
Phase 1/2 Trial of CLAG-M with Dose-Escalated Mitoxantrone in Combination with Fractionated-Dose Gemtuzumab Ozogamicin for Newly Diagnosed Acute Myeloid Leukemia and Other High-Grade Myeloid Neoplasms



Abstract
:Simple Summary
Abstract
1. Introduction
2. Patients and Methods
2.1. Study Population
2.2. Disease and Response Classification
2.3. Treatment Plan
2.4. Comparison of CLAG-M + GO with CLAG-M
2.5. Statistical Considerations
3. Results
3.1. Study Cohort and Treatment
3.2. Phase 1
3.3. RP2D Cohort
3.4. Grade 3–4 Toxicities and Sinusoidal Obstruction Syndrome/Veno-Occlusive Disease (SOS/VOD)
3.5. Duration of Cytopenias
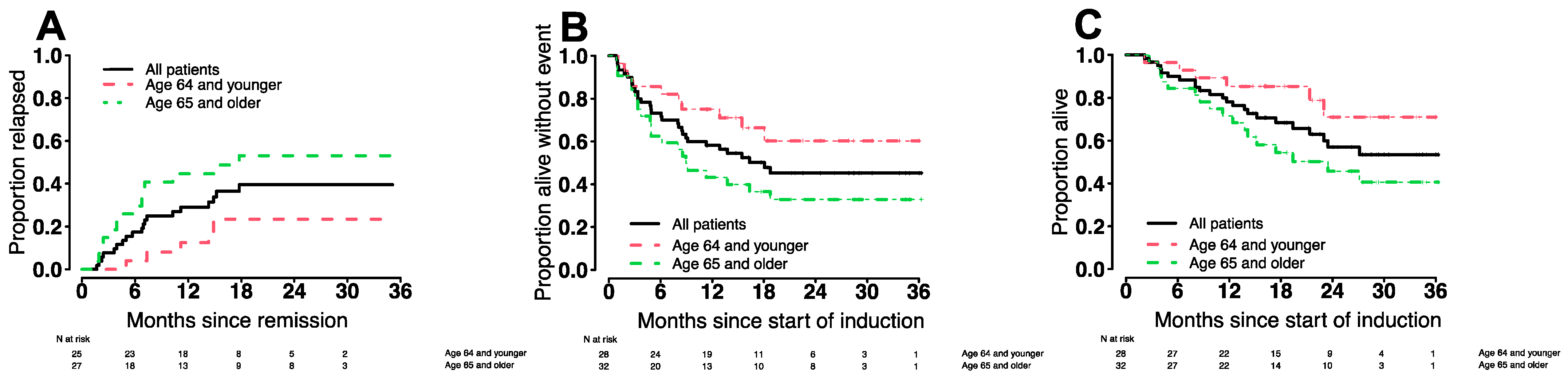
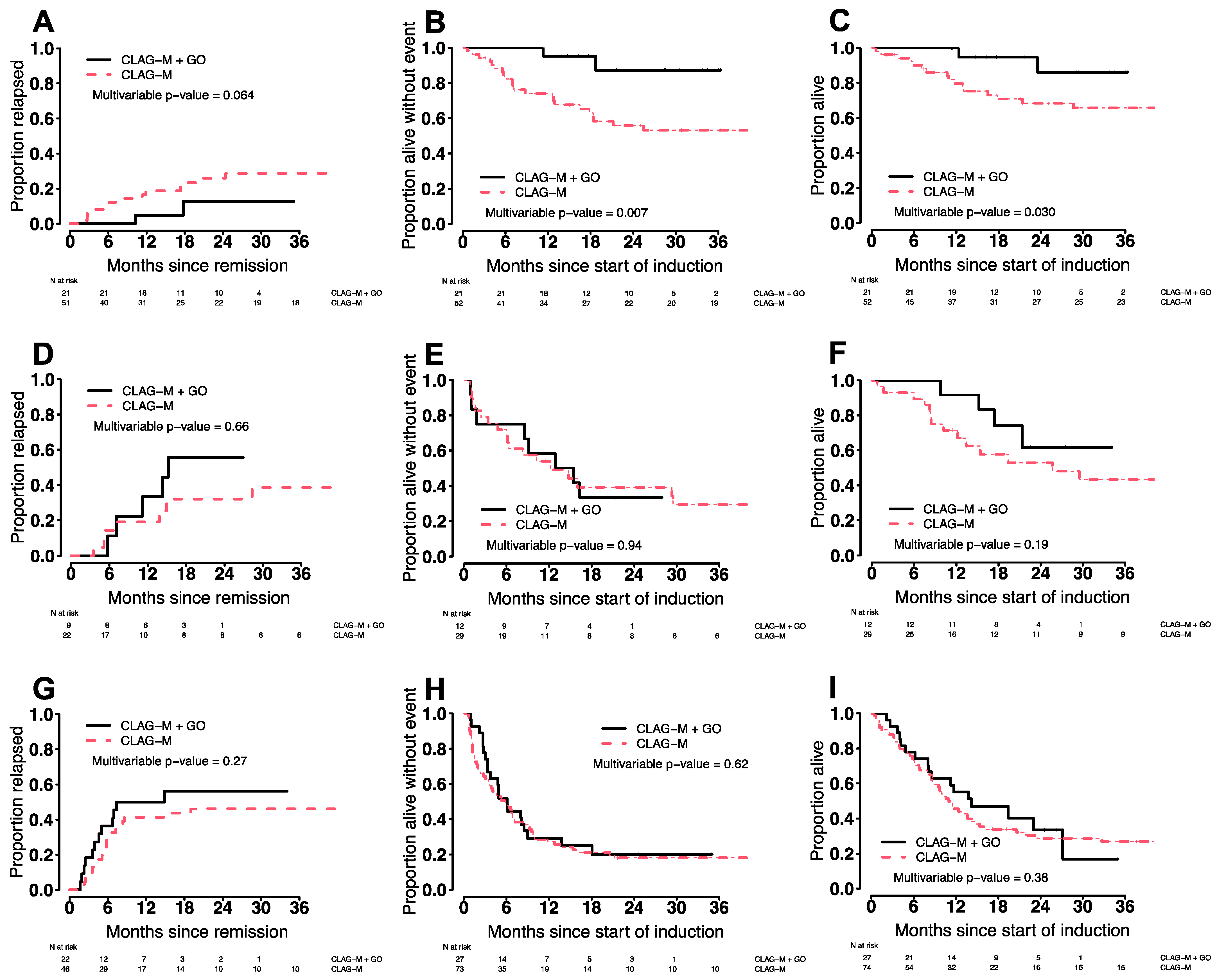
3.6. Treatment Outcomes in Various Patient Subgroups
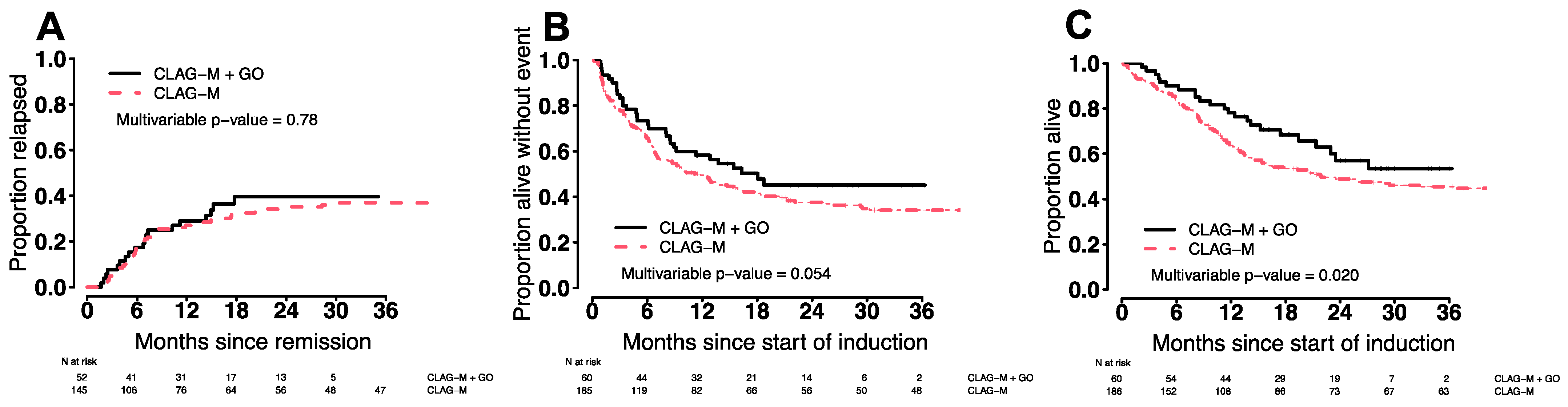
3.7. Comparison of CLAG-M/GO3 vs. CLAG-M
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Burnett, A.K.; Hills, R.K.; Milligan, D.; Kjeldsen, L.; Kell, J.; Russell, N.H.; Yin, J.A.; Hunter, A.; Goldstone, A.H.; Wheatley, K. Identification of patients with acute myeloblastic leukemia who benefit from the addition of gemtuzumab ozogamicin: Results of the MRC AML15 trial. J. Clin. Oncol. 2011, 29, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Delaunay, J.; Recher, C.; Pigneux, A.; Witz, F.; Vey, N.; Blanchet, O.; Lefebvre, P.; Luquet, I.; Guillerme, I.; Volteau, C.; et al. Addition of gemtuzumab ozogamycin to chemotherapy improves event-free survival but not overall survival of AML patients with intermediate cytogenetics not eligible for allogeneic transplantation. Results of the GOELAMS AML 2006 IR study [abstract]. Blood 2011, 118, 37–38. [Google Scholar] [CrossRef]
- Castaigne, S.; Pautas, C.; Terré, C.; Raffoux, E.; Bordessoule, D.; Bastie, J.N.; Legrand, O.; Thomas, X.; Turlure, P.; Reman, O.; et al. Effect of gemtuzumab ozogamicin on survival of adult patients with de-novo acute myeloid leukaemia (ALFA-0701): A randomised, open-label, phase 3 study. Lancet 2012, 379, 1508–1516. [Google Scholar] [CrossRef]
- Burnett, A.K.; Hills, R.K.; Hunter, A.E.; Milligan, D.; Kell, W.J.; Wheatley, K.; Yin, J.; McMullin, M.F.; Dignum, H.; Bowen, D.; et al. The addition of gemtuzumab ozogamicin to low-dose Ara-C improves remission rate but does not significantly prolong survival in older patients with acute myeloid leukaemia: Results from the LRF AML14 and NCRI AML16 pick-a-winner comparison. Leukemia 2013, 27, 75–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlenk, R.F.; Paschka, P.; Krzykalla, J.; Weber, D.; Kapp-Schwoerer, S.; Gaidzik, V.I.; Leis, C.; Fiedler, W.; Kindler, T.; Schroeder, T.; et al. Gemtuzumab ozogamicin in NPM1-mutated acute myeloid leukemia: Early results from the prospective randomized AMLSG 09-09 phase III study. J. Clin. Oncol. 2020, 38, 623–632. [Google Scholar] [CrossRef]
- Godwin, C.D.; Gale, R.P.; Walter, R.B. Gemtuzumab ozogamicin in acute myeloid leukemia. Leukemia 2017, 31, 1855–1868. [Google Scholar] [CrossRef]
- Hills, R.K.; Castaigne, S.; Appelbaum, F.R.; Delaunay, J.; Petersdorf, S.; Othus, M.; Estey, E.H.; Dombret, H.; Chevret, S.; Ifrah, N.; et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: A meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. 2014, 15, 986–996. [Google Scholar] [CrossRef] [Green Version]
- Halpern, A.B.; Othus, M.; Huebner, E.M.; Scott, B.L.; Becker, P.S.; Percival, M.M.; Hendrie, P.C.; Gardner, K.M.; Chen, T.L.; Buckley, S.A.; et al. Phase 1/2 trial of GCLAM with dose-escalated mitoxantrone for newly diagnosed AML or other high-grade myeloid neoplasms. Leukemia 2018, 32, 2352–2362. [Google Scholar] [CrossRef]
- Halpern, A.B.; Walter, R.B. CLAG-M with dose-escalated mitoxantrone for adults with acute myeloid leukemia. Oncotarget 2018, 9, 36543–36544. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Walter, R.B.; Othus, M.; Borthakur, G.; Ravandi, F.; Cortes, J.E.; Pierce, S.A.; Appelbaum, F.R.; Kantarjian, H.A.; Estey, E.H. Prediction of early death after induction therapy for newly diagnosed acute myeloid leukemia with pretreatment risk scores: A novel paradigm for treatment assignment. J. Clin. Oncol. 2011, 29, 4417–4423. [Google Scholar] [CrossRef] [PubMed]
- Grimwade, D.; Hills, R.K.; Moorman, A.V.; Walker, H.; Chatters, S.; Goldstone, A.H.; Wheatley, K.; Harrison, C.J.; Burnett, A.K. Refinement of cytogenetic classification in acute myeloid leukemia: Determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood 2010, 116, 354–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, R.B.; Gooley, T.A.; Wood, B.L.; Milano, F.; Fang, M.; Sorror, M.L.; Estey, E.H.; Salter, A.I.; Lansverk, E.; Chien, J.W.; et al. Impact of pretransplantation minimal residual disease, as detected by multiparametric flow cytometry, on outcome of myeloablative hematopoietic cell transplantation for acute myeloid leukemia. J. Clin. Oncol. 2011, 29, 1190–1197. [Google Scholar] [CrossRef] [Green Version]
- Araki, D.; Wood, B.L.; Othus, M.; Radich, J.P.; Halpern, A.B.; Zhou, Y.; Mielcarek, M.; Estey, E.H.; Appelbaum, F.R.; Walter, R.B. Allogeneic hematopoietic cell transplantation for acute myeloid leukemia: Is it time to move toward a minimal residual disease-based definition of complete remission. J. Clin. Oncol. 2016, 34, 329–336. [Google Scholar] [CrossRef]
- Wood, B.L. Acute myeloid leukemia minimal residual disease detection: The difference from normal approach. Curr. Protoc. Cytom. 2020, 93, e73. [Google Scholar] [CrossRef]
- Paras, G.; Morsink, L.M.; Othus, M.; Milano, F.; Sandmaier, B.M.; Zarling, L.C.; Palmieri, R.; Schoch, G.; Davis, C.; Bleakley, M.; et al. Conditioning intensity and peritransplant flow cytometric MRD dynamics in adult AML. Blood 2022, 139, 1694–1706. [Google Scholar] [CrossRef]
- Chen, X.; Xie, H.; Wood, B.L.; Walter, R.B.; Pagel, J.M.; Becker, P.S.; Sandhu, V.K.; Abkowitz, J.L.; Appelbaum, F.R.; Estey, E.H. Relation of clinical response and minimal residual disease and their prognostic impact on outcome in acute myeloid leukemia. J. Clin. Oncol. 2015, 33, 1258–1264. [Google Scholar] [CrossRef]
- Godwin, C.D.; McDonald, G.B.; Walter, R.B. Sinusoidal obstruction syndrome following CD33-targeted therapy in acute myeloid leukemia. Blood 2017, 129, 2330–2332. [Google Scholar] [CrossRef] [Green Version]
- Cortes, J.E.; de Lima, M.; Dombret, H.; Estey, E.H.; Giralt, S.A.; Montesinos, P.; Röllig, C.; Venditti, A.; Wang, E.S. Prevention, recognition, and management of adverse events associated with gemtuzumab ozogamicin use in acute myeloid leukemia. J. Hematol. Oncol. 2020, 13, 137. [Google Scholar] [CrossRef] [PubMed]
- McDonald, G.B.; Sharma, P.; Matthews, D.E.; Shulman, H.M.; Thomas, E.D. Venocclusive disease of the liver after bone marrow transplantation: Diagnosis, incidence, and predisposing factors. Hepatology 1984, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Burnett, A.; Cavenagh, J.; Russell, N.; Hills, R.; Kell, J.; Jones, G.; Nielsen, O.J.; Khwaja, A.; Thomas, I.; Clark, R.; et al. Defining the dose of gemtuzumab ozogamicin in combination with induction chemotherapy in acute myeloid leukemia: A comparison of 3 mg/m2 with 6 mg/m2 in the NCRI AML17 Trial. Haematologica 2016, 101, 724–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Parameter | All Patients (n = 66) | Phase 1 Cohort (n = 18) | RP2D Cohort (n = 60) | RP2D, Age < 65 (n = 28) | RP2D, Age ≥ 65 (n = 32) |
|---|---|---|---|---|---|
| Age, median (range), years | 65 (19–80) | 66 (29–78) | 65 (19–80) | 51 (19–64) | 71 (65–80) |
| Male gender, n (%) | 34 (52%) | 6 (33%) | 32 (53%) | 17 (61%) | 15 (47%) |
| Disease | |||||
| AML | 53 (80%) | 14 (78%) | 48 (80%) | 22 (85%) | 26 (81%) |
| With recurrent genetic abnormalities | 25 (38%) | 5 (28%) | 23 (38%) | 11 (39%) | 12 (38%) |
| With myelodysplasia-related changes | 14 (21%) ‡ | 4 (22%) ‡ | 11 (18%) | 3 (11%) | 8 (25%) |
| Treatment-related myeloid neoplasm | 5 (8%) | 3 (17%) | 5 (8%) | 2 (7%) | 3 (9%) |
| AML, not otherwise specified | 9 (14%) | 2 (11%) | 9 (15%) | 6 (21%) | 3 (9%) |
| MDS-EB-2 | 9 (14%) | 4 (22%) | 8 (13%) | 4 (14%) | 4 (13%) |
| CMML-2 | 3 (5%) | 0 (0%) | 3 (5%) | 2 (7%) | 1 (3%) |
| Other high-grade myeloid neoplasm | 1 (2%) | 0 (0%) | 1 (2%) | 0 (0%) | 1 (3%) |
| Secondary disease * | 27 (41%) | 8 (44%) | 24 (40%) | 6 (21%) | 18 (56%) |
| Median TRM score (range) | 3.5 (0.02–11.8) | 3.9 (0.14–10.4) | 3.5 (0.02–11.8) | 1.8 (0.14–9) | 4.4 (0.02–11.8) |
| Performance status, n (%) | |||||
| 0 | 14 (21%) | 3 (17%) | 12 (20%) | 9 (32%) | 3 (9%) |
| 1 | 45 (68%) | 14 (78%) | 42 (70%) | 17 (61%) | 25 (78%) |
| 2 | 7 (11%) | 1 (6%) | 6 (10%) | 2 (7%) | 4 (13%) |
| Cytogenetic/molecular risk, n (%) ** | |||||
| Favorable | 22 (33%) | 7 (39%) | 21 (35%) | 12 (43%) | 9 (28%) |
| Intermediate | 15 (23%) | 5 (28%) | 12 (20%) | 7 (25%) | 5 (16%) |
| Adverse | 29 (44%) | 6 (33%) | 27 (45%) | 9 (32%) | 18 (56%) |
| Mutational status, n (%) | |||||
| FLT3-ITD | |||||
| Wild type | 54 (82%) | 15 (83%) | 49 (82%) | 24 (86%) | 25 (78%) |
| Mutated | 7 (11%) | 1 (6%) | 7 (12%) | 2 (7%) | 5 (16%) |
| Unknown | 5 (8%) | 2 (11%) | 4 (7%) | 2 (7%) | 2 (6%) |
| NPM1 | |||||
| Wild type | 44 (67%) | 12 (67%) | 39 (65%) | 19 (68%) | 20 (63%) |
| Mutated | 17 (26%) | 4 (22%) | 17 (28%) | 7 (25%) | 10 (31%) |
| Unknown | 5 (8%) | 2 (11%) | 4 (7%) | 2 (7%) | 2 (6%) |
| TP53 | |||||
| Wild type | 46 (70%) | 13 (72%) | 42 (70%) | 21 (75%) | 21 (66%) |
| Mutated | 8 (12%) | 2 (11%) | 7 (12%) | 4 (14%) | 3 (9%) |
| Unknown | 12 (18%) | 3 (17%) | 11 (18%) | 3 (11%) | 8 (25%) |
| RUNX1 | |||||
| Wild type | 46 (70%) | 13 (72%) | 42 (70%) | 23 (82%) | 19 (59%) |
| Mutated | 8 (12%) | 2 (11%) | 7 (12%) | 2 (7%) | 5 (16%) |
| Unknown | 12 (18%) | 3 (17%) | 11 (18%) | 3 (11%) | 8 (25%) |
| Laboratory findings at screening, median (range) | |||||
| Total WBC count (×109/L) | 6.6 (0.8–157) | 6.3 (0.8–157) | 6.9 (0.8–157) | 7.2 (0.8–157) | 6.1 (1.3–64) |
| Absolute neutrophil count (×109/L) | 1.2 (0–23) | 1.1 (0.03–6.9) | 1.2 (0–23) | 1.2 (0–20) | 1.2 (0.1–23) |
| Peripheral blood blasts (%) | 21 (0–94) | 27 (0–87) | 23 (0–94) | 28 (0–90) | 20 (0–94) |
| Hemoglobin (g/dL) | 8.9 (6.5–13.3) | 9.2 (6.8–13.2) | 8.9 (6.5–13.3) | 8.8 (6.8–11.9) | 8.9 (6.5–13.3) |
| Platelets (×109/L) | 59 (6–820) | 50 (12–413) | 62 (6–820) | 50 (8–413) | 77 (6–820) |
| Serum creatinine (mg/dL) | 0.8 (0.5–1.8) | 0.8 (0.5–1.7) | 0.8 (0.5–1.8) | 0.7 (0.5–1.2) | 0.9 (0.5–1.8) |
| Total bilirubin (mg/dL) | 0.5 (0.3–1.7) | 0.5 (0.3–1.7) | 0.5 (0.3–1.7) | 0.4 (0.3–1.7) | 0.6 (0.3–1.2) |
| AST (U/L) | 18 (9–72) | 17 (11–72) | 18 (9–72) | 17 (9–72) | 20 (10–39) |
| ALT (U/L) | 15 (5–138) | 13 (5–120) | 16 (5–138) | 16 (8–138) | 16 (5–42) |
| Subsequent allogeneic HCT | 28 (42%) | 8 (44%) | 25 (42%) | 14 (50%) | 11 (34%) |
| Dose Level | GO (D0 to D5) | Patients (n) | Best Response | Dose-Limiting Toxicities |
|---|---|---|---|---|
| 1 (GO1) | 3 mg/m2 on day 1 | 6 | 1 unevaluable * 1 CRi MRDneg 4 CR MRDneg | 1 grade 3 left ventricular systolic dysfunction |
| 2 (GO3) | 3 mg/m2 on days 1, 4, and 7 | 12 | 6 CR MRDneg 2 CR MRDpos 1 CRi MRDneg 1 MLFS MRDneg 1 Aplasia ** MRDneg 1 RD | 1 grade 4 aminotransferase level increase 1 grade 3 posterior reversible encephalopathy syndrome 1 grade 3 intracranial hemorrhage |
| Response, n (%) | All Patients (n = 66) | RP2D Cohort (n = 60) | RP2D, Age < 65 (n = 28) | RP2D, Age ≥ 65 (n = 32) |
|---|---|---|---|---|
| CR | ||||
| MRDneg | 42 (64%) | 38 (63%) | 20 (71%) | 18 (56%) |
| MRDpos | 7 (11%) | 7 (12%) | 4 (14%) | 3 (9%) |
| CRi | ||||
| MRDneg | 8 (12%) | 7 (12%) | 1 (4%) | 6 (19%) |
| CR/CRi | 57 (86%) | 52 (87%) | 25 (89%) | 27 (84%) |
| MLFS | ||||
| MRDneg | 2 (3%) | 2 (3%) | 1 (4%) | 1 (3%) |
| Aplasia | ||||
| MRDneg | 2 (3%) | 2 (3%) | 1 (4%) | 1 (3%) |
| Resistant disease | 3 (5%) | 3 (5%) | 1 (4%) | 2 (6%) |
| Death from indeterminate cause | 1 ‡ (2%) | 1 ‡ (2%) | 0 | 1 (3%) |
| Unevaluable for response | 1 § (2%) | 0 (0%) | 0 (0%) | 0 (0%) |
| 8-week mortality | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) |
| Overall Survival | Event-Free Survival | Cumulative Incidence of Relapse | ||||
|---|---|---|---|---|---|---|
| 6 Months | 12 Months | 6 Months | 12 Months | 6 Months | 12 Months | |
| All patients | 90% (83–98%) | 76% (66–89%) | 73% (63–85%) | 58% (47–72%) | 18% (12–23%) | 27% (21–33%) |
| By age | ||||||
| <65 years | 96% (84–100%) | 85% (73–100%) | 86% (74–100%) | 75% (61–93%) | 16% (10–23%) | 21% (14–29%) |
| ≥65 years | 84% (73–98%) | 72% (58–89%) | 63% (48–82%) | 43% (29–65%) | 20% (12–28%) | 34% (24–44%) |
| By disease risk * | ||||||
| Favorable | 100% | 100% | 100% | 95% (87–100%) | 7% (3–15%) | 14% (7–24%) |
| Intermediate | 100% | 92% (78–100%) | 75% (54–100%) | 58% (36–94%) | 12% (4–26%) | 22% (9–38%) |
| Adverse | 78% (64–95%) | 55% (39–78%) | 52% (36–75%) | 29% (16–53%) | 32% (21–43%) | 44% (32–55%) |
| Adverse Events by System Organ Class | Phase 1 Cohort (n = 18) | RP2D Cohort (n = 60) |
|---|---|---|
| Blood and lymphatic system disorders * | ||
| Disseminated intravascular coagulation | − | 4 (7%) |
| Febrile neutropenia | 15 (83%) | 50 (83%) |
| Infections and infestations | ||
| Catheter-related infection | − | 3 (5%) |
| Encephalitis | − | 1 (2%) |
| Enterocolitis | 2 (11%) | 1 (2%) |
| Lung infection | 1 (6%) | 7 (12%) |
| Sepsis | 6 (33%) | 23 (38%) |
| Skin infection | − | 1 (2%) |
| Urinary tract infection | 1 (6%) | 2 (3%) |
| Cardiac disorders | ||
| Atrial fibrillation | − | 1 (2%) |
| Cardiac arrest | − | 1 (2%) |
| Cardiomyopathy | 3 (17%) | 5 (8%) |
| Edema | − | 1 (2%) |
| Hypertension | 2 (11%) | 4 (7%) |
| Hypotension | 1 (6%) | − |
| Pericardial effusion | − | 1 (2%) |
| Myocardial infarction | − | 1 (2%) |
| Gastrointestinal disorders | ||
| Colonic perforation | − | 1 (2%) |
| Dysphagia | − | 2 (3%) |
| Esophagitis | − | 1 (2%) |
| Gastrointestinal hemorrhage | 1 (6%) | 5 (8%) |
| Mucositis | − | 3 (5%) |
| Vomiting | 1 (6%) | 1 (2%) |
| Investigations | ||
| Alanine aminotransferase increased | 2 (11%) | 3 (5%) |
| Aspartate aminotransferase increased | 2 (11%) | 5 (8%) |
| Blood bilirubin increased | − | 1 (2%) |
| Metabolism and nutrition disorders | ||
| Acidosis | − | 1 (2%) |
| Anorexia | − | 1 (2%) |
| Hyperglycemia | − | 4 (7%) |
| Hyperphosphatemia | − | 1 (2%) |
| Hypocalcemia | − | 2 (3%) |
| Hypokalemia | 1 (6%) | 1 (2%) |
| Hypophosphatemia | − | 1 (2%) |
| Tumor lysis | 2 (11%) | 6 (10%) |
| Nervous system disorders | ||
| Ataxia | − | 1 (2%) |
| Dysarthria | − | 1 (2%) |
| Intracranial hemorrhage | 1 (6%) | 2 (3%) |
| Reversible posterior encephalopathy syndrome | 1 (6%) | 1 (2%) |
| Stroke | − | 1 (2%) |
| Syncope | − | 1 (2%) |
| Renal and urinary disorders | ||
| Acute kidney injury | − | 1 (2%) |
| Respiratory, thoracic, and mediastinal disorders | ||
| Hypoxia | − | 4 (7%) |
| Pulmonary edema/effusion | 1 (6%) | 5 (8%) |
| Pulmonary hemorrhage/hemoptysis | − | 2 (3%) |
| Respiratory failure | − | 2 (3%) |
| Skin and subcutaneous tissue disorders | ||
| Maculo-papular rash | − | 1 (2%) |
| Regimen | CLAG-M/GO3 (n = 60) | CLAG-M (n = 186) | p-Value |
|---|---|---|---|
| Median age, years (range) | 65 (19–80) | 64 (19–84) | 0.18 |
| Disease, n (%) | 0.49 | ||
| AML | 48 (80%) | 158 (85%) | |
| MDS-EB-2 | 8 (13%) | 21 (11%) | |
| Other | 4 (7%) | 7 (4%) | |
| Secondary disease *, n (%) | 24 (40%) | 54 (29%) | 0.15 |
| Disease risk (ELN2017), n (%) | 0.94 | ||
| Favorable | 21 (35%) | 52 (34%) | |
| Intermediate | 12 (20%) | 29 (19%) | |
| Adverse | 27 (45%) | 74 (48%) | |
| Insufficient data | 0 | 31 | |
| TRM score (range) | 3.5 (0.02–11.8) | 3.0 (0.01–12.3) | 0.54 |
| Performance status, n (%) | 0.51 | ||
| 0 | 12 (20%) | 45 (24%) | |
| 1 | 42 (70%) | 129 (69%) | |
| 2 | 6 (10%) | 10 (5%) | |
| 3 | 0 (0%) | 2 (1%) | |
| CR, n (%) | 1 | ||
| MRDneg | 38 (63%) | 116 (62%) | |
| MRDpos | 7 (12%) | 20 (11%) | |
| CRi, n (%) | 0.21 | ||
| MRDneg | 7 (12) | 6 (3%) | |
| MRDpos | 0 (0%) | 3 (2%) | |
| CR/CRi, n (%) | 52 (87%) | 145 (78%) | 0.19 |
| MLFS or marrow aplasia, n (%) | 4 (7%) | 15 (8%) | 1 |
| Resistant disease, n (%) | 3 (5%) | 17 (9%) | 0.42 |
| Death from indeterminate cause, n (%) | 1 (2%) | 9 (5%) | 0.46 |
| Early mortality, n (%) | |||
| 4-week | 0 (0%) | 6 (3%) | 0.34 |
| 8-week | 0 (%) | 13 (7%) | 0.042 |
| Days to ANC ≥1000/µL, median (range) ** | 32 (22–51) | 28 (18–60) | 0.001 |
| Days to platelets ≥100,000/µL, median (range) ** | 31 (21–48) | 26 (18–54) | <0.001 |
| Subsequent allogeneic HCT within 2 years, n (%) | 25 (42%) | 94 (51%) | 0.24 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godwin, C.D.; Rodríguez-Arbolí, E.; Othus, M.; Halpern, A.B.; Appelbaum, J.S.; Percival, M.-E.M.; Hendrie, P.C.; Oehler, V.G.; Keel, S.B.; Abkowitz, J.L.; et al. Phase 1/2 Trial of CLAG-M with Dose-Escalated Mitoxantrone in Combination with Fractionated-Dose Gemtuzumab Ozogamicin for Newly Diagnosed Acute Myeloid Leukemia and Other High-Grade Myeloid Neoplasms. Cancers 2022, 14, 2934. https://doi.org/10.3390/cancers14122934
Godwin CD, Rodríguez-Arbolí E, Othus M, Halpern AB, Appelbaum JS, Percival M-EM, Hendrie PC, Oehler VG, Keel SB, Abkowitz JL, et al. Phase 1/2 Trial of CLAG-M with Dose-Escalated Mitoxantrone in Combination with Fractionated-Dose Gemtuzumab Ozogamicin for Newly Diagnosed Acute Myeloid Leukemia and Other High-Grade Myeloid Neoplasms. Cancers. 2022; 14(12):2934. https://doi.org/10.3390/cancers14122934
Chicago/Turabian StyleGodwin, Colin D., Eduardo Rodríguez-Arbolí, Megan Othus, Anna B. Halpern, Jacob S. Appelbaum, Mary-Elizabeth M. Percival, Paul C. Hendrie, Vivian G. Oehler, Siobán B. Keel, Janis L. Abkowitz, and et al. 2022. "Phase 1/2 Trial of CLAG-M with Dose-Escalated Mitoxantrone in Combination with Fractionated-Dose Gemtuzumab Ozogamicin for Newly Diagnosed Acute Myeloid Leukemia and Other High-Grade Myeloid Neoplasms" Cancers 14, no. 12: 2934. https://doi.org/10.3390/cancers14122934
APA StyleGodwin, C. D., Rodríguez-Arbolí, E., Othus, M., Halpern, A. B., Appelbaum, J. S., Percival, M.-E. M., Hendrie, P. C., Oehler, V. G., Keel, S. B., Abkowitz, J. L., Cooper, J. P., Cassaday, R. D., Estey, E. H., & Walter, R. B. (2022). Phase 1/2 Trial of CLAG-M with Dose-Escalated Mitoxantrone in Combination with Fractionated-Dose Gemtuzumab Ozogamicin for Newly Diagnosed Acute Myeloid Leukemia and Other High-Grade Myeloid Neoplasms. Cancers, 14(12), 2934. https://doi.org/10.3390/cancers14122934