
Complex Physical Structure of Complete Mitochondrial Genome of Quercus acutissima (Fagaceae): A Significant Energy Plant

 ,
,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Sequencing
2.2. Genome Assembly and Annotation
2.3. Analysis of RSCU and Repeated Sequences
2.4. Detection of Genome Recombination
2.5. Chloroplast to Mitochondrion DNA Transformation and RNA Editing Prediction
2.6. Synteny and Phylogenetic Analysis
3. Results
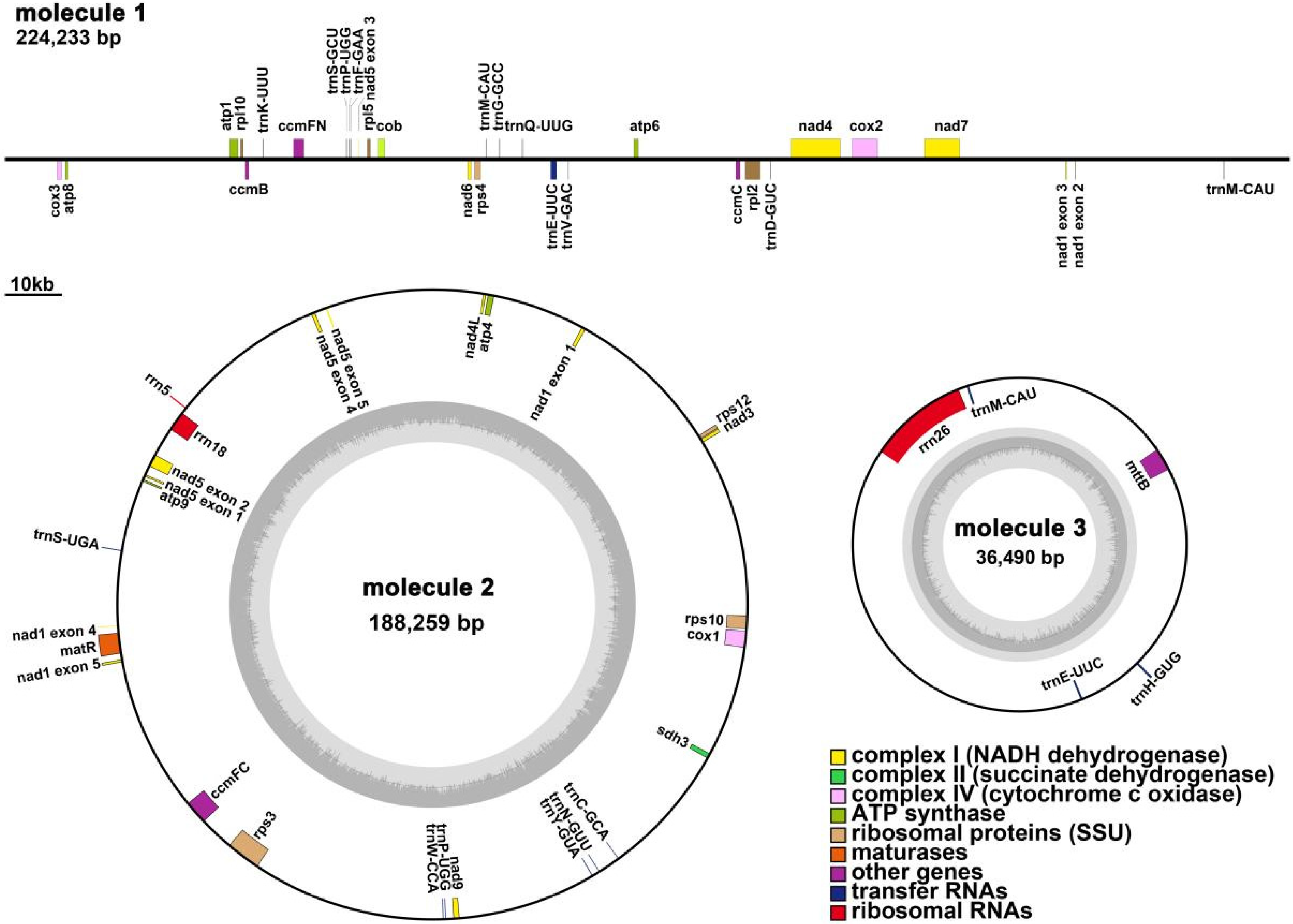
3.1. Q. acutissima Mitogenome Features
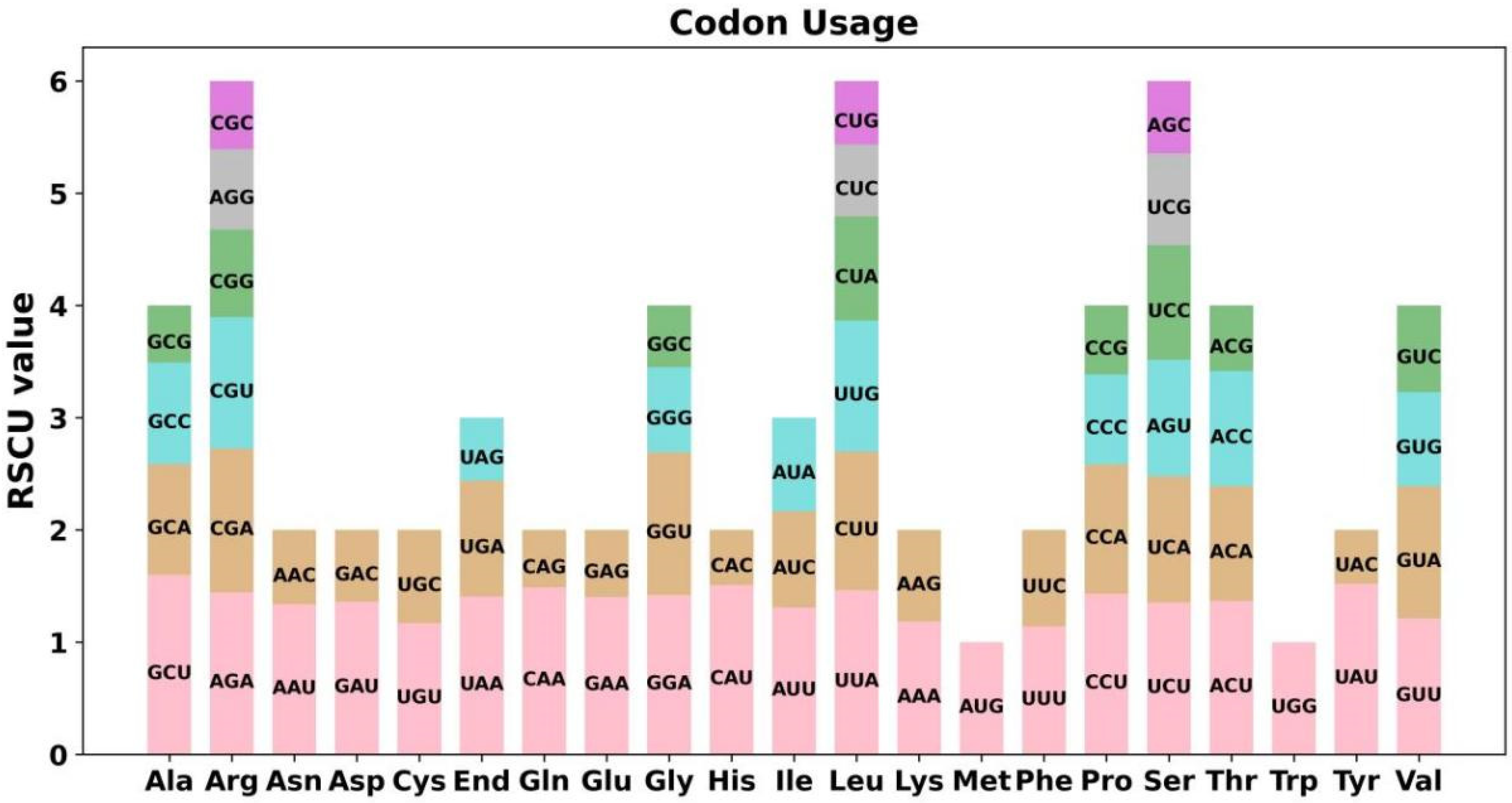
3.2. PCGs Codon Usage Analysis
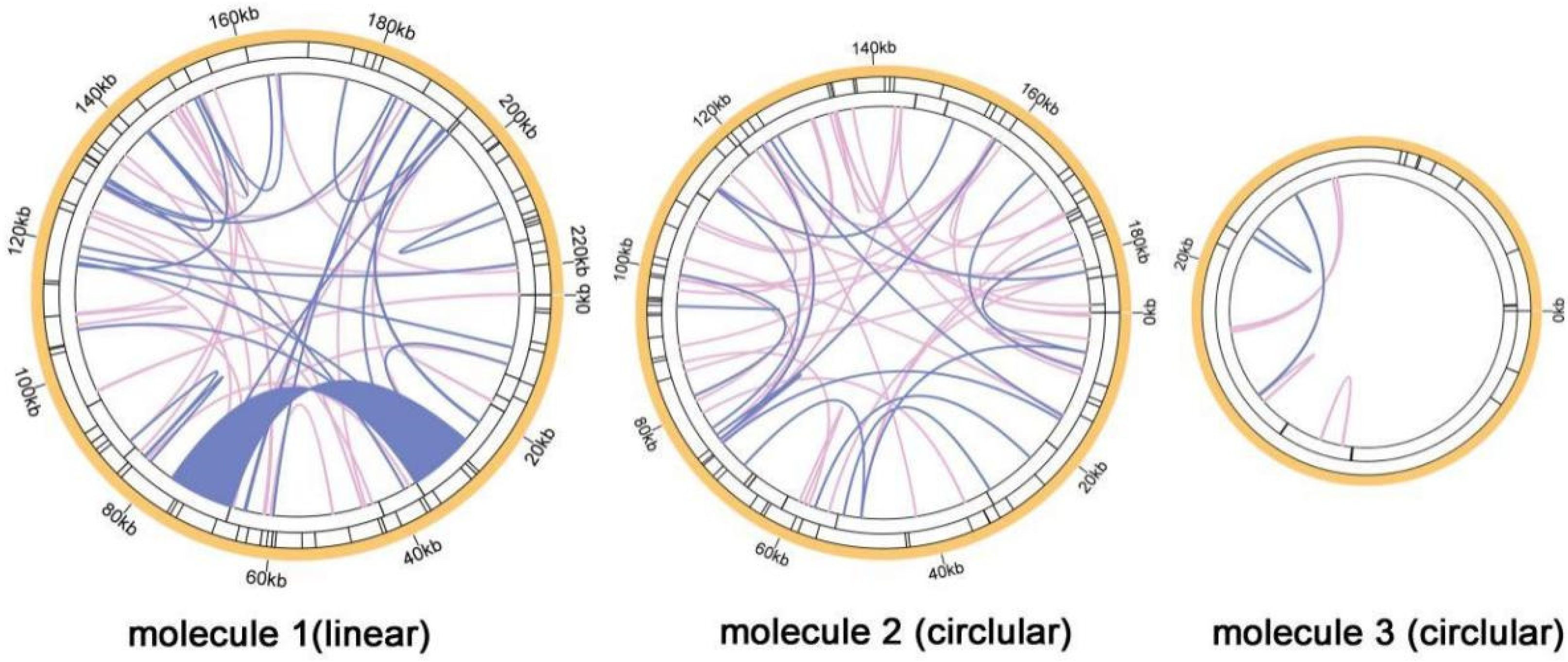
3.3. Q. acutissima Mitogenome Repeats Analysis
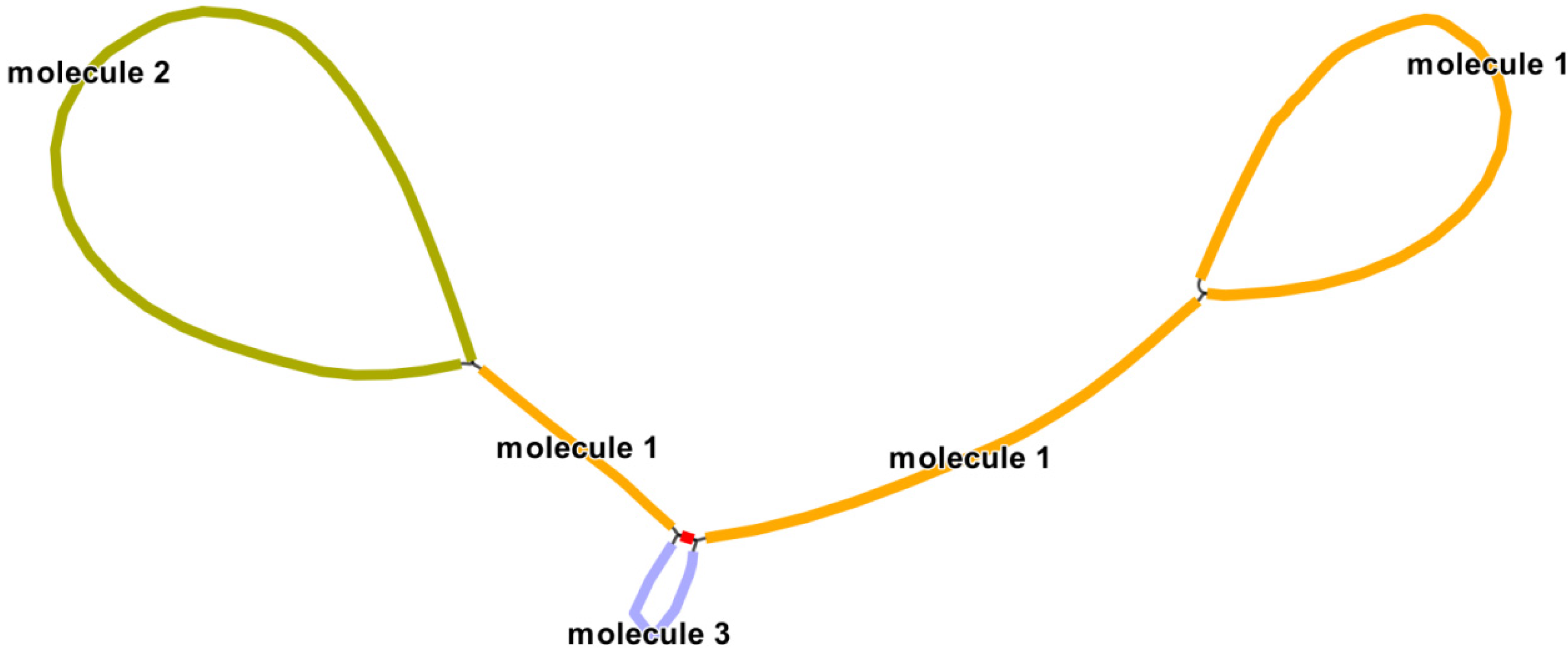
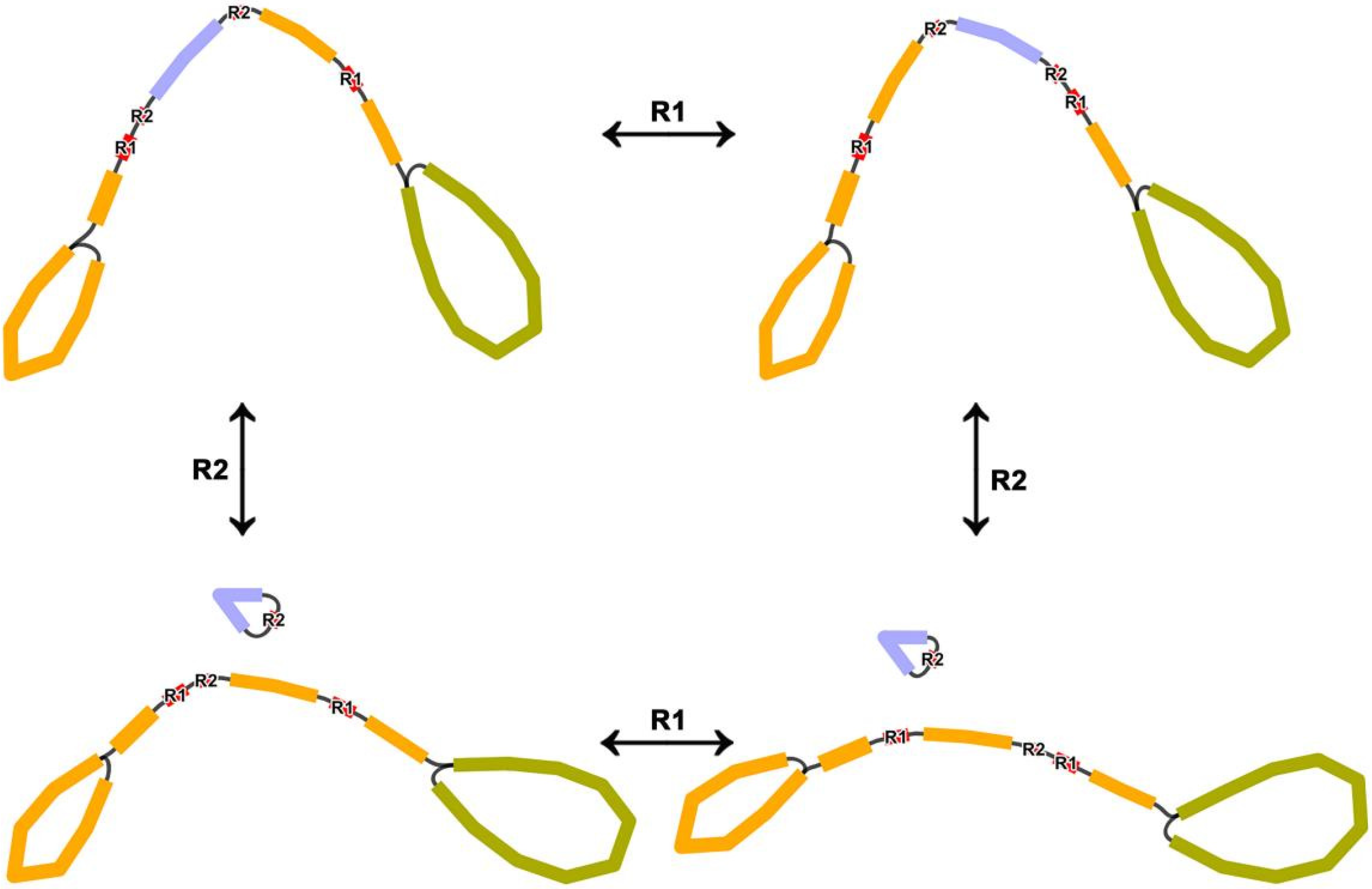
3.4. Detection of Genome Recombination
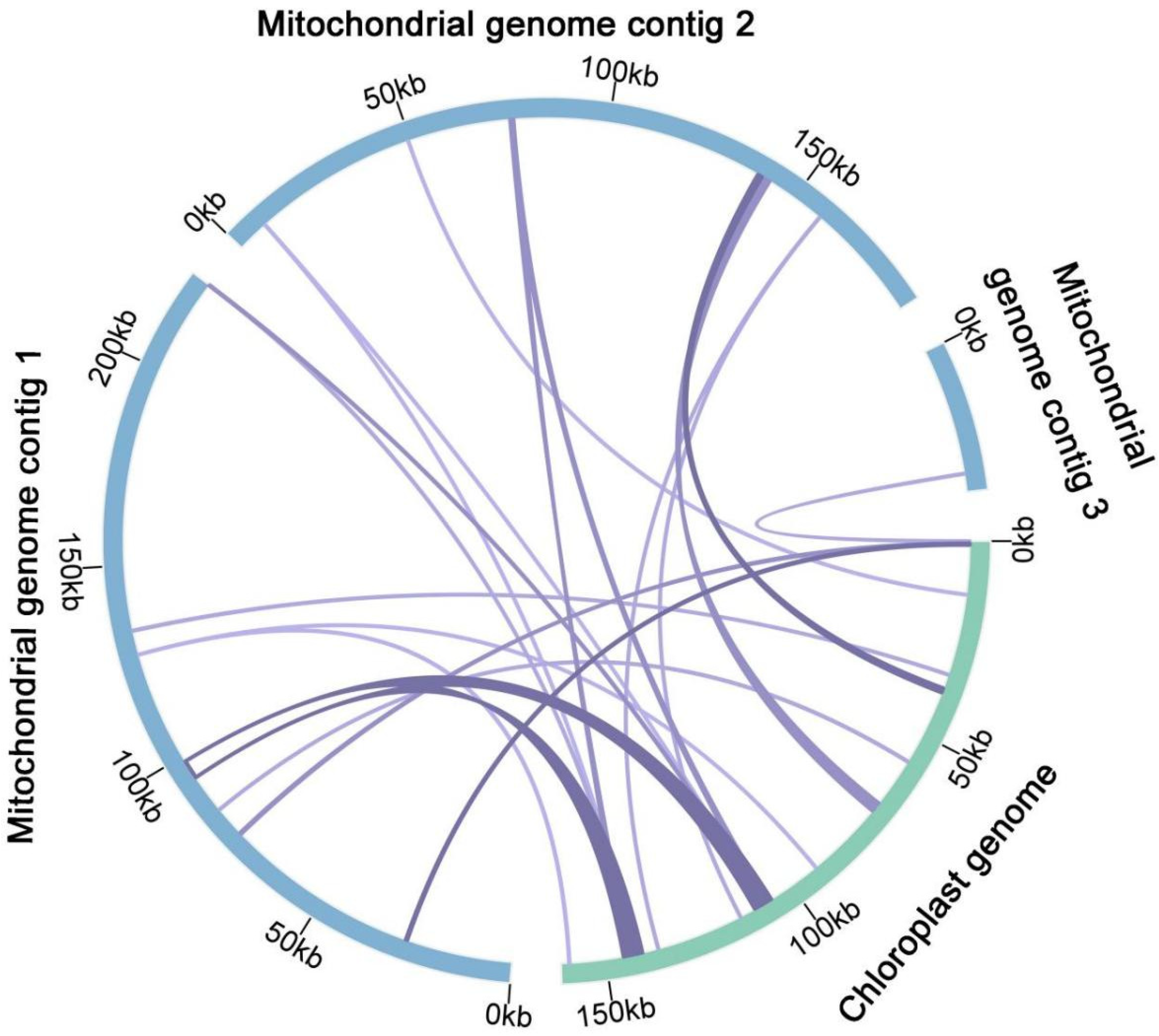
3.5. Chloroplast to Mitochondrion DNA Transformation
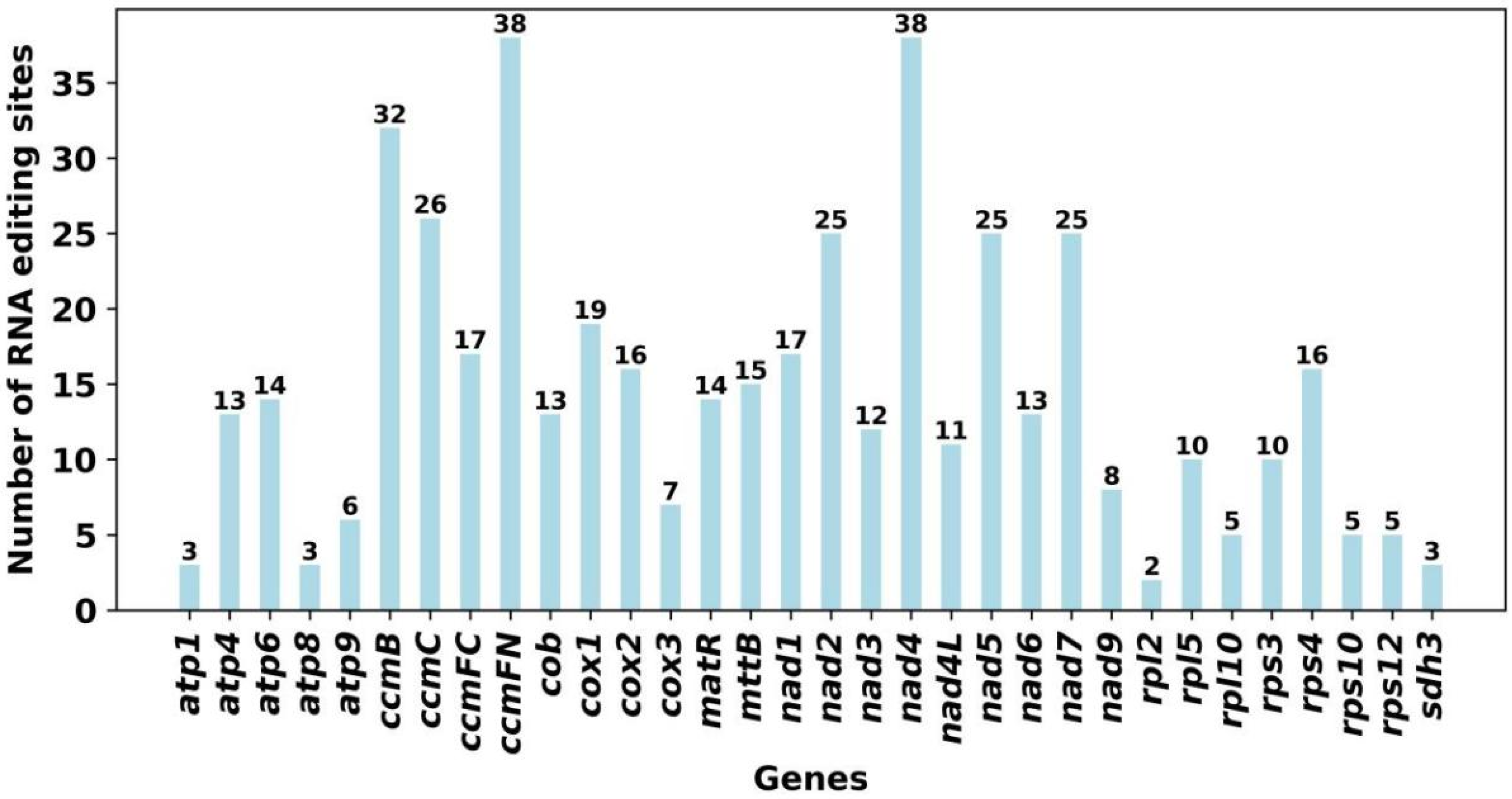
3.6. The Prediction of RNA Editing
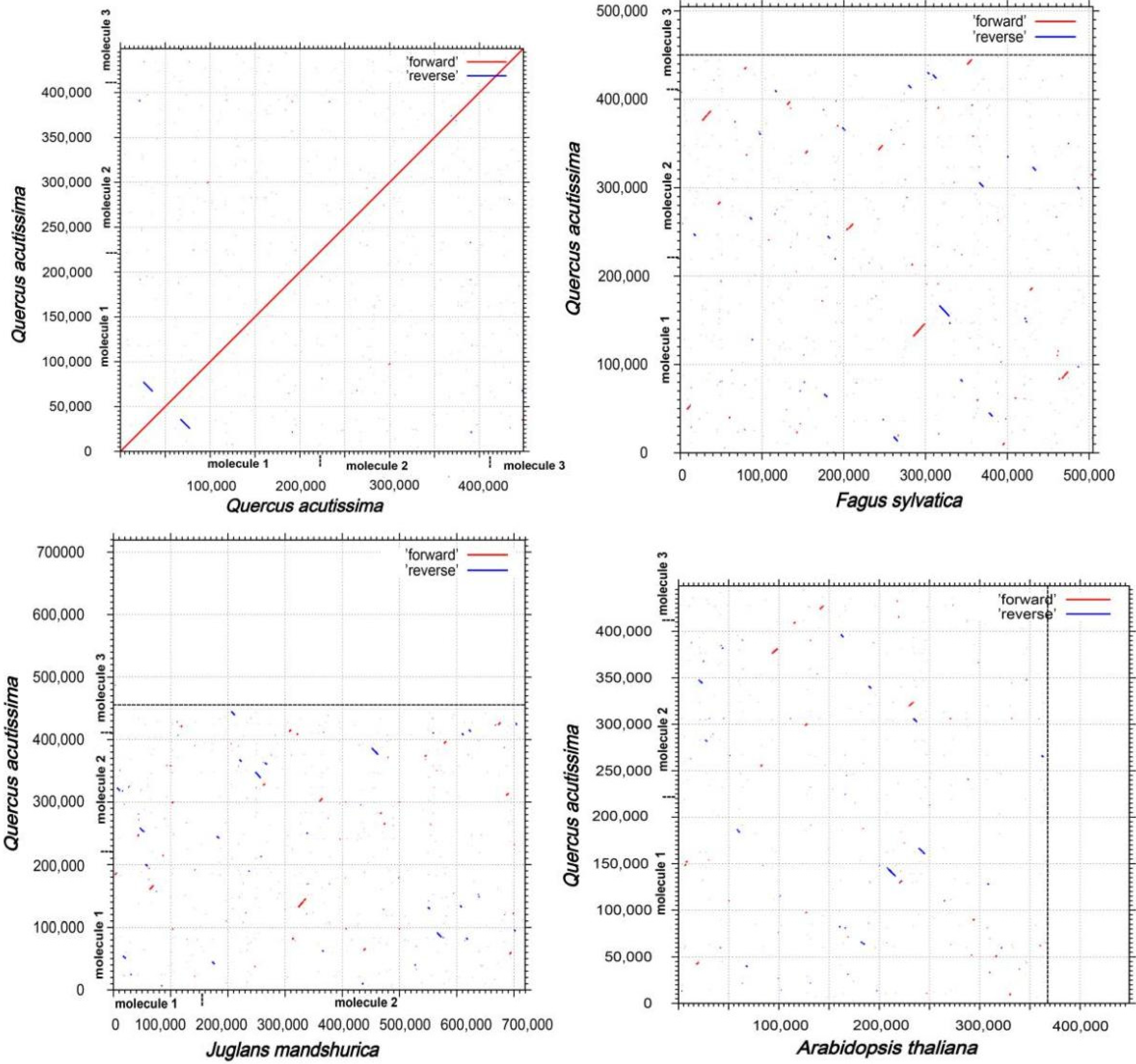
3.7. Synteny and Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, H. Study on Population Genetics and Demographic History of the Three Closely Related Species of Section Aegilops Occurred in China; Northwest University: Xi’an, China, 2018. [Google Scholar]
- Zhang, X.; Li, Y.; Fang, Y. Geographical Distribution and Prediction of Potential Ranges of Quercus acutissima in China. Acta Bot. Boreali-Occident. Sin. 2014, 34, 1685–1692. [Google Scholar]
- Ge, L.; Cheng, X.; Duan, X.; Yu, M.; Liu, Z. Effects of fertilization on the carbon density of sawtooth oak plantations and the soil respi-ration in dormant period. Chin. J. Ecol. 2012, 31, 248–253. [Google Scholar]
- Li, Y. Genetic Structure and Evolutionary History of Chinese Oak Species in Quercus Section Cerris; Nanjing Forestry University: Nanjing, China, 2019. [Google Scholar]
- Tan, Z.Y.; Tang, H.F. Cultivation technology of native species of Quercus acutissima. For. Ecol. 2022, 1, 38–39. [Google Scholar]
- Ye, N.; Wang, X.; Li, J.; Bi, C.; Xu, Y.; Wu, D.; Ye, Q. Assembly and comparative analysis of complete mitochondrial genome sequence of an economic plant Salix suchowensis. PeerJ 2017, 5, e3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birky, C.W. Uniparental inheritance of mitochondrial and chloroplast genes: Mechanisms and evolution. Proc. Natl. Acad. Sci. USA 1995, 92, 11331–11338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogihara, Y.; Yamazaki, Y.; Murai, K.; Kanno, A.; Terachi, T.; Shiina, T.; Miyashita, N.; Nasuda, S.; Nakamura, C.; Mori, N.; et al. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 2005, 33, 6235–6250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Reed, J.C. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar] [CrossRef] [PubMed]
- van Loo, G.; Saelens, X.; van Gurp, M.; MacFarlane, M.; Martin, S.J.; Vandenabeele, P. The role of mitochondrial factors in apoptosis: A Russian roulette with more than one bullet. Cell Death Differ. 2002, 9, 1031–1042. [Google Scholar] [CrossRef]
- Bonora, M.; De Marchi, E.; Patergnani, S.; Suski, J.M.; Celsi, F.; Bononi, A.; Giorgi, C.; Marchi, S.; Rimessi, A.; Duszynski, J.; et al. Tumor necrosis factor-α impairs oligodendroglial differentiation through a mitochondria-dependent process. Cell Death Differ. 2014, 21, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; He, X.; Priyadarshani, S.V.G.N.; Wang, Y.; Ye, L.; Shi, C.; Ye, K.; Zhou, Q.; Luo, Z.; Deng, F.; et al. Assembly and comparative analysis of the complete mitogenomes of Suaeda Glauca. BMC Genom. 2021, 22, 167. [Google Scholar] [CrossRef]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.S.; Elsas II, L.J.; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Lu, N.; Xu, Y.; He, C.; Lu, Z. Characterization and Analysis of the Mitochondrial Genome of Common Bean (Phaseolus vulgaris) by Comparative Genomic Approaches. Int. J. Mol. Sci. 2020, 21, 3778. [Google Scholar] [CrossRef] [PubMed]
- Skippington, E.; Barkman, T.J.; Rice, D.W.; Palmer, J.D. Miniaturized mitogenome of the parasitic plant Viscum scurruloideum is extremely divergent and dynamic and has lost all nad genes. Proc. Natl. Acad. Sci. USA 2015, 112, E3515–E3524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, D.B.; Alverson, A.J.; Chuckalovcak, J.P.; Wu, M.; McCauley, D.E.; Palmer, J.D.; Taylor, D.R. Rapid Evolution of Enormous, Multichromosomal Genomes in Flowering Plant Mitochondria with Exceptionally High Mutation Rates. PLoS Biol. 2012, 10, e1001241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mower, J.P.; Sloan, D.B.; Alverson, A.J. Plant Mitochondrial Genome Diversity: The Genomics Revolution; Springer: Heidelberg, Austria, 2012; Volume 1, pp. 123–144. [Google Scholar]
- Burger, G.; Gray, M.W.; Franz Lang, B. Mitochondrial genomes: Anything goes. Trends Genet. 2003, 19, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Christiane, F.; Mark, C.; Yan, G.; Barry, M. The maize mitochondrial genome: Dynamic, yet functional. Trends Genet. 1995, 11, 228–235. [Google Scholar]
- Wang, S.B.; Li, D.W.; Yao, X.H.; Song, Q.W.; Wang, Z.P.; Zhang, Q.; Zhong, C.; Liu, Y.; Huang, H. Evolution and diversification of kiwifruit mitogenomes through extensive whole-genome rearrangement and mosaic loss of intergenic sequences in a highly variable region. Genome Biol. Evol. 2019, 11, 1192–1206. [Google Scholar] [CrossRef]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLOS Genet. 2019, 15, e1008373. [Google Scholar] [CrossRef] [Green Version]
- Jackman, S.D.; Lauren, C.; Warren, R.L.; Heather, K.; Eva, T.; MacLeod, T.; Pleasance, S.; Pandoh, P.; Zhao, Y.; Coope, R.J.; et al. Complete mitochondrial genome of a gymnosperm, Sitka spruce (Picea sitchensis), indicates a complex physical structure. Genome Biol. Evol. 2020, 12, 1174–1179. [Google Scholar] [CrossRef]
- Petit, R.J.; Csaikl, U.M.; Bordacs, S.; Burg, K.; Coart, E.; Cottrell, J.; Dam, B.; Deans, J.D.; Dumolin-Lapegue, S.; Fineschi, S.; et al. Corrigendum to “Chloroplast DNA variation in European white oaks phylogeography and patterns of diversity based on data from over 2600 populations”. For. Ecol. Manag. 2003, 176, 595–599. [Google Scholar] [CrossRef]
- Meng, X. Study on Phylogeography and Population Genetics Structure in Quercus Acutissima Carr; Northwest University: Xi’an, China, 2017. [Google Scholar]
- Li, X.; Li, Y.; Zang, M.; Li, M.; Fang, Y. Complete Chloroplast Genome Sequence and Phylogenetic Analysis of Quercus acutissima. Int. J. Mol. Sci. 2018, 19, 2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; dePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, T.; Pascal, L.; Tommaso, P.; Elena, S.U.J.; Axel, F.; Ralph, B.; Stephan, G. GeSeq-versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef] [Green Version]
- Lewis, S.E.; Searle, S.; Harris, N.; Gibson, M.; Iyer, V.; Richter, J.; Wiel, C.; Bayraktaroglu, L.; Birney, E.; Crosby, M.A. Apollo: A sequence annotation editor. Genome Biol. 2002, 3, research0082. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Li, W.X.; Jakovlić, I.; Zou, H.; Zhang, J.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7. 0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Meltzer, P.; Davis, S. RCircos: An R package for Circos 2D track plots. BMC Bioinform. 2013, 14, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.Q.; Wu, X.; Huang, L.F.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef] [PubMed]
- Mower, J.P. The PREP suite: Predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 2009, 37, W253–W259. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.-H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Hiesel, R.; Haseler, A.V.; Brennicke, A. Plant Mitochondrial Nucleic Acid Sequences as a Tool for Phylogenetic Analysis. Proc. Natl. Acad. Sci. USA 1994, 91, 634–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unseld, M.; Marienfeld, J.R.; Brandt, P.; Brennicke, A. The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat. Genet. 1997, 15, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Notsu, Y.; Masood, S.; Nishikawa, T.; Kubo, N.; Akiduki, G.; Nakazono, M.; Hirai, A.; Kadowaki, K. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: Frequent DNA sequence acquisition and loss during the evolution of fowering plants. Mol. Gen. Genom. 2002, 268, 434–445. [Google Scholar] [CrossRef] [PubMed]
- Clifton, S.W.; Minx, P.; Fauron, C.M.-R.; Gibson, M.; Allen, J.O.; Sun, H.; Thompson, M.; Barbazuk, W.B.; Kanuganti, S.; Tayloe, C.; et al. Sequence and Comparative Analysis of the Maize NB Mitochondrial Genome. Plant Physiol. 2004, 136, 3486–3503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morley, S.A.; Nielsen, B.L. Plant mitochondrial DNA. Front. Biosci. 2017, 22, 1023–1032. [Google Scholar]
- Oldenburg, D.J.; Bendich, A.J. DNA maintenance in plastids and mitochondria of plants. Front. Plant Sci. 2015, 6, 883. [Google Scholar] [CrossRef] [Green Version]
- Wynn, E.L.; Christensen, A.C. Repeats of Unusual Size in Plant Mitochondrial Genomes: Identification, Incidence and Evolution. G3-Genes Genom. Genet. 2018, 9, 549–559. [Google Scholar] [CrossRef] [Green Version]
- Backert, S.; Nielsen, B.L.; Borner, T. The mystery of the rings: Structure and replication of mitochondrial genomes from higher plants. Trends Plant Sci. 1997, 2, 477–483. [Google Scholar] [CrossRef]
- Bi, Q.; Li, D.; Zhao, Y.; Wang, M.; Li, Y.; Liu, X.; Wang, L.; Yu, H. Complete mitochondrial genome of Quercus variabilis (Fagales, Fagaceae). Mitochondrial DNA Part B 2019, 4, 3927–3928. [Google Scholar] [CrossRef] [Green Version]
- Salojarvi, J.; Smolander, O.P.; Nieminen, K.; Rajaraman, S.; Safronov, O.; Safdari, P.; Lamminmäki, A.; Immanen, J.; Lan, T.; Tanskanen, J.; et al. Genome sequencing and population genomic analyses provide insights into the adaptive landscape of silver birch. Nat. Genet. 2017, 49, 904–912. [Google Scholar] [CrossRef]
- Mader, M.; Schroeder, H.; Schott, T.; Schning-Stierand, K.; Montalvão, A.P.L.; Liesebach, H.; Liesebach, M.; Fussi, B.; Kersten, B. Mitochondrial genome of Fagus sylvatica L. as a source for taxonomic marker development in the fagales. Plants 2020, 9, 1274. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Wang, Y.; Li, S.; Wen, J.; Zhu, L.; Yan, K.; Du, Y.; Ren, J.; Li, S.; Chen, Z.; et al. Assembly and comparative analysis of the first complete mitochondrial genome of Acer truncatum Bunge: A woody oil-tree species producing nervonic acid. BMC Plant Biol. 2022, 22, 29. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wariss, H.M.; Tao, L.; Zhang, R.; Yun, Q.; Hollingsworth, P.; Dao, Z.; Luo, G.; Guo, H.; Ma, Y.; et al. De novo genome assembly of the endangered Acer yangbiense, a plant species with extremely small populations endemic to Yunnan Province, China. GigaScience 2019, 8, giz085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, D.; Wu, Z.; Sharbrough, J. Correction of persistent errors in Arabidopsis reference mitochondrial genomes. Plant Cell 2018, 30, 525–527. [Google Scholar] [CrossRef] [Green Version]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef]
- Guo, W.; Grewe, F.; Fan, W.; Young, G.J.; Knoop, V.; Palmer, J.D.; Mower, J.P. Ginkgo and Welwitschia Mitogenomes reveal extreme contrasts in gymnosperm mitochondrial evolution. Mol. Biol. Evol. 2016, 33, 1448–1460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Wang, W.; Yao, Y.; Sun, Q. Mitochondrial RNase H1 activity regulates R-loop homeostasis to maintain genome integrity and enable early embryogenesis in Arabidopsis. PLoS Biol. 2021, 19, e3001357. [Google Scholar] [CrossRef] [PubMed]
- Odahara, M.; Nakamura, K.; Sekine, Y.; Oshima, T. Ultra-deep sequencing reveals dramatic alteration of organellar genomes in Physcomitrella patens due to biased asymmetric recombination. Commun. Biol. 2021, 4, 633. [Google Scholar] [CrossRef]
- Varré, J.-S.; D’Agostino, N.; Touzet, P.; Gallina, S.; Tamburino, R.; Cantarella, C.; Ubrig, E.; Cardi, T.; Drouard, L.; Gualberto, J.M.; et al. Complete Sequence, Multichromosomal Architecture and Transcriptome Analysis of the Solanum tuberosum Mitochondrial Genome. Int. J. Mol. Sci. 2019, 20, 4788. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Zhao, C.; Chen, F.; Liu, Y.; Zhang, S.; Wu, H.; Zhang, L.; Liu, Y. The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genom. 2018, 19, 614. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Shan, Y.; Pei, X.; Yong, S.; Liu, C.; Yu, J. Assembly of the complete mitochondrial genome of an endemic plant, Scutellaria tsinyunensis, revealed the existence of two conformations generated by a repeat-mediated recombination. Planta 2021, 254, 36. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, J.; Ma, Y.; Kou, L.; Wei, J.; Wang, W. The complete mitochondrial genome of okra (Abelmoschus esculentus): Using nanopore long reads to investigate gene transfer from chloroplast genomes and rearrangements of mitochondrial DNA molecules. BMC Genom. 2022, 23, 481. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Li, J.; Zhao, Q.; Liang, Y.; Yu, J. Assembly of the Complete Mitochondrial Genome of Chinese Plum (Prunus salicina): Characterization of Genome Recombination and RNA Editing Sites. Genes 2021, 12, 1970. [Google Scholar] [CrossRef] [PubMed]
- Bi, C.; Paterson, A.H.; Wang, X.; Xu, Y.; Wu, D.; Qu, Y.; Jiang, A.; Ye, Q.; Ye, N. Analysis of the complete mitochondrial genome sequence of the diploid cotton Gossypium raimondii by comparative genomics approaches. BioMed Res. Int. 2016, 2016, 5040598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
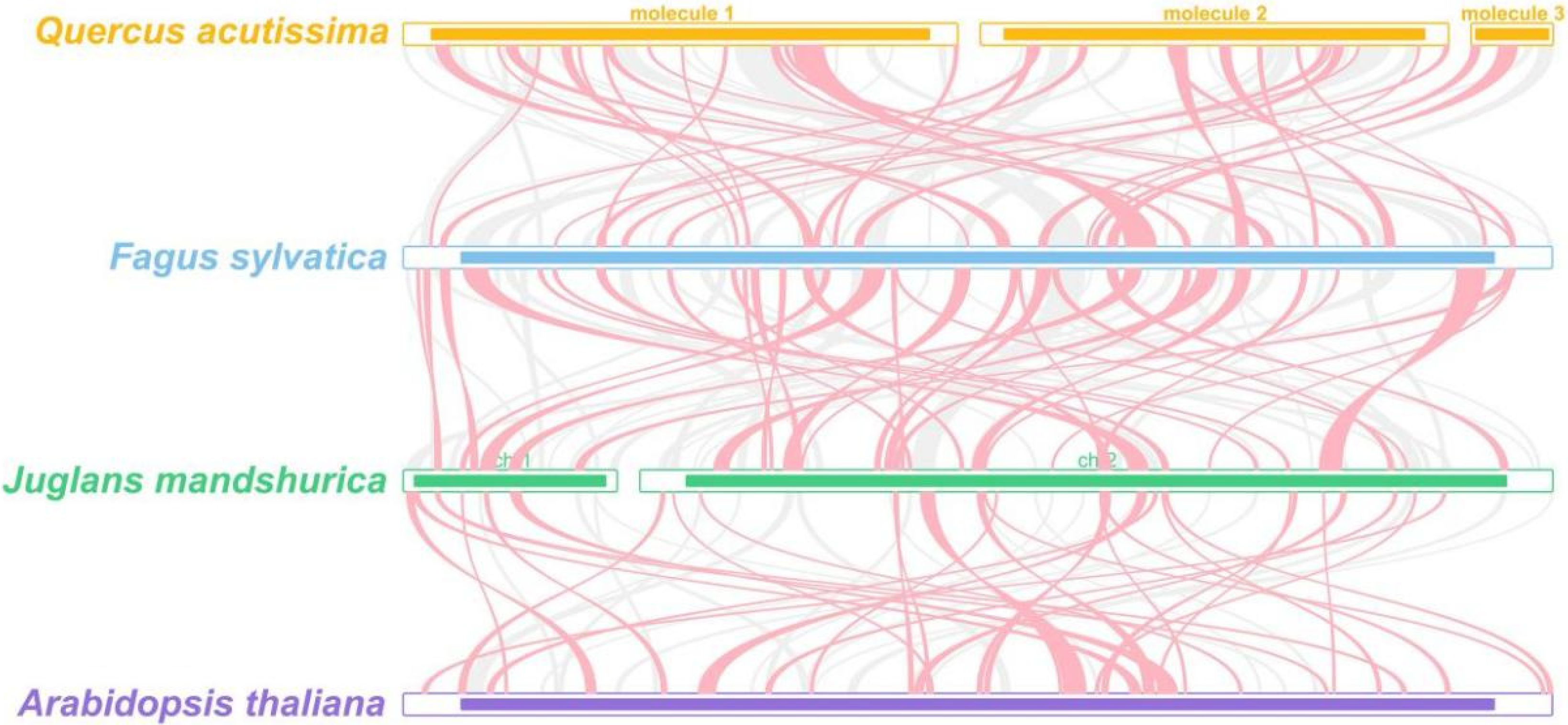
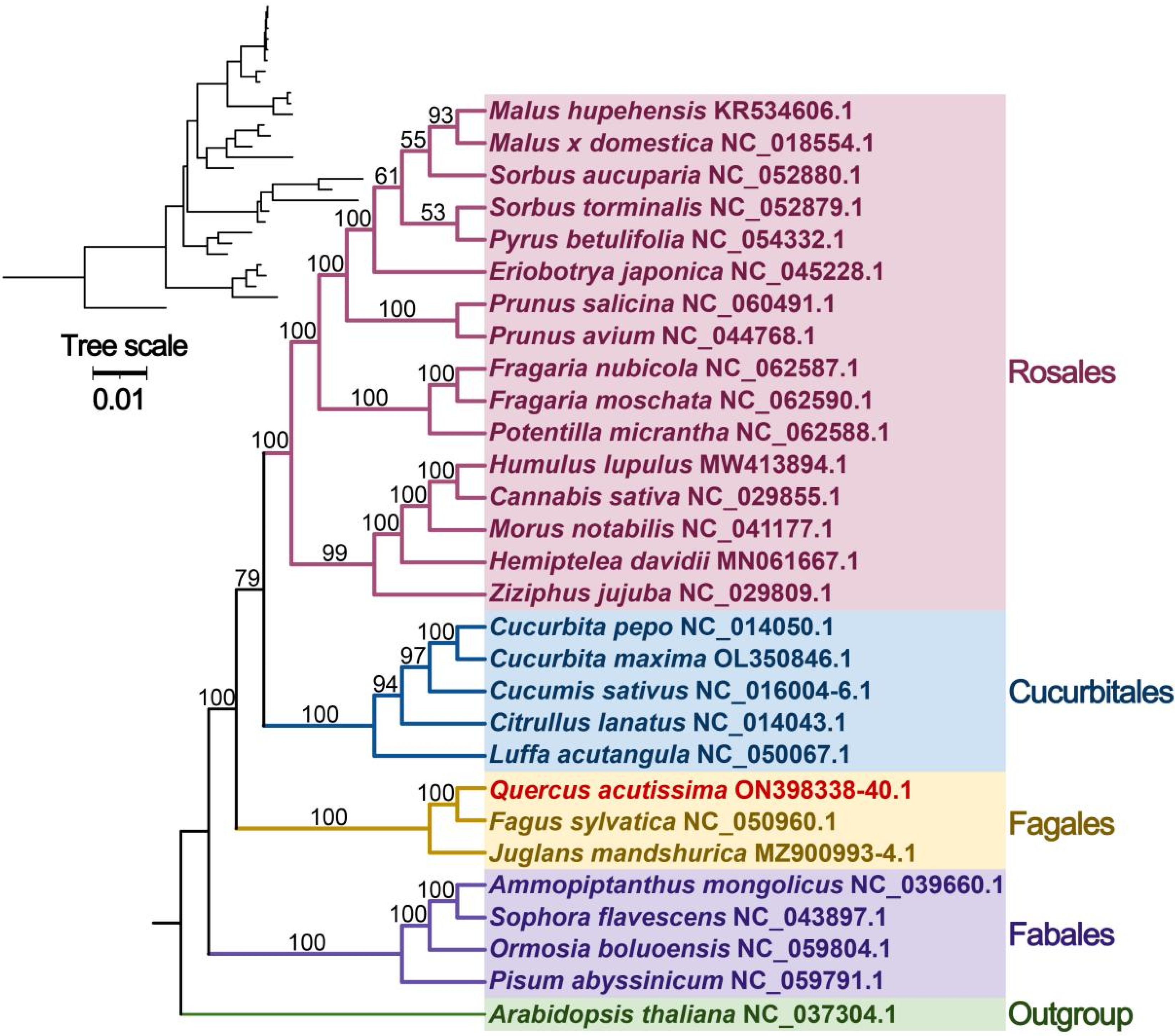

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Repeat | Length (bp) | Location | Reads Support Major Conformation | Reads Support Alternative Conformation |
|---|---|---|---|---|
| R1 | 10,578 | molecule 1: 35,975–25,398 | 19 | 8 |
| molecule 1: 66,867–77,444 | ||||
| R2 | 1679 | molecule 1: 68,252–66,574 | 224 | 182 |
| molecule 2: 34,939–36,490; 1–127 |
| Alignment Length | Identity% | Mis-match | Gap Openings | CP Start | CP End | Mt Start | Mt End | MTPT Annotation | |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4760 | 99.979 | 1 | 0 | 106,031 | 110,790 | 98,643 | 93,884 | Complete (trnV-GAC, rrn16S, trnI-GAU, trnA-UGC), Partial (rrn23S) |
| 2 | 4760 | 99.979 | 1 | 0 | 140,802 | 145,561 | 93,884 | 98,643 | Partial (rrn23S), Complete (trnA-UGC, trnI-GAU, rrn16S, trnV-GAC) |
| 3 | 354 | 97.74 | 8 | 0 | 620 | 973 | 26,955 | 26,602 | Partial (psbA) |
| 4 | 354 | 97.74 | 8 | 0 | 620 | 973 | 75,887 | 76,240 | Partial (psbA) |
| 5 | 127 | 100 | 0 | 0 | 108,462 | 108,588 | 224,233 | 224,107 | Partial (trnI-GAU) |
| 6 | 127 | 100 | 0 | 0 | 143,004 | 143,130 | 224,107 | 224,233 | Partial (trnI-GAU) |
| 7 | 77 | 98.701 | 1 | 0 | 33,701 | 33,777 | 133,648 | 133,724 | Complete (trnD-GUC) |
| 8 | 75 | 94.667 | 4 | 0 | 57,523 | 57,597 | 83,943 | 84,017 | Complete (trnM-CAU) |
| 9 | 77 | 89.61 | 6 | 2 | 159,056 | 159,130 | 127,587 | 127,511 | Complete (trnI-CAU) |
| 10 | 77 | 89.61 | 6 | 2 | 92,462 | 92,536 | 127,511 | 127,587 | Complete (trnI-CAU) |
| 11 | 28 | 100 | 0 | 0 | 1015 | 1042 | 26,579 | 26,606 | Partial (psbA) |
| 12 | 28 | 100 | 0 | 0 | 1015 | 1042 | 76,263 | 76,236 | Partial (psbA) |
| 13 | 1149 | 99.739 | 3 | 0 | 37,300 | 38,448 | 140,389 | 139,241 | Partial (psbD, psbC) |
| 14 | 1017 | 77.778 | 150 | 46 | 71,581 | 72,585 | 141,644 | 142,596 | Complete (petL, petG, trnW-CCA, trnP-UGG) |
| 15 | 495 | 83.03 | 73 | 8 | 70,186 | 70,678 | 140,403 | 140,888 | Partial (psbE) |
| 16 | 889 | 73.903 | 177 | 42 | 106,819 | 107,682 | 76,204 | 75,346 | Partial (rrn16S) |
| 17 | 889 | 73.903 | 177 | 42 | 143,910 | 144,773 | 75,346 | 76,204 | Partial (rrn16S) |
| 18 | 83 | 98.795 | 1 | 0 | 136,687 | 136,769 | 157,954 | 157,872 | Complete (trnN-GUU) |
| 19 | 83 | 98.795 | 1 | 0 | 114,823 | 114,905 | 157,872 | 157,954 | Complete (trnN-GUU) |
| 20 | 55 | 96.364 | 2 | 0 | 143,130 | 143,184 | 8165 | 8111 | Partial (trnI-GAU) |
| 21 | 55 | 96.364 | 2 | 0 | 108,408 | 108,462 | 8111 | 8165 | Partial (trnI-GAU) |
| 22 | 38 | 100 | 0 | 0 | 13,356 | 13,393 | 49,527 | 49,490 | Partial (atpA) |
| 23 | 91 | 96.703 | 2 | 1 | 14 | 103 | 31,975 | 31,885 | Complete (trnH-GUG) |
| Total | 15,688 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Guo, H.; Zhu, J.; Qu, K.; Chen, Y.; Guo, Y.; Ding, P.; Yang, H.; Xu, T.; Jing, Q.; et al. Complex Physical Structure of Complete Mitochondrial Genome of Quercus acutissima (Fagaceae): A Significant Energy Plant. Genes 2022, 13, 1321. https://doi.org/10.3390/genes13081321
Liu D, Guo H, Zhu J, Qu K, Chen Y, Guo Y, Ding P, Yang H, Xu T, Jing Q, et al. Complex Physical Structure of Complete Mitochondrial Genome of Quercus acutissima (Fagaceae): A Significant Energy Plant. Genes. 2022; 13(8):1321. https://doi.org/10.3390/genes13081321
Chicago/Turabian StyleLiu, Dan, Haili Guo, Jingle Zhu, Kai Qu, Ying Chen, Yingtian Guo, Ping Ding, Haiping Yang, Ting Xu, Qi Jing, and et al. 2022. "Complex Physical Structure of Complete Mitochondrial Genome of Quercus acutissima (Fagaceae): A Significant Energy Plant" Genes 13, no. 8: 1321. https://doi.org/10.3390/genes13081321
APA StyleLiu, D., Guo, H., Zhu, J., Qu, K., Chen, Y., Guo, Y., Ding, P., Yang, H., Xu, T., Jing, Q., Han, S., Li, W., & Tong, B. (2022). Complex Physical Structure of Complete Mitochondrial Genome of Quercus acutissima (Fagaceae): A Significant Energy Plant. Genes, 13(8), 1321. https://doi.org/10.3390/genes13081321