Microwave-Assisted Acid Activation of Clays Composed of 2:1 Clay Minerals: A Comparative Study





Abstract
:1. Introduction
2. Materials and Methods
2.1. Starting Materials
2.2. Microwave-Assisted Acid Treatment (MAT)
2.3. Analytical Methodology
3. Results and Discussion
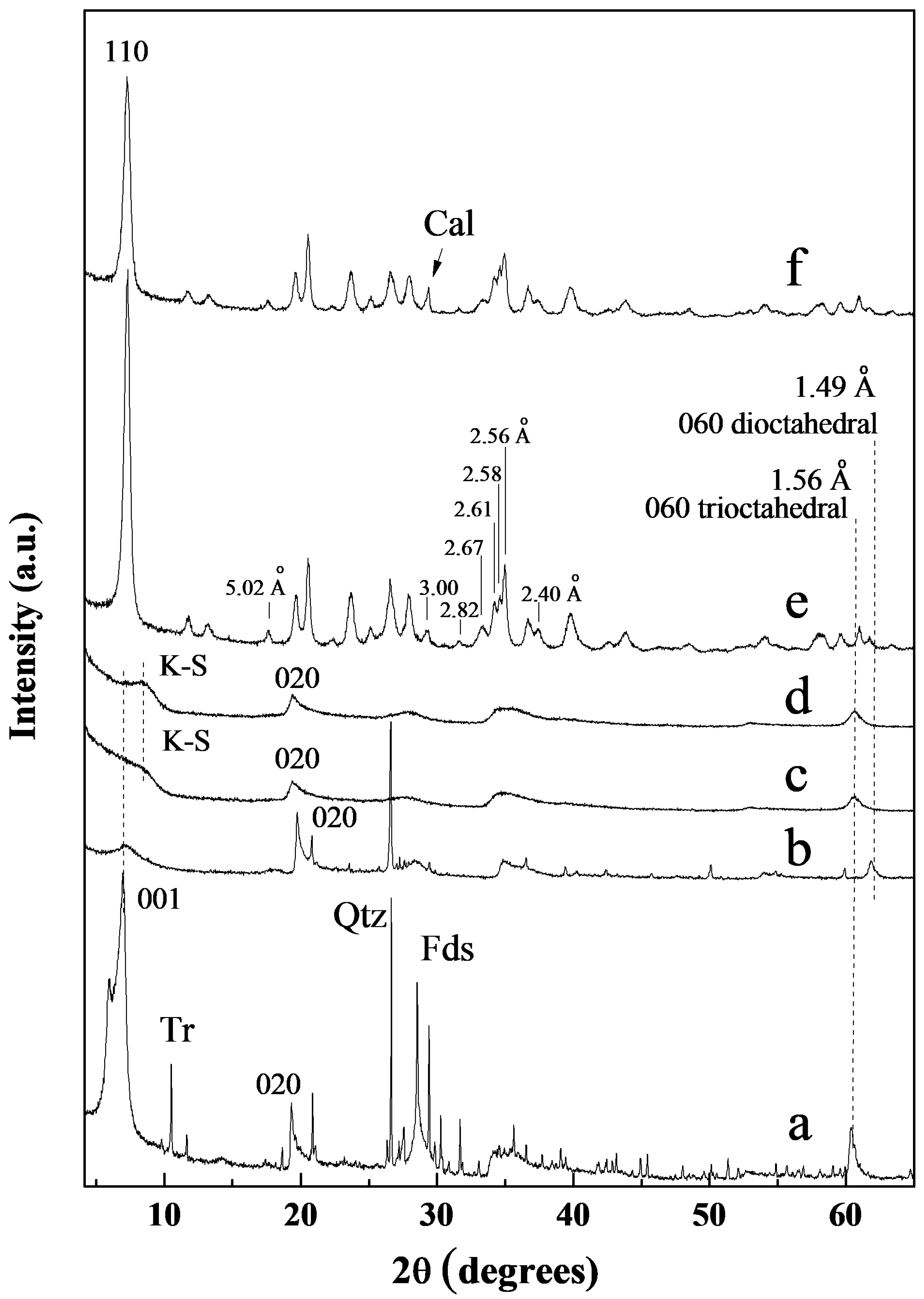
3.1. X-Ray Diffraction of the Starting Minerals
3.1.1. Bentonite
3.1.2. Kerolitic Clay
3.1.3. Sepiolite
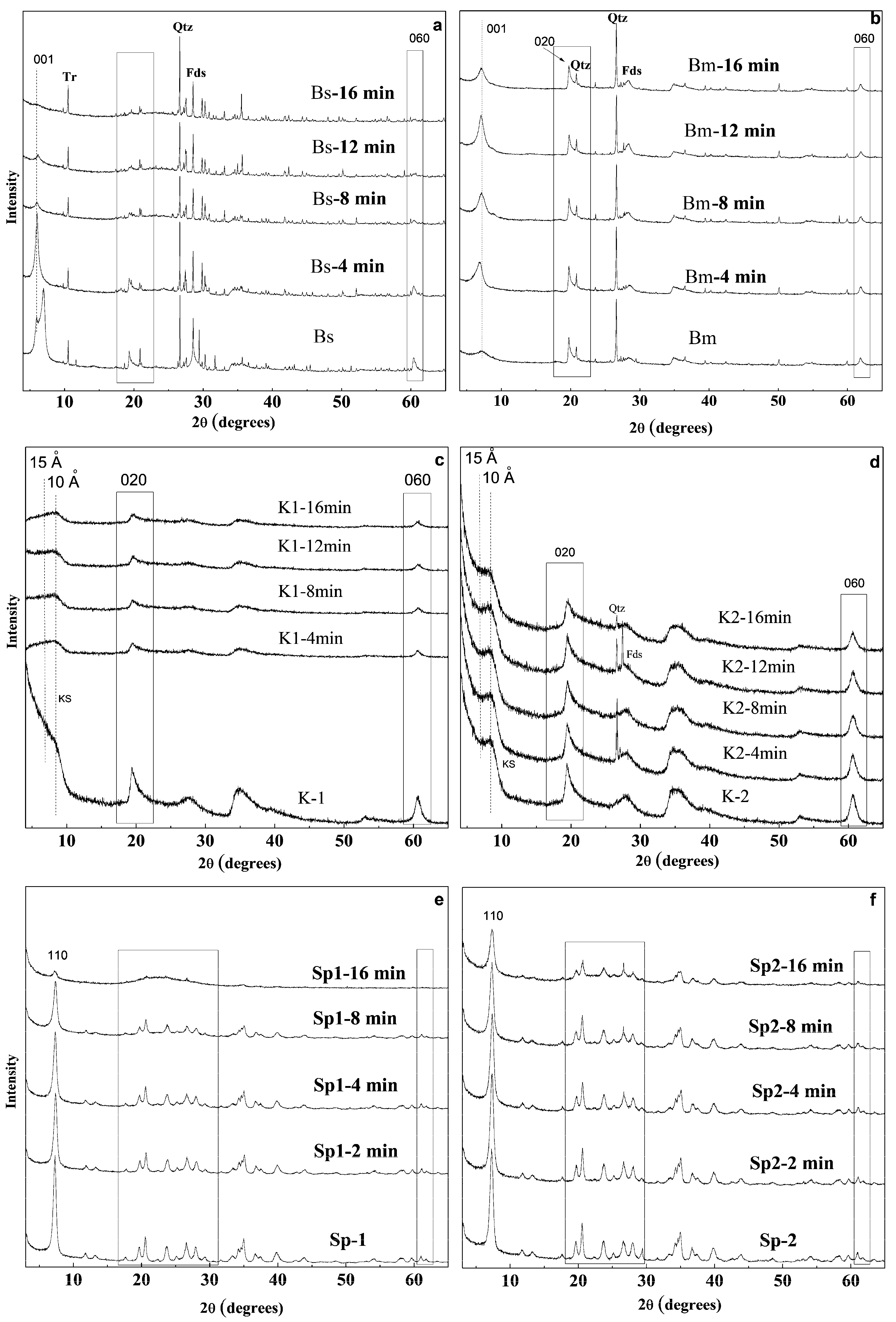
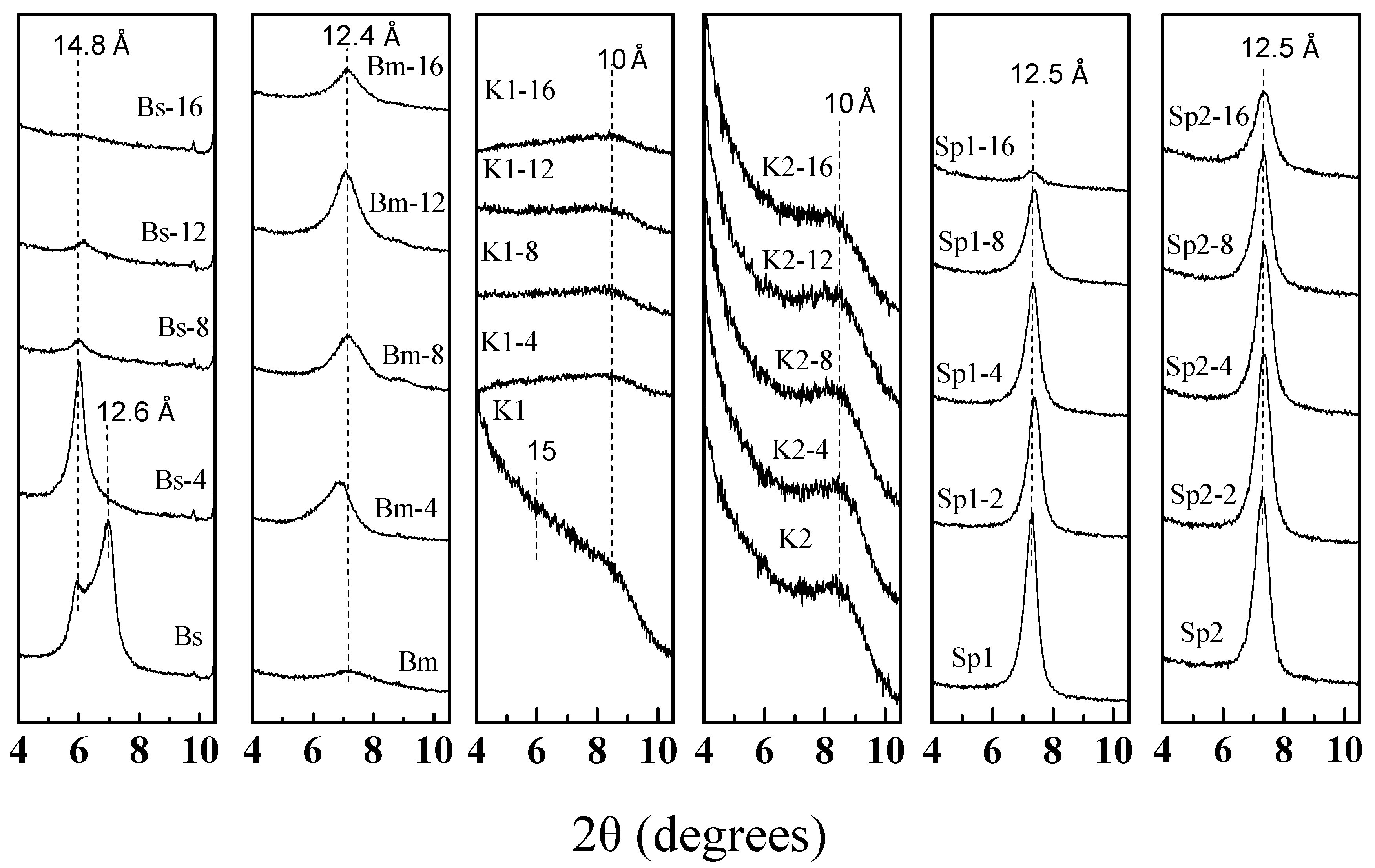
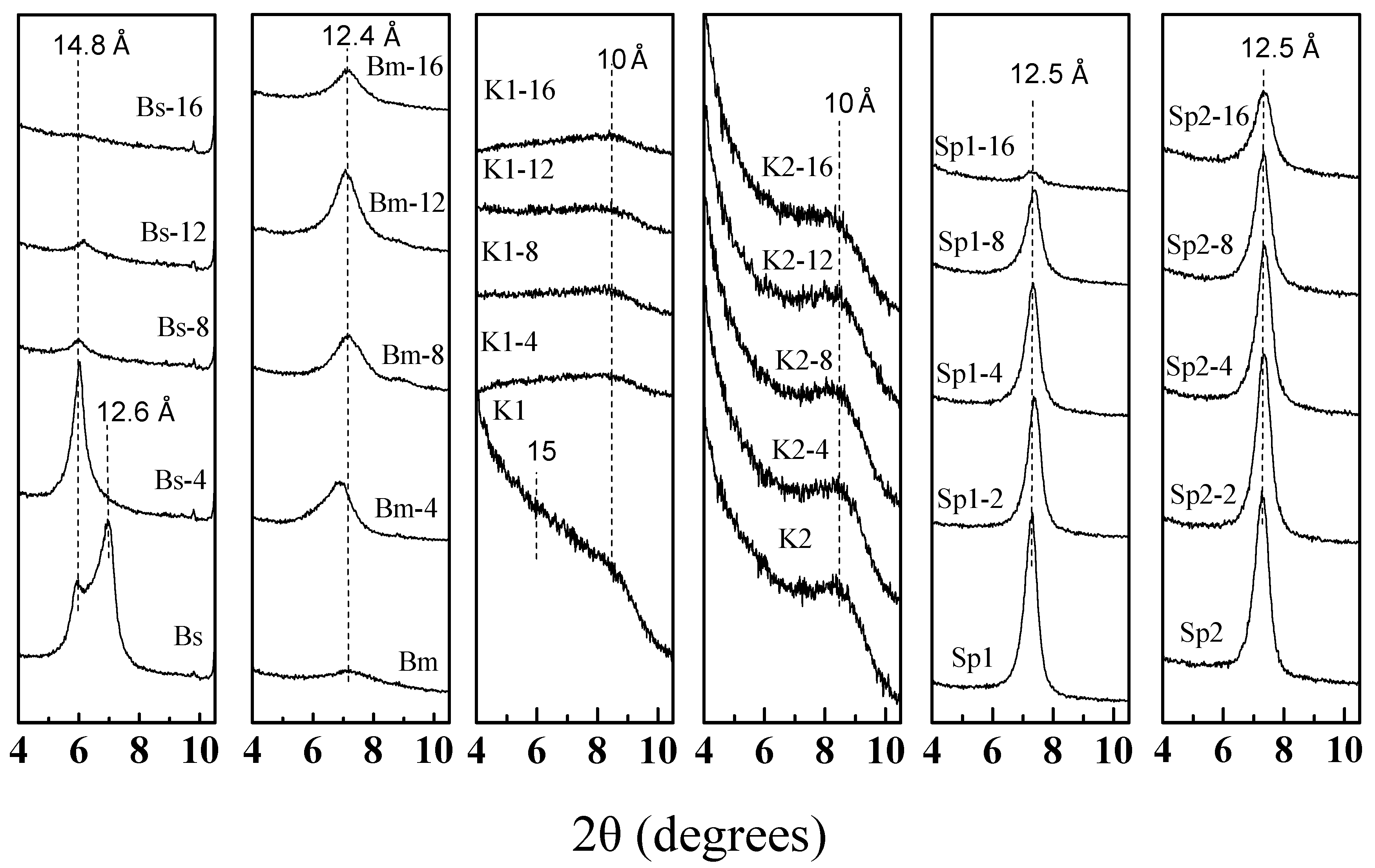
3.2. X-Ray Diffraction of the Microwave-Assisted Acid-Treated Samples
3.2.1. Bentonite
3.2.2. Kerolitic Clay
3.2.3. Sepiolite
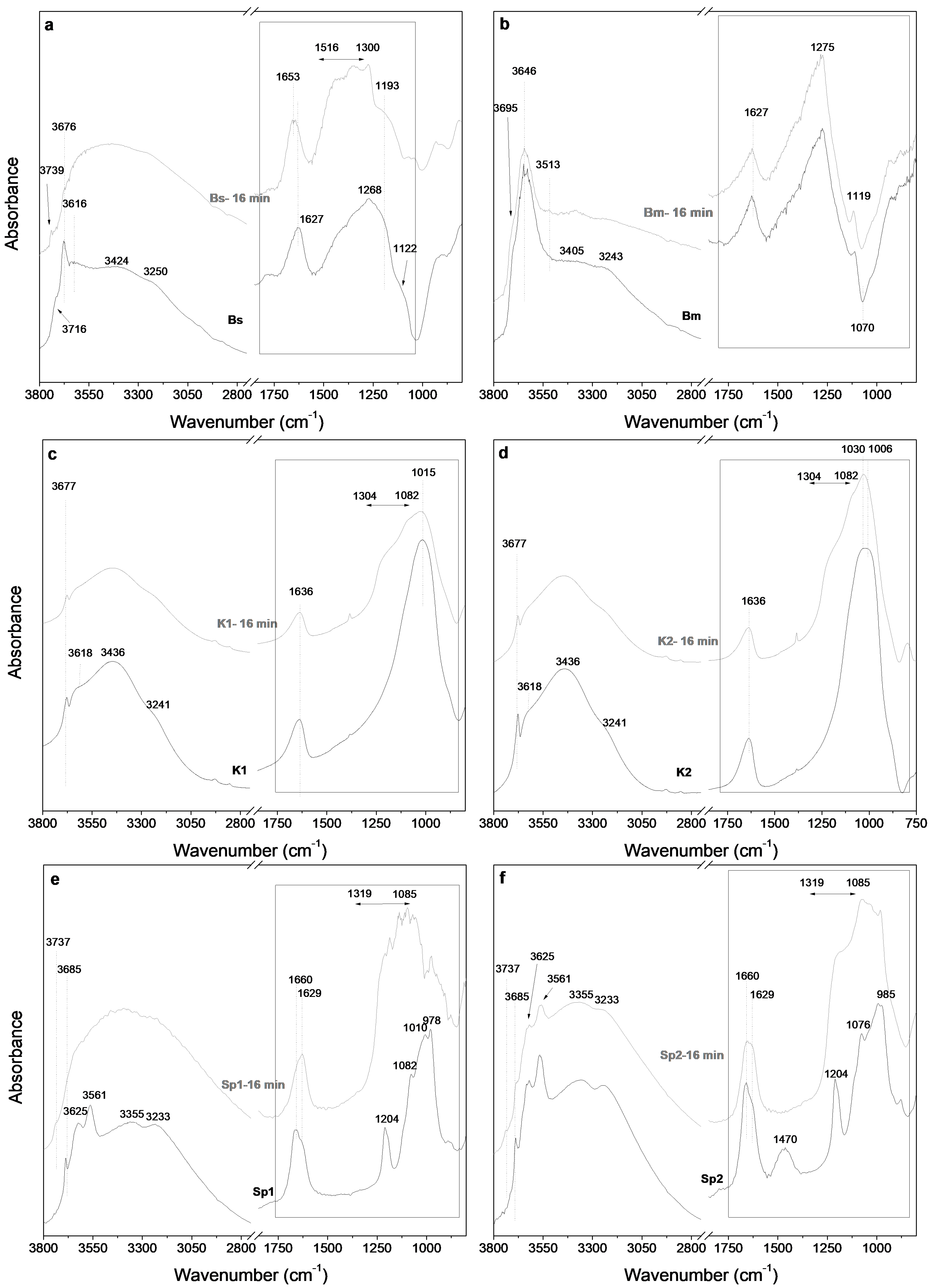
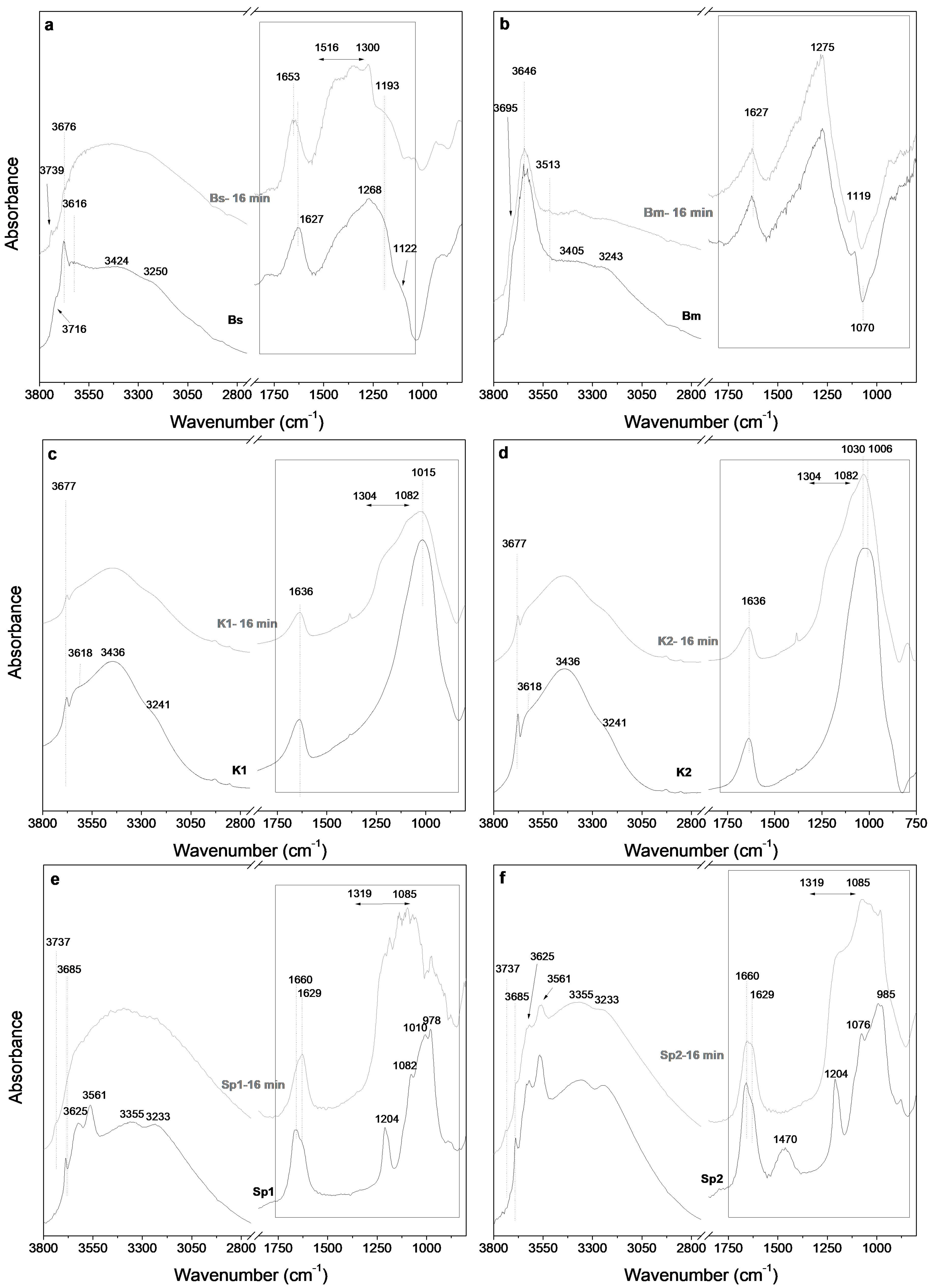
3.3. Fourier Transform Infrared Spectroscopy Study of the Starting Samples and the Microwave-Assisted Acid-Treated Samples
3.3.1. Bentonite
3.3.2. Kerolitic Clay
3.3.3. Sepiolite
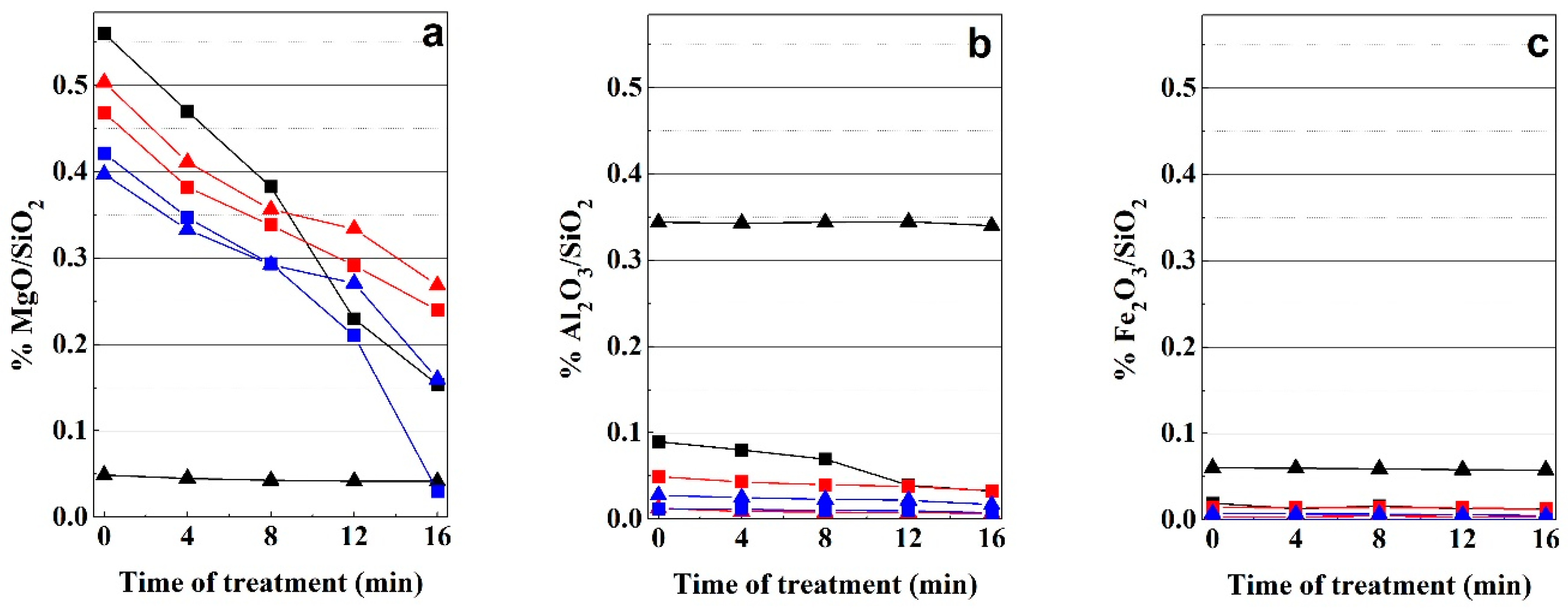
3.4. Evolution of the Chemical Composition
3.4.1. Bentonite
3.4.2. Kerolitic Clay
3.4.3. Sepiolite
3.5. SEM Examination
3.5.1. Bentonite
3.5.2. Kerolitic Clay
3.5.3. Sepiolite
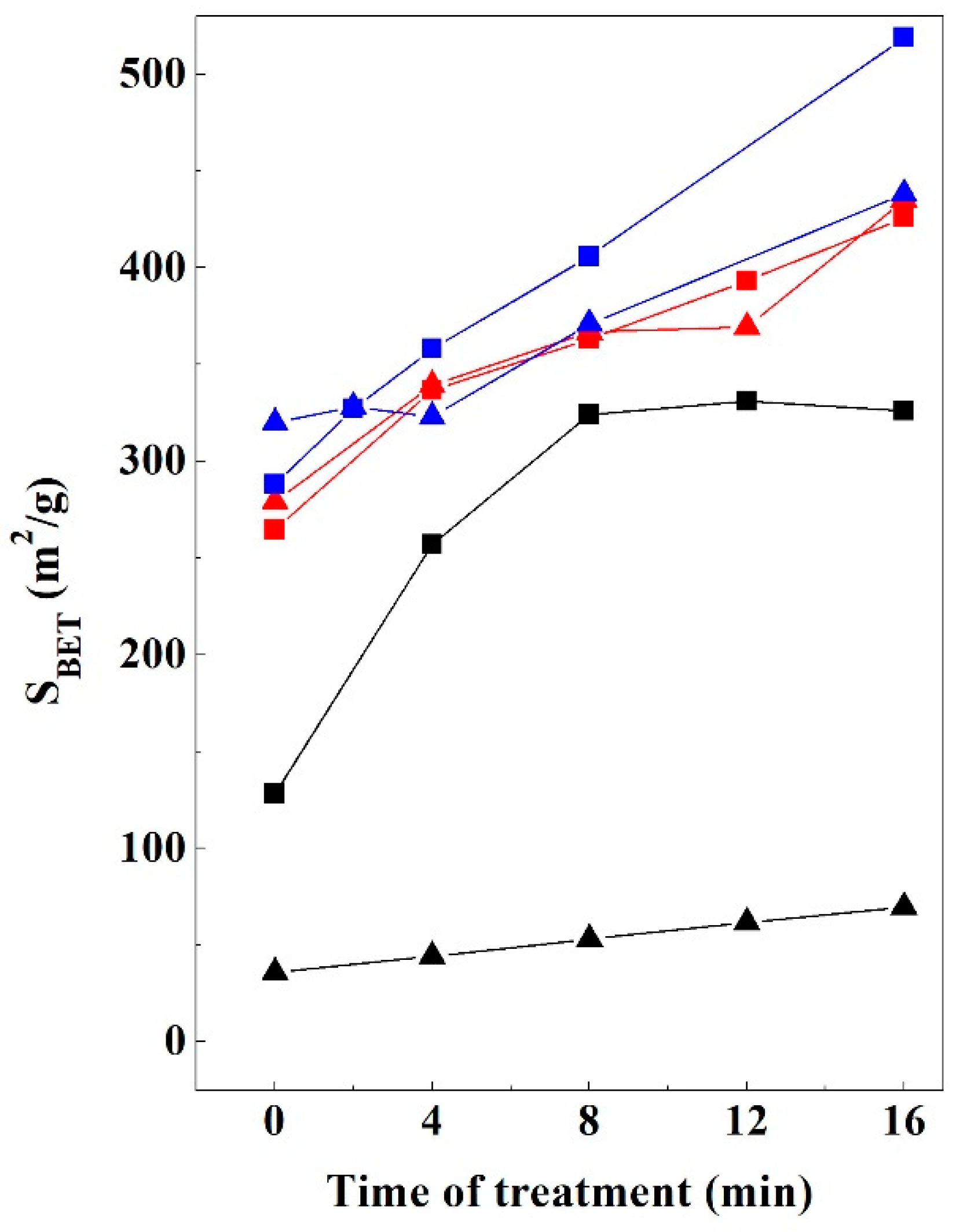
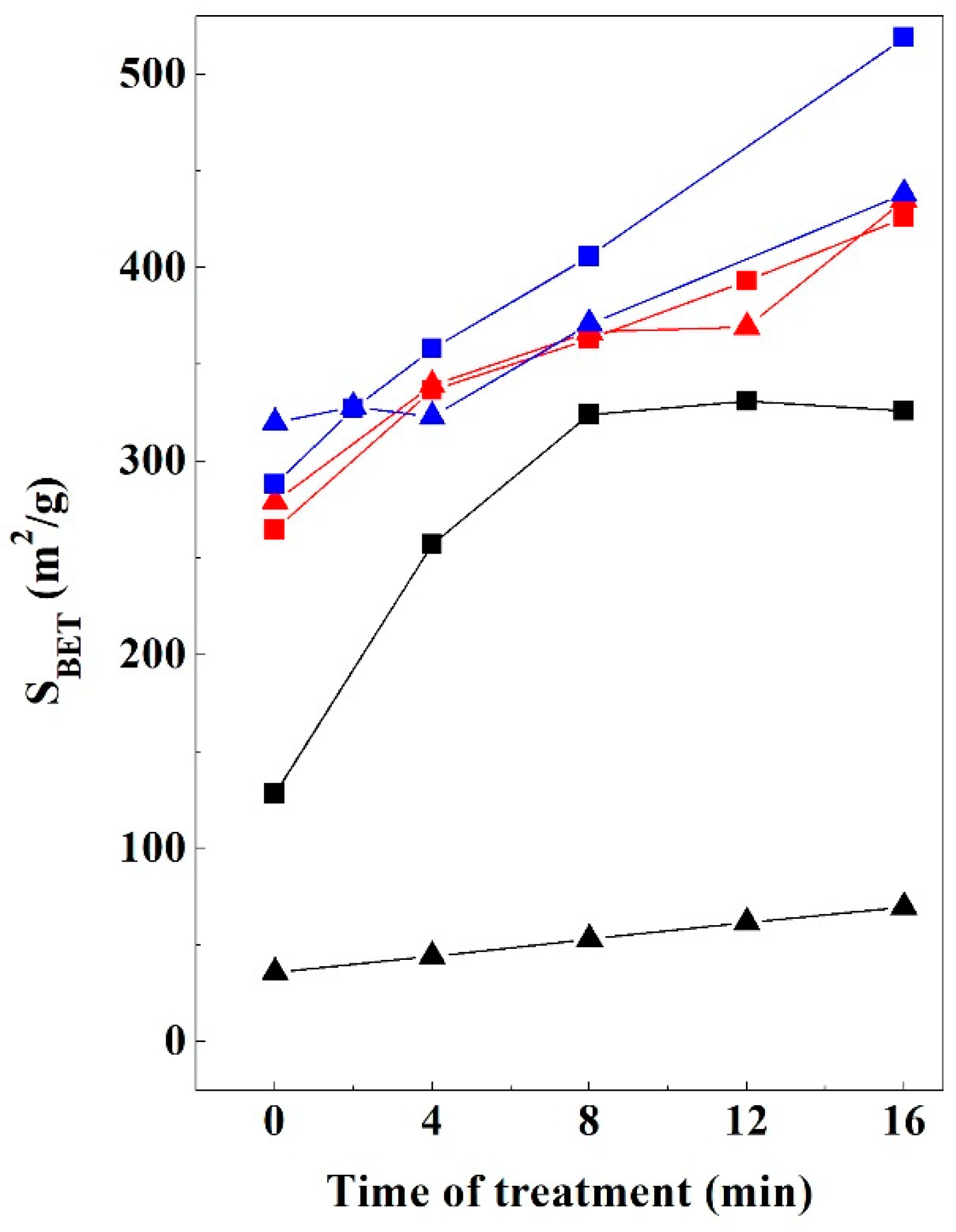
3.6. Textural Properties Evolution
3.6.1. Bentonite
3.6.2. Kerolitic Clay
3.6.3. Sepiolite
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Guggenheim, S.; Adams, J.M.; Bain, D.C.; Bergaya, F.; Brigatti, M.F.; Drits, V.A.; Formoso, M.L.L.; Galán, E.; Kogure, T.; Stanjek, H. Summary of recommendations of nomenclature committees relevant to clay mineralogy: Report of the Association Internationale pour l’Etude des Argiles (AIPEA) Nomenclature Committee for 2006. Clays Clay Miner. 2006, 54, 761–772. [Google Scholar] [CrossRef]
- Guggenheim, S.; Adams, J.M.; Bain, D.C.; Bergaya, F.; Brigatti, M.F.; Drits, V.A.; Formoso, M.L.L.; Galán, E.; Kogure, T.; Stanjek, H. Summary of recommendations of nomenclature committees relevant to clay mineralogy: Report of the Association Internationale pour l’Etude des Argiles (AIPEA) Nomenclature Committee for 2006. Clay Miner. 2006, 41, 863–878. [Google Scholar] [CrossRef]
- Brindley, G.W.; Brown, G. X-ray diffraction procedures for clay mineral identification. In Crystal Structures of Clay Minerals and Their X-Ray Identification; Brindley, G.W., Brown, G., Eds.; Mineralogical Society: London, UK, 1980; pp. 305–356. ISBN 9780903056373. [Google Scholar]
- Eberl, D.D.; Jones, B.F.; Khoury, H.N. Mixed layer kerolite-stevensite from the Amargosa Desert, Nevada. Clays Clay Miner. 1982, 30, 321–326. [Google Scholar] [CrossRef]
- Martin de Vidales, J.L.; Pozo, M.; Alia, J.M.; Garcia Navarro, F.; Rull, F. Kerolite-stevensite mixed-layers from the Madrid Basin, Central Spain. Clay Miner. 1991, 26, 329–342. [Google Scholar] [CrossRef]
- Pozo, M.; Casas, J. Origin of kerolite and associated Mg clays in palustrine-lacustrine environments. The Esquivias deposit (Neogene Madrid Basin, Spain). Clay Miner. 1999, 34, 395–418. [Google Scholar] [CrossRef]
- Dekov, V.M.; Cuadros, J.; Shanks, W.C.; Koski, R.A. Deposition of talc-kerolite-smectite-smectite at seafloor hydrothermal vent fields: Evidence from mineralogical, geochemical and oxygen isotope studies. Chem. Geol. 2008, 247, 171–194. [Google Scholar] [CrossRef]
- Steudel, A.; Friedrich, F.; Schuhmann, R.; Friedrich, R.; Ruf, U.; Sohling, U.; Emmerich, K. Characterization of a fine-grained interstratification of turbostratic talc and saponite. Minerals 2017, 7, 5. [Google Scholar] [CrossRef]
- Miller, A.K.; Guggenheim, S.; Van Groos, A.F.K. Bond energy of adsorbed and interlayer water kerolite dehydration at elevated pressures. Clays Clay Miner. 1991, 39, 127–130. [Google Scholar] [CrossRef]
- Brindley, G.W.; Sempels, R.E. Preparation and properties of some hydroxy-aluminium beidellites. Clay Miner. 1977, 12, 229–237. [Google Scholar] [CrossRef]
- Murray, H.H. Traditional and new applications for kaolin, smectite, and palygorskite: A general overview. Appl. Clay Sci. 2000, 17, 207–221. [Google Scholar] [CrossRef]
- González-Pradas, E.; Socias-Viciana, M.; Saifi, M.; Ureña-Amate, M.D.; Flores-Cespedes, F.; Fernández-Pérez, M.; Villafrenca-Sánchez, M. Adsorption of chloridazon from aqueous solution on heat and acid treated sepiolites. Water Res. 2005, 39, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Tan, Ö.; Yilmaz, L.; Zaimoglu, A.S. Variation of some engineering properties of clays with heat treatment. Mater. Lett. 2004, 58, 1176–1179. [Google Scholar] [CrossRef]
- Kara, M.; Yuzer, H.; Sabah, E.; Celik, M.S. Adsorption of cobalt from aqueous solutions onto sepiolite. Water Res. 2003, 37, 224–232. [Google Scholar] [CrossRef]
- Dekany, I.; Turi, L.; Fonseca, A.; Nagy, B.J. The structure of acid treated sepiolites: Small-angle X-ray scattering and multi MAS-NMR investigations. Appl. Clay Sci. 1999, 14, 141–160. [Google Scholar] [CrossRef]
- Balci, S. Thermal decomposition of sepiolite and variations in pore structure with and without acid pre-treatment. J. Chem. Technol. Biotechnol. 1996, 66, 72–78. [Google Scholar] [CrossRef]
- Jiménez-López, A.; López-González, J.D.; Ramírez-Sáenz, A.; Rodríguez-Reinoso, F.; Valenzuela-Calahorro, C.; Zurita-Herrera, L. Evolution of surface area in a sepiolite as a function of acid and heat treatments. Clay Miner. 1978, 13, 375–385. [Google Scholar] [CrossRef]
- Inukai, K.; Miyawaki, R.; Tomura, S.; Shimosaka, K.; Irkec, T. Purification of Turkish sepiolite through hydrochloric acid treatment. Appl. Clay Sci. 1994, 9, 11–29. [Google Scholar] [CrossRef]
- Aznar, A.J.; Gutiérrez, E.; Díaz, P.; Álvarez, A.; Poncelet, G. Silica from sepiolite: Preparation, textural properties, and use as support to catalyst. Microporous Mater. 1996, 6, 105–114. [Google Scholar] [CrossRef]
- Sabah, E.; Turan, M.; Çelik, M.S. Adsorption mechanism of canonic surfactants onto acid and heat-activated sepiolites. Water Res. 2002, 36, 3957–3964. [Google Scholar] [CrossRef]
- Vico, L.I. Acid-base behaviour and Cu2+ and Zn2+ complexation properties of the sepiolite/water interface. Chem. Geol. 2003, 198, 213–222. [Google Scholar] [CrossRef]
- Kendall, T. Smectite Clays. In Industrial Clays; Kendall, T., Ed.; Industrial Minerals Information Ltd.: London, UK, 1996; pp. 1–12. [Google Scholar]
- Čičel, B.; Komadel, P. Structural formulae of layer silicates. In Quantitative Methods in Soil Mineralogy; Luxmoore, R.J., Ed.; Soil Science Society of America, Inc.: Madison, WI, USA, 1994; pp. 114–136. [Google Scholar]
- Kaviratna, H.; Pinnavaia, T.J. Acid hydrolysis of octahedral Mg2+ Sites in 2:1 layered silicates: An assessment of edge attack and gallery access mechanisms. Clays Clay Miner. 1994, 42, 717–723. [Google Scholar] [CrossRef]
- Komadel, P.; Madejová, J. Acid activation of clay minerals. In Handbook of Clay Science; Bergaya, F., Theng, B.K.G., Lagaly, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 1, pp. 263–287. [Google Scholar]
- Rice, N.M.; Strong, L.W. The leaching oflateritic nickel ores in hydrochloric acid. Can. Metall. Quart. 1974, b13, 485–493. [Google Scholar] [CrossRef]
- Komadel, P.; Schmidt, D.; Madejová, J.; Čičel, B. Alteration of smectites by treatments with hydrochloric acid and sodium carbonate solutions. Appl. Clay Sci. 1990, 5, 113–122. [Google Scholar] [CrossRef]
- Komadel, P.; Bujdák, J.; Madejová, J.; Šucha, V.; Elsass, F. Effect of non-swelling layers on the dissolution of reduced-charge montmorillonite in hydrochloric acid. Clay Miner. 1996, 31, 333–345. [Google Scholar] [CrossRef]
- Vicente-Rodríguez, M.A.; López González, J.D.; Bañares-Muñoz, M.A. Preparation of microporous solids by acid treatment of a saponite. Microporous Mater. 1995, 4, 251–264. [Google Scholar] [CrossRef]
- Tkáč, I.; Komadel, P.; Müller, D. Acid-treated montmorillonites-A study by 29Si and 27Al MAS NMR. Clay Miner. 1994, 29, 11–19. [Google Scholar] [CrossRef]
- Breen, C.; Madejová, J.; Komadel, P. Characterization of moderately acid-treated, size-fractionated montmorillonites using IR and MAS NMR spectroscopy and thermal analysis. J. Mater. Chem. 1995, 5, 469–474. [Google Scholar] [CrossRef]
- Adams, J.M.; McCabe, R.W. Clay Minerals as Catalysts. In Handbook of Clay Science; Bergaya, F., Theng, B.K.G., Lagaly, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 1, pp. 541–581. [Google Scholar]
- García Carmona, J.; Rodriguez Clemente, R.; Gómez Morales, J. Comparative preparation of microporous VPI-5 using conventional and microwave heating techniques. Zeolites 1997, 18, 340–346. [Google Scholar] [CrossRef]
- Choudhary, V.R.; Tillu, V.H.; Narkhede, V.S.; Borate, H.B.; Wakharkar, R.D. Microwave assisted solvent-free synthesis of dihydropyrimidinones by Biginelli reaction over Si-MCM-41 supported FeCl3 catalyst. Catal Commun. 2003, 4, 449–453. [Google Scholar] [CrossRef]
- Deksnys, T.P.; Menezes, R.R.; Fagury-Neto, E.; Kiminami, R.H.G.A. Synthesis of Al2O3/SiC in microwave oven: Study of the processing parameters. Cerâmica 2005, 51, 343–348. [Google Scholar] [CrossRef]
- Deng, S.G.; Lin, Y.S. Microwave heating synthesis of supported sorbents. Chem. Eng. Sci. 1997, 52, 1563–1575. [Google Scholar] [CrossRef]
- Roy, R.; Komarneni, S.; Yang, J.L. Controlled microwave heating and melting of gels. J. Am. Ceram. Soc. 1985, 68, 392–395. [Google Scholar] [CrossRef]
- Korichi, S.; Elias, A.; Mefti, A. Characterization of smectite after acid activation with microwave irradiation. Appl. Clay Sci. 2009, 42, 432–438. [Google Scholar] [CrossRef]
- Franco, F.; Pozo, M.; Cecilia, J.A.; Benítez-Guerrero, M.; Pozo, E.; Martín Rubí, J.A. Microwave assisted acid treatment of sepiolite: The role of composition and “crystallinity”. Appl. Clay Sci. 2014, 102, 15–27. [Google Scholar] [CrossRef]
- Franco, F.; Pozo, M.; Cecilia, J.A.; Benítez-Guerrero, M.; Lorente, M. Effectiveness of microwave assisted acid treatment on dioctahedral and trioctahedral smectites. The influence of octahedral composition. Appl. Clay Sci. 2016, 120, 70–80. [Google Scholar] [CrossRef]
- Martín de Vidales, J.L.; Pozo, M.; Medina, J.A.; Leguey, S. Formación de sepiolita-paligorskita en litofacies lutítico-carbonáticas en el sector de Borox-Esquivias (Cuenca de Madrid). Estud. Geol. 1988, 44, 7–18. [Google Scholar] [CrossRef]
- Santiago Buey, C.; Suárez Barrios, M.; García Romero, E.; Doval Montoya, M. Mg-Rich Smectite “Precursor” Phase in the Tagus Basin, Spain. Clays Clay Miner. 2000, 48, 366–373. [Google Scholar] [CrossRef]
- Pozo, M.; Calvo, J.P. Madrid Basin (Spain): A natural lab for the formation and evolution of magnesian clay minerals. In Magnesian Clays: Characterization Origin and Applications; Pozo, M., Galán, E., Theng, B.K.G., Lagaly, G., Eds.; AIPEA Pub.: Rio de Janeiro, Brazil, 2015. [Google Scholar]
- Galán, E.; Pozo, M. Palygorskite and sepiolite deposits in continental environments. Description, genetic patterns and sedimentary settings. In Developments in Palygorskite–Sepiolite Research. A New Outlook on These Nanomaterials. Developments in Clay Science; Galán, E., Singer, A., Eds.; Elsevier: Amsterdam, The Netherlands, 2011; Volume 3, pp. 125–173. [Google Scholar]
- Lippens, B.C.; De Boer, J.H. Studies on pore systems in catalysts: V. The t method. J. Catal. 1965, 4, 319–323. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Ceram. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Galán, E.; Aparicio, P. Methodology for the identification and characterization of magnesian clays. In Magnesian Clays: Characterization, Origin and Applications; AIPEA Educational, Series; Pozo, M., Galán, E., Eds.; Digilabs: Bari, Italy, 2015; Volume 2, pp. 63–121. [Google Scholar]
- Sánchez del Río, M.; García Romero, E.; Martínez Criado, G. Variability in sepiolite: Diffraction studies. Am. Miner. 2011, 96, 1443–1454. [Google Scholar] [CrossRef]
- Pozo, M.; Calvo, J.P.; Pozo, E.; Moreno, A. Genetic constraints on crystallinity, thermal behaviour and surface area of sepiolite from the Cerro de los Batallones deposit (Madrid Basin, Spain). Appl. Clay Sci. 2014, 91, 30–45. [Google Scholar] [CrossRef]
- Van der Marel, H.W.; Beutelspacher, H. Atlas of Infrared Spectroscopy of Clay Minerals and Their Admixtures; Elsevier: Amsterdam, The Netherlands, 1976; Volume 396. [Google Scholar]
- Kloprogge, J.T.; Frost, R.L. Study of the thermal behavior of rectorite by in-situ infrared emission spectroscopy. N. Jb. Miner. Mh. 2000, 4, 145–157. [Google Scholar]
- Bisio, C.; Boccaleri, E.; Marchese, L.; Superti, G.B.; Pastore, H.O.; Thommes, M. Understanding physico-chemical properties of saponite synthetic clays. Microporous Mesoporous Mater. 2008, 107, 90–101. [Google Scholar] [CrossRef]
- Janin, A.; Maache, M.; Lavalley, J.C.; Joly, J.F.; Raatz, F.; Szydlowski, N. FT-IR study of the silanol groups in dealuminated HY zeolites: Nature of the extraframe debris. Zeolites 1991, 11, 391–396. [Google Scholar] [CrossRef]
- Weitkamp, J. Zeolites and catalysis. Solid State Ion. 2000, 131, 175–188. [Google Scholar] [CrossRef]
- Zviagina, B.B.; McCarty, D.K.; Środoń, J.; Drits, V.A. Interpretation of infrared spectra of dioctahedral smectites in the region of OH-stretching vibrations. Clays Clay Miner. 2004, 52, 399–410. [Google Scholar] [CrossRef]
- Madejová, J. FTIR techniques in clay mineral studies. Vib. Spectrosc. 2003, 31, 1–10. [Google Scholar] [CrossRef]
- Farmer, V.C. The Infrared Spectra of Minerals; Mineralogical Society: London, UK, 1974; p. 539. [Google Scholar]
- Anderson, T.H.; Weaver, F.W.; Owen, N.L. Anomalies in diffuse reflectance infrared spectra of wood and wood polymers. J. Mol. Struct. 1991, 249, 257–275. [Google Scholar] [CrossRef]
- Madejová, J.; Bujdák, J.; Janek, M.; Komadel, P. Comparative FT-IR study of structural modifications during acid treatment of dioctahedral smectites and hectorite. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1998, 54, 1397–1406. [Google Scholar] [CrossRef]
- Valentín, J.L.; López-Manchado, M.A.; Rodríguez, A.; Posadas, P.; Ibarra, L. Novel anhydrous unfolded structure by heating of acid pre-treated sepiolite. Appl. Clay Sci. 2007, 36, 245–255. [Google Scholar] [CrossRef]
- Frost, R.L.; Locos, O.B.; Ruan, H.; Kloprogge, J.T. Near-infrared and mid-infrared spectroscopic study of sepiolites and palygorskites. Vib. Spectrosc. 2001, 27, 1–13. [Google Scholar] [CrossRef]
- Frost, R.L.; Cash, G.A.; Kloprogge, J.T. ‘Rocky Mountain leather’, sepiolite and attapulgite-an infrared emission spectroscopic study. Vib. Spectrosc. 1998, 16, 173–184. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | % SiO2 | % Al2O3 | % MgO | % Fe2O3 | % Na2O | % K2O | % CaO | % MnO | % P2O5 | % TiO2 |
|---|---|---|---|---|---|---|---|---|---|---|
| Bs | 53.73 | 4.82 | 30.08 | 1.03 | 2.76 | 0.55 | 5.34 | 0.03 | 0.11 | 0.53 |
| Bs-16 min | 77.11 | 2.51 | 16.49 | 0.98 | 0.00 | 0.71 | 5.59 | 0.02 | 0.03 | 0.83 |
| Bm | 65.75 | 22.61 | 3.21 | 3.96 | 1.67 | 0.60 | 1.65 | 0.02 | 0.09 | 0.15 |
| Bm-16 min | 68.52 | 23.30 | 2.85 | 3.97 | 0.30 | 0.47 | 0.20 | 0.01 | 0.04 | 0.15 |
| K1 | 53.94 | 2.66 | 25.26 | 0.77 | 0.09 | 0.59 | 0.32 | 0.01 | 0.05 | 0.15 |
| K1-16 min | 67.75 | 2.22 | 16.24 | 0.88 | 0.07 | 0.64 | 0.12 | 0.02 | 0.04 | 0.19 |
| K2 | 55.00 | 0.70 | 27.70 | 0.20 | 0.03 | 0.15 | 0.20 | 0.01 | 0.04 | 0.10 |
| K2-16 min | 66.49 | 0.45 | 17.84 | 0.23 | 0.03 | 0.13 | 0.23 | 0.01 | 0.03 | 0.09 |
| Sp1 | 56.89 | 0.69 | 23.97 | 0.02 | 0.01 | 0.20 | 0.59 | 0.05 | 0.04 | 0.03 |
| Sp1-16 min | 81.77 | 0.59 | 2.40 | 0.03 | 0.02 | 0.23 | 0.60 | 0.04 | 0.04 | 0.08 |
| Sp2 | 56.41 | 1.56 | 22.39 | 0.44 | 0.03 | 0.22 | 0.82 | 0.05 | 0.04 | 0.22 |
| Sp2-16 min | 70.48 | 1.18 | 11.57 | 0.36 | 0.04 | 0.21 | 0.06 | 0.05 | 0.04 | 0.07 |
| SBET | Stext | Stmicro | SBET | Stext | Stmicro | ||
| Sample | (m2·g−1) | (m2·g−1) | (m2·g−1) | Sample | (m2·g−1) | (m2·g−1) | (m2·g−1) |
| Degasification Temperature | 200 °C | Degasification Temperature | 200 °C | ||||
| M | 49 | 25 | 24 | K1 | 265 | 101 | 163 |
| M-4 min | 49 | 22 | 27 | K1-4 min | 337 | 139 | 198 |
| M-8 min | 56 | 28 | 28 | K1-8 min | 363 | 155 | 209 |
| M12 min | 61 | 32 | 29 | K1-12 min | 393 | 159 | 234 |
| M-16 min | 62 | 33 | 29 | K1-16 min | 426 | 195 | 230 |
| S | 128 | 76 | 52 | K2 | 279 | 167 | 112 |
| S-4 min | 257 | 128 | 129 | K2-4 min | 339 | 236 | 104 |
| S-8 min | 324 | 138 | 186 | K2-8 min | 367 | 241 | 126 |
| S-12 min | 331 | 114 | 217 | K2-12 min | 369 | 248 | 121 |
| S-16 min | 326 | 106 | 221 | K2-16 min | 435 | 266 | 170 |
| SBET | Stext | Stmicro | SBET | Stext | Stmicro | Best SBET (m2·g−1) | |
| Sample | (m2·g−1) | (m2·g−1) | (m2·g−1) | (m2·g−1) | (m2·g−1) | (m2·g−1) | |
| Degasification Temperature | 200 °C | 120 °C | |||||
| Sp1 | 117 | 102 | 15 | 288 | 117 | 171 | 288 |
| Sp1-2 min | 175 | 132 | 43 | 327 | 161 | 166 | 327 |
| Sp1-4 min | 244 | 150 | 94 | 358 | 173 | 185 | 358 |
| Sp1-8 min | 378 | 191 | 187 | 406 | 222 | 184 | 406 |
| Sp1-16 min | 519 | 437 | 82 | 437 | 348 | 89 | 519 |
| Sp2 | 133 | 111 | 22 | 320 | 127 | 193 | 320 |
| Sp2-2 min | 154 | 133 | 21 | 328 | 170 | 158 | 328 |
| Sp2-4 min | 323 | 187 | 136 | 280 | 174 | 106 | 323 |
| Sp2-8 min | 371 | 174 | 197 | 371 | 176 | 141 | 371 |
| Sp2-16 min | 438 | 197 | 241 | 396 | 205 | 191 | 438 |
| Type of Clay | Sample | Main Characteristic | XRD Modification | FTIR Amorphous Si–O Bands | % Octahedral Cations Loss | ΔSBET (m2/g) | %ΔSBET |
|---|---|---|---|---|---|---|---|
| Bentonite | Bs | trioctahedral smectite | 020 and 060 reflections disappears | YES | 71 | 198 | 155 |
| Bm | dioctahedral smectite | 020 and 060 reflections appears to be unaffected | NO | 3 | 13 | 27 | |
| Kerolitic clay | K1 | stevensite > kerolite | 020 and 060 reflections strongly decreased | YES | 49 | 161 | 61 |
| K2 | stevensite < kerolite | 020 and 060 reflections slightly decreased | YES | 47 | 156 | 56 | |
| Sepiolite | Sp1 | fibres: thinner, longer | reflections between 18 and 32° 2θ disappear and replaced with a broad band | YES | 93 | 231 | 80 |
| Sp2 | fibres: wider, shorter | reflections between 18 and 32° 2θ slightly decrease | YES | 60 | 118 | 37 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cecilia, J.A.; Pardo, L.; Pozo, M.; Bellido, E.; Franco, F. Microwave-Assisted Acid Activation of Clays Composed of 2:1 Clay Minerals: A Comparative Study. Minerals 2018, 8, 376. https://doi.org/10.3390/min8090376
Cecilia JA, Pardo L, Pozo M, Bellido E, Franco F. Microwave-Assisted Acid Activation of Clays Composed of 2:1 Clay Minerals: A Comparative Study. Minerals. 2018; 8(9):376. https://doi.org/10.3390/min8090376
Chicago/Turabian StyleCecilia, Juan Antonio, Laura Pardo, Manuel Pozo, Eva Bellido, and Francisco Franco. 2018. "Microwave-Assisted Acid Activation of Clays Composed of 2:1 Clay Minerals: A Comparative Study" Minerals 8, no. 9: 376. https://doi.org/10.3390/min8090376
APA StyleCecilia, J. A., Pardo, L., Pozo, M., Bellido, E., & Franco, F. (2018). Microwave-Assisted Acid Activation of Clays Composed of 2:1 Clay Minerals: A Comparative Study. Minerals, 8(9), 376. https://doi.org/10.3390/min8090376