Building Blood Vessels—One Rho GTPase at a Time


Abstract
1. Introduction
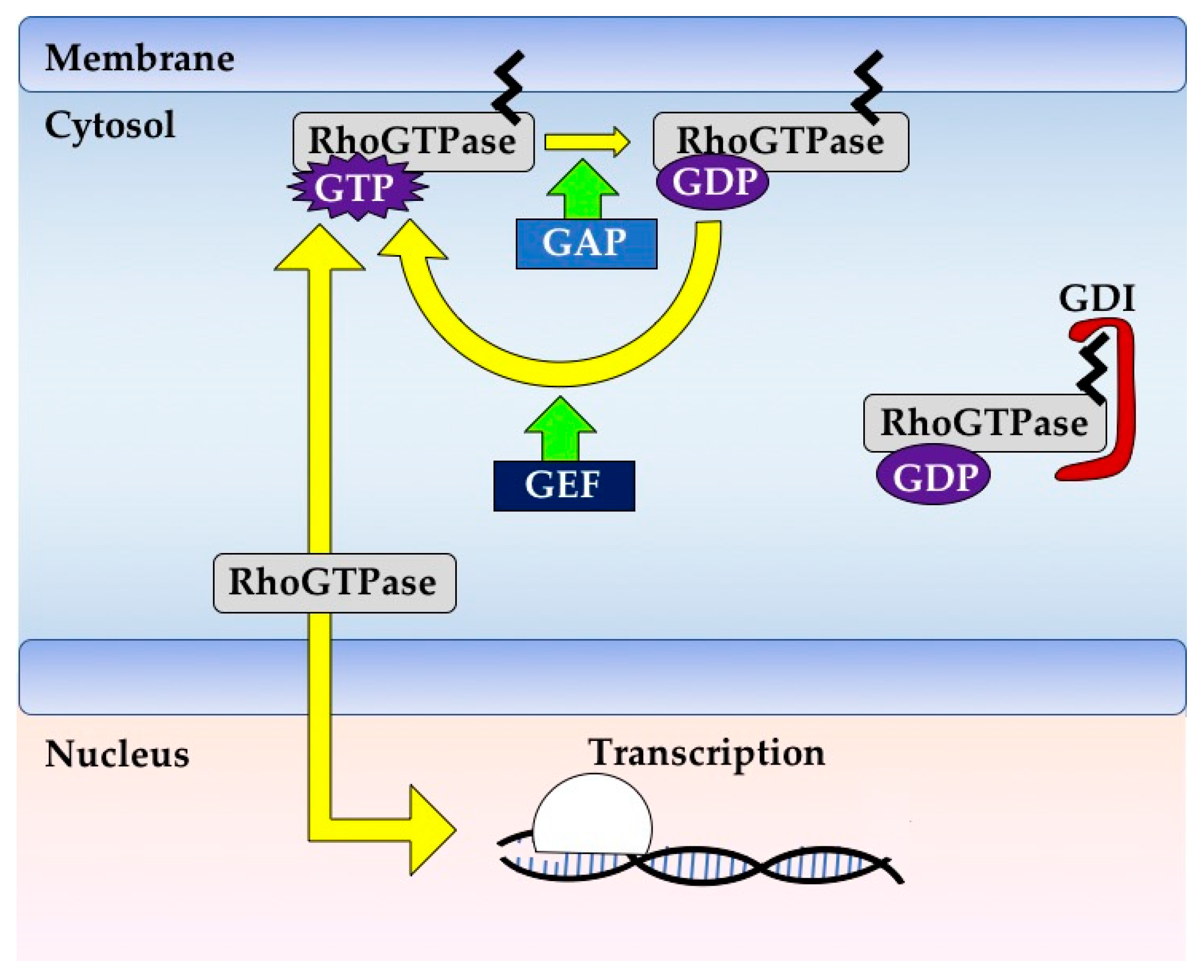
1.1. GTPases Are Powerful Biomolecular Switches
1.2. GTPase Regulation Is Fine-Tuned and Context-Dependent
2. Rho GTPases: A Diverse Family of Molecules
3. In Vitro Models: The Foundations of Rho GTPase Biology
3.1. RhoJ Signaling
3.2. Rac1 Signaling
3.3. Cdc42 Signaling
3.4. Rho Subfamily Signaling
4. Vasculogenesis In Vivo
4.1. Rho GTPases Control Cellular Processes Underlying Vasculogenesis
4.1.1. Cell–Cell Adhesion Formation
4.1.2. Junctional Remodeling
4.1.3. Cell Contractility and Expansion
5. Angiogenesis
5.1. Rho GTPases Required for Angiogenesis
5.1.1. RhoA Signaling
5.1.2. RhoB Signaling
5.1.3. RhoJ Signaling
5.1.4. Rac2 Signaling
5.1.5. Rnd3 Signaling
5.2. Rho GTPases Control Cellular Processes Underlying Angiogenesis
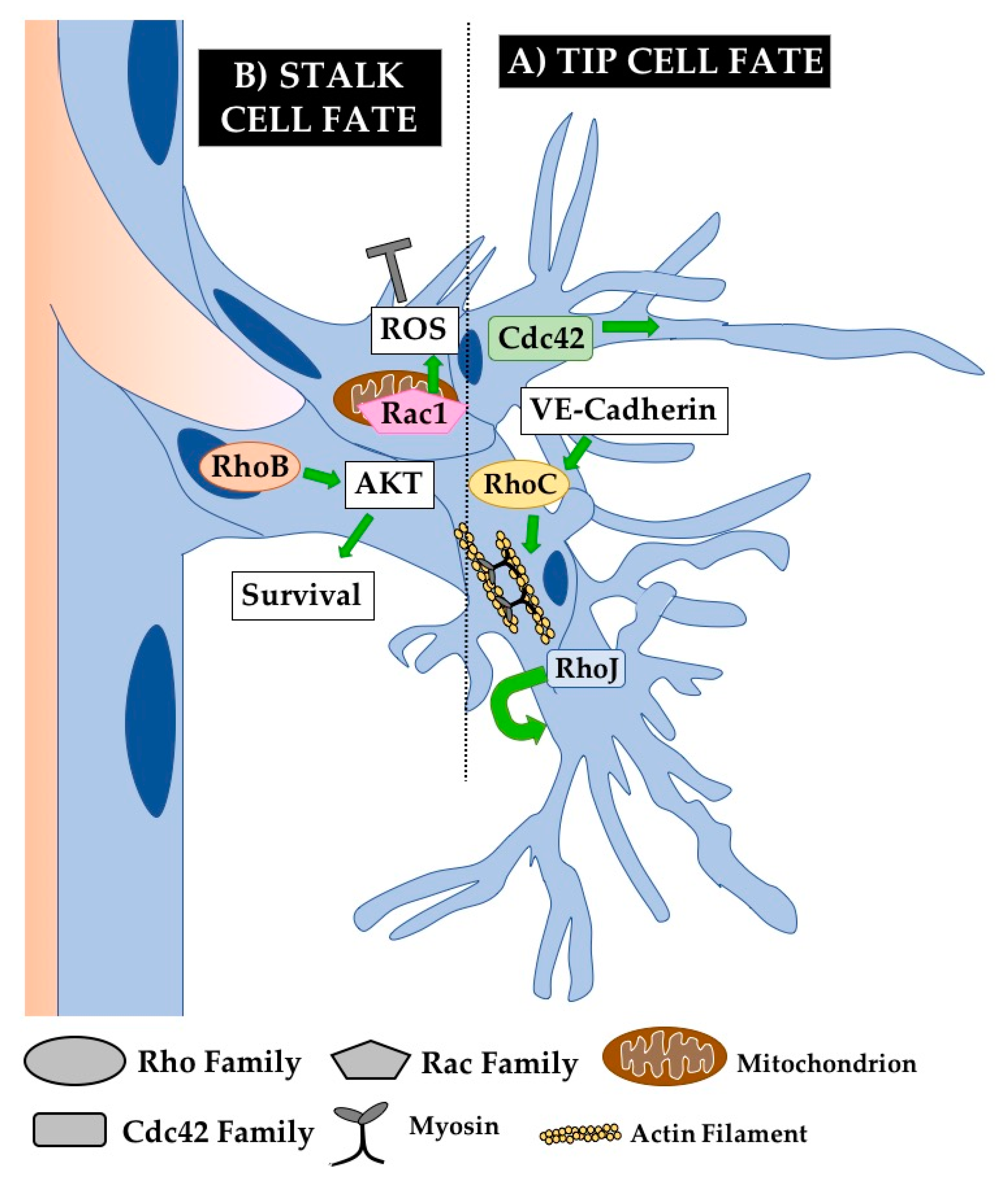
5.2.1. Tip Cell Defects
5.2.2. Sprouting Defects
6. Barrier Function in Mature Vessels
6.1. Barrier Stabilization
6.2. Barrier Destabilization
6.3. Barrier Recovery from Damage
7. Rho GTPases in Disease
7.1. Cerebral Cavernous Malformations (CCMs)
7.2. Complications of Diabetes
7.3. Anaphylaxis
7.4. Tumor Angiogenesis
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xu, K.; Cleaver, O. Tubulogenesis during blood vessel formation. Semin. Cell Dev. Biol. 2011, 22, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, O.; Krieg, P.A. Molecular Mechanisms of Vascular Development. In Heart Development, 1st ed.; Harvey, R.P., Rosenthal, N., Eds.; Academic Press: San Diego, CA, USA, 1999; pp. 221–244. [Google Scholar]
- Carmeliet, P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. [Google Scholar] [CrossRef]
- Herbert, S.P.; Stainier, D.Y. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat. Rev. Mol. Cell Biol. 2011, 12, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.J.; Fleming, P.A. Vasculogenesis in the day 6.5 to 9.5 mouse embryo. Blood 2000, 95, 1671–1679. [Google Scholar] [PubMed]
- Claesson-Welsh, L. Vascular permeability—The essentials. Upsala J. Med. Sci. 2015, 120, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Wennerberg, K.; Der, C.J. Rho-family GTPases: it’s not only Rac and Rho (and I like it). J. Cell Sci. 2004, 117, 1301–1312. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Stoeckle, C.; Geering, B.; Yousefi, S.; Rozman, S.; Andina, N.; Benarafa, C.; Simon, H.U. RhoH is a negative regulator of eosinophilopoiesis. Cell Death Differ. 2016, 23, 1961–1972. [Google Scholar] [CrossRef]
- Jahner, D.; Hunter, T. The ras-related gene rhoB is an immediate-early gene inducible by v-Fps, epidermal growth factor, and platelet-derived growth factor in rat fibroblasts. Mol. Cell. Biol. 1991, 11, 3682–3690. [Google Scholar] [CrossRef]
- Adini, I.; Rabinovitz, I.; Sun, J.F.; Prendergast, G.C.; Benjamin, L.E. RhoB controls Akt trafficking and stage-specific survival of endothelial cells during vascular development. Genes Dev. 2003, 17, 2721–2732. [Google Scholar] [CrossRef]
- Shi, D.; Qi, M.; Zhou, L.; Li, X.; Ni, L.; Li, C.; Yuan, T.; Wang, Y.; Chen, Y.; Hu, C.; et al. Endothelial Mitochondrial Preprotein Translocase Tomm7-Rac1 Signaling Axis Dominates Cerebrovascular Network Homeostasis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2665–2677. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, O.; Poland, M.; Gebbink, M.; Oomen, L.; Moolenaar, W.H. Dissociation of LPA-induced cytoskeletal contraction from stress fiber formation by differential localization of RhoA. J. Cell Sci. 1997, 110 (Pt 19), 2417–2427. [Google Scholar]
- Masiero, M.; Simoes, F.C.; Han, H.D.; Snell, C.; Peterkin, T.; Bridges, E.; Mangala, L.S.; Wu, S.Y.; Pradeep, S.; Li, D.; et al. A core human primary tumor angiogenesis signature identifies the endothelial orphan receptor ELTD1 as a key regulator of angiogenesis. Cancer Cell 2013, 24, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Vega, F.M.; Ridley, A.J. Rho GTPases in cancer cell biology. FEBS Lett. 2008, 582, 2093–2101. [Google Scholar] [CrossRef] [PubMed]
- Pijuan-Sala, B.; Griffiths, J.A.; Guibentif, C.; Hiscock, T.W.; Jawaid, W.; Calero-Nieto, F.J.; Mulas, C.; Ibarra-Soria, X.; Tyser, R.C.V.; Ho, D.L.L.; et al. A single-cell molecular map of mouse gastrulation and early organogenesis. Nature 2019, 566, 490–495. [Google Scholar] [CrossRef]
- Faure, S.; Fort, P. Atypical RhoV and RhoU GTPases control development of the neural crest. Small GTPases 2015, 6, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Troeger, A.; Williams, D.A. Hematopoietic-specific Rho GTPases Rac2 and RhoH and human blood disorders. Exp. Cell Res. 2013, 319, 2375–2383. [Google Scholar] [CrossRef] [PubMed]
- Tabula Muris Consortium; Overall Coordination; Logistical Coordination; Organ Collection and Processing; Library Preparation and Sequencing; Computational Data Analysis; Cell Type Annotation; Writing Group; Supplemental Text Writing Group; Principal Investigators. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature 2018, 562, 367–372. [Google Scholar] [CrossRef]
- Roberts, P.J.; Mitin, N.; Keller, P.J.; Chenette, E.J.; Madigan, J.P.; Currin, R.O.; Cox, A.D.; Wilson, O.; Kirschmeier, P.; Der, C.J. Rho Family GTPase modification and dependence on CAAX motif-signaled posttranslational modification. J. Biol. Chem. 2008, 283, 25150–25163. [Google Scholar] [CrossRef] [PubMed]
- Shutes, A.; Berzat, A.C.; Chenette, E.J.; Cox, A.D.; Der, C.J. Biochemical analyses of the Wrch atypical Rho family GTPases. Methods Enzymol 2006, 406, 11–26. [Google Scholar] [CrossRef]
- Kay, B.K.; Williamson, M.P.; Sudol, M. The importance of being proline: The interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. 2000, 14, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Berzat, A.C.; Buss, J.E.; Chenette, E.J.; Weinbaum, C.A.; Shutes, A.; Der, C.J.; Minden, A.; Cox, A.D. Transforming activity of the Rho family GTPase, Wrch-1, a Wnt-regulated Cdc42 homolog, is dependent on a novel carboxyl-terminal palmitoylation motif. J. Biol. Chem. 2005, 280, 33055–33065. [Google Scholar] [CrossRef] [PubMed]
- Berthold, J.; Schenkova, K.; Ramos, S.; Miura, Y.; Furukawa, M.; Aspenstrom, P.; Rivero, F. Characterization of RhoBTB-dependent Cul3 ubiquitin ligase complexes—Evidence for an autoregulatory mechanism. Exp. Cell Res. 2008, 314, 3453–3465. [Google Scholar] [CrossRef]
- Chang, F.K.; Sato, N.; Kobayashi-Simorowski, N.; Yoshihara, T.; Meth, J.L.; Hamaguchi, M. DBC2 is essential for transporting vesicular stomatitis virus glycoprotein. J. Mol. Biol. 2006, 364, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, E.J.; Calero, M.; Sridevi, K.; Pfeffer, S.R. RhoBTB3: A Rho GTPase-family ATPase required for endosome to Golgi transport. Cell 2009, 137, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, A.; Ping, Q.; Carpenter, C.L. RhoBTB2 is a substrate of the mammalian Cul3 ubiquitin ligase complex. Genes Dev. 2004, 18, 856–861. [Google Scholar] [CrossRef] [PubMed]
- Nobes, C.D.; Lauritzen, I.; Mattei, M.G.; Paris, S.; Hall, A.; Chardin, P. A new member of the Rho family, Rnd1, promotes disassembly of actin filament structures and loss of cell adhesion. J. Cell Biol. 1998, 141, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Foster, R.; Hu, K.Q.; Lu, Y.; Nolan, K.M.; Thissen, J.; Settleman, J. Identification of a novel human Rho protein with unusual properties: GTPase deficiency and in vivo farnesylation. Mol. Cell. Biol. 1996, 16, 2689–2699. [Google Scholar] [CrossRef]
- Chardin, P. Function and regulation of Rnd proteins. Nat. Rev. Mol. Cell Biol. 2006, 7, 54–62. [Google Scholar] [CrossRef]
- Riento, K.; Guasch, R.M.; Garg, R.; Jin, B.; Ridley, A.J. RhoE binds to ROCK I and inhibits downstream signaling. Mol. Cell. Biol. 2003, 23, 4219–4229. [Google Scholar] [CrossRef]
- Aspenstrom, P.; Fransson, A.; Saras, J. Rho GTPases have diverse effects on the organization of the actin filament system. Biochem. J. 2004, 377, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Suehiro, J.; Kanki, Y.; Makihara, C.; Schadler, K.; Miura, M.; Manabe, Y.; Aburatani, H.; Kodama, T.; Minami, T. Genome-wide approaches reveal functional vascular endothelial growth factor (VEGF)-inducible nuclear factor of activated T cells (NFAT) c1 binding to angiogenesis-related genes in the endothelium. J. Biol. Chem. 2014, 289, 29044–29059. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Ma, L.; Parrini, M.C.; Mao, X.; Lopez, M.; Wu, C.; Marks, P.W.; Davidson, L.; Kwiatkowski, D.J.; Kirchhausen, T.; et al. Cdc42 is required for PIP(2)-induced actin polymerization and early development but not for cell viability. Curr. Biol. 2000, 10, 758–765. [Google Scholar] [CrossRef]
- Barry, D.M.; Xu, K.; Meadows, S.M.; Zheng, Y.; Norden, P.R.; Davis, G.E.; Cleaver, O. Cdc42 is required for cytoskeletal support of endothelial cell adhesion during blood vessel formation in mice. Development 2015, 142, 3058–3070. [Google Scholar] [CrossRef] [PubMed]
- Takase, H.; Matsumoto, K.; Yamadera, R.; Kubota, Y.; Otsu, A.; Suzuki, R.; Ishitobi, H.; Mochizuki, H.; Kojima, T.; Takano, S.; et al. Genome-wide identification of endothelial cell-enriched genes in the mouse embryo. Blood 2012, 120, 914–923. [Google Scholar] [CrossRef]
- Wilson, E.; Leszczynska, K.; Poulter, N.S.; Edelmann, F.; Salisbury, V.A.; Noy, P.J.; Bacon, A.; Rappoport, J.Z.; Heath, J.K.; Bicknell, R.; et al. RhoJ interacts with the GIT-PIX complex and regulates focal adhesion disassembly. J. Cell Sci. 2014, 127, 3039–3051. [Google Scholar] [CrossRef]
- Sugihara, K.; Nakatsuji, N.; Nakamura, K.; Nakao, K.; Hashimoto, R.; Otani, H.; Sakagami, H.; Kondo, H.; Nozawa, S.; Aiba, A.; et al. Rac1 is required for the formation of three germ layers during gastrulation. Oncogene 1998, 17, 3427–3433. [Google Scholar] [CrossRef]
- Tan, W.; Palmby, T.R.; Gavard, J.; Amornphimoltham, P.; Zheng, Y.; Gutkind, J.S. An essential role for Rac1 in endothelial cell function and vascular development. FASEB J. 2008, 22, 1829–1838. [Google Scholar] [CrossRef]
- Nohata, N.; Uchida, Y.; Stratman, A.N.; Adams, R.H.; Zheng, Y.; Weinstein, B.M.; Mukouyama, Y.S.; Gutkind, J.S. Temporal-specific roles of Rac1 during vascular development and retinal angiogenesis. Dev. Biol. 2016, 411, 183–194. [Google Scholar] [CrossRef]
- De, P.; Peng, Q.; Traktuev, D.O.; Li, W.; Yoder, M.C.; March, K.L.; Durden, D.L. Expression of RAC2 in endothelial cells is required for the postnatal neovascular response. Exp Cell Res 2009, 315, 248–263. [Google Scholar] [CrossRef]
- Corbetta, S.; Gualdoni, S.; Albertinazzi, C.; Paris, S.; Croci, L.; Consalez, G.G.; de Curtis, I. Generation and characterization of Rac3 knockout mice. Mol. Cell. Biol. 2005, 25, 5763–5776. [Google Scholar] [CrossRef] [PubMed]
- Vigorito, E.; Bell, S.; Hebeis, B.J.; Reynolds, H.; McAdam, S.; Emson, P.C.; McKenzie, A.; Turner, M. Immunological function in mice lacking the Rac-related GTPase RhoG. Mol. Cell. Biol. 2004, 24, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.; Grimm-Gunter, E.M.; Joshi, P.; Rivero, F. Expression analysis of mouse Rhobtb3 using a LacZ reporter and preliminary characterization of a knockout strain. Histochem. Cell Biol. 2014, 142, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Mikelis, C.M.; Simaan, M.; Ando, K.; Fukuhara, S.; Sakurai, A.; Amornphimoltham, P.; Masedunskas, A.; Weigert, R.; Chavakis, T.; Adams, R.H.; et al. RhoA and ROCK mediate histamine-induced vascular leakage and anaphylactic shock. Nat. Commun. 2015, 6, 6725. [Google Scholar] [CrossRef] [PubMed]
- Gerald, D.; Adini, I.; Shechter, S.; Perruzzi, C.; Varnau, J.; Hopkins, B.; Kazerounian, S.; Kurschat, P.; Blachon, S.; Khedkar, S.; et al. RhoB controls coordination of adult angiogenesis and lymphangiogenesis following injury by regulating VEZF1-mediated transcription. Nat. Commun. 2013, 4, 2824. [Google Scholar] [CrossRef] [PubMed]
- Hakem, A.; Sanchez-Sweatman, O.; You-Ten, A.; Duncan, G.; Wakeham, A.; Khokha, R.; Mak, T.W. RhoC is dispensable for embryogenesis and tumor initiation but essential for metastasis. Genes Dev. 2005, 19, 1974–1979. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, T.; Lin, X.; Yue, X.; Wang, Q.; Wang, G.; Fu, Q.; Ai, X.; Chiang, D.Y.; Miyake, C.Y.; et al. Genetic deletion of Rnd3/RhoE results in mouse heart calcium leakage through upregulation of protein kinase A signaling. Circ. Res. 2015, 116, e1–e10. [Google Scholar] [CrossRef]
- Lin, X.; Liu, B.; Yang, X.; Yue, X.; Diao, L.; Wang, J.; Chang, J. Genetic deletion of Rnd3 results in aqueductal stenosis leading to hydrocephalus through up-regulation of Notch signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 8236–8241. [Google Scholar] [CrossRef]
- Goggs, R.; Savage, J.S.; Mellor, H.; Poole, A.W. The small GTPase Rif is dispensable for platelet filopodia generation in mice. PLoS ONE 2013, 8, e54663. [Google Scholar] [CrossRef]
- Kishimoto, M.; Matsuda, T.; Yanase, S.; Katsumi, A.; Suzuki, N.; Ikejiri, M.; Takagi, A.; Ikawa, M.; Kojima, T.; Kunishima, S.; et al. Rhof promotes murine marginal zone B cell development. Nagoya J. Med. Sci. 2014, 76, 293–305. [Google Scholar]
- Davis, G.E.; Black, S.M.; Bayless, K.J. Capillary morphogenesis during human endothelial cell invasion of three-dimensional collagen matrices. In Vitro Cell. Dev. Biol. Anim. 2000, 36, 513–519. [Google Scholar] [CrossRef]
- Staton, C.A.; Reed, M.W.; Brown, N.J. A critical analysis of current in vitro and in vivo angiogenesis assays. Int. J. Exp. Pathol. 2009, 90, 195–221. [Google Scholar] [CrossRef] [PubMed]
- Viemann, D.; Goebeler, M.; Schmid, S.; Nordhues, U.; Klimmek, K.; Sorg, C.; Roth, J. TNF induces distinct gene expression programs in microvascular and macrovascular human endothelial cells. J. Leukoc. Biol. 2006, 80, 174–185. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K.; Just, I. Clostridial Rho-inhibiting protein toxins. Curr. Top. Microbiol. Immunol. 2005, 291, 113–145. [Google Scholar]
- Donovan, D.; Brown, N.J.; Bishop, E.T.; Lewis, C.E. Comparison of three in vitro human ‘angiogenesis’ assays with capillaries formed in vivo. Angiogenesis 2001, 4, 113–121. [Google Scholar] [CrossRef]
- Kaur, S.; Leszczynska, K.; Abraham, S.; Scarcia, M.; Hiltbrunner, S.; Marshall, C.J.; Mavria, G.; Bicknell, R.; Heath, V.L. RhoJ/TCL regulates endothelial motility and tube formation and modulates actomyosin contractility and focal adhesion numbers. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 657–664. [Google Scholar] [CrossRef]
- Yuan, L.; Sacharidou, A.; Stratman, A.N.; Le Bras, A.; Zwiers, P.J.; Spokes, K.; Bhasin, M.; Shih, S.C.; Nagy, J.A.; Molema, G.; et al. RhoJ is an endothelial cell-restricted Rho GTPase that mediates vascular morphogenesis and is regulated by the transcription factor ERG. Blood 2011, 118, 1145–1153. [Google Scholar] [CrossRef]
- Kim, C.; Yang, H.; Fukushima, Y.; Saw, P.E.; Lee, J.; Park, J.S.; Park, I.; Jung, J.; Kataoka, H.; Lee, D.; et al. Vascular RhoJ is an effective and selective target for tumor angiogenesis and vascular disruption. Cancer Cell 2014, 25, 102–117. [Google Scholar] [CrossRef]
- Fukushima, Y.; Okada, M.; Kataoka, H.; Hirashima, M.; Yoshida, Y.; Mann, F.; Gomi, F.; Nishida, K.; Nishikawa, S.; Uemura, A. Sema3E-PlexinD1 signaling selectively suppresses disoriented angiogenesis in ischemic retinopathy in mice. J. Clin. Investig. 2011, 121, 1974–1985. [Google Scholar] [CrossRef]
- Bayless, K.J.; Davis, G.E. The Cdc42 and Rac1 GTPases are required for capillary lumen formation in three-dimensional extracellular matrices. J. Cell Sci. 2002, 115, 1123–1136. [Google Scholar]
- El Atat, O.; Fakih, A.; El-Sibai, M. RHOG Activates RAC1 through CDC42 Leading to Tube Formation in Vascular Endothelial Cells. Cells 2019, 8, 171. [Google Scholar] [CrossRef] [PubMed]
- Abraham, S.; Scarcia, M.; Bagshaw, R.D.; McMahon, K.; Grant, G.; Harvey, T.; Yeo, M.; Esteves, F.O.; Thygesen, H.H.; Jones, P.F.; et al. A Rac/Cdc42 exchange factor complex promotes formation of lateral filopodia and blood vessel lumen morphogenesis. Nat. Commun. 2015, 6, 7286. [Google Scholar] [CrossRef]
- Lamalice, L.; Houle, F.; Jourdan, G.; Huot, J. Phosphorylation of tyrosine 1214 on VEGFR2 is required for VEGF-induced activation of Cdc42 upstream of SAPK2/p38. Oncogene 2004, 23, 434–445. [Google Scholar] [CrossRef]
- Qi, Y.; Liu, J.; Wu, X.; Brakebusch, C.; Leitges, M.; Han, Y.; Corbett, S.A.; Lowry, S.F.; Graham, A.M.; Li, S. Cdc42 controls vascular network assembly through protein kinase Ciota during embryonic vasculogenesis. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1861–1870. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ju, L.; Zhou, Z.; Jiang, B.; Lou, Y.; Guo, X. Autocrine VEGF and IL-8 Promote Migration via Src/Vav2/Rac1/PAK1 Signaling in Human Umbilical Vein Endothelial Cells. Cell. Physiol. Biochem. 2017, 41, 1346–1359. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Shi, W.; Wu, J.; Xu, C.; Wang, J.; Shao, Y.; Wu, X.; Zhang, Z. Endothelial Rac1 is essential for hematogenous metastasis to the lung. Oncotarget 2015, 6, 17501–17513. [Google Scholar] [CrossRef][Green Version]
- van Nieuw Amerongen, G.P.; Koolwijk, P.; Versteilen, A.; van Hinsbergh, V.W. Involvement of RhoA/Rho kinase signaling in VEGF-induced endothelial cell migration and angiogenesis in vitro. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Agrawal, S.; Vasanji, A.; Drazba, J.; Sarkaria, S.; Xie, J.; Welch, C.M.; Liu, M.; Anand-Apte, B.; Horowitz, A. Rab13-dependent trafficking of RhoA is required for directional migration and angiogenesis. J. Biol. Chem. 2011, 286, 23511–23520. [Google Scholar] [CrossRef]
- Shih, Y.P.; Yuan, S.Y.; Lo, S.H. Down-regulation of DLC1 in endothelial cells compromises the angiogenesis process. Cancer Lett. 2017, 398, 46–51. [Google Scholar] [CrossRef]
- Howe, G.A.; Addison, C.L. RhoB controls endothelial cell morphogenesis in part via negative regulation of RhoA. Vasc. Cell 2012, 4, 1. [Google Scholar] [CrossRef]
- Gottesbuhren, U.; Garg, R.; Riou, P.; McColl, B.; Brayson, D.; Ridley, A.J. Rnd3 induces stress fibres in endothelial cells through RhoB. Biol. Open 2013, 2, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Liu, Y.; Lin, Q.; Li, J.; Druso, J.E.; Antonyak, M.A.; Meininger, C.J.; Zhang, S.L.; Dostal, D.E.; Guan, J.L.; et al. Deletion of Cdc42 enhances ADAM17-mediated vascular endothelial growth factor receptor 2 shedding and impairs vascular endothelial cell survival and vasculogenesis. Mol. Cell. Biol. 2013, 33, 4181–4197. [Google Scholar] [CrossRef] [PubMed]
- Barry, D.M.; Koo, Y.; Norden, P.R.; Wylie, L.A.; Xu, K.; Wichaidit, C.; Azizoglu, D.B.; Zheng, Y.; Cobb, M.H.; Davis, G.E.; et al. Rasip1-Mediated Rho GTPase Signaling Regulates Blood Vessel Tubulogenesis via Nonmuscle Myosin II. Circ. Res. 2016, 119, 810–826. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Sacharidou, A.; Fu, S.; Chong, D.C.; Skaug, B.; Chen, Z.J.; Davis, G.E.; Cleaver, O. Blood vessel tubulogenesis requires Rasip1 regulation of GTPase signaling. Dev. Cell 2011, 20, 526–539. [Google Scholar] [CrossRef] [PubMed]
- Norden, P.R.; Kim, D.J.; Barry, D.M.; Cleaver, O.B.; Davis, G.E. Cdc42 and k-Ras Control Endothelial Tubulogenesis through Apical Membrane and Cytoskeletal Polarization: Novel Stimulatory Roles for GTPase Effectors, the Small GTPases, Rac2 and Rap1b, and Inhibitory Influence of Arhgap31 and Rasa1. PLoS ONE 2016, 11, e0147758. [Google Scholar] [CrossRef] [PubMed]
- Cardama, G.A.; Gonzalez, N.; Maggio, J.; Menna, P.L.; Gomez, D.E. Rho GTPases as therapeutic targets in cancer (Review). Int. J. Oncol. 2017, 51, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Blanco, R.; Gerhardt, H. VEGF and Notch in tip and stalk cell selection. Cold Spring Harb. Perspect. Med. 2013, 3, a006569. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Bryan, B.A.; Dennstedt, E.; Mitchell, D.C.; Walshe, T.E.; Noma, K.; Loureiro, R.; Saint-Geniez, M.; Campaigniac, J.P.; Liao, J.K.; D’Amore, P.A. RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. FASEB J. 2010, 24, 3186–3195. [Google Scholar] [CrossRef]
- Wheeler, A.P.; Ridley, A.J. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp. Cell Res. 2004, 301, 43–49. [Google Scholar] [CrossRef]
- Hoang, M.V.; Whelan, M.C.; Senger, D.R. Rho activity critically and selectively regulates endothelial cell organization during angiogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 1874–1879. [Google Scholar] [CrossRef] [PubMed]
- Martucciello, S.; Lavric, M.; Toth, B.; Korponay-Szabo, I.; Nadalutti, C.; Myrsky, E.; Rauhavirta, T.; Esposito, C.; Sulic, A.M.; Sblattero, D.; et al. RhoB is associated with the anti-angiogenic effects of celiac patient transglutaminase 2-targeted autoantibodies. J. Mol. Med. 2012, 90, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Almonte-Baldonado, R.; Bravo-Nuevo, A.; Gerald, D.; Benjamin, L.E.; Prendergast, G.C.; Laury-Kleintop, L.D. RhoB antibody alters retinal vascularization in models of murine retinopathy. J. Cell. Biochem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kusuhara, S.; Fukushima, Y.; Fukuhara, S.; Jakt, L.M.; Okada, M.; Shimizu, Y.; Hata, M.; Nishida, K.; Negi, A.; Hirashima, M.; et al. Arhgef15 promotes retinal angiogenesis by mediating VEGF-induced Cdc42 activation and potentiating RhoJ inactivation in endothelial cells. PLoS ONE 2012, 7, e45858. [Google Scholar] [CrossRef] [PubMed]
- Yue, X.; Lin, X.; Yang, T.; Yang, X.; Yi, X.; Jiang, X.; Li, X.; Li, T.; Guo, J.; Dai, Y.; et al. Rnd3/RhoE Modulates Hypoxia-Inducible Factor 1alpha/Vascular Endothelial Growth Factor Signaling by Stabilizing Hypoxia-Inducible Factor 1alpha and Regulates Responsive Cardiac Angiogenesis. Hypertension 2016, 67, 597–605. [Google Scholar] [CrossRef]
- Fantin, A.; Lampropoulou, A.; Gestri, G.; Raimondi, C.; Senatore, V.; Zachary, I.; Ruhrberg, C. NRP1 Regulates CDC42 Activation to Promote Filopodia Formation in Endothelial Tip Cells. Cell Rep. 2015, 11, 1577–1590. [Google Scholar] [CrossRef] [PubMed]
- Dubrac, A.; Genet, G.; Ola, R.; Zhang, F.; Pibouin-Fragner, L.; Han, J.; Zhang, J.; Thomas, J.L.; Chedotal, A.; Schwartz, M.A.; et al. Targeting NCK-Mediated Endothelial Cell Front-Rear Polarity Inhibits Neovascularization. Circulation 2016, 133, 409–421. [Google Scholar] [CrossRef]
- Sakabe, M.; Fan, J.; Odaka, Y.; Liu, N.; Hassan, A.; Duan, X.; Stump, P.; Byerly, L.; Donaldson, M.; Hao, J.; et al. YAP/TAZ-CDC42 signaling regulates vascular tip cell migration. Proc. Natl. Acad. Sci. USA 2017, 114, 10918–10923. [Google Scholar] [CrossRef]
- Lavina, B.; Castro, M.; Niaudet, C.; Cruys, B.; Alvarez-Aznar, A.; Carmeliet, P.; Bentley, K.; Brakebusch, C.; Betsholtz, C.; Gaengel, K. Defective endothelial cell migration in the absence of Cdc42 leads to capillary-venous malformations. Development 2018, 145. [Google Scholar] [CrossRef]
- Hoeppner, L.H.; Sinha, S.; Wang, Y.; Bhattacharya, R.; Dutta, S.; Gong, X.; Bedell, V.M.; Suresh, S.; Chun, C.; Ramchandran, R.; et al. RhoC maintains vascular homeostasis by regulating VEGF-induced signaling in endothelial cells. J. Cell Sci. 2015, 128, 3556–3568. [Google Scholar] [CrossRef]
- Sabatel, C.; Malvaux, L.; Bovy, N.; Deroanne, C.; Lambert, V.; Gonzalez, M.L.; Colige, A.; Rakic, J.M.; Noel, A.; Martial, J.A.; et al. MicroRNA-21 exhibits antiangiogenic function by targeting RhoB expression in endothelial cells. PLoS ONE 2011, 6, e16979. [Google Scholar] [CrossRef] [PubMed]
- Vader, P.; van der Meel, R.; Symons, M.H.; Fens, M.H.; Pieters, E.; Wilschut, K.J.; Storm, G.; Jarzabek, M.; Gallagher, W.M.; Schiffelers, R.M.; et al. Examining the role of Rac1 in tumor angiogenesis and growth: A clinically relevant RNAi-mediated approach. Angiogenesis 2011, 14, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Wallez, Y.; Huber, P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim. Biophys. Acta 2008, 1778, 794–809. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, C.; Ridley, A.J. Endothelial cell-cell adhesion and signaling. Exp. Cell Res. 2017, 358, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.Y.; Rigor, R.R. Regulation of Endothelial Barrier Function; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Marcos-Ramiro, B.; Garcia-Weber, D.; Barroso, S.; Feito, J.; Ortega, M.C.; Cernuda-Morollon, E.; Reglero-Real, N.; Fernandez-Martin, L.; Duran, M.C.; Alonso, M.A.; et al. RhoB controls endothelial barrier recovery by inhibiting Rac1 trafficking to the cell border. J. Cell Biol. 2016, 213, 385–402. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhang, X.; Buscher, K.; Wang, Y.; Wang, H.; Di Russo, J.; Li, L.; Lutke-Enking, S.; Zarbock, A.; Stadtmann, A.; et al. Endothelial Basement Membrane Laminin 511 Contributes to Endothelial Junctional Tightness and Thereby Inhibits Leukocyte Transmigration. Cell Rep. 2017, 18, 1256–1269. [Google Scholar] [CrossRef]
- Ortega, M.C.; Santander-Garcia, D.; Marcos-Ramiro, B.; Barroso, S.; Cox, S.; Jimenez-Alfaro, I.; Millan, J. Activation of Rac1 and RhoA Preserve Corneal Endothelial Barrier Function. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6210–6222. [Google Scholar] [CrossRef] [PubMed]
- Polacheck, W.J.; Kutys, M.L.; Yang, J.; Eyckmans, J.; Wu, Y.; Vasavada, H.; Hirschi, K.K.; Chen, C.S. A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 2017, 552, 258–262. [Google Scholar] [CrossRef]
- Daneshjou, N.; Sieracki, N.; van Nieuw Amerongen, G.P.; Conway, D.E.; Schwartz, M.A.; Komarova, Y.A.; Malik, A.B. Rac1 functions as a reversible tension modulator to stabilize VE-cadherin trans-interaction. J. Cell Biol. 2015, 209, 181. [Google Scholar] [CrossRef]
- Timmerman, I.; Heemskerk, N.; Kroon, J.; Schaefer, A.; van Rijssel, J.; Hoogenboezem, M.; van Unen, J.; Goedhart, J.; Gadella, T.W., Jr.; Yin, T.; et al. A local VE-cadherin and Trio-based signaling complex stabilizes endothelial junctions through Rac1. J. Cell Sci. 2015, 128, 3514. [Google Scholar] [CrossRef]
- Lv, J.; Zeng, J.; Guo, F.; Li, Y.; Xu, M.; Cheng, Y.; Zhang, L.; Cai, S.; Chen, Y.; Zheng, Y.; et al. Endothelial Cdc42 deficiency impairs endothelial regeneration and vascular repair after inflammatory vascular injury. Respir. Res. 2018, 19, 27. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, S.; Al-Ahmad, A.J.; Gassmann, M.; Ogunshola, O.O. Hypoxia selectively disrupts brain microvascular endothelial tight junction complexes through a hypoxia-inducible factor-1 (HIF-1) dependent mechanism. J. Cell. Physiol. 2014, 229, 1096–1105. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, J.; Chen, B.; Fan, L. A Positive Feedback Loop of Profilin-1 and RhoA/ROCK1 Promotes Endothelial Dysfunction and Oxidative Stress. Oxid. Med. Cell. Longev. 2018, 2018, 4169575. [Google Scholar] [CrossRef] [PubMed]
- Wojciak-Stothard, B.; Zhao, L.; Oliver, E.; Dubois, O.; Wu, Y.; Kardassis, D.; Vasilaki, E.; Huang, M.; Mitchell, J.A.; Harrington, L.S.; et al. Role of RhoB in the regulation of pulmonary endothelial and smooth muscle cell responses to hypoxia. Circ. Res. 2012, 110, 1423–1434. [Google Scholar] [CrossRef] [PubMed]
- Kouklis, P.; Konstantoulaki, M.; Vogel, S.; Broman, M.; Malik, A.B. Cdc42 regulates the restoration of endothelial barrier function. Circ. Res. 2004, 94, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Ene, C.; Kaul, A.; Kim, L. Natural history of cerebral cavernous malformations. Handb. Clin. Neurol. 2017, 143, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Richardson, B.T.; Dibble, C.F.; Borikova, A.L.; Johnson, G.L. Cerebral cavernous malformation is a vascular disease associated with activated RhoA signaling. Biol. Chem. 2013, 394, 35–42. [Google Scholar] [CrossRef]
- Stockton, R.A.; Shenkar, R.; Awad, I.A.; Ginsberg, M.H. Cerebral cavernous malformations proteins inhibit Rho kinase to stabilize vascular integrity. J. Exp. Med. 2010, 207, 881–896. [Google Scholar] [CrossRef]
- Whitehead, K.J.; Chan, A.C.; Navankasattusas, S.; Koh, W.; London, N.R.; Ling, J.; Mayo, A.H.; Drakos, S.G.; Jones, C.A.; Zhu, W.; et al. The cerebral cavernous malformation signaling pathway promotes vascular integrity via Rho GTPases. Nat. Med. 2009, 15, 177–184. [Google Scholar] [CrossRef]
- Shi, Y.; Vanhoutte, P.M. Macro- and microvascular endothelial dysfunction in diabetes. J. Diabetes 2017, 9, 434–449. [Google Scholar] [CrossRef]
- Han, J.; Weisbrod, R.M.; Shao, D.; Watanabe, Y.; Yin, X.; Bachschmid, M.M.; Seta, F.; Janssen-Heininger, Y.M.W.; Matsui, R.; Zang, M.; et al. The redox mechanism for vascular barrier dysfunction associated with metabolic disorders: Glutathionylation of Rac1 in endothelial cells. Redox Biol. 2016, 9, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Bruder-Nascimento, T.; Callera, G.E.; Montezano, A.C.; He, Y.; Antunes, T.T.; Nguyen Dinh Cat, A.; Tostes, R.C.; Touyz, R.M. Vascular injury in diabetic db/db mice is ameliorated by atorvastatin: Role of Rac1/2-sensitive Nox-dependent pathways. Clin. Sci. 2015, 128, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Lu, L.; Chen, W.; Chen, H.; Xu, X.; Zheng, Z. RhoA/mDia-1/profilin-1 signaling targets microvascular endothelial dysfunction in diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2015, 253, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Kazerounian, S.; Gerald, D.; Huang, M.; Chin, Y.R.; Udayakumar, D.; Zheng, N.; O’Donnell, R.K.; Perruzzi, C.; Mangiante, L.; Pourat, J.; et al. RhoB differentially controls Akt function in tumor cells and stromal endothelial cells during breast tumorigenesis. Cancer Res. 2013, 73, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wu, F.; Fang, F.; Tao, Y.; Yang, L. RhoC is essential for angiogenesis induced by hepatocellular carcinoma cells via regulation of endothelial cell organization. Cancer Sci. 2008, 99, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Nowak-Sliwinska, P.; Alitalo, K.; Allen, E.; Anisimov, A.; Aplin, A.C.; Auerbach, R.; Augustin, H.G.; Bates, D.O.; van Beijnum, J.R.; Bender, R.H.F.; et al. Consensus guidelines for the use and interpretation of angiogenesis assays. Angiogenesis 2018, 21, 425–532. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rho GTPase | Expressed in ECs between E6.5–E8.5? | Expressed in adult ECs? | Full Body or EC-Specific KO Available? |
|---|---|---|---|
| Cdc42 | Yes | Yes | Yes (both) |
| RhoQ (TC10) | Yes | No | No |
| RhoJ | Yes | Yes | Yes |
| RhoU (Wrch-1) | Yes | No | No |
| RhoV (Chp) | No | No | No |
| Rac1 | Yes | Yes | Yes (both) |
| Rac2 | No | No | Yes |
| Rac3 | Yes | No | Yes |
| RhoG | Yes | Yes | Yes |
| RhoBTB1 | Yes | Yes | No |
| RhoBTB2 | Yes | Yes | No |
| RhoBTB3 | Yes | No | Yes |
| RhoH | No | No | Yes |
| RhoA | Yes | Yes | Yes |
| RhoB | Yes | Yes | Yes |
| RhoC | Yes | Yes | Yes |
| Rnd1 | Yes | Yes | No |
| Rnd2 | Yes | No | No |
| Rnd3 (RhoE) | Yes | Yes | Yes |
| RhoD | Yes | No | No |
| RhoF (Rif) | Yes | No | Yes |
| Rho GTPase | Cre Driver | Phenotype | Citation |
|---|---|---|---|
| Cdc42 | Full KO | Embryonic lethal E7.5, with obvious defects as early as E5.5 | [34] |
| Tie2-Cre, | Embryonic lethal by E9–10; angioblast coalescence and lumenogenesis are blocked | [35] | |
| Cdh5-CreERT2 | Deleted at E11.5—widespread hemorrhaging, failure of EC polarization and lumenogenesis, defects in vessel integrity, actin organization, and cell–ECM adhesion; Deleted from Post-natal day (P) 0–4 —required for angiogenic growth in retina but not for existing vessel stability | [35] | |
| RhoQ | N/A | No information available | |
| RhoJ | Full KO | Mice viable and fertile; delay in radial growth of retinal vasculature and an increase in empty sleeves | [36] |
| Full KO | Mice viable; decrease in tumor angiogenesis | [37] | |
| RhoU | N/A | N/A | |
| Rac1 | Full KO | Embryonic lethal by E9.5 | [38] |
| Tie2-Cre | Embryonic lethal by E9.5–10.5—improper development of major vessels and lack of small branched vessels | [39] | |
| Cdh5-CreERT2 | Embryonic deletion (E10.5)—vessel hemorrhaging and decreased vascular area and branch points; Postnatal deletion (P1–P3)—decreased vascular area and branch points, defective angiogenic sprouting, decreased vertical blood vessel sprouting in retina | [40] | |
| Rac2 | Full KO | Mice viable and fertile; decrease in sprouting from aortic ring assay, decrease in vascularization of ischemic hindlimb and Matrigel plug assay | [41] |
| Rac3 | Full KO | Mice viable and fertile; ECs not studied | [42] |
| RhoG | Full KO | Mice viable and fertile; ECs not studied | [43] |
| RhoBTB1 | N/A | N/A | |
| RhoBTB2 | N/A | N/A | |
| RhoBTB3 | Full KO | Some lethality (homozygous weanlings present at 9.2%), mice are viable with reduced size | [44] |
| RhoA | Cdh5-CreERT2 | Knockout at 4–6 weeks postnatal increases vessel barrier function and prevents passive cutaneous anaphylaxis | [45] |
| RhoB | Full KO | Mice viable and fertile with reduced size; defective angiogenesis in postnatal retina with tip cells lacking cytoplasmic extensions; decrease in angiogenesis in response to wounding, decrease in pathological angiogenesis in retina after hypoxia | [11,46] |
| RhoC | Full KO | Mice viable and fertile, ECs not studied | [47] |
| Rnd1 | N/A | N/A | |
| Rnd2 | N/A | N/A | |
| Rnd3 (RhoE) | Full KO | Heterozygote mice are viable but prone to heart failure after pressure overload and are predisposed to hemodynamic stress; heterozygote mice present dilated cardiomyopathy with heart failure and impaired angiogenesis; one report of full KO causes hydrocephaly; another report of full KO causes embryonic lethality from cardiac arrhythmia | [48,49] |
| RhoD | N/A | N/A | |
| RhoF (Rif) | Full KO | Mice viable, no external abnormalities, ECs not studied | [50,51] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barlow, H.R.; Cleaver, O. Building Blood Vessels—One Rho GTPase at a Time. Cells 2019, 8, 545. https://doi.org/10.3390/cells8060545
Barlow HR, Cleaver O. Building Blood Vessels—One Rho GTPase at a Time. Cells. 2019; 8(6):545. https://doi.org/10.3390/cells8060545
Chicago/Turabian StyleBarlow, Haley Rose, and Ondine Cleaver. 2019. "Building Blood Vessels—One Rho GTPase at a Time" Cells 8, no. 6: 545. https://doi.org/10.3390/cells8060545
APA StyleBarlow, H. R., & Cleaver, O. (2019). Building Blood Vessels—One Rho GTPase at a Time. Cells, 8(6), 545. https://doi.org/10.3390/cells8060545