werpachowska, I.; Tobolczyk, J.; Kaczmarski, M. Serum Concentration of Adiponectin, Leptin and Resistin in Obese Children with Non-Alcoholic Fatty Liver Disease. Adv. Med. Sci. 2009, 54, 177–182. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ramanathan, R.; Ali, A.H.; Ibdah, J.A. Mitochondrial Dysfunction Plays Central Role in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7280. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, R.P.; Sheldon, R.D.; Rector, R.S. The Emerging Role of Hepatocellular eNOS in Non-Alcoholic Fatty Liver Disease Development. Front. Physiol. 2020, 11, 767. [Google Scholar] [CrossRef]
- Udomsinprasert, W.; Honsawek, S.; Poovorawan, Y. Adiponectin as a Novel Biomarker for Liver Fibrosis. World J. Hepatol. 2018, 10, 708–718. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, C.; Wang, Y.; He, X.; Shen, C.; Cao, W.; Zhou, J.; Zhen, Z. Adiponectin inhibits the activation of hepatic stellate cells induced by TGFb1 via up-regulating the expression of eNOS. Zhonghua Gan Zang Bing Za Zhi 2011, 19, 917–922. [Google Scholar] [CrossRef]
- Adachi, M.; Brenner, D.A. High Molecular Weight Adiponectin Inhibits Proliferation of Hepatic Stellate Cells via Activation of Adenosine Monophosphate-Activated Protein Kinase. Hepatology 2008, 47, 677–685. [Google Scholar] [CrossRef]
- Kato, H.; Kashiwagi, H.; Shiraga, M.; Tadokoro, S.; Kamae, T.; Ujiie, H.; Honda, S.; Miyata, S.; Ijiri, Y.; Yamamoto, J.; et al. Adiponectin Acts as an Endogenous Antithrombotic Factor. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-E.; Kim, J.-S.; Jo, M.-J.; Cho, E.; Ahn, S.-Y.; Kwon, Y.-J.; Ko, G.-J. The Roles and Associated Mechanisms of Adipokines in Development of Metabolic Syndrome. Molecules 2022, 27, 334. [Google Scholar] [CrossRef] [PubMed]
- Lowe, G.; Rumley, A. The Relevance of Coagulation in Cardiovascular Disease: What Do the Biomarkers Tell Us? Thromb. Haemost. 2014, 112, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, P.P.; Tofler, G.H. Hemostatic Risk Factors for Cardiovascular Disease. In Triggering of Acute Coronary Syndromes: Implications for Prevention; Willich, S.N., Muller, J.E., Eds.; Developments in Cardiovascular Medicine; Springer: Dordrecht, The Netherlands, 1996; pp. 135–151. ISBN 978-94-009-1577-0. [Google Scholar]
- Zhang, H.; Niu, Y.; Gu, H.; Lu, S.; Zhang, W.; Li, X.; Yang, Z.; Qin, L.; Su, Q. Low Serum Adiponectin Is a Predictor of Progressing to Nonalcoholic Fatty Liver Disease. J. Clin. Lab. Anal. 2018, 33, e22709. [Google Scholar] [CrossRef]
- Marsche, G.; Zelzer, S.; Meinitzer, A.; Kern, S.; Meissl, S.; Pregartner, G.; Weghuber, D.; Almer, G.; Mangge, H. Adiponectin Predicts High-Density Lipoprotein Cholesterol Efflux Capacity in Adults Irrespective of Body Mass Index and Fat Distribution. J. Clin. Endocrinol. Metab. 2017, 102, 4117–4123. [Google Scholar] [CrossRef]
- Lee, S.; Kwak, H.-B. Role of Adiponectin in Metabolic and Cardiovascular Disease. J. Exerc. Rehabil. 2014, 10, 54–59. [Google Scholar] [CrossRef]
- Hui, X.; Lam, K.S.; Vanhoutte, P.M.; Xu, A. Adiponectin and Cardiovascular Health: An Update. Br. J. Pharmacol. 2012, 165, 574–590. [Google Scholar] [CrossRef]
- Bruun, J.M.; Lihn, A.S.; Verdich, C.; Pedersen, S.B.; Toubro, S.; Astrup, A.; Richelsen, B. Regulation of Adiponectin by Adipose Tissue-Derived Cytokines: In Vivo and in Vitro Investigations in Humans. Am. J. Physiol.—Endocrinol. Metab. 2003, 285, E527–E533. [Google Scholar] [CrossRef]
- Simons, P.J.; van den Pangaart, P.S.; Aerts, J.M.F.G.; Boon, L. Pro-Inflammatory Delipidizing Cytokines Reduce Adiponectin Secretion from Human Adipocytes without Affecting Adiponectin Oligomerization. J. Endocrinol. 2006, 192, 289–299. [Google Scholar] [CrossRef]
- Olie, R.H.; van der Meijden, P.E.J.; ten Cate, H. The Coagulation System in Atherothrombosis: Implications for New Therapeutic Strategies. Res. Pract. Thromb. Haemost. 2018, 2, 188–198. [Google Scholar] [CrossRef]
- Szmitko, P.E.; Wang, C.-H.; Weisel, R.D.; de Almeida, J.R.; Anderson, T.J.; Verma, S. New Markers of Inflammation and Endothelial Cell Activation. Circulation 2003, 108, 1917–1923. [Google Scholar] [CrossRef] [PubMed]
- Tomas, L.; Prica, F.; Schulz, C. Trafficking of Mononuclear Phagocytes in Healthy Arteries and Atherosclerosis. Front. Immunol. 2021, 12, 718432. [Google Scholar] [CrossRef]
- Xu, X.; Lu, L.; Dong, Q.; Li, X.; Zhang, N.; Xin, Y.; Xuan, S. Research Advances in the Relationship between Nonalcoholic Fatty Liver Disease and Atherosclerosis. Lipids Health Dis. 2015, 14, 158. [Google Scholar] [CrossRef] [PubMed]
- Lovren, F.; Pan, Y.; Quan, A.; Szmitko, P.E.; Singh, K.K.; Shukla, P.C.; Gupta, M.; Chan, L.; Al-Omran, M.; Teoh, H.; et al. Adiponectin Primes Human Monocytes into Alternative Anti-Inflammatory M2 Macrophages. Am. J. Physiol.—Heart Circ. Physiol. 2010, 299, H656–H663. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.M. Leptin and the Endocrine Control of Energy Balance. Nat. Metab. 2019, 1, 754–764. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, A.G.; Crujeiras, A.B.; Casanueva, F.F.; Carreira, M.C. Leptin, Obesity, and Leptin Resistance: Where Are We 25 Years Later? Nutrients 2019, 11, 2704. [Google Scholar] [CrossRef]
- Francisco, V.; Pino, J.; Campos-Cabaleiro, V.; Ruiz-Fernández, C.; Mera, A.; Gonzalez-Gay, M.A.; Gómez, R.; Gualillo, O. Obesity, Fat Mass and Immune System: Role for Leptin. Front. Physiol. 2018, 9, 640. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Aronis, K.N.; Kountouras, J.; Raptis, D.D.; Vasiloglou, M.F.; Mantzoros, C.S. Circulating Leptin in Non-Alcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Diabetologia 2016, 59, 30–43. [Google Scholar] [CrossRef]
- Buis, D.T.P.; Christen, T.; Smit, R.a.J.; de Mutsert, R.; Jukema, J.W.; Cannegieter, S.C.; Lijfering, W.M.; Rosendaal, F.R. The Association between Leptin Concentration and Blood Coagulation: Results from the NEO Study. Thromb. Res. 2020, 188, 44–48. [Google Scholar] [CrossRef]
- Guagnano, M.T.; Romano, M.; Falco, A.; Nutini, M.; Marinopiccoli, M.; Manigrasso, M.R.; Basili, S.; Davì, G. Leptin Increase Is Associated with Markers of the Hemostatic System in Obese Healthy Women. J. Thromb. Haemost. 2003, 1, 2330–2334. [Google Scholar] [CrossRef]
- Napoleone, E.; Di santo, A.; Amore, C.; Baccante, G.; Di febbo, C.; Porreca, E.; De gaetano, G.; Donati, M.B.; Lorenzet, R. Leptin Induces Tissue Factor Expression in Human Peripheral Blood Mononuclear Cells: A Possible Link between Obesity and Cardiovascular Risk? J. Thromb. Haemost. 2007, 5, 1462–1468. [Google Scholar] [CrossRef]
- Singh, P.; Peterson, T.E.; Barber, K.R.; Kuniyoshi, F.S.; Jensen, A.; Hoffmann, M.; Shamsuzzaman, A.S.M.; Somers, V.K. Leptin Upregulates the Expression of Plasminogen Activator Inhibitor-1 in Human Vascular Endothelial Cells. Biochem. Biophys. Res. Commun. 2010, 392, 47–52. [Google Scholar] [CrossRef]
- Bełtowski, J. Role of Leptin in Blood Pressure Regulation and Arterial Hypertension. J. Hypertens. 2006, 24, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Konstantinides, S.; Schäfer, K.; Koschnick, S.; Loskutoff, D.J. Leptin-Dependent Platelet Aggregation and Arterial Thrombosis Suggests a Mechanism for Atherothrombotic Disease in Obesity. J. Clin. Investig. 2001, 108, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Elbatarny, H.S.; Maurice, D.H. Leptin-Mediated Activation of Human Platelets: Involvement of a Leptin Receptor and Phosphodiesterase 3A-Containing Cellular Signaling Complex. Am. J. Physiol.—Endocrinol. Metab. 2005, 289, E695–E702. [Google Scholar] [CrossRef] [PubMed]
- Payne, G.A.; Tune, J.D.; Knudson, J.D. Leptin-Induced Endothelial Dysfunction: A Target for Therapeutic Interventions. Curr. Pharm. Des. 2014, 20, 603–608. [Google Scholar] [CrossRef]
- Schäfer, K.; Konstantinides, S. Mechanisms Linking Leptin to Arterial and Venous Thrombosis: Potential Pharmacological Targets. Curr. Pharm. Des. 2014, 20, 635–640. [Google Scholar] [CrossRef]
- Amitani, M.; Asakawa, A.; Amitani, H.; Inui, A. The Role of Leptin in the Control of Insulin-Glucose Axis. Front. Neurosci. 2013, 7, 51. [Google Scholar] [CrossRef]
- Iikuni, N.; Lam, Q.L.K.; Lu, L.; Matarese, G.; La Cava, A. Leptin and Inflammation. Curr. Immunol. Rev. 2008, 4, 70–79. [Google Scholar] [CrossRef]
- Jiménez-Cortegana, C.; García-Galey, A.; Tami, M.; del Pino, P.; Carmona, I.; López, S.; Alba, G.; Sánchez-Margalet, V. Role of Leptin in Non-Alcoholic Fatty Liver Disease. Biomedicines 2021, 9, 762. [Google Scholar] [CrossRef]
- Ding, N.; Liu, B.; Song, J.; Bao, S.; Zhen, J.; Lv, Z.; Wang, R. Leptin Promotes Endothelial Dysfunction in Chronic Kidney Disease through AKT/GSK3β and β-Catenin Signals. Biochem. Biophys. Res. Commun. 2016, 480, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Knudson, J.D.; Payne, G.A.; Borbouse, L.; Tune, J.D. Leptin and Mechanisms of Endothelial Dysfunction and Cardiovascular Disease. Curr. Hypertens. Rep. 2008, 10, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Knudson, J.D.; Dincer, U.D.; Zhang, C.; Swafford, A.N.; Koshida, R.; Picchi, A.; Focardi, M.; Dick, G.M.; Tune, J.D. Leptin Receptors Are Expressed in Coronary Arteries, and Hyperleptinemia Causes Significant Coronary Endothelial Dysfunction. Am. J. Physiol.—Heart Circ. Physiol. 2005, 289, H48–H56. [Google Scholar] [CrossRef] [PubMed]
- Manuel-Apolinar, L.; López-Romero, R.; Zarate, A.; Damasio, L.; Ruiz, M.; Castillo-Hernández, C.; Guevara, G.; Mera-Jiménez, E. Leptin Mediated ObRb Receptor Increases Expression of Adhesion Intercellular Molecules and Cyclooxygenase 2 on Murine Aorta Tissue Inducing Endothelial Dysfunction. Int. J. Clin. Exp. Med. 2013, 6, 192–196. [Google Scholar] [PubMed]
- Afarid, M.; Attarzadeh, A.; Farvardin, M.; Ashraf, H. The Association of Serum Leptin Level and Anthropometric Measures with the Severity of Diabetic Retinopathy in Type 2 Diabetes Mellitus. Med. Hypothesis Discov. Innov. Ophthalmol. 2018, 7, 156–162. [Google Scholar] [PubMed]
- Maberley, D.; Cui, J.Z.; Matsubara, J.A. Vitreous Leptin Levels in Retinal Disease. Eye 2006, 20, 801–804. [Google Scholar] [CrossRef]
- Shukla, U.V.; Tripathy, K. Diabetic Retinopathy. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Jiang, Y.; Fan, H.; Xie, J.; Xu, Y.; Sun, X. Association between Adipocytokines and Diabetic Retinopathy: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2023, 14, 1271027. [Google Scholar] [CrossRef]
- Suganami, E.; Takagi, H.; Ohashi, H.; Suzuma, K.; Suzuma, I.; Oh, H.; Watanabe, D.; Ojima, T.; Suganami, T.; Fujio, Y.; et al. Leptin Stimulates Ischemia-Induced Retinal Neovascularization: Possible Role of Vascular Endothelial Growth Factor Expressed in Retinal Endothelial Cells. Diabetes 2004, 53, 2443–2448. [Google Scholar] [CrossRef]
- Verhamme, P.; Hoylaerts, M.F. Hemostasis and Inflammation: Two of a Kind? Thromb. J. 2009, 7, 15. [Google Scholar] [CrossRef]
- van der Poll, T.; de Jonge, E.; ten Cate An, H. Cytokines as Regulators of Coagulation. In Madame Curie Bioscience Database [Internet]; Landes Bioscience: Austin, TX, USA, 2013. [Google Scholar]
- Agrawal, S.; Gollapudi, S.; Su, H.; Gupta, S. Leptin Activates Human B Cells to Secrete TNF-α, IL-6, and IL-10 via JAK2/STAT3 and p38MAPK/ERK1/2 Signaling Pathway. J. Clin. Immunol. 2011, 31, 472–478. [Google Scholar] [CrossRef]
- Tazawa, R.; Uchida, K.; Fujimaki, H.; Miyagi, M.; Inoue, G.; Sekiguchi, H.; Murata, K.; Takata, K.; Kawakubo, A.; Takaso, M. Elevated Leptin Levels Induce Inflammation through IL-6 in Skeletal Muscle of Aged Female Rats. BMC Musculoskelet. Disord. 2019, 20, 199. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.; Fink, L.M.; Hauer-Jensen, M. Cytokines in Coagulation and Thrombosis: A Preclinical and Clinical Review. Blood Coagul. Fibrinolysis 2002, 13, 105. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Park, S.; Cho, H.; Kim, J.H.; Choi, H. Adipokine Human Resistin Promotes Obesity-Associated Inflammatory Intervertebral Disc Degeneration via pro-Inflammatory Cytokine Cascade Activation. Sci. Rep. 2022, 12, 8936. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, P. The Role of Adipokines in Chronic Inflammation. Immunotargets Ther. 2016, 5, 47–56. [Google Scholar] [CrossRef]
- Tripathi, D.; Kant, S.; Pandey, S.; Ehtesham, N.Z. Resistin in Metabolism, Inflammation, and Disease. FEBS J. 2020, 287, 3141–3149. [Google Scholar] [CrossRef] [PubMed]
- Gierej, P.; Gierej, B.; Kalinowski, P.; Wróblewski, T.; Paluszkiewicz, R.; Kobryń, K.; Ziarkiewicz-Wróblewska, B. Expression of Resistin in the Liver of Patients with Non-Alcoholic Fatty Liver Disease. Pol. J. Pathol. 2017, 68, 225–233. [Google Scholar] [CrossRef] [PubMed]
- da Silva, T.E.; Costa-Silva, M.; Correa, C.G.; Denardin, G.; Alencar, M.L.A.; Coelho, M.S.P.H.; Muraro-Wildner, L.; Luiza-Bazzo, M.; González-Chica, D.A.; Dantas-Correa, E.B.; et al. Clinical Significance of Serum Adiponectin and Resistin Levels in Liver Cirrhosis. Ann. Hepatol. 2018, 17, 286–299. [Google Scholar] [CrossRef]
- Dong, Z.-X.; Su, L.; Brymora, J.; Bird, C.; Xie, Q.; George, J.; Wang, J.-H. Resistin Mediates the Hepatic Stellate Cell Phenotype. World J. Gastroenterol. 2013, 19, 4475–4485. [Google Scholar] [CrossRef]
- Garcia, C.C.; Piotrkowski, B.; Baz, P.; Poncino, D.; Benavides, J.; Colombato, L.; Toso, M.L.R.; Yantorno, S.; Descalzi, V.; Gondolesi, G.E.; et al. A Decreased Response to Resistin in Mononuclear Leukocytes Contributes to Oxidative Stress in Nonalcoholic Fatty Liver Disease. Dig. Dis. Sci. 2022, 67, 3006–3016. [Google Scholar] [CrossRef]
- Su, K.; Li, Y.; Zhang, D.; Yuan, J.; Zhang, C.; Liu, Y.; Song, L.; Lin, Q.; Li, M.; Dong, J. Relation of Circulating Resistin to Insulin Resistance in Type 2 Diabetes and Obesity: A Systematic Review and Meta-Analysis. Front. Physiol. 2019, 10, 1399. [Google Scholar] [CrossRef]
- Berger, A. Resistin: A New Hormone That Links Obesity with Type 2 Diabetes. BMJ 2001, 322, 193. [Google Scholar]
- Gupta, V.; Singh, A.K.; Gupta, V.; Kumar, S.; Srivastava, N.; Jafar, T.; Pant, A.B. Association of Circulating Resistin with Metabolic Risk Factors in Indian Females Having Metabolic Syndrome. Toxicol. Int. 2011, 18, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.Q.; Zhang, Q.; Peng, Y.B.; Chen, M.; Lin, X.P.; Wu, J.H.; Cai, C.H.; Mei, Y.F.; Jin, H. Resistin Level Is Positively Correlated with Thrombotic Complications in Southern Chinese Metabolic Syndrome Patients. J. Endocrinol. Investig. 2011, 34, e36–e42. [Google Scholar] [CrossRef] [PubMed]
- Pablo-Moreno, J.A.D.; Serrano, L.J.; Revuelta, L.; Sánchez, M.J.; Liras, A. The Vascular Endothelium and Coagulation: Homeostasis, Disease, and Treatment, with a Focus on the Von Willebrand Factor and Factors VIII and V. Int. J. Mol. Sci. 2022, 23, 8283. [Google Scholar] [CrossRef] [PubMed]
- Kougias, P.; Chai, H.; Lin, P.H.; Lumsden, A.B.; Yao, Q.; Chen, C. Adipocyte-Derived Cytokine Resistin Causes Endothelial Dysfunction of Porcine Coronary Arteries. J. Vasc. Surg. 2005, 41, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.M.; Katz, P.S.; Farias, M.; Morris, M.; James, J.; Knudson, J.D.; Tune, J.D. Resistin Impairs Endothelium-Dependent Dilation to Bradykinin, but Not Acetylcholine, in the Coronary Circulation. Am. J. Physiol.—Heart Circ. Physiol. 2006, 291, H2997–H3002. [Google Scholar] [CrossRef] [PubMed]
- Mu, H.; Ohashi, R.; Yan, S.; Chai, H.; Yang, H.; Lin, P.; Yao, Q.; Chen, C. Adipokine Resistin Promotes in Vitro Angiogenesis of Human Endothelial Cells. Cardiovasc. Res. 2006, 70, 146–157. [Google Scholar] [CrossRef]
- Verma, S.; Li, S.-H.; Wang, C.-H.; Fedak, P.W.M.; Li, R.-K.; Weisel, R.D.; Mickle, D.A.G. Resistin Promotes Endothelial Cell Activation. Circulation 2003, 108, 736–740. [Google Scholar] [CrossRef]
- Hsu, W.-Y.; Chao, Y.-W.; Tsai, Y.-L.; Lien, C.-C.; Chang, C.-F.; Deng, M.-C.; Ho, L.-T.; Kwok, C.F.; Juan, C.-C. Resistin Induces Monocyte–Endothelial Cell Adhesion by Increasing ICAM-1 and VCAM-1 Expression in Endothelial Cells via p38MAPK-Dependent Pathway. J. Cell. Physiol. 2011, 226, 2181–2188. [Google Scholar] [CrossRef]
- Jamaluddin, M.S.; Yan, S.; Lü, J.; Liang, Z.; Yao, Q.; Chen, C. Resistin Increases Monolayer Permeability of Human Coronary Artery Endothelial Cells. PLoS ONE 2013, 8, e84576. [Google Scholar] [CrossRef]
- Ayer, J.G.; Song, C.; Steinbeck, K.; Celermajer, D.S.; Freedman, S.B. Increased Tissue Factor Activity in Monocytes from Obese Young Adults. Clin. Exp. Pharmacol. Physiol. 2010, 37, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Jiang, J.; Lü, J.-M.; Chai, H.; Wang, X.; Lin, P.H.; Yao, Q. Resistin Decreases Expression of Endothelial Nitric Oxide Synthase through Oxidative Stress in Human Coronary Artery Endothelial Cells. Am. J. Physiol.—Heart Circ. Physiol. 2010, 299, H193–H201. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.; Sanz-Rosa, D.; Emerson, M. Distinct Role and Location of the Endothelial Isoform of Nitric Oxide Synthase in Regulating Platelet Aggregation in Males and Females in Vivo. Eur. J. Pharmacol. 2011, 651, 152–158. [Google Scholar] [CrossRef]
- Wang, H.; Chen, D.; Cao, J.; He, Z.; Zhu, B.; Long, M. High Serum Resistin Level May Be an Indicator of the Severity of Coronary Disease in Acute Coronary Syndrome. Chin. Med. Sci. J. 2009, 24, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Qiao, S.; Hou, Q.; Yuan, J. Plasma Resistin Is Increased in Patients with Unstable Angina. Chin. Med. J. 2007, 120, 871. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, J.-Y.; He, P.-P.; Yu, X.-H.; Tang, C.-K. Resistin: Potential Biomarker and Therapeutic Target in Atherosclerosis. Clin. Chim. Acta 2021, 512, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Ebihara, T.; Matsumoto, H.; Matsubara, T.; Togami, Y.; Nakao, S.; Matsuura, H.; Onishi, S.; Kojima, T.; Sugihara, F.; Okuzaki, D.; et al. Resistin Associated with Cytokines and Endothelial Cell Adhesion Molecules Is Related to Worse Outcome in COVID-19. Front. Immunol. 2022, 13, 830061. [Google Scholar] [CrossRef]
- Calabro, P.; Samudio, I.; Willerson, J.T.; Yeh, E.T.H. Resistin Promotes Smooth Muscle Cell Proliferation Through Activation of Extracellular Signal–Regulated Kinase 1/2 and Phosphatidylinositol 3-Kinase Pathways. Circulation 2004, 110, 3335–3340. [Google Scholar] [CrossRef]
- Acquarone, E.; Monacelli, F.; Borghi, R.; Nencioni, A.; Odetti, P. Resistin: A Reappraisal. Mech. Ageing Dev. 2019, 178, 46–63. [Google Scholar] [CrossRef]
- Rasineni, K.; Kubik, J.L.; Knight, K.L.; Hall, L.; Casey, C.A.; Kharbanda, K.K. Ghrelin Regulates Adipose Tissue Metabolism: Role in Hepatic Steatosis. Chem. Biol. Interact. 2020, 322, 109059. [Google Scholar] [CrossRef]
- Castorina, S.; Barresi, V.; Luca, T.; Privitera, G.; De Geronimo, V.; Lezoche, G.; Cosentini, I.; Di Vincenzo, A.; Barbatelli, G.; Giordano, A.; et al. Gastric Ghrelin Cells in Obese Patients Are Hyperactive. Int. J. Obes. 2021, 45, 184–194. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin Is a Growth-Hormone-Releasing Acylated Peptide from Stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Kangawa, K. Ghrelin: Structure and Function. Physiol. Rev. 2005, 85, 495–522. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Li, Z.; Wang, Z.; Li, Y.; Zhao, J.; Mulholland, M.; Zhang, W. Ghrelin Contributes to Protection of Hepatocellular Injury Induced by Ischaemia/Reperfusion. Liver Int. 2014, 34, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Moreno, M.; Chaves, J.F.; Sancho-Bru, P.; Ramalho, F.; Ramalho, L.N.; Mansego, M.L.; Ivorra, C.; Dominguez, M.; Conde, L.; Millán, C.; et al. Ghrelin Attenuates Hepatocellular Injury and Liver Fibrogenesis in Rodents and Influences Fibrosis Progression in Humans. Hepatology 2010, 51, 974. [Google Scholar] [CrossRef]
- Cetin, E.; Kanbur, M.; Cetin, N.; Eraslan, G.; Atasever, A. Hepatoprotective Effect of Ghrelin on Carbon Tetrachloride-Induced Acute Liver Injury in Rats. Regul. Pept. 2011, 171, 1–5. [Google Scholar] [CrossRef]
- Arıcı, O.F.; Cetin, N. Protective Role of Ghrelin against Carbon Tetrachloride (CCl₄)-Induced Coagulation Disturbances in Rats. Regul. Pept. 2011, 166, 139–142. [Google Scholar] [CrossRef]
- Golestan Jahromi, M.; Nabavizadeh, F.; Vahedian, J.; Nahrevanian, H.; Dehpour, A.-R.; Zare-Mehrjardi, A. Protective Effect of Ghrelin on Acetaminophen-Induced Liver Injury in Rat. Peptides 2010, 31, 2114–2117. [Google Scholar] [CrossRef]
- Mao, Y.; Zhang, S.; Yu, F.; Li, H.; Guo, C.; Fan, X. Ghrelin Attenuates Liver Fibrosis through Regulation of TGF-Β1 Expression and Autophagy. Int. J. Mol. Sci. 2015, 16, 21911–21930. [Google Scholar] [CrossRef]
- Li, Y.; Hai, J.; Li, L.; Chen, X.; Peng, H.; Cao, M.; Zhang, Q. Administration of Ghrelin Improves Inflammation, Oxidative Stress, and Apoptosis during and after Non-Alcoholic Fatty Liver Disease Development. Endocrine 2013, 43, 376–386. [Google Scholar] [CrossRef]
- Yuan, F.; Zhang, Q.; Dong, H.; Xiang, X.; Zhang, W.; Zhang, Y.; Li, Y. Effects of Des-Acyl Ghrelin on Insulin Sensitivity and Macrophage Polarization in Adipose Tissue. J. Transl. Int. Med. 2021, 9, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhang, H.; Guo, W.; Yu, L. Potential Role of Ghrelin in the Regulation of Inflammation. FASEB J. 2022, 36, e22508. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Lin, L.; Yue, J.; Wu, C.-S.; Guo, C.A.; Wang, R.; Yu, K.-J.; Devaraj, S.; Murano, P.; Chen, Z.; et al. Suppression of Ghrelin Exacerbates HFCS-Induced Adiposity and Insulin Resistance. Int. J. Mol. Sci. 2017, 18, 1302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-R.; Fan, X.-M. Ghrelin-Ghrelin O-Acyltransferase System in the Pathogenesis of Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2015, 21, 3214–3222. [Google Scholar] [CrossRef] [PubMed]
- Tabaeian, S.P.; Mahmoudi, T.; Sabzikarian, M.; Rezamand, G.; Dabiri, R.; Nobakht, H.; Asadi, A.; Farahani, H.; Mansour-Ghanaei, F.; Zali, M.R. The Leu72Met (Rs696217 G>T) Polymorphism of the Ghrelin Gene Might Be a Protective Factor for Nonalcoholic Fatty Liver Disease. J. Gastrointest. Liver Dis. 2021, 30, 233–239. [Google Scholar] [CrossRef]
- Rezamand, G.; Mahmoudi, T.; Tabaeian, S.P.; Farahani, H.; Shahinmehr, F.; Nobakht, H.; Dabiri, R.; Asadi, A.; Mansour-Ghanaei, F.; Zali, M.R. The “GG” Genotype of Rs26802 Variant in the Ghrelin Gene Is a Potential Protective Factor against Nonalcoholic Fatty Liver Disease. Physiol. Int. 2021, 108, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Ghelardoni, S.; Carnicelli, V.; Frascarelli, S.; Ronca-Testoni, S.; Zucchi, R. Ghrelin Tissue Distribution: Comparison between Gene and Protein Expression. J. Endocrinol. Investig. 2006, 29, 115–121. [Google Scholar] [CrossRef]
- Kleinz, M.J.; Maguire, J.J.; Skepper, J.N.; Davenport, A.P. Functional and Immunocytochemical Evidence for a Role of Ghrelin and Des-Octanoyl Ghrelin in the Regulation of Vascular Tone in Man. Cardiovasc. Res. 2006, 69, 227–235. [Google Scholar] [CrossRef]
- Baldanzi, G.; Filigheddu, N.; Cutrupi, S.; Catapano, F.; Bonissoni, S.; Fubini, A.; Malan, D.; Baj, G.; Granata, R.; Broglio, F.; et al. Ghrelin and Des-Acyl Ghrelin Inhibit Cell Death in Cardiomyocytes and Endothelial Cells through ERK1/2 and PI 3-Kinase/AKT. J. Cell Biol. 2002, 159, 1029–1037. [Google Scholar] [CrossRef]
- Gupta, S.; Mitra, A. Heal the Heart through Gut (Hormone) Ghrelin: A Potential Player to Combat Heart Failure. Heart Fail. Rev. 2021, 26, 417–435. [Google Scholar] [CrossRef]
- Zhang, G.; Yin, X.; Qi, Y.; Pendyala, L.; Chen, J.; Hou, D.; Tang, C. Ghrelin and Cardiovascular Diseases. Curr. Cardiol. Rev. 2010, 6, 62–70. [Google Scholar] [CrossRef]
- Eid, R.A.; Alkhateeb, M.A.; Eleawa, S.; Al-Hashem, F.H.; Al-Shraim, M.; El-Kott, A.F.; Zaki, M.S.A.; Dallak, M.A.; Aldera, H. Cardioprotective Effect of Ghrelin against Myocardial Infarction-Induced Left Ventricular Injury via Inhibition of SOCS3 and Activation of JAK2/STAT3 Signaling. Basic Res. Cardiol. 2018, 113, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, Q.; Pang, J. The Effects and Mechanisms of Ghrelin upon Angiogenesis in Human Coronary Artery Endothelial Cells under Hypoxia. Peptides 2023, 160, 170921. [Google Scholar] [CrossRef] [PubMed]
- Ahluwalia, A.; Li, A.; Cheng, G.; Deng, X.; Tarnawski, A.S. Reduced Ghrelin in Endothelial Cells Plays Important Mechanistic Role in Aging-Related Impairment of Angiogenesis. J. Physiol. Pharmacol. 2009, 60, 29–34. [Google Scholar] [PubMed]
- Li, W.G.; Gavrila, D.; Liu, X.; Wang, L.; Gunnlaugsson, S.; Stoll, L.L.; McCormick, M.L.; Sigmund, C.D.; Tang, C.; Weintraub, N.L. Ghrelin Inhibits Proinflammatory Responses and Nuclear Factor-kappaB Activation in Human Endothelial Cells. Circulation 2004, 109, 2221–2226. [Google Scholar] [CrossRef]
- Wang, L.; Chen, Q.; Li, G.; Ke, D. Ghrelin Stimulates Angiogenesis via GHSR1a-Dependent MEK/ERK and PI3K/Akt Signal Pathways in Rat Cardiac Microvascular Endothelial Cells. Peptides 2012, 33, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Narsinh, K.; Zhao, L.; Sun, D.; Wang, D.; Zhang, Z.; Sun, Z.; Zhang, R.; Wang, H.; Cao, F. Effects and Mechanisms of Ghrelin on Cardiac Microvascular Endothelial Cells in Rats. Cell Biol. Int. 2011, 35, 135–140. [Google Scholar] [CrossRef]
- Tesauro, M.; Schinzari, F.; Iantorno, M.; Rizza, S.; Melina, D.; Lauro, D.; Cardillo, C. Ghrelin Improves Endothelial Function in Patients with Metabolic Syndrome. Circulation 2005, 112, 2986–2992. [Google Scholar] [CrossRef]
- Tesauro, M.; Schinzari, F.; Rovella, V.; Di Daniele, N.; Lauro, D.; Mores, N.; Veneziani, A.; Cardillo, C. Ghrelin Restores the Endothelin 1/Nitric Oxide Balance in Patients with Obesity-Related Metabolic Syndrome. Hypertension 2009, 54, 995–1000. [Google Scholar] [CrossRef]
- Aird, W.C. Endothelium and Haemostasis. Hamostaseologie 2015, 35, 11–16. [Google Scholar] [CrossRef]
- Verhamme, P.; Hoylaerts, M.F. The Pivotal Role of the Endothelium in Haemostasis and Thrombosis. Acta Clin. Belg. 2006, 61, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Solovey, A.; Kollander, R.; Chang Milbauer, L.; Abdulla, F.; Chen, Y.; Kelm, R.J., Jr.; Hebbel, R.P. Endothelial Nitric Oxide Synthase and Nitric Oxide Regulate Endothelial Tissue Factor Expression in Vivo in the Sickle Transgenic Mouse. Am. J. Hematol. 2010, 85, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Ruiz, A.; Montes, R.; Velasco, F.; López-Pedrera, C.; Páramo, J.A.; Orbe, J.; Hermida, J.; Rocha, E. Regulation by Nitric Oxide of Endotoxin-Induced Tissue Factor and Plasminogen Activator Inhibitor-1 in Endothelial Cells. Thromb. Haemost. 2002, 88, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lu, Z.-J.; Yang, Y.; Lu, G.-P.; Chen, W.-M.; Zhang, L.-E. Suppression of Plasminogen Activator Inhibitor-1 by Inhaled Nitric Oxide Attenuates the Adverse Effects of Hyperoxia in a Rat Model of Acute Lung Injury. Thromb. Res. 2015, 136, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Swiatkowska, M.; Cierniewska-Cieslak, A.; Pawlowska, Z.; Cierniewski, C.S. Dual Regulatory Effects of Nitric Oxide on Plasminogen Activator Inhibitor Type 1 Expression in Endothelial Cells. Eur. J. Biochem. 2000, 267, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.; Park, E.J.; Siragusa, M.; Frohlich, F.; Mahfuzul Haque, M.; Pascale, J.V.; Heberlein, K.R.; Isakson, B.E.; Stuehr, D.J.; Sessa, W.C. Unbiased Proteomics Identifies Plasminogen Activator Inhibitor-1 as a Negative Regulator of Endothelial Nitric Oxide Synthase. Proc. Natl. Acad. Sci. USA 2020, 117, 9497–9507. [Google Scholar] [CrossRef] [PubMed]
- Khatib, N.; Gaidhane, S.; Gaidhane, A.M.; Khatib, M.; Simkhada, P.; Gode, D.; Zahiruddin, Q.S. Ghrelin: Ghrelin as a Regulatory Peptide in Growth Hormone Secretion. J. Clin. Diagn. Res. 2014, 8, MC13–MC17. [Google Scholar] [CrossRef]
- Miljic, D.; Miljic, P.; Doknic, M.; Pekic, S.; Djurovic, M.; Colovic, M.; Popovic, V. Changes in Prothrombin and Activated Partial Thromboplastin Time during Replacement Therapy with Human Recombinant Growth Hormone in Growth Hormone Deficient Adults. Hormones 2006, 5, 187–191. [Google Scholar] [CrossRef][Green Version]
- Bužga, M.; Švagera, Z.; Tomášková, H.; Hauptman, K.; Holéczy, P. Metabolic Effects of Sleeve Gastrectomy and Laparoscopic Greater Curvature Plication: An 18-Month Prospective, Observational, Open-Label Study. Obes. Surg. 2017, 27, 3258–3266. [Google Scholar] [CrossRef]
- Mazzone, C.; Pezzino, S.; Sofia, M.; Litrico, G.; Sarvà, I.; Agosta, M.; La Greca, G.; Latteri, S. Scientific and Public Interest in Bariatric Surgery for Obesity: The Italian Scenario. Gastrointest. Disord. 2023, 5, 438–454. [Google Scholar] [CrossRef]
- Morsy, M.D. Hemostatic Effect of Acylated Ghrelin in Control and Sleeve Gastrectomy-Induced Rats: Mechanisms of Action. Arch. Physiol. Biochem. 2020, 126, 31–40. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pezzino, S.; Luca, T.; Castorina, M.; Puleo, S.; Latteri, S.; Castorina, S. Role of Perturbated Hemostasis in MASLD and Its Correlation with Adipokines. Life 2024, 14, 93. https://doi.org/10.3390/life14010093
Pezzino S, Luca T, Castorina M, Puleo S, Latteri S, Castorina S. Role of Perturbated Hemostasis in MASLD and Its Correlation with Adipokines. Life. 2024; 14(1):93. https://doi.org/10.3390/life14010093
Chicago/Turabian StylePezzino, Salvatore, Tonia Luca, Mariacarla Castorina, Stefano Puleo, Saverio Latteri, and Sergio Castorina. 2024. "Role of Perturbated Hemostasis in MASLD and Its Correlation with Adipokines" Life 14, no. 1: 93. https://doi.org/10.3390/life14010093
APA StylePezzino, S., Luca, T., Castorina, M., Puleo, S., Latteri, S., & Castorina, S. (2024). Role of Perturbated Hemostasis in MASLD and Its Correlation with Adipokines. Life, 14(1), 93. https://doi.org/10.3390/life14010093