Abstract
Accurate calculation of rate constants for gas-phase OH-oxidation reactions of fluorinated compounds is crucial for the understanding of atmospheric processes that are subject of the Kigali Agreement. Here, we have determined two such rate constants for two hydrofluoroketones, HFK-447mcc and HFK-465mc. The calculations were performed with a cost-effective multiconformer transition state theory protocol coupled with the constrained transition state randomization sampling method. The calculated rate constants of and at 298.15 K imply an atmospheric lifetime of 10 years and 1 year, respectively. To our knowledge, these rate constants have never been determined experimentally or theoretically, and the similarity between the ratios of these two rate constants and of the well-studied acetone and diethyl ketone suggest the validity of our approach toward obtaining accurate rate constants and branching ratios.
1. Introduction
After it became clear that CFC compounds were directly connected to ozone depletion and global warming [1,2,3,4,5], theoretical and experimental efforts in the search for environmental-friendly CFC replacements have been vastly increased. This field of research gained extra relevance after October 2016, when almost 200 nations adopted the Kigali Amendment to the Montreal Protocol [6]. The ratification of this protocol urges countries to phase down production and the use of hydrofluorocarbons (HFCs), thus reinforcing the urgency of finding environmentally friendly alternatives, as these second-generation replacements have [7] elevated values of global warming potential (GWP). One of the most important parameters defining the GWP of a compound is the value of its atmospheric lifetime [8]. One potential solution for third-generation replacements in industrial and commercial applications [9] is provided by fluorine-containing oxygenated volatile organic compounds (OVOCs), since their atmospheric lifetimes are frequently decreased due to the presence of oxygen-containing functional groups. These functional groups promote long-range activating effects with respect to hydrogen-atom abstraction at sites remote from the substituent group, increasing the atmospheric reactivity when compared with the corresponding non-oxygenated fluorinated compounds [10,11,12]. Compounds such as fluorinated alcohols, aldehydes, ethers and ketones have a variety of industrial uses [9] and can gradually substitute HFCs, such as required by the Kigali Amendment to the Montreal Protocol.
Specifically, the atmospheric chemistry of ketones is important because they can be emitted to the atmosphere from anthropogenic (e.g., chemical industries) and biogenic (e.g., plants) sources, while they can also be generated in situ by the oxidation of other VOCs in the atmosphere [11,12]. The main atmospheric degradation processes of ketones are photolysis (ketones absorb radiation in the ultraviolet region around 280 nm due to the transition of the carbonyl group) and reaction with OH radicals, with the former being more relevant at higher altitudes [10,12]. Tropospheric oxidation reactions with OH radicals typically proceed via hydrogen atom abstraction [10,12,13,14], with the reactivity of alkyl bonds at the -position being enhanced with respect to the bonds at the -position due to the deactivation/activation of the hydrogen atom by the carbonyl group at the / positions, respectively [15,16]. In other words, the rate coefficients for the alkanones generally exceed those of the equivalent alkanes [11]. Large alkyl groups will lead to hydrogen abstraction from the various sites with the respective alkoxy radicals reacting with and NO and generating a large variety of products [10,12].
Because of the recognized environment-friendly effect of the carbonyl group on the atmospheric lifetimes of ketones, hydrofluoroketones (HFKs) are considered to be potential third-generation replacements for CFCs and have been used as industrial synthesis reagents and fire extinguishing agents [9]. Naturally, for a particular HFK to be seriously considered as a potential replacement, it is necessary to determine if it has the required physicochemical properties and technical specifications. An example of such analysis has been performed on four HFKs [17]: HFK-447mcc, HFK-465mc, HFK-354pc and HFK-549mccc. The mentioned study concluded that HFK-354pc is thermally unstable, therefore rendering this particular HFK non-viable as a third-generation replacement option. Moreover, and to our knowledge, there are no experimental or theoretical investigations concerning the atmospheric lifetime of any of the remaining three HFKs. Such studies would provide an indication of the atmospheric impact of these three HFKs, and, assuming that they could all be efficiently used for the same purpose, it could also provide crucial information to potential stakeholders on which is the greener option (greener in the sense of Green Chemistry principle number 10 [18,19], design for degradation). Consequently, it is therefore important to gather relevant information about the atmospheric lifetime of these HFKs.
Considering their two main degradation pathways, the global lifetime of ketones can be approximated as [8,20]
where represents the HFK atmospheric lifetime resulting from the photolytic process with rate constant J and is the atmospheric lifetime associated with a pseudo-first-order loss process with a corresponding rate constant. However, in this study, we will discard the calculation of as we will be focusing only on the reaction between HFKs and the OH radical, for which the respective atmospheric lifetimes can be obtained by
Although photolysis is generally considered to be more relevant at higher altitudes [10,12] (larger overlap between the absorption band and the available actinic flux [21]), the complete approach would require the calculation of the photolysis rate constant J in order to determine and solve Equation (1), which is outside the scope of this investigation. Alternatively, we will provide a brief overview of what we can expect from the photolysis process. For example, the atmospheric lifetimes for 2-butanone, 2-pentanone and 2-hexanone range between 1.5 and 11 days [22], while the respective lifetimes associated with values (overhead Sun, corresponding to a solar zenith angle of 0 degrees, see Figure VIII-D-8 of Ref. [22]) range from 2.5 to 5.5 days. Under the mentioned conditions, the photolysis and OH-oxidation processes clearly compete. Interestingly, the fluorination of ketones leads to a bathochromic shift in the UV absorption bands of the carbonyl groups [23,24], enabling their overlap with actinic radiation at lower altitudes. Recent photolysis studies of several fluorinated ketones [23,24] under outdoor atmospheric conditions point to lifetimes in the range of a few days to two weeks.
In this study, our calculations will focus solely on HFK-447mcc () and HFK-465mc (), excluding HFK-549mccc (). The reason for this is that the size of the CxF2x+1 group usually has little to no effect on the
reactivity of halogenated OVOCs toward OH radicals [11], and therefore, HFK-447mcc and
HFK-549mccc should have very similar rate constants. The main goal of this study will
then be to calculate the kOH rate constants for the oxidation reactions between two HFKs
(HFK-447mcc and HFK-465mc) and OH radicals
This will be completed by using multiconformer transition state theory (MC-TST) [25,26,27,28,29,30,31,32] coupled with a newly developed transition state sampling method denominated constrained transition state randomization (CTSR) [33]. Our theoretical approach will be summarized in the next section, which is followed by a discussion of the results. The conclusions are gathered in the final section.
2. Methodology
2.1. Theoretical Background
The reaction between HFKs and is mechanistically similar to other radical-molecule reactions of atmospheric interest [34], including reactions between OVOCs and [10,13,14]. Such reaction paths feature loose complexes in both the reactant and product channels connected by a hydrogen-abstraction transition state (TS) with the underlying mechanism consisting of three steps: (1) formation of the prereaction complex (PRC) from the isolated OVOC + OH radical reactants, (2) formation of the product complex (PC) from the PRC, and (3) formation of the final products (OVOC radical and water) from the PC. For the specific case of an HFK, one obtains
If one assumes that the PRC concentration remains constant over time (steady-state assumption [35], with ), the effective rate constant for reaction (3) can be calculated as [36,37,38,39]
In the context of tropospheric reactions, it is usually found that the reverse rate constants for step 1 are much larger than the rate constant values for the irreversible unimolecular step 2 (the conditions for which are discussed in detail elsewhere [30,36,37,38,39,40,41]), which simplifies the overall rate constant to . Under such conditions, the system is assumed to be fully equilibrated and thermalized at the high-pressure limit (equilibrium-state assumption [40]), and therefore, the reactants, the PRC and the TS of step 2 can be characterized by Boltzmann equilibrium distributions. These conditions should be checked before performing the calculations, particularly if the hydrogen-abstraction TSs are submerged below the reactants. This can be achieved by calculating the Gibbs free energies along the reaction path and verifying their relative values. In many cases, the Gibbs free energy profile along the reaction coordinate will resemble the profile represented in Figure 5b of Ref. [40], providing a strong indication that will be considerably larger than and and that both the steady-state and equilibrium assumptions will be satisfied for the reaction under study [40]. In that case, is identified as the thermal equilibrium constant for the fast pre-equilibrium between the isolated reactants and the PRC, which can be determined by employing the usual statistical thermodynamic tools [36,37,38,39]. Similarly, can also be calculated by resorting to thermal TST, simplifying our TST-based rate coefficient to . This approach, which we will utilize in this investigation, is frequently used to describe reactions between OVOCs and , and it has the advantage of avoiding the need to deal with the PRCs, since their partition functions and energies cancel out in the product.
Alternatively, our recent version of the MC-TST/CTSR protocol [33,41] considers the approach followed by Petit and Harvey [27], where both the numerator and denominator of Equation (7) are multiplied by , yielding
Here, is the forward rate coefficient for the formation of the PRC and can be identified as our working equation for the calculation of the MC-TST rate constants. Naturally, the rate constant is unique for each specific reaction under analysis. However, since these rate coefficients are almost independent of the collider, we used [27] a value of for collision with acetone of () obtained via experimental rate constants [42,43,44,45] for the quenching of vibrationally excited .
2.2. Computational Methods
We will now proceed to the calculation of . Our cost-effective protocol [30,33,41] considers N conformers for each HFK reactant and M possible hydrogen abstraction TSs, with the rate coefficient being written as
Here, is the standard concentration at a temperature of 298.15 K and a pressure of 1 atmosphere, while and h are the Boltzmann and Planck’s constants, respectively. The Q values are the total partition functions for , the ith HFK reactant and jth TS, which are evaluated as the product of the translational, electronic, vibrational and rotational partition functions () under the rigid rotor and harmonic oscillator approximation. For each HFK or a TS conformer, every total partition function is weighted by its respective or thermal weight factor, which depends on the relative and zero-point corrected energies of conformations and , respectively. In addition, , , and are the rotational symmetry numbers and reaction path degeneracy parameters of conformations and , respectively. The calculation of the rotational symmetry numbers is performed by first determining the respective point group symmetry with a brute force symmetry analyzer [46] and then associating the obtained group symmetry with the correct factor by examining Table 2 of Ref. [26]. The parameter is calculated [26,28,29,30] with the formula where the subscript 0 indicates a lowest energy conformer. The tunneling correction is calculated [28,29,30] via the Eckart formula [47] using both the imaginary frequency characterizing and the zero-point corrected energies of the PRC and PC minima that are connected to through the respective intrinsic reaction coordinate path.
The above parameters are obtained through our cost-effective MC-TST/CTSR protocol [30,33,41], which we will briefly recapitulate. The protocol is initiated with the calculation of the reactant conformers with the use of confab [48], which is a knowledge-based conformer generation tool that utilizes force fields (for our protocol, we are employing the MMFF94 [49] force field) and a database of allowed torsion angles. These conformations are then minimized with the same force field as in the confab calculation by employing the Obminimize program. Both programs are available in the Open Babel [50] chemical toolbox. The minimized structures are then used as starting geometries for optimization at the M08-HX/pcseg-2 [51,52] level of theory. A similarity evaluation (SE) between all generated conformers is then performed with a superimposing algorithm [53]. The purpose of the SE is to identify identical structures and enantiomers in our pool of optimized reactants, which we can then safely eliminate, with the remaining structures constituting the final set of N distinguishable reactants.
The next step concerns the TS sampling, which was performed with the new CTSR method [33]. The CTSR code first generates random OH-molecule structures which are then subjected to constraints targeting an optimization of TSs at the PM3 level of theory. The three constraints for the HFK + TSs were the following: (1) the distance between the oxygen atom and the hydrogen atom under attack must be in the interval [1.2, 1.4] Å; (2) the distance between the oxygen atom and all reactants’ non-hydrogen atoms must be greater than 1.70 Å and (3) the distance between the hydrogen atom and all reactants’ hydrogen atoms must be greater than 0.70 Å. Guess structures were then generated by the CTSR code and subsequently used as input geometry in PM3 saddle-point optimizations. The converged PM3 calculations were then analyzed, with the coordinates corresponding to the TSs yielding an imaginary frequency within a specific interval being subjected to an SE step. The surviving geometries were used as guess structures for saddle-point optimizations at the M08-HX/pcseg-2 level, with the geometries of the successful optimizations being again analyzed in terms of their imaginary frequencies. The geometries filtered by the previous step were then put through another SE, so that the final set of unique TSs was obtained. It should be stated that the M08-HX functional was chosen due to its quality in describing hydrogen abstraction barrier heights, yielding, respectively, mean unsigned errors of 1.00 and 0.73 for the DBH76 and HTBH38 data sets involving this particular type of barrier heights [51]. In this study, M08-HX was the best performing functional, surpassing M06-2X, which is routinely used in atmospheric chemistry studies of the same type. Moreover, we recently [54] made a rough estimate of 0.6 for the uncertainty associated with , which led to rate constants and error bars with the approximate values of . It should also be noted that although our protocol relies on many approximations and on error cancellation [30,41], the quality of its calculated rate constants is quite encouraging [33,41] when taking into account the complexity of the studied systems. The electronic structure calculations necessary to the determination of the parameters entering Equation (9) were performed with the GAMESS package [55]. Finally, the value of was refined with single-point energy calculations at the M08-HX/apcseg-2 level.
3. Results and Discussion
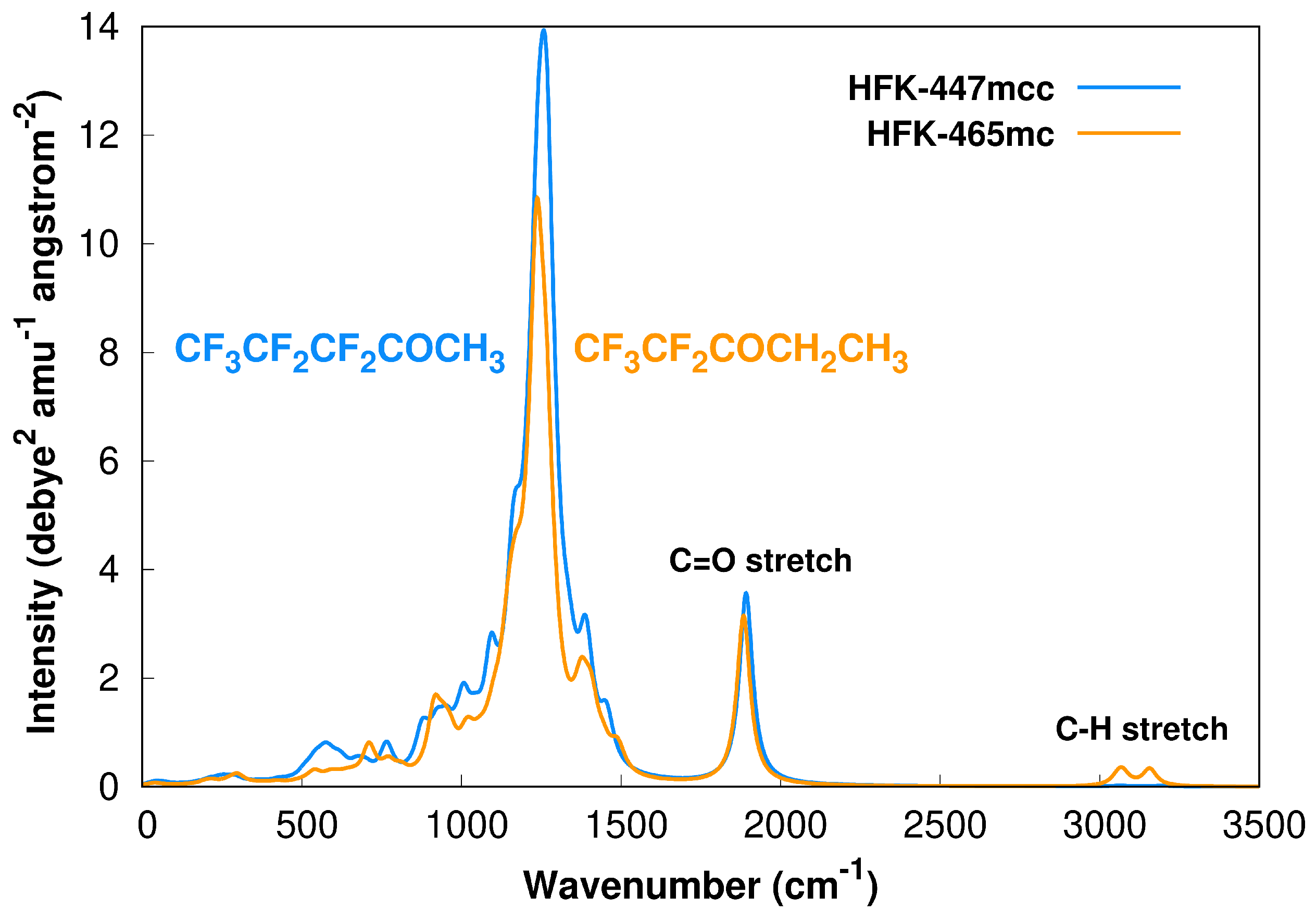
We start by showing Figure 1, where we present the simulated IR spectra of both reactants, HFK-447mcc and HFK465mc, based on the M08-HX/pcseg-2 harmonic calculations and including the contribution of all conformers through the respective Boltzmann weighting at 298.15 K, which was calculated from the Gibbs energies. Each spectrum was obtained by convolution of the calculated vibrational frequencies () with a Lorentzian profile given by
where N is the number of atoms in each molecule, is the calculated vibrational frequency with a corresponding intensity and W is the full width at half-maximum, which we empirically selected as .
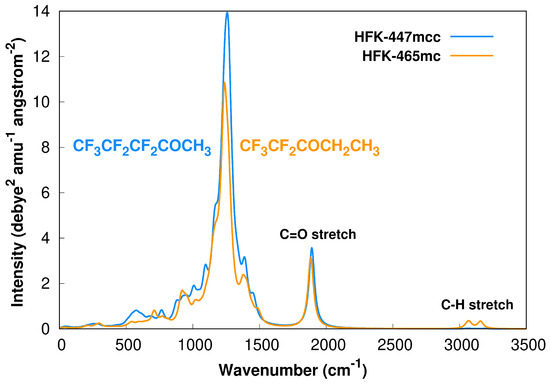
Figure 1.
Boltzmann weighted (298.15 K) harmonic M08-HX/pcseg-2 calculated IR spectra of HFK-447mcc and HFK-465mc, including all obtained conformers for both species.
For saturated aliphatic ketones, the and stretch bands are usually located at 1715 and [2800,3000] , respectively. For the studied ketones, our calculations predict these bands to be found at slightly higher wavenumbers, as represented in Figure 1. However, it should be noted that although the M08-HX/pcseg-2 model chemistry is a good choice for MC-TST rate constants [30,33,41,56], it might not be the best theoretical method to employ if one is pursuing an accurate prediction of the vibrational frequencies [57].
3.1. OH Oxidation of HFK-447mcc
Starting the analysis of our results with the , our confab-based [48] conformer search [30,41] relying on the MMFF94 [49] force field and followed by M08-HX/pcseg-2 optimizations found 20 unique conformers of symmetry (each one belonging to an enantiomeric pair by definition), with the 15 lowest energy ones having a relative energy in the interval [0.00,0.45] and representing approximately 90% of the total fractional population of reactants. Application of the CTSR sampling procedure yielded 90 distinguishable hydrogen abstraction TSs of symmetry, with the barrier having a value of 4.2 . At this point, it becomes advisable to test the validity of our theoretical kinetic approach by investigating if the steady-state and equilibrium assumptions [40] are applicable to the reaction under scrutiny. For this, we have gathered the Gibbs free energies for the reactants, , and the associated PRC and PC minima at the M08-HX/pcseg-2 level of theory. The results for the reactants, PRC, and PC, are, respectively, 0.0, 4.7, 12.6 and . These values indicate that the Gibbs free energy profile along the reaction coordinate is similar to the one shown in Figure 5b of Ref. [40], indicating that will most likely be considerably larger than and , guaranteeing that both the steady-state and equilibrium assumptions are satisfied for the reaction between HFK-447mcc and at 298.15 K.
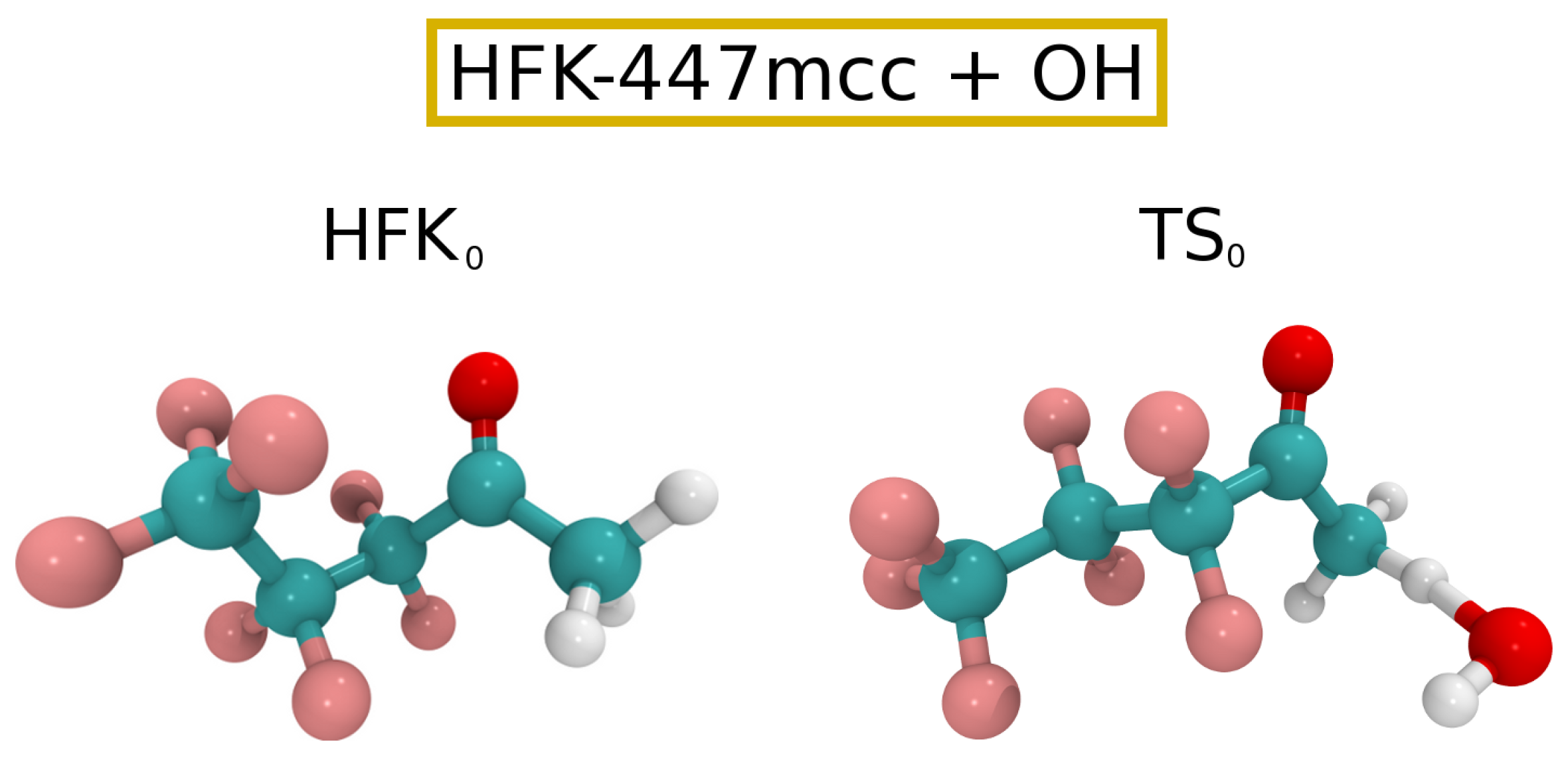
The Eckart tunneling correction yielded a value of 6.3, with the final rate constant for the reaction between HFK-447mcc and being . Such a rate constant implies a atmospheric lifetime of approximately 10 years, considering a value of for the global average concentration of OH radicals [58]. Additionally, we have extended our calculations to a temperature interval relevant for tropospheric chemistry [21,59,60], namely , and fitted the corresponding data to an Arrhenius–Kooij equation [59,61,62] to give . Cartesian coordinates for reactants and TSs can be found in the Supplementary Information, while Figure 2 shows ball and stick drawings of the lowest energy HFK-447mcc and corresponding TS ( and , respectively).
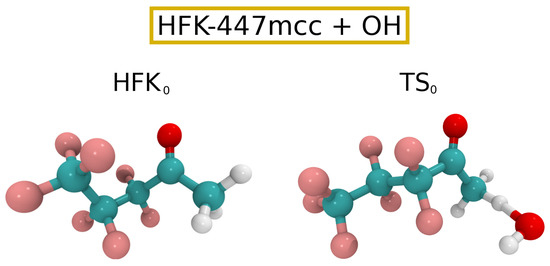
Figure 2.
Ball and stick drawings for the lowest energy HFK-447mcc reactant and respective for reaction (3).
3.2. OH Oxidation of HFK-465mc
Proceeding to the , our calculations found 16 unique conformers of symmetry, with the seven lowest energy ones having a relative energy in the interval [0.00,0.75] and representing slightly more than 90% of the total fractional population of reactants. Application of the CTSR sampling procedure yielded 52 distinguishable hydrogen abstraction TSs of symmetry, with the barrier being calculated as 2.1 . For this reaction, the relative Gibbs free energies for the reactants, PRC, and PC minima at the M08-HX/pcseg-2 level of theory are, respectively: 0.0, 4.2, 10.8 and . As in the case above, this strongly suggests that both the steady-state and equilibrium assumptions are satisfied for the reaction between HFK-465mc and at 298.15 K.
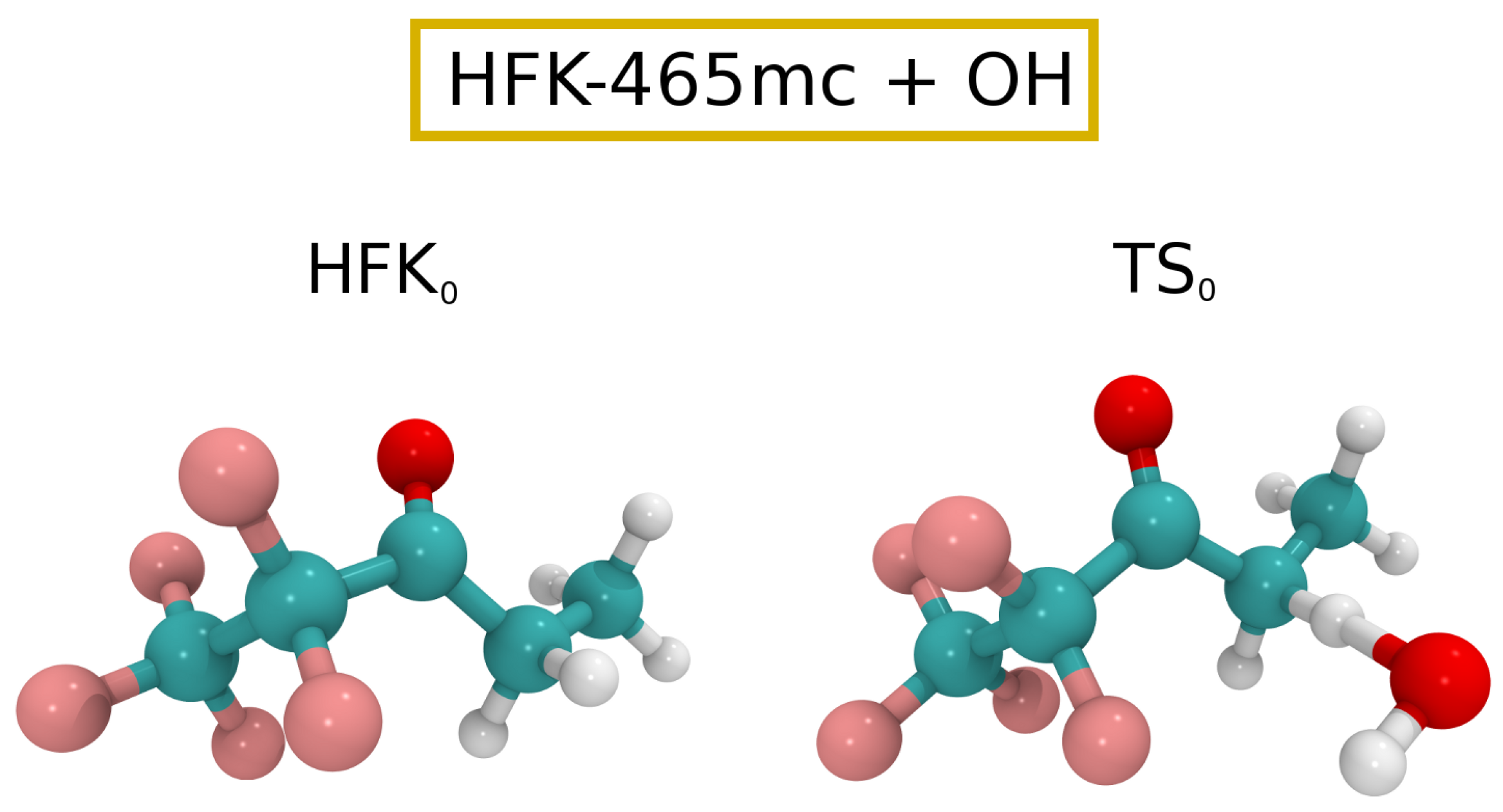
The Eckart tunneling correction yielded a value of 2.7, with the final rate constant for the reaction between HFK-465mc and being . Such a rate constant implies a atmospheric lifetime of approximately 1 year. Moreover, the fit of the theoretical data points in the interval to the Arrhenius–Kooij equation leads to . Cartesian coordinates for reactants and TSs can be found in the Supplementary Information, while Figure 3 shows ball and stick drawings of the lowest energy HFK-465mc and corresponding TS ( and , respectively).
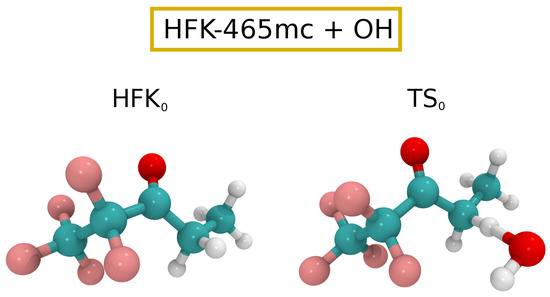
Figure 3.
Ball and stick drawings for the lowest energy HFK-465mc reactant and respective for reaction (3).
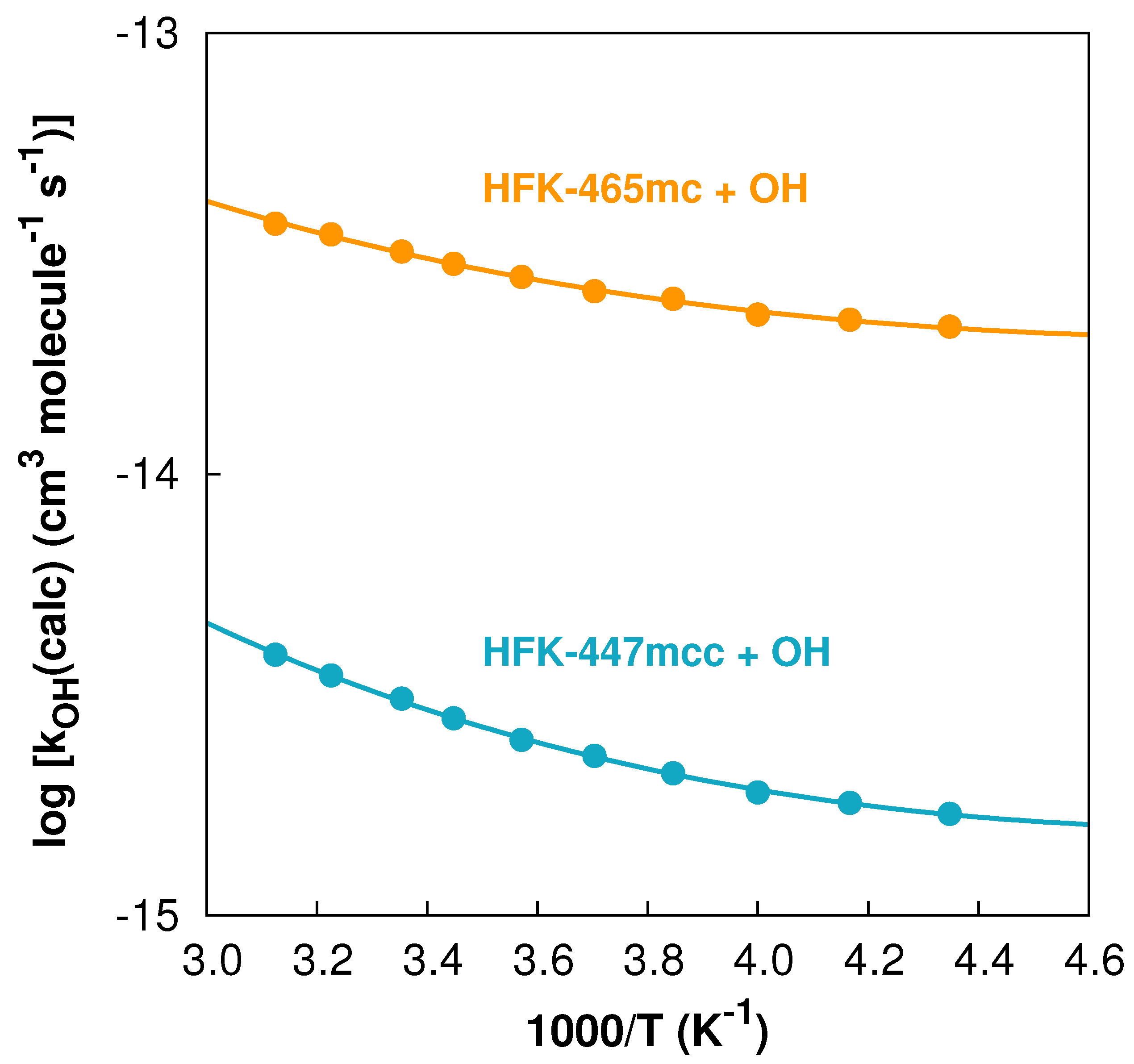
In Figure 4, we present the points calculated in the temperature interval together with the Arrhenius–Kooij fits to those points for both reactions. Although both curves exhibit some curvature, it is clearly noticeable that the curvature of the HFK-447mcc + OH reaction is more pronounced mainly because of the differences in the tunneling correction factors, which increase with decreasing temperature: and .
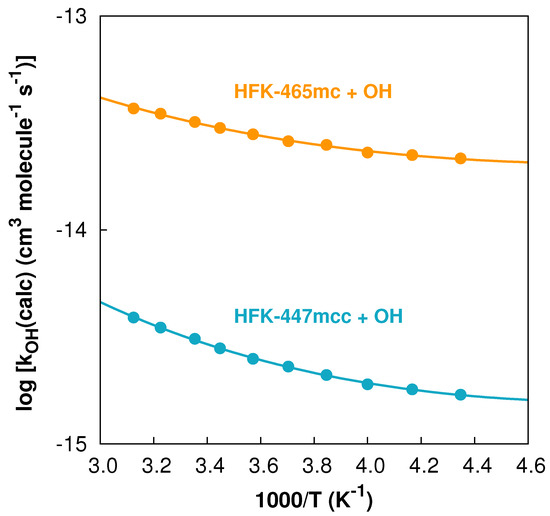
Figure 4.
Arrhenius–Kooij fits (solid lines) and data points (solid circles) for both reactions in the temperature interval.
The reaction between HFK-465mc and can proceed through two different abstraction sites, as the OH radical can abstract a hydrogen atom in two distinct locations of the : at the group (-position) and at the group (-position). The calculated branching ratios for the two abstraction sites are: and . Interestingly, these values coincide with the branching ratios calculated for the corresponding unsubstituted diethyl ketone (DEK, ) with the use of structure–activity relationships (SARs). Using the additivity method [10,11,16,63] with the recommended values of for the reaction of OH with ketones (see Table 4 of Ref. [10] or Table V-B-20 of Ref. [11]), we can determine as
which yields a value of and corresponding branching ratios of and . Note that the recommended value [11] for this rate coefficient is with an uncertainty of .
Moreover, it is also observed that the ratio between the calculated OH-oxidation rate constants of HFK-465mc and HFK-447mcc is approximately 10, which reflects the different reactivity of the and alkyl groups. An almost equivalent ratio is obtained if the same type of comparison is performed with unsubstituted ketones: the ratio between the recommended values [11] for the OH-oxidation rate constants of DEK and acetone (, with a rate constant of ) is approximately 11. In the absence of experimental rate coefficients concerning the HFK-447mcc and HFK-465mc molecules, such an agreement may be interpreted as an indication that despite all approximations, the MC-TST/CTSR protocol [30,33,41] correctly captures essential reactivity trends.
4. Conclusions
In this study, we have calculated the rate constants for the reactions between two HFKs (HFK-447mcc and HFK-465mc) and the OH radical, which is the most important oxidant in the atmosphere. The results, which were obtained via theoretical calculations employing our MC-TST/CTSR protocol, show that the rate constant for the reaction involving HFK-465mc is ten times larger than the rate constant involving HFK-447mcc, which results in an atmospheric lifetime of approximately 1 and 10 years for HFK-465mc and HFK-447mcc, respectively. These results indicate that photolysis will probably be the main atmospheric sink for HFK-447mcc and HFK-465mc. Although there are no experimental studies to compare our results to, the observed reactivity pattern and branching ratios resemble the ones observed in well-tested systems with the same reactive alkyl groups, such as acetone and diethyl ketone. Despite the fact that these are good indications and that our MC-TST protocol consistently yields accurate rate constants for reactions between fluorinated compounds and the OH radical, a full assessment of these two rate constants can only be made in the future if and when the corresponding experimental rate coefficients are determined.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/atmos13081256/s1, Cartesian coordinates for all unique conformers used in the calculation of both MC-TST rate constants present in the manuscript.
Funding
The Coimbra Chemistry Centre-Institute of Molecular Sciences (CQC-IMS) is supported by the Portuguese Foundation for Science and Technology (FCT) through projects UIDB/QUI/0313/2020 and UIDP/QUI/0313/2020 and LA/P/0056/2020 (national funds). L.P.V. acknowledges FCT for an Assistant Researcher grant.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available alongside the article and the Supplementary Materials.
Acknowledgments
L.P.V. acknowledges Dorothee Birke for proofreading this manuscript.
Conflicts of Interest
The author declares no conflict of interest.
References
- Molina, M.J.; Rowland, F.S. Stratospheric Sink for Chlorofluoromethanes: Chlorine Atom-Catalysed Destruction of Ozone. Nature 1974, 249, 810–812. [Google Scholar] [CrossRef]
- Farman, J.D.; Gardiner, B.G.; Shanklin, J.D. Large Losses of Total Ozone in Antarctica Reveal Seasonal ClOx/NOx Interaction. Nature 1985, 315, 207–210. [Google Scholar] [CrossRef]
- United Nations: Montreal Protocol on Substances that Deplete the Ozone Layer-Adjustments and Amendment. Int. Leg. Mater. 1993, 32, 874–887. [CrossRef]
- Lazarou, Y.G.; Papagiannakopoulos, P. Theoretical Investigation of the Thermochemistry of Hydrofluoroethers. Chem. Phys. Lett. 1999, 301, 19–28. [Google Scholar] [CrossRef]
- McGivern, B.P. Conference of the Parties to the Framework Convention on Climate Change: Kyoto Protocol. Int. Leg. Mater. 1998, 37, 22–43. [Google Scholar] [CrossRef]
- Heath, E.A. Amendment to the Montreal Protocol on Substances that Deplete the Ozone Layer (Kigali Amendment). Int. Leg. Mater. 2017, 56, 193–205. [Google Scholar] [CrossRef] [Green Version]
- UNEP. HFCs: A Critical Link in Protecting Climate and the Ozone Layer; United Nations Environment Programme (UNEP): 2011; 36p. Available online: https://www.unep.org/resources/report/hfcs-critical-link-protecting-climate-and-ozone-layer (accessed on 6 August 2022).
- Hodnebrog, Ø.; Etminan, M.; Fuglestvedt, J.S.; Marston, G.; Myhre, G.; Nielsen, C.J.; Shine, K.P.; Wallington, T.J. Global Warming Potentials and Radiative Efficiencies of Halocarbons and Related Compounds: A Comprehensive Review. Rev. Geophys. 2013, 51, 300–378. [Google Scholar] [CrossRef] [Green Version]
- Wallington, T.J.; Andersen, M.P.S.; Nielsen, O.J. Atmospheric Chemistry of Halogenated Organic Compounds. In Advances in Atmospheric Chemistry; World Scientific: Singapore, 2017; Volume 1, pp. 305–402. [Google Scholar]
- Mellouki, A.; Le Bras, G.; Sidebottom, H. Kinetics and Mechanisms of the Oxidation of Oxygenated Organic Compounds in the Gas Phase. Chem. Rev. 2003, 103, 5077–5096. [Google Scholar] [CrossRef]
- Calvert, J.G.; Mellouki, A.; Orlando, J.J.; Pilling, M.J.; Wallington, T.J. The Mechanisms of Atmospheric Oxidation of the Oxygenates, 1st ed.; Oxford University Press: New York, NY, USA, 2011. [Google Scholar]
- Mellouki, A.; Wallington, T.J.; Chen, J. Atmospheric Chemistry of Oxygenated Volatile Organic Compounds: Impacts on Air Quality and Climate. Chem. Rev. 2015, 115, 3984–4014. [Google Scholar] [CrossRef]
- Vereecken, L.; Francisco, J.S. Theoretical Studies of Atmospheric Reaction Mechanisms in the Troposphere. Chem. Soc. Rev. 2012, 41, 6259–6293. [Google Scholar] [CrossRef]
- Vereecken, L.; Glowacki, D.R.; Pilling, M.J. Theoretical Chemical Kinetics in Tropospheric Chemistry: Methodologies and Applications. Chem. Rev. 2015, 115, 4063–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atkinson, R.; Aschmann, S.M.; Carter, W.P.L.; Pitts, J.N., Jr. Rate Constants for the Gas-Phase Reaction of OH Radicals with a Series of Ketones at 299 ± 2 K. Int. J. Chem. Kinet. 1982, 14, 839–847. [Google Scholar] [CrossRef]
- Le Calvé, S.; Hitier, D.; Le Bras, G.; Mellouki, A. Kinetic Studies of OH Reactions with a Series of Ketones. J. Phys. Chem. A 1998, 102, 4579–4584. [Google Scholar] [CrossRef]
- Otake, K.; Yasumoto, M.; Yamada, Y.; Murata, J.; Urata, S. Critical Parameters and Vapor Pressure Measurements of Potential Replacements for Chlorofluorocarbons - Four Hydrofluoroketones and a Hydrofluoroamine. J. Chem. Eng. Data 2003, 48, 1380–1383. [Google Scholar] [CrossRef]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Erythropel, H.C.; Zimmerman, J.B.; de Winter, T.M.; Petitjean, L.; Melnikov, F.; Lam, C.H.; Lounsbury, A.W.; Mellor, K.E.; Janković, N.Z.; Tu, Q.; et al. The Green ChemisTREE: 20 Years After Taking Root With the 12 Principles. Green Chem. 2018, 20, 1929–1961. [Google Scholar] [CrossRef]
- Kurylo, M.J.; Orkin, V.L. Determination of Atmospheric Lifetimes via the Measurement of OH Radical Kinetics. Chem. Rev. 2003, 103, 5049–5076. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J.; Pitts, J.N., Jr. Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, 1st ed.; Academic Press: San Diego, CA, USA, 2000. [Google Scholar]
- Calvert, J.G.; Orlando, J.J.; Stockwell, W.R.; Wallington, T.J. The Mechanisms of Reactions Influencing Atmospheric Ozone, 1st ed.; Oxford University Press: New York, NY, USA, 2015. [Google Scholar]
- Díaz-de-Mera, Y.; Aranda, A.; Notario, A.; Rodríguez, A.; Rodríguez, D.; Bravo, I. Photolysis Study of Fluorinated Ketones Under Natural Sunlight Conditions. Phys. Chem. Chem. Phys. 2015, 17, 22991–22998. [Google Scholar] [CrossRef]
- Ren, Y.; Bernard, F.; Daële, V.; Mellouki, A. Atmospheric Fate and Impact of Perfluorinated Butanone and Pentanone. Environ. Sci. Technol. 2019, 53, 8862–8871. [Google Scholar] [CrossRef]
- Vereecken, L.; Peeters, J. The 1,5-H-Shift in 1-Butoxy: A Case Study in the Rigorous Implementation of Transition State Theory for a Multirotamer System. J. Chem. Phys. 2003, 119, 5159–5170. [Google Scholar] [CrossRef]
- Fernández-Ramos, A.; Ellingson, B.A.; Meana-Pañeda, R.; Marques, J.M.C.; Truhlar, D.G. Symmetry Numbers and Chemical Reaction Rates. Theor. Chem. Acc. 2007, 118, 813–826. [Google Scholar] [CrossRef] [Green Version]
- Petit, A.S.; Harvey, J.N. Atmospheric Hydrocarbon Activation by the Hydroxyl Radical: A Simple yet Accurate Computational Protocol for Calculating Rate Coefficients. Phys. Chem. Chem. Phys. 2012, 14, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Rissanen, M.P.; Kurtén, T.; Sipilä, M.; Thornton, J.A.; Kangasluoma, J.; Sarnela, N.; Junninen, H.; Jørgensen, S.; Schallhart, S.; Kajos, M.K.; et al. The Formation of Highly Oxidized Multifunctional Products in the Ozonolysis of Cyclohexene. J. Am. Chem. Soc. 2014, 136, 15596–15606. [Google Scholar] [CrossRef] [PubMed]
- Møller, K.H.; Otkjaer, R.V.; Hyttinen, N.; Kurtén, T.; Kjaergaard, H.G. Cost-Effective Implementation of Multiconformer Transition State Theory for Peroxy Radical Hydrogen Shift Reactions. J. Phys. Chem. A 2016, 120, 10072–10087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viegas, L.P. Exploring the Reactivity of Hydrofluoropolyethers toward OH Through a Cost-Effective Protocol for Calculating Multiconformer Transition State Theory Rate Constants. J. Phys. Chem. A 2018, 122, 9721–9732. [Google Scholar] [CrossRef]
- Novelli, A.; Vereecken, L.; Bohn, B.; Dorn, H.; Gkatzelis, G.L.; Hofzumahaus, A.; Holland, F.; Reimer, D.; Rohrer, F.; Rosanka, S.; et al. Importance of Isomerization Reactions for OH Radical Regeneration from the Photo-Oxidation of Isoprene Investigated in the Atmospheric Simulation Chamber SAPHIR. Atmos. Chem. Phys. 2020, 20, 3333–3355. [Google Scholar] [CrossRef] [Green Version]
- Vereecken, L.; Nozière, B. H Migration in Peroxy Radicals Under Atmospheric Conditions. Atmos. Chem. Phys. 2020, 20, 7429–7458. [Google Scholar] [CrossRef]
- Viegas, L.P. Gas-Phase OH-Oxidation of 2-Butanethiol: Multiconformer Transition State Theory Rate Constant with Constrained Transition State Randomization. Chem. Phys. Lett. 2022, 803, 139829. [Google Scholar] [CrossRef]
- Hansen, J.C.; Francisco, J.S. Radical-Molecule Complexes: Changing Our Perspective on the Molecular Mechanisms of Radical-Molecule Reactions and Their Impact on Atmospheric Chemistry. ChemPhysChem 2002, 3, 833–840. [Google Scholar] [CrossRef]
- Raines, R.T.; Hansen, D.E. An Intuitive Approach to Steady-State Kinetics. J. Chem. Educ. 1988, 65, 757–759. [Google Scholar] [CrossRef]
- Singleton, D.L.; Cvetanović, R.J. Temperature Dependence of the Reaction of Oxygen Atoms with Olefins. J. Am. Chem. Soc. 1976, 98, 6812–6819. [Google Scholar] [CrossRef]
- Alvarez-Idaboy, J.R.; Mora-Diez, N.; Vivier-Bunge, A. A Quantum Chemical and Classical Transition State Theory Explanation of Negative Activation Energies in OH Addition To Substituted Ethenes. J. Am. Chem. Soc. 2000, 122, 3715–3720. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R.; Bravo-Pérez, G.; Ruiz-Santoyo, M.E. Gas Phase Reactions of C1–C4 Alcohols with the OH Radical: A Quantum Mechanical Approach. Phys. Chem. Chem. Phys. 2002, 4, 4648–4662. [Google Scholar] [CrossRef]
- Cruz-Torres, A.; Galano, A. On the Mechanism of Gas-Phase Reaction of C1–C3 Aliphatic Thiols + OH Radicals. J. Phys. Chem. A 2007, 111, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Xiao, F.; Sun, X.; Li, Z.; Li, X. Theoretical Study of Radical-Molecule Reactions with Negative Activation Energies in Combustion: Hydroxyl Radical Addition to Alkenes. ACS Omega 2020, 5, 12777–12788. [Google Scholar] [CrossRef]
- Viegas, L.P. Simplified Protocol for the Calculation of Multiconformer Transition State Theory Rate Constants Applied to Tropospheric OH-Initiated Oxidation Reactions. J. Phys. Chem. A 2021, 125, 4499–4512. [Google Scholar] [CrossRef]
- Smith, I.W.M. Collisional Energy Transfer, Intramolecular Vibrational Relaxation and Unimolecular Reactions. J. Chem. Soc. Faraday Trans. 1997, 93, 3741–3750. [Google Scholar] [CrossRef]
- Smith, I.W.M.; Ravishankara, A.R. Role of Hydrogen-Bonded Intermediates in the Bimolecular Reactions of the Hydroxyl Radical. J. Phys. Chem. A 2002, 106, 4798–4807. [Google Scholar] [CrossRef]
- McCabe, D.C.; Brown, S.S.; Gilles, M.K.; Talukdar, R.K.; Smith, I.W.M.; Ravishankara, A.R. Kinetics of the Removal of OH(v = 1) and OD(v = 1) by HNO3 and DNO3 from 253 to 383 K. J. Phys. Chem. A 2003, 107, 7762–7769. [Google Scholar] [CrossRef]
- Talukdar, R.K.; Gierczak, T.; McCabe, D.C.; Ravishankara, A.R. Reaction of Hydroxyl Radical with Acetone. 2. Products and Reaction Mechanism. J. Phys. Chem. A 2003, 107, 5021–5032. [Google Scholar] [CrossRef]
- Patchkovskii, S. Brute Force Symmetry Analyzer. Available online: https://github.com/nquesada/symmetry (accessed on 6 August 2022).
- Eckart, C. The Penetration of a Potential Barrier by Electrons. Phys. Rev. 1930, 35, 1303–1309. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Vandermeersch, T.; Flynn, C.J.; Maguire, A.R.; Hutchison, G.R. Confab-Systematic Generation of Diverse Low-Energy Conformers. J. Cheminf. 2011, 3, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halgren, T.A. Merck Molecular Force Field. I. Basis, Form, Scope, Parameterization, and Performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Truhlar, D.G. Exploring the Limit of Accuracy of the Global Hybrid Meta Density Functional for Main-Group Thermochemistry, Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2008, 4, 1849–1868. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F. Unifying General and Segmented Contracted Basis Sets. Segmented Polarization Consistent Basis Sets. J. Chem. Theory Comput. 2014, 10, 1074–1085. [Google Scholar] [CrossRef]
- Marques, J.M.C.; Llanio-Trujillo, J.L.; Abreu, P.E.; Pereira, F.B. How Different Are Two Chemical Structures? J. Chem. Inf. Model. 2010, 50, 2129–2140. [Google Scholar] [CrossRef]
- Viegas, L.P.; Jensen, F. Reactivity of α,ω-Dihydrofluoropolyethers toward OH Predicted by Multiconformer Transition State Theory and the Interacting Quantum Atoms Approach. J. Phys. Chem. A 2020, 124, 3460–3470. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General Atomic and Molecular Electronic Structure System. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Viegas, L.P. Assessment of Model Chemistries for Hydrofluoropolyethers: A DFT/M08-HX Benchmark Study. Int. J. Quantum Chem. 2017, 117, e25381. [Google Scholar] [CrossRef]
- Palafox, M.A. DFT Computations on Vibrational Spectra: Scaling Procedures to Improve the Wavenumbers. Phys. Sci. Rev. 2018, 3, 0184. [Google Scholar] [CrossRef]
- Prinn, R.G.; Huang, J.; Weiss, R.F.; Cunnold, D.M.; Fraser, P.J.; Simmonds, P.G.; McCulloch, A.; Harth, C.; Salameh, P.; O’Doherty, S.; et al. Evidence for Substantial Variations of Atmospheric Hydroxyl Radicals in the Past Two Decades. Science 2001, 292, 1882–1888. [Google Scholar] [CrossRef] [PubMed]
- McGillen, M.R.; Carter, W.P.L.; Mellouki, A.; Orlando, J.J.; Picquet-Varrault, B.; Wallington, T.J. Database for the Kinetics of the Gas-Phase Atmospheric Reactions of Organic Compounds. Earth Syst. Sci. Data 2020, 12, 1203–1216. [Google Scholar] [CrossRef]
- Altarawneh, M. A Closer Look Into the Contribution of Atmospheric Gas-Phase Pathways in the Formation of Perfluorocarboxylic Acids. Atmos. Pol. Res. 2021, 12, 101255. [Google Scholar] [CrossRef]
- Kooij, D.M. Über die Zersetzung des Gasförmigen Phosphorwasserstoffs. Z. Phys. Chem. 1893, 12, 155–161. [Google Scholar] [CrossRef]
- Laidler, K.J. The Development of the Arrhenius Equation. J. Chem. Ed. 1984, 61, 494–498. [Google Scholar] [CrossRef]
- Ren, Y.; Baramoussi, E.M.E.; Daële, V.; Mellouki, A. Atmospheric Chemistry of Ketones: Reaction of OH Radicals with 2-Methyl-3-Pentanone, 3-Methyl-2-Pentanone and 4-Methyl-2-Pentanone. Sci. Total Environ. 2021, 780, 146249. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).