MiR-216b/Smad3/BCL-2 Axis Is Involved in Smoking-Mediated Drug Resistance in Non-Small Cell Lung Cancer


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. CSC Treatment Regulates Tumor Suppressor and Oncogenic MiRNAs Identified by Microarray Analyses
2.2. CSC Induces MiR-216b and Reduces Smad3 Expressions in Lung Epithelial Cells
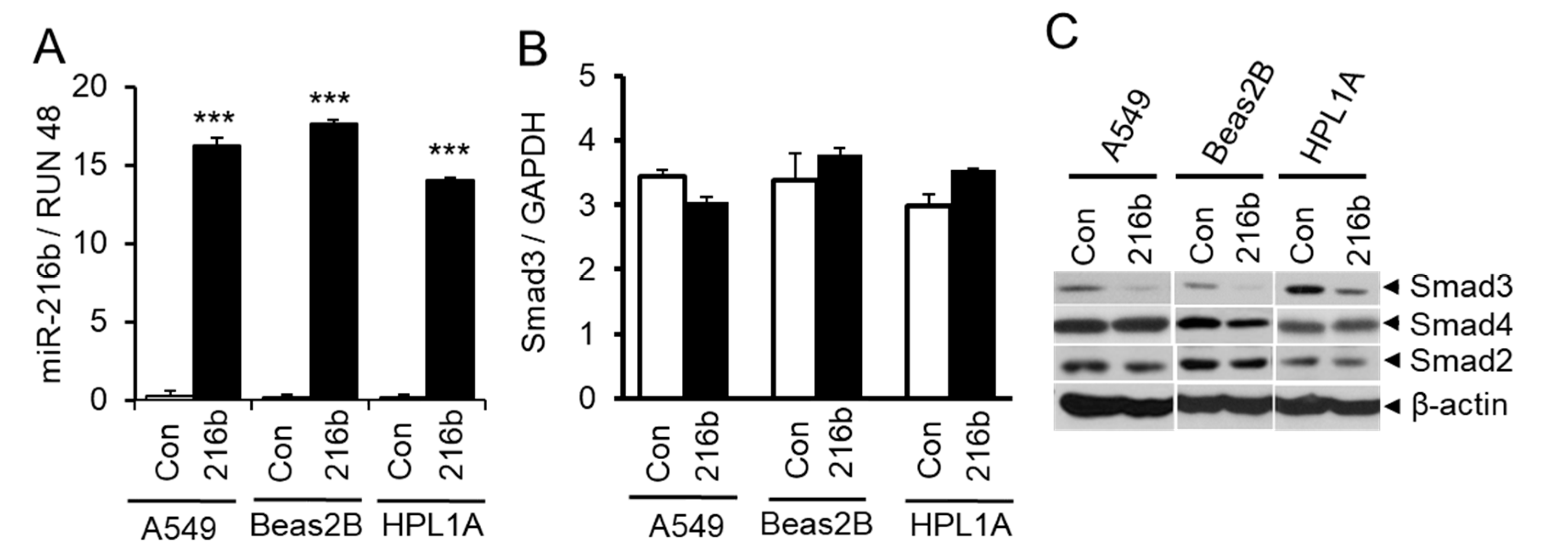
2.3. MiR-216b Inhibits the Expression of Smad3 in Lung Epithelial Cells
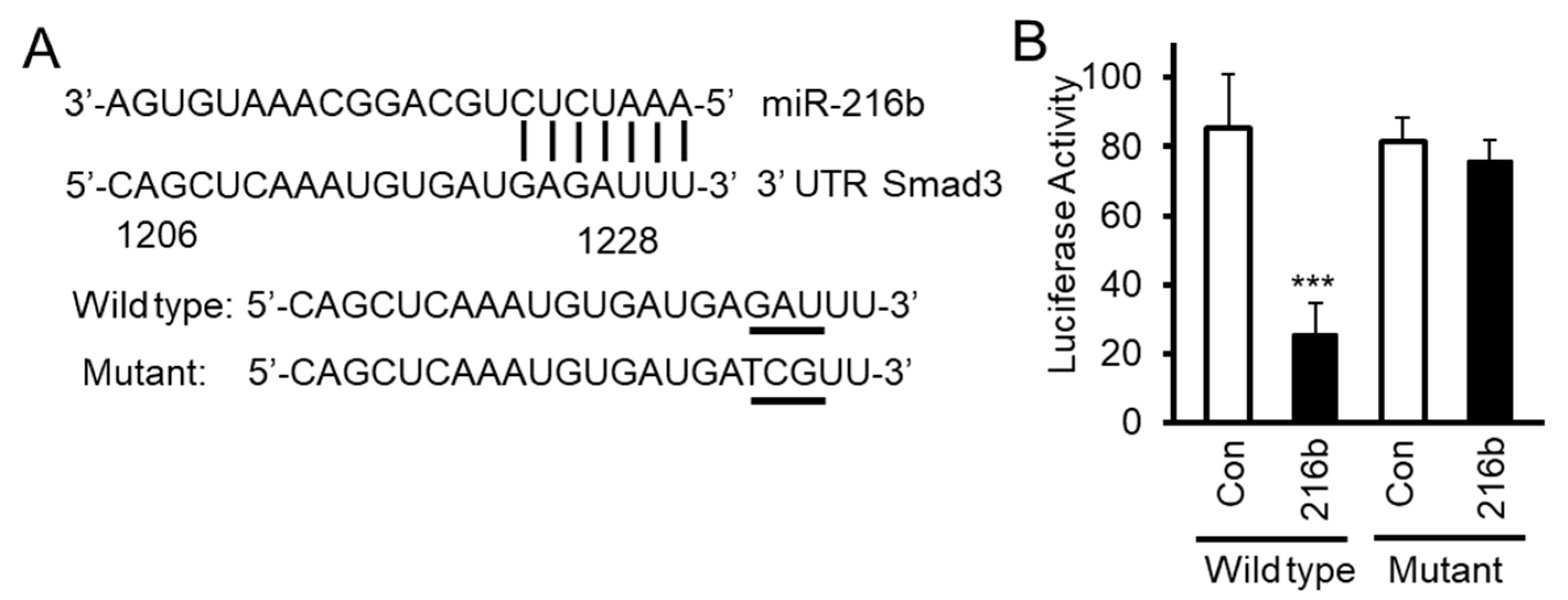
2.4. MiR-216b Inhibits Expression of Smad3 by Binding to Its 3′UTR
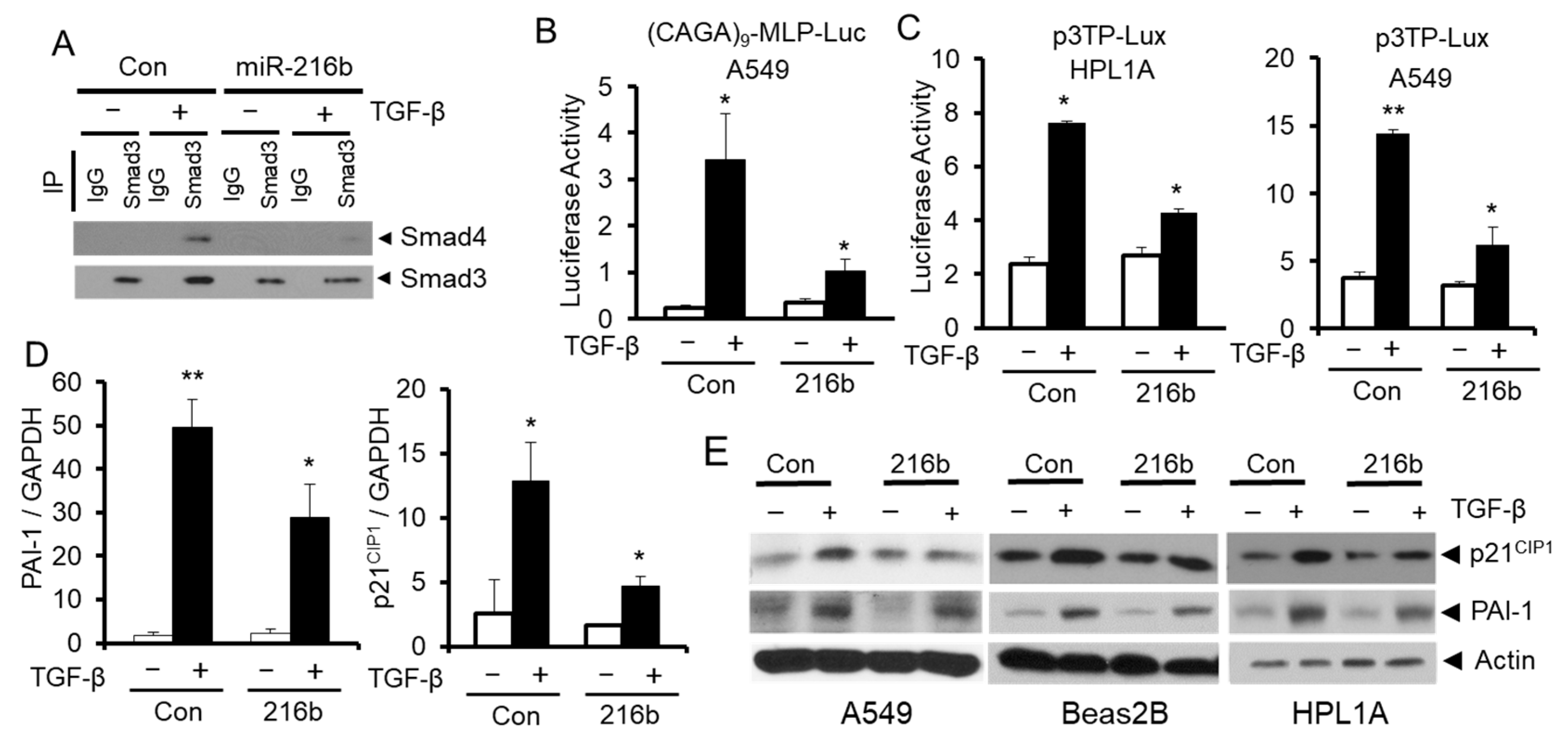
2.5. MiR-216b Attenuates TGF-β Signaling by Downregulating Smad3 Expression
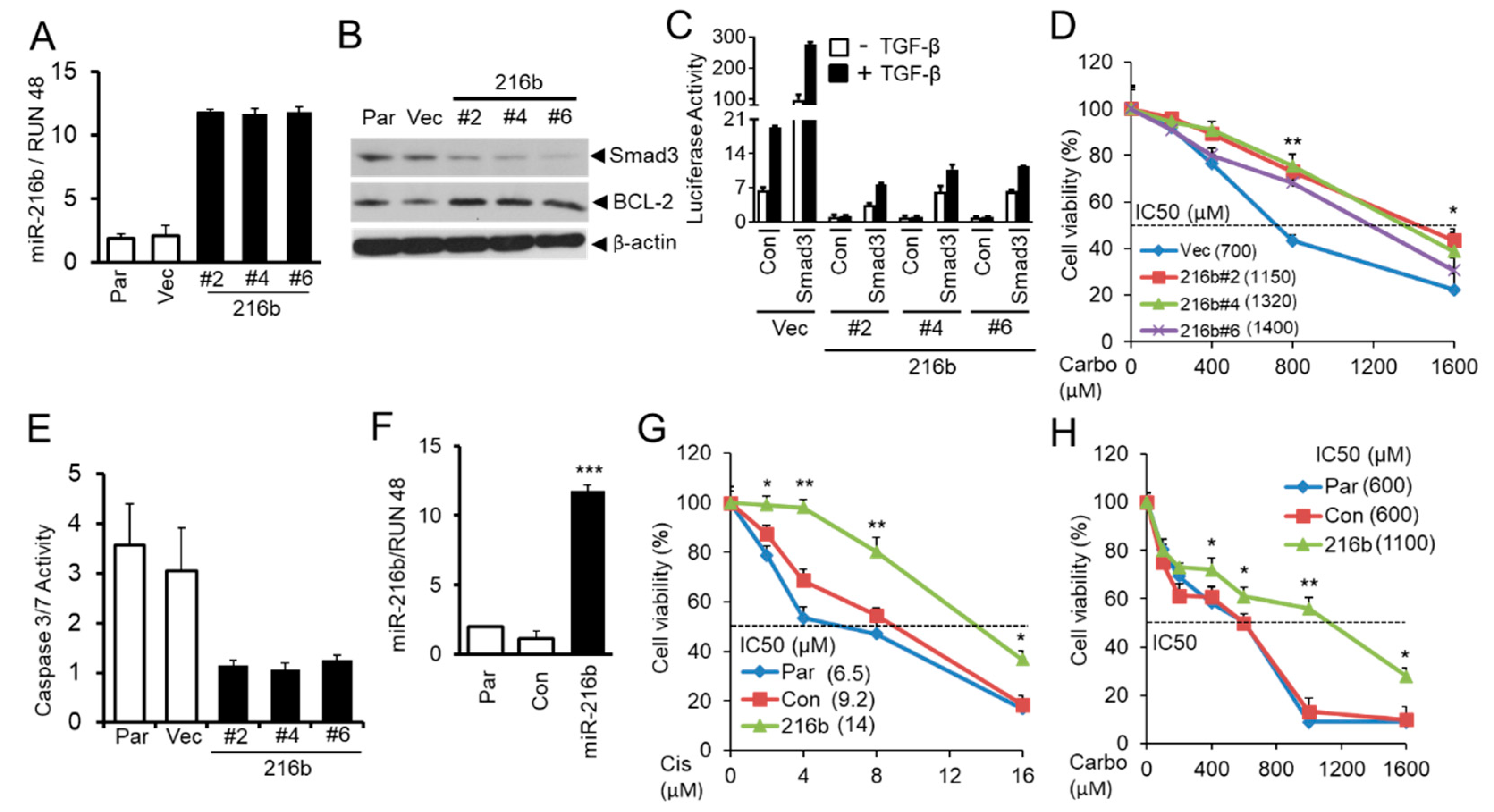
2.6. MiR-216b Increases BCL-2 Expression and Promotes Resistance of Lung Cancer Cells to Anti-cancer Drugs by Downregulating Smad3 Expression
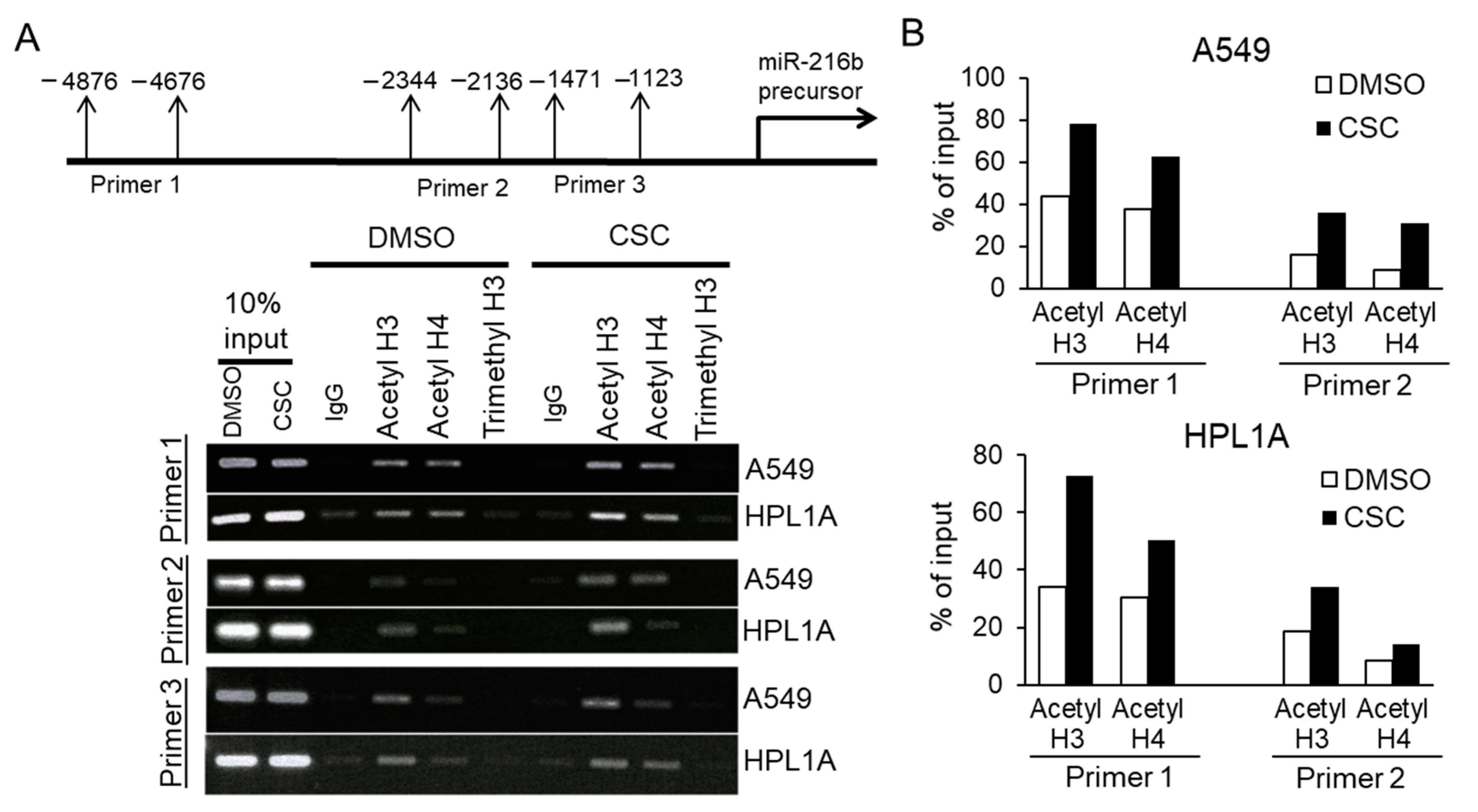
2.7. Aberrant Histone Acetylation Contributes to Elevated MiR-216b Expression in CSC Treated Lung Epithelial Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Tissues
4.2. Reagents and Antibodies
4.3. Immunoblot Analysis
4.4. Immunoprecipitation
4.5. Plasmids
4.6. Quantitative Real-time PCR Analysis
4.7. Luciferase Reporter Assay
4.8. Chromatin Immunoprecipitation (ChIP) Assay
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics. CA Cancer J. Clin. 2020, 20, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Bade, B.C.; Cruz, C.S.D. Lung Cancer 2020: Epidemiology, Etiology, and Prevention. Clin. Chest Med. 2020, 41, 1–24. [Google Scholar] [CrossRef] [PubMed]
- National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health. The Health Consequences of Smoking—50 Years of Progress: A Report of the Surgeon General; Centers for Disease Control and Prevention (US): Atlanta, GA, USA, 2014; pp. 1–10.
- Hecht, S.S. Lung carcinogenesis by tobacco smoke. Int. J. Cancer 2012, 131, 2724–2732. [Google Scholar] [CrossRef]
- Wistuba, I.I.; Lam, S.; Behrens, C.; Virmani, A.K.; Fong, K.M.; LeRiche, J.; Samet, J.M.; Srivastava, S.; Minna, J.D.; Gazdar, A.F. Molecular damage in the bronchial epithelium of current and former smokers. J. Natl. Cancer Inst. 1997, 89, 1366–1373. [Google Scholar] [CrossRef] [PubMed]
- Spira, A.; Beane, J.E.; Shah, V.; Steiling, K.; Liu, G.; Schembri, F.; Gilman, G.; Dumas, Y.; Calner, P.; Sebastiani, P.; et al. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat. Med. 2007, 13, 361–366. [Google Scholar] [CrossRef]
- Sundar, I.K.; Nevid, M.Z.; Friedman, A.E.; Rahman, I. Cigarette smoke induces distinct histone modifications in lung cells: Implications for the pathogenesis of COPD and lung cancer. J. Proteome Res. 2014, 13, 982–996. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Akhurst, R.J. Differentiation plasticity regulated by TGF-beta family proteins in development and disease. Nat. Cell Biol. 2007, 9, 1000–1004. [Google Scholar] [CrossRef]
- Massagué, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Nagaraj, N.S.; Datta, P.K. Targeting the transforming growth factor-beta signaling pathway in human cancer. Expert Opin. Investig. Drugs 2010, 19, 77–91. [Google Scholar] [CrossRef]
- Nagatake, M.; Takagi, Y.; Osada, H.; Uchida, K.; Mitsudomi, T.; Saji, S.; Shimokata, K.; Takahashi, T.; Takahashi, T. Somatic in vivo alterations of the DPC4 gene at 18q21 in human lung cancers. Cancer Res. 1996, 56, 2718–2720. [Google Scholar]
- Uchida, K.; Nagatake, M.; Osada, H.; Yatabe, Y.; Kondo, M.; Mitsudomi, T.; Masuda, A.; Takahashi, T.; Takahashi, T. Somatic in vivo alterations of the JV18-1 gene at 18q21 in human lung cancers. Cancer Res. 1996, 56, 5583–5585. [Google Scholar] [PubMed]
- Hougaard, S.; Nørgaard, P.; Abrahamsen, N.; Moses, H.L.; Spang-Thomsen, M.; Poulsen, H.S. Inactivation of the transforming growth factor beta type II receptor in human small cell lung cancer cell lines. Br. J. Cancer 1999, 79, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Osada, H.; Tatematsu, Y.; Masuda, A.; Saito, T.; Sugiyama, M.; Yanagisawa, K.; Takahashi, T. Heterogeneous transforming growth factor (TGF)-beta unresponsiveness and loss of TGF-beta receptor type II expression caused by histone deacetylation in lung cancer cell lines. Cancer Res. 2001, 61, 8331–8339. [Google Scholar] [PubMed]
- Samanta, D.; Kaufman, J.; Carbone, D.P.; Datta, P.K. Long-term smoking mediated down-regulation of Smad3 induces resistance to carboplatin in non-small cell lung cancer. Neoplasia 2012, 14, 644–655. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Montano, M. MicroRNAs: miRRORS of health and disease. Transl. Res. 2011, 157, 157–162. [Google Scholar] [CrossRef][Green Version]
- Nana-Sinkam, S.P.; Karsies, T.; Riscili, B.; Ezzie, M.; Piper, M. Lung microRNA: From development to disease. Expert Rev. Respir. Med. 2009, 3, 373–385. [Google Scholar] [CrossRef]
- Dacic, S.; Kelly, L.; Shuai, Y.; Nikiforova, M.N. MiRNA expression profiling of lung adenocarcinomas: Correlation with mutational status. Mod. Pathol. 2010, 23, 1577–1582. [Google Scholar] [CrossRef]
- Du, L.; Pertsemlidis, A. MicroRNAs and lung cancer: Tumors and 22-mers. Cancer Metastasis Rev. 2010, 29, 109–122. [Google Scholar] [CrossRef]
- Angulo, M.; Lecuona, E.; Sznajder, J.I. Role of MicroRNAs in Lung Disease. Arch. Bronconeumol. 2012, 48, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Wang, R.; Strulovici-Barel, Y.; Salit, J.; Staudt, M.R.; Ahmed, J.; Tilley, A.E.; Yee-Levin, J.; Hollmann, C.; Harvey, B.G.; et al. Persistence of smoking-induced dysregulation of miRNA expression in the small airway epithelium despite smoking cessation. PLoS ONE 2015, 10, e0120824. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.V.; Fabbri, M.; Gitlitz, B.J.; Laird-Offringa, I.A. Epigenetic therapy in lung cancer. Front. Oncol. 2013, 3, 135. [Google Scholar] [CrossRef]
- Vendetti, F.P.; Rudin, C.M. Epigenetic therapy in non-small-cell lung cancer: Targeting DNA methyltransferases and histone deacetylases. Expert Opin. Biol. Ther. 2013, 13, 1273–1285. [Google Scholar] [CrossRef] [PubMed]
- Izzotti, A.; Calin, G.A.; Arrigo, P.; Steele, V.E.; Croce, C.M.; De Flora, S. Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J. 2009, 23, 806–812. [Google Scholar] [CrossRef]
- Izzotti, A.; Larghero, P.; Longobardi, M.; Cartiglia, C.; Camoirano, A.; Steele, V.E.; De Flora, S. Dose-responsiveness and persistence of microRNA expression alterations induced by cigarette smoke in mouse lung. Mutat. Res. 2011, 717, 9–16. [Google Scholar] [CrossRef]
- Izzotti, A.; Pulliero, A. Molecular damage and lung tumors in cigarette smoke-exposed mice. Ann. N. Y. Acad. Sci. 2015, 1340, 75–83. [Google Scholar] [CrossRef]
- Willinger, C.M.; Rong, J.; Tanriverdi, K.; Courchesne, P.L.; Huan, T.; Wasserman, G.A.; Lin, H.; Dupuis, J.; Joehanes, R.; Jones, M.R.; et al. MicroRNA Signature of Cigarette Smoking and Evidence for a Putative Causal Role of MicroRNAs in Smoking-Related Inflammation and Target Organ Damage. Circ. Cardiovasc. Genet. 2017, 10, e001678. [Google Scholar] [CrossRef]
- Schembri, F.; Sridhar, S.; Perdomo, C.; Gustafson, A.M.; Zhang, X.; Ergun, A.; Lu, J.; Liu, G.; Zhang, X.; Bowers, J.; et al. MicroRNAs as modulators of smoking-induced gene expression changes in human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 2319–2324. [Google Scholar] [CrossRef]
- Szulakowski, P.; Crowther, A.J.; Jiménez, L.A.; Donaldson, K.; Mayer, R.; Leonard, T.B.; MacNee, W.; Drost, E.M. The effect of smoking on the transcriptional regulation of lung inflammation in patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2006, 174, 41–50. [Google Scholar] [CrossRef]
- Marwick, J.A.; Kirkham, P.A.; Stevenson, C.S.; Danahay, H.; Giddings, J.; Butler, K.; Donaldson, K.; Macnee, W.; Rahman, I. Cigarette smoke alters chromatin remodeling and induces proinflammatory genes in rat lungs. Am. J. Respir. Cell Mol. Biol. 2004, 31, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Davis-Dusenbery, B.N.; Chan, M.C.; Reno, K.E.; Weisman, A.S.; Layne, M.D.; Lagna, G.; Hata, A. Down-regulation of Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-beta and bone morphogenetic protein 4. J. Biol. Chem. 2011, 286, 28097–28110. [Google Scholar] [CrossRef]
- Liu, G.; Friggeri, A.; Yang, Y.; Milosevic, J.; Ding, Q.; Thannickal, V.J.; Kaminski, N.; Abraham, E. MiR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 2010, 207, 1589–1597. [Google Scholar] [CrossRef] [PubMed]
- Sivadas, V.P.; Kannan, S. The microRNA networks of TGFβ signaling in cancer. Tumor Biol. 2014, 35, 2857–2869. [Google Scholar] [CrossRef] [PubMed]
- Dews, M.; Fox, J.L.; Hultine, S.; Sundaram, P.; Wang, W.; Liu, Y.Y.; Furth, E.; Enders, G.H.; El-Deiry, W.; Schelter, J.M.; et al. The myc-miR-17~92 axis blunts TGF{beta} signaling and production of multiple TGF{beta}-dependent antiangiogenic factors. Cancer Res. 2010, 70, 8233–8246. [Google Scholar] [CrossRef]
- Dews, M.; Tan, G.S.; Hultine, S.; Raman, P.; Choi, J.; Duperret, E.K.; Lawler, J.; Bass, A.; Thomas-Tikhonenko, A. Masking epistasis between MYC and TGF-β pathways in antiangiogenesis-mediated colon cancer suppression. J. Natl. Cancer Inst. 2014, 106, dju043. [Google Scholar] [CrossRef]
- Yang, S.; Cho, Y.J.; Jin, L.; Yuan, G.; Datta, A.; Buckhaults, P.; Datta, P.K. An epigenetic auto-feedback loop regulates TGF-β type II receptor expression and function in NSCLC. Oncotarget 2015, 6, 33237–33252. [Google Scholar] [CrossRef]
- Deng, M.; Tang, H.; Zhou, Y.; Zhou, M.; Xiong, W.; Zheng, Y.; Ye, Q.; Zeng, X.; Liao, Q.; Guo, X.; et al. MiR-216b suppresses tumor growth and invasion by targeting KRAS in nasopharyngeal carcinoma. J. Cell Sci. 2011, 124, 2997–3005. [Google Scholar] [CrossRef]
- Liu, F.Y.; Zhou, S.J.; Deng, Y.L.; Zhang, Z.Y.; Zhang, E.L.; Wu, Z.Y.; Huang, Z.Y.; Chen, X.P. MiR-216b is involved in pathogenesis and progression of hepatocellular carcinoma through HBx-miR-216b-IGF2BP2 signaling pathway. Cell Death Dis. 2015, 6, e1670. [Google Scholar] [CrossRef]
- van den Berg, A.; Mols, J.; Han, J. RISC-Target Interaction: Cleavage and Translational Suppression. Biochim. Biophys. Acta 2008, 1779, 668–677. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, U.A.; Krammer, P.H. Upregulation of Bcl-2 Is Involved in the Mediation of Chemotherapy Resistance in Human Small Cell Lung Cancer Cell Lines. Int. J. Cancer 2002, 97, 584–592. [Google Scholar] [CrossRef]
- Fennell, D.A. Bcl-2 as a Target for Overcoming Chemoresistance in Small-Cell Lung Cancer. Clin. Lung Cancer 2003, 4, 307–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, J.; Zhang, Q.; Wei, L. Downregulation of serum exosomal miR-216b predicts unfavorable prognosis in patients with non-small cell lung cancer. Cancer Biomark. 2020, 27, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Halder, S.K.; Kashikar, N.D.; Cho, Y.J.; Datta, A.; Gorden, D.L.; Datta, P.K. Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology 2010, 138, 969–980. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Rayman, J.B.; Dynlacht, B.D. Analysis of promoter binding by the E2F and pRB families in vivo: Distinct E2F proteins mediate activation and repression. Genes Dev. 2000, 14, 804–816. [Google Scholar]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vu, T.; Yang, S.; Datta, P.K. MiR-216b/Smad3/BCL-2 Axis Is Involved in Smoking-Mediated Drug Resistance in Non-Small Cell Lung Cancer. Cancers 2020, 12, 1879. https://doi.org/10.3390/cancers12071879
Vu T, Yang S, Datta PK. MiR-216b/Smad3/BCL-2 Axis Is Involved in Smoking-Mediated Drug Resistance in Non-Small Cell Lung Cancer. Cancers. 2020; 12(7):1879. https://doi.org/10.3390/cancers12071879
Chicago/Turabian StyleVu, Trung, Shanzhong Yang, and Pran K. Datta. 2020. "MiR-216b/Smad3/BCL-2 Axis Is Involved in Smoking-Mediated Drug Resistance in Non-Small Cell Lung Cancer" Cancers 12, no. 7: 1879. https://doi.org/10.3390/cancers12071879
APA StyleVu, T., Yang, S., & Datta, P. K. (2020). MiR-216b/Smad3/BCL-2 Axis Is Involved in Smoking-Mediated Drug Resistance in Non-Small Cell Lung Cancer. Cancers, 12(7), 1879. https://doi.org/10.3390/cancers12071879