
Dietary Carnosine Supplementation in Healthy Human Volunteers: A Safety, Tolerability, Plasma and Brain Concentration Study
 ,
,  , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Participants
2.2. Study Protocol
2.3. Safety and Tolerability
2.4. Analysis of Plasma Carnosine, Beta-Alanine and L-Histidine
2.5. Preparation of Calibration Standards and Quality Controls
2.6. Extraction of Carnosine and Metabolites from Plasma Samples
2.7. Liquid Chromatography Mass Spectrometry (LC-MS)
2.8. Brain Proton MRI Spectroscopy for L-Carnosine
2.9. Pharmacokinetics and Statistical Analysis
3. Results
3.1. Safety and Tolerability
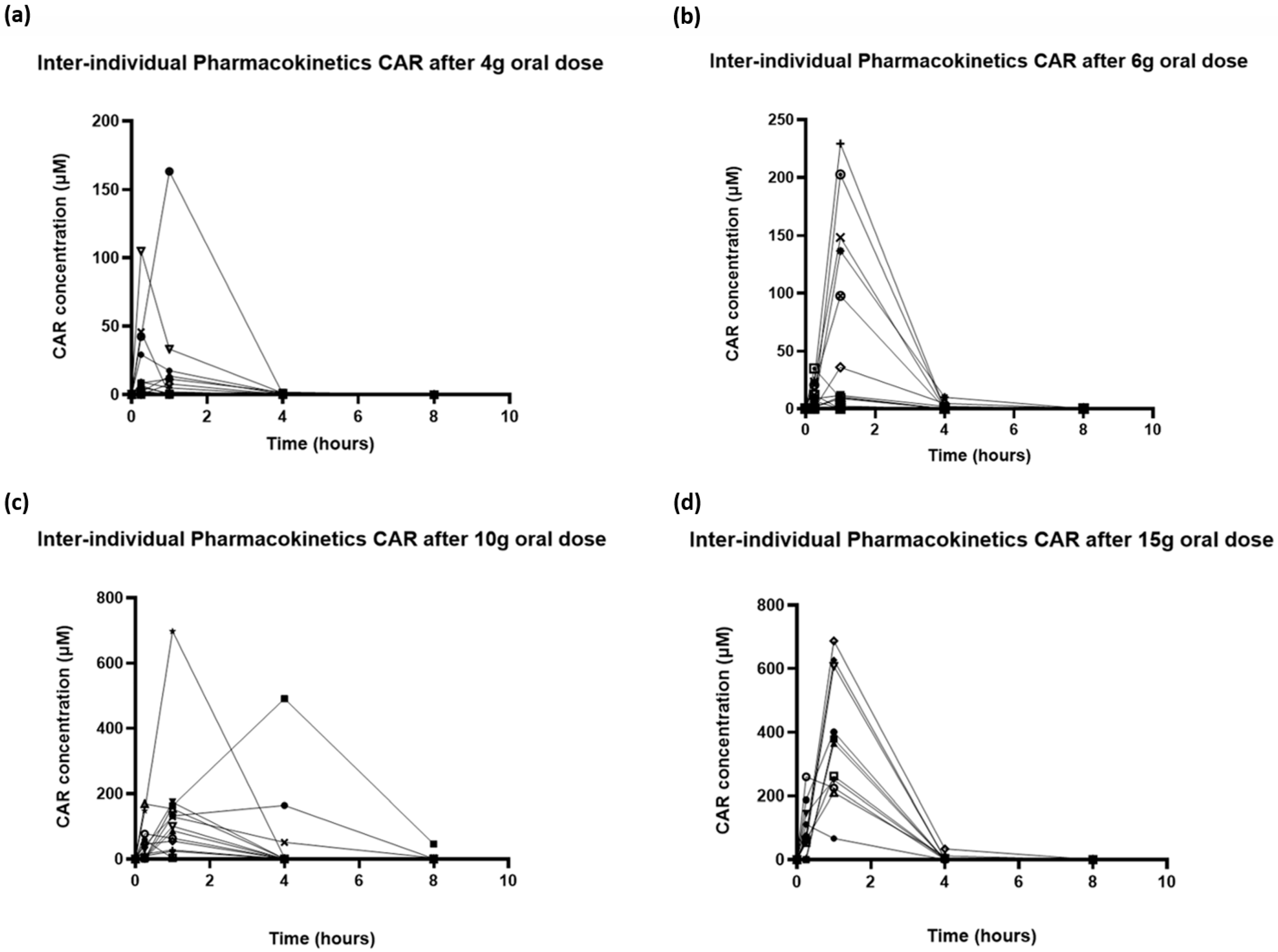
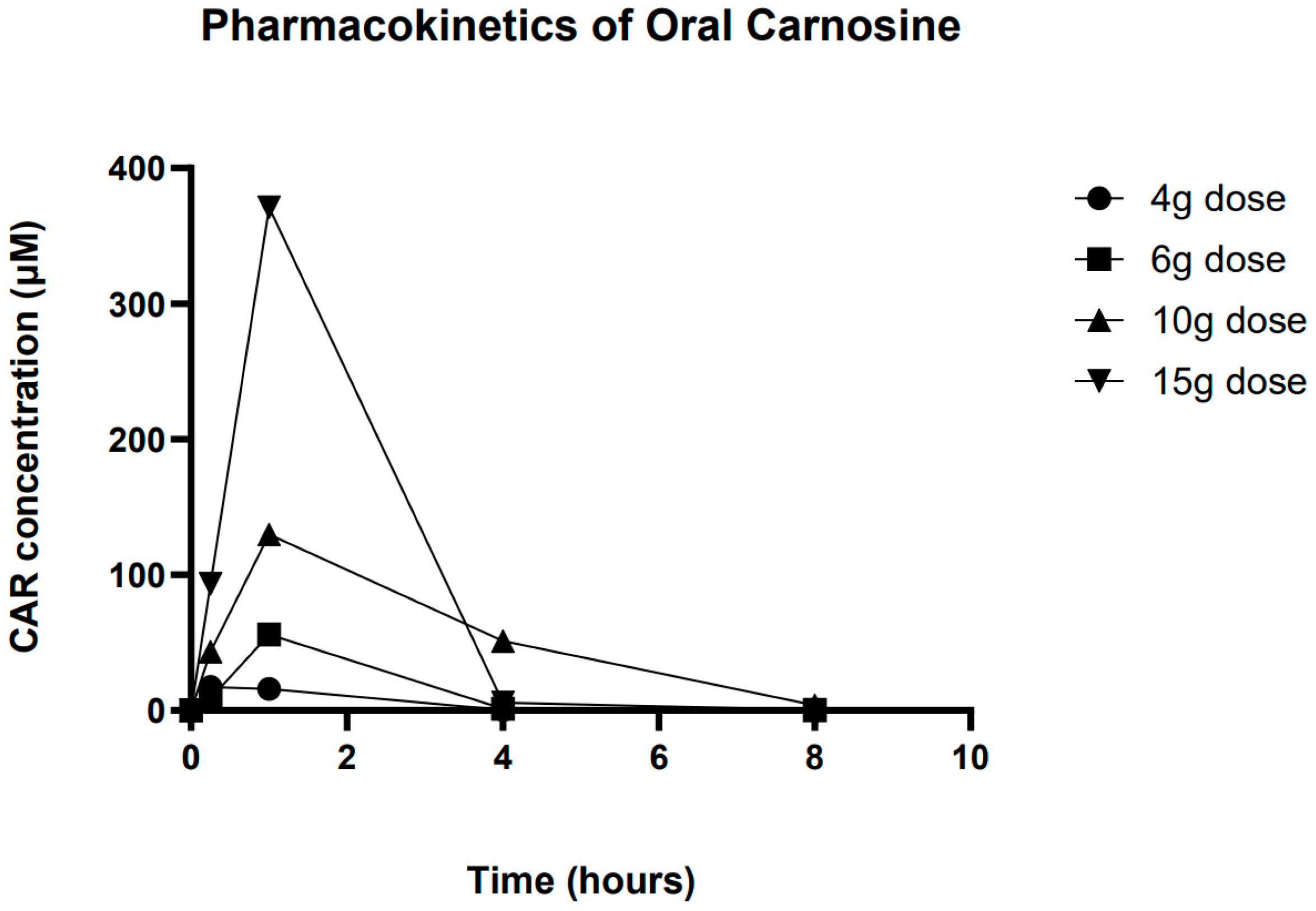
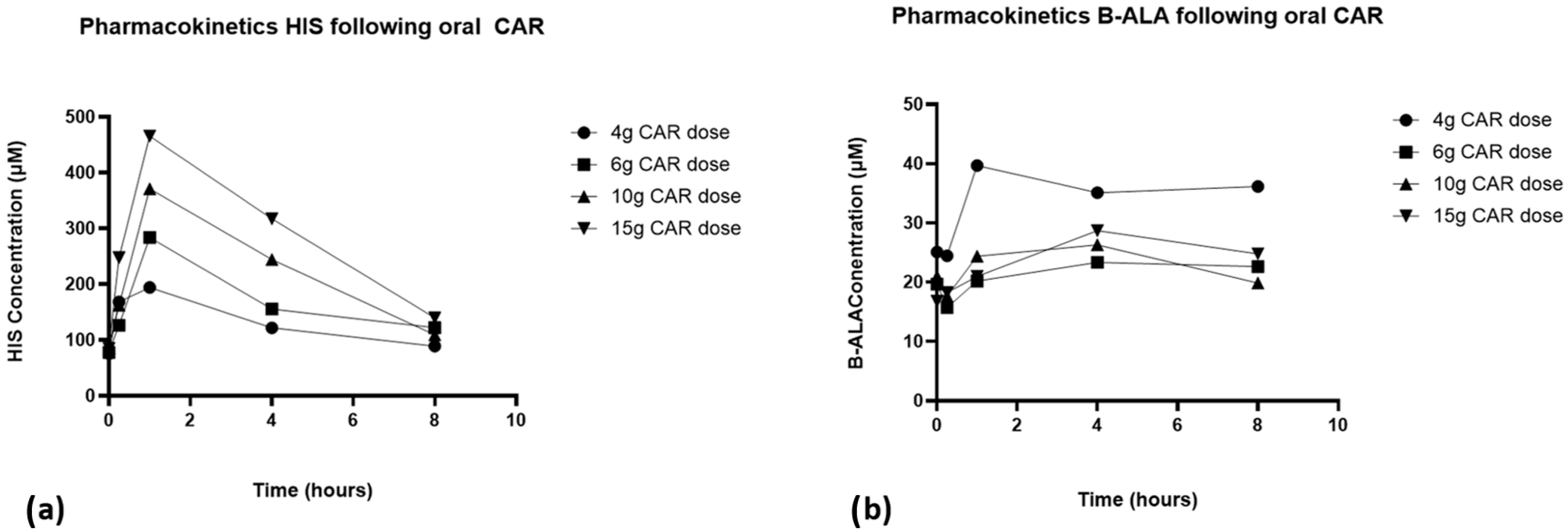
3.2. Pharmacokinetics of Carnosine
3.3. L-Histidine and Beta-Alanine
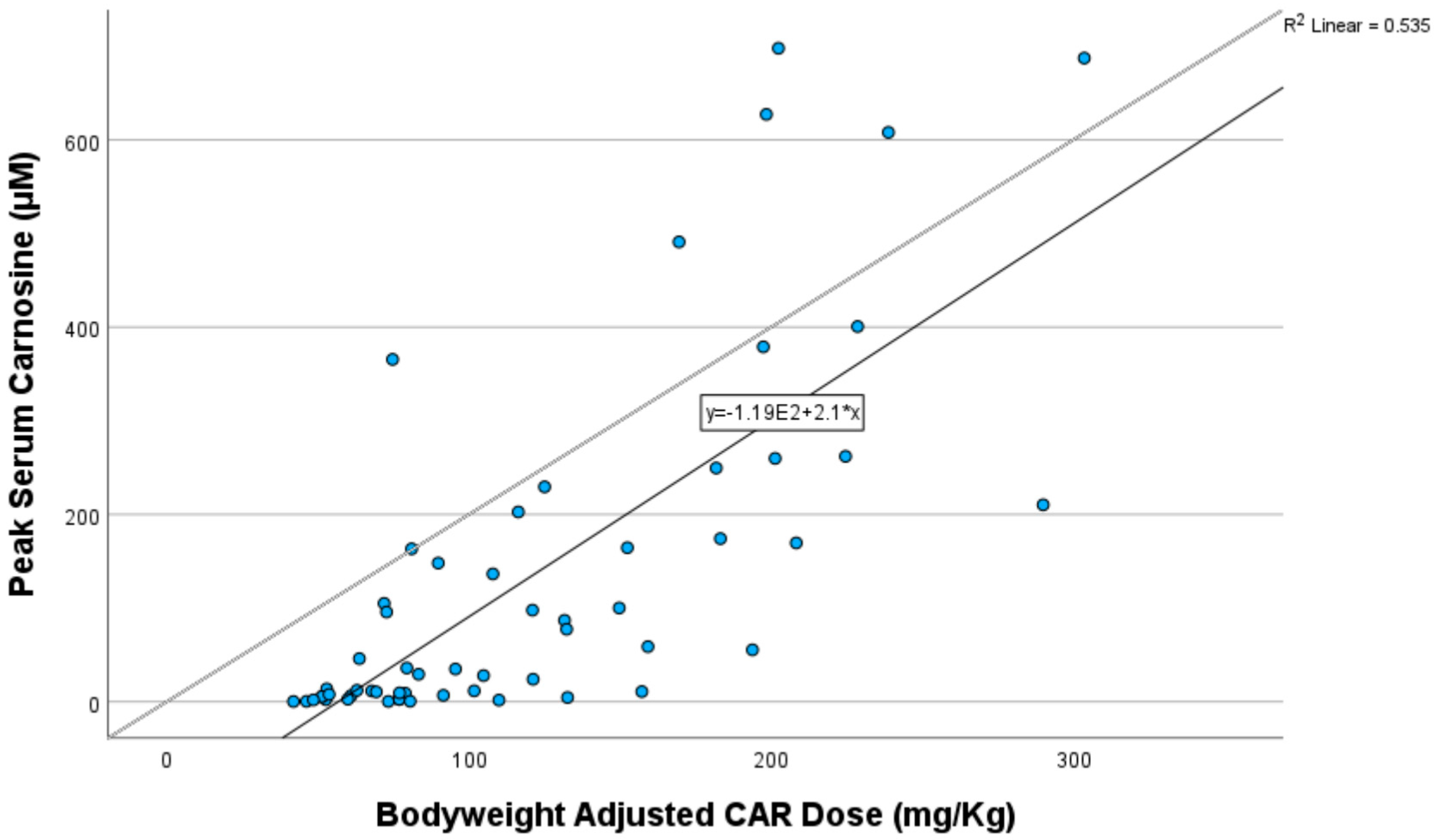
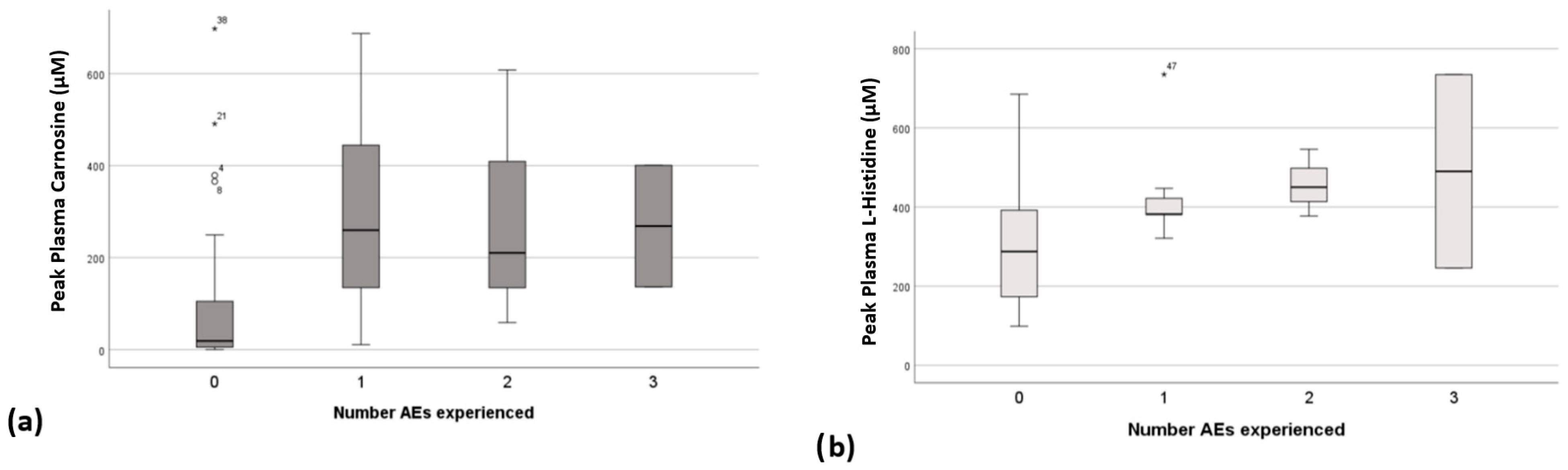
3.4. Relationship Between Peak Plasma Concentrations, CAR Dose and Adverse Events
3.5. Blood Pressure
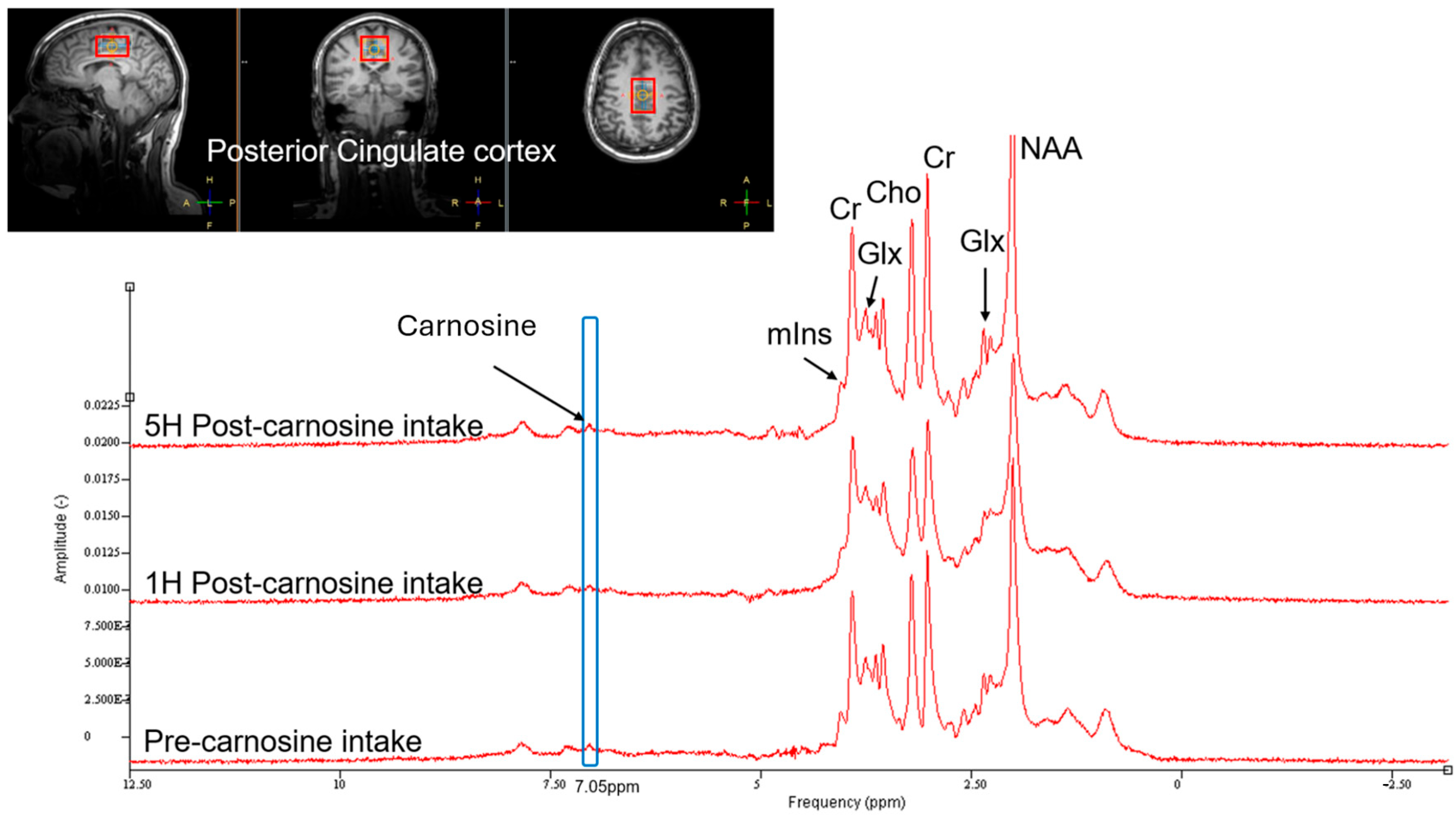
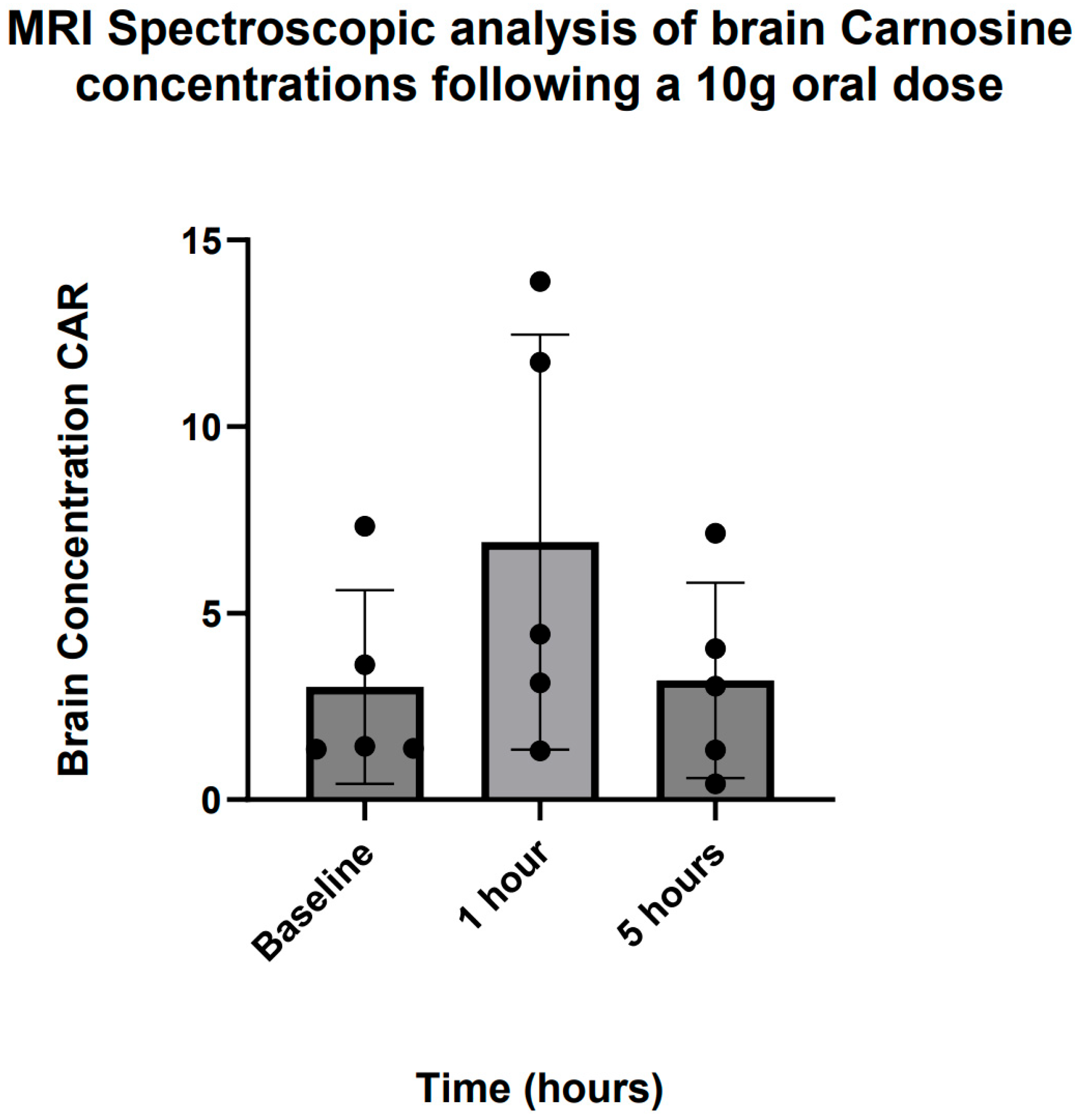
3.6. Brain Carnosine Concentrations
4. Discussion
4.1. Safety and Tolerability
4.2. Blood Pressure Effects
4.3. Brain Penetration
4.4. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boldyrev, A.; Aldini, G.; Derave, W. Physiology and Pathophysiology of Carnosine. Physiol. Rev. 2013, 93, 1803–1845. [Google Scholar] [CrossRef]
- Guilherme, G.A.; Sale, C.; Jones, R.L. Carnosine in health and disease. Eur. J. Sport Sci. 2019, 19, 30–39. [Google Scholar]
- Hoffman, J.R.; Stout, J.R.; Harris, R.C.; Moran, D.S. β-Alanine supplementation and military performance. Amino Acids 2015, 47, 2463–2474. [Google Scholar] [CrossRef] [PubMed]
- Alsheblak, M.M.; Elsherbiny, N.M.; El-Karef, A.; El-Shishtawy, M.M. Protective effects of L-carnosine on CCl4 -induced hepatic injury in rats. Eur. Cytokine Netw. 2016, 27, 6–15. [Google Scholar] [CrossRef]
- Aldini, G.; de Courten, B.; Regazzoni, L.; Gilardoni, E.; Ferrario, G.; Baron, G.; Altomare, A.; D’Amato, A.; Vistoli, G.; Carini, M. Understanding the antioxidant and carbonyl sequestering activity of carnosine: Direct and indirect mechanisms. Free Radic. Res. 2020, 55, 321–330. [Google Scholar] [CrossRef]
- Son, D.O.; Satsu, H.; Kiso, Y.; Totsuka, M.; Shimizu, M. Inhibitory effect of carnosine on interleukin-8 production in intestinal epithelial cells through translational regulation. Cytokine 2008, 42, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, M.; Tanaka, K.-I.; Kato-Negishi, M. Zinc, carnosine, and neurodegenerative diseases. Nutrients 2018, 10, 147. [Google Scholar] [CrossRef]
- Kim, E.H.; Kim, E.S.; Shin, D.; Kim, D.; Choi, S.; Shin, Y.J.; Kim, K.A.; Noh, D.; Caglayan, A.B.; Rajanikant, G.K.; et al. Carnosine Protects against Cerebral Ischemic Injury by Inhibiting Matrix-Metalloproteinases. Int. J. Mol. Sci. 2021, 22, 7495. [Google Scholar] [CrossRef]
- Wang, J.P.; Yang, Z.T.; Liu, C.; He, Y.H.; Zhao, S.S. L-carnosine inhibits neuronal cell apoptosis through signal transducer and activator of transcription 3 signaling pathway after acute focal cerebral ischemia. Brain Res. 2013, 1507, 125–133. [Google Scholar] [CrossRef]
- Aloisi, A.; Barca, A.; Romano, A.; Guerrieri, S.; Storelli, C.; Rinaldi, R.; Verri, T. Anti-aggregating effect of the naturally occurring dipeptide carnosine on aβ1-42 fibril formation. PLoS ONE 2013, 8, e68159. [Google Scholar] [CrossRef]
- Afshin-Majd, S.; Khalili, M.; Roghani, M.; Mehranmehr, N.; Baluchnejadmojarad, T. Carnosine exerts neuroprotective effect against 6-hydroxydopamine toxicity in hemiparkinsonian rat. Mol. Neurobiol. 2015, 51, 1064–1070. [Google Scholar] [CrossRef]
- Boldyrev, A.; Fedorova, T.; Stepanova, M.; Dobrotvorskaya, I.; Kozlova, E.; Boldanova, N.; Bagyeva, G.; Ivanova-Smolenskaya, I.; Illarioshkin, S. Carnisone increases efficiency of DOPA therapy of Parkinson’s disease: A pilot study. Rejuvenation Res. 2008, 11, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Szcześniak, D.; Budzeń, S.; Kopeć, W.; Rymaszewska, J.; Szczȩśniak, D. Anserine and carnosine supplementation in the elderly: Effects on cognitive functioning and physical capacity. Arch. Gerontol. Geriatr. 2014, 59, 485–490. [Google Scholar] [CrossRef]
- Caruso, G.; Caraci, F.; Jolivet, R.B. Pivotal role of carnosine in the modulation of brain cells activity: Multimodal mechanism of action and therapeutic potential in neurodegenerative disorders. Prog. Neurobiol. 2019, 175, 35–53. [Google Scholar] [CrossRef] [PubMed]
- Chmielewska, K.; Vittorio, S.; Gervasoni, S.; Dzierzbicka, K.; Inkielewicz-Stepniak, I.; Vistoli, G. Human carnosinases: A brief history, medicinal relevance, and in silico analyses. Drug Discov. Today 2024, 29, 103860. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.J.; Volpe, S.L.; Decker, E.A. Quantitation of carnosine in humans plasma after dietary consumption of beef. J. Agric. Food Chem. 2005, 53, 4736–4739. [Google Scholar] [CrossRef]
- Evaeraert, I.; Taes, Y.; De Heer, E.; Baelde, H.; Zutinic, A.; Yard, B.; Sauerhöfer, S.; Vanhee, L.; Delanghe, J.; Aldini, G.; et al. Low plasma carnosinase activity promotes carnosinemia after carnosine ingestion in humans. Am. J. Physiol. Renal Physiol. 2012, 302, F1537–F1544. [Google Scholar] [CrossRef]
- Frankenfield, D.C.; Rowe, W.A.; Smith, J.S.; Cooney, R.N. Validation of several established equations for resting metabolic rate in obese and nonobese people. J. Am. Diet. Assoc. 2003, 103, 1152–1159. [Google Scholar] [CrossRef] [PubMed]
- ICH Harmonised Tripartite Guideline. Clinical Safety Data Management: Definitions and Standards for Expedited Reporting V4. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. 1994. Available online: https://database.ich.org/sites/default/files/E2A_Guideline.pdf (accessed on 19 January 2022).
- Benovic, P.; Sokol, J.; Purdesova, A.; Maliarova, M. Biological properties and methods for determination of carnosine. Monatsh Chem. 2023, 154, 1045–1060. [Google Scholar] [CrossRef]
- Yeum, K.J.; Orioli, M.; Regazzoni, L.; Carini, M.; Rasmussen, H.; Russell, R.M.; Aldini, G. Profiling histidine dipeptides in plasma and urine after ingesting beef, chicken or chicken broth in humans. Amino Acids 2009, 38, 847–858. [Google Scholar] [CrossRef]
- Frahm, J.; Merbold, K.; Hanicke, W. Localized proton spectroscopy using stimulated echoes. J. Magn. Reson. 1987, 72, 502–508. [Google Scholar] [CrossRef]
- Petroff, O.A.; Mattson, R.H.; Behar, K.L.; Hyder, F.; Rothman, D.L. Vigabatrin increases human brain homocarnosine and improves seizure control. Ann. Neurol. 1998, 44, 948–952. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.L.; Illingworth, K.M.; Kelleher, J.; Wood, D. Intestinal absorption of the intact peptide carnosine in man, and comparison with intestinal permeability of lactulose. J. Physiol. 1991, 439, 411–422. [Google Scholar] [CrossRef]
- Bell, S.; Hariharan, R.; Laud, P.; Majid, A.; de Courten, B. Histidine-containing dipeptide supplementation improves delayed recall: A systematic review and meta-analysis. Nutr. Rev. 2023, 82, 1372–1385. [Google Scholar] [CrossRef]
- Salanti, G.; Chaimani, A.; Furukawa, T.A.; Higgins, J.P.T.; Ogawa, Y.; Cipriani, A.; Egger, M. Impact of placebo arms on outcomes in antidepressant trials: Systematic review and meta-regression analysis. Int. J. Epidemiol. 2018, 47, 1454–1464. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Araminia, B.; Shalbafan, M.; Mortezaei, A.; Shirazi, E.; Ghaffari, S.; Sahebolzamani, E.; Mortazavi, S.H.; Shariati, B.; Ardebili, M.E.; Aqamolaei, A.; et al. L-Carnosine combination therapy for major depressive disorder: A randomized, double-blind, placebo-controlled trial. J. Affect. Disord. 2020, 267, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Baye, E.; Ukropec, J.; de Courten, M.P.J.; Mousa, A.; Kurdiova, T.; Johnson, J.; Wilson, K.; Plebanski, M.; Aldini, G.; Ukropcova, B.; et al. Carnosine Supplementation Improves Serum Resistin Concentrations in Overweight or Obese Otherwise Healthy Adults: A Pilot Randomized Trial. Nutrients 2018, 10, 1258. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Thalacker-Mercer, A.E.; Gheller, M.E. Benefits and Adverse Effects of Histidine Supplementation. J. Nutr. 2020, 150 (Suppl. 1), 2588S–2592S. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.H.; Signorile, J.F.; Perry, A.C.; Jacobs, K.A.; Myers, N.D. Beta-Alanine does not enhance the effects of resistance training in older adults. J. Diet. Suppl. 2018, 15, 860–870. [Google Scholar] [CrossRef]
- Stout, J.R.; Cramer, J.T.; Zoeller, R.F.; Torok, D.; Costa, P.; Hoffman, J.R.; Harris, R.C.; O’Kroy, J. Effects of beta-alanine supplementation on the onsent of neuromuscular fatigue and ventilatory threshold in women. Amino Acids 2007, 32, 381–386. [Google Scholar] [CrossRef]
- Schön, M.; Mousa, A.; Berk, M.; Chia, W.L.; Ukropec, J.; Majid, A.; Ukropcová, B.; de Courten, B. The Potential of Carnosine in Brain-Related Disorders: A Comprehensive Review of Current Evidence. Nutrients 2019, 11, 1196. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ririe, D.G.; Roberts, P.R.; Shouse, M.N.; Zaloga, G.P. Vasodilatory actions of the dietary peptide carnosine. Nutrition 2000, 16, 168–172. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, A.; Miller, D.J. Analysis of an H1 receptor-mediated, zincpotentiated vasoconstrictor action of the histidyl dipeptide carnosine in rabbit saphenous vein. Br. J. Pharmacol. 1998, 125, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Mazza, M.; Marano, G.; Traversi, G.; Bria, P.; Mazza, S. Primary cerebral blood flow deficiency and Alzheimer’s disease: Shadows and lights. J. Alzheimers Dis. 2011, 23, 375–389. [Google Scholar] [CrossRef] [PubMed]
- Hisatsune, T.; Kaneko, J.; Kurashige, H.; Cao, Y.; Satsu, H.; Totsuka, M.; Katakura, Y.; Imabayashi, E.; Matsuda, H. Effect of Anserine/Carnosine Supplementation on Verbal Episodic Memory in Elderly People. J. Alzheimer’s Dis. 2016, 50, 149–159. [Google Scholar] [CrossRef]
- Xia, X.; Qiu, C.; Rizzuto, D.; Fratiglioni, L.; Dai, L.; Laukka, E.J.; Grande, G.; Vetrano, D.L. Role of Orthostatic Hypotension in the Development of Dementia in People with and Without Cardiovascular Disease. Hypertension 2023, 80, 1474–1483. [Google Scholar] [CrossRef]
- Saadati, S.; Cameron, J.; Menon, K.; Hodge, A.; Lu, Z.X.; de Courten, M.; Feehan, J.; de Courten, B. Carnosine Did Not Affect Vascular and Metabolic Outcomes in Patients with Prediabetes and Type 2 Diabetes: A 14-Week Randomized Controlled Trial. Nutrients 2023, 15, 4835. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- da Era Silva, V.; Painelli, V.d.S.; Shinjo, S.K.; Pereira, W.R.; Cilli, E.M.; Sale, C.; Gualano, B.; Otaduy, M.C.; Artioli, G.G. Magnetic Resonance Spectroscopy as a Non-invasive Method to Quantify Muscle Carnosine in Humans: A Comprehensive Validity Assessment. Sci. Rep. 2020, 10, 4908. [Google Scholar] [CrossRef]
- Solis, M.Y.; Cooper, S.; Hobson, R.M.; Artioli, G.G.; Otaduy, M.C.; Roschel, H.; Robertson, J.; Martin, D.; SPainelli, V.; Harris, R.C.; et al. Effects of beta-alanine supplementation on brain homocarnosine/carnosine signal and cognitive function: An exploratory study. PLoS ONE 2015, 10, e0123857. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lin, L.; Shi, L.; Chu, H.; Murad, M.H. The magnitude of small-study effects in the Cochrane Database of Systematic Reviews: An empirical study of nearly 30,000 meta-analyses. BMJ Evid. Based Med. 2020, 25, 27–32. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- European Medicines Agency. ICH Guideline M10 on Bioanalytical Method Validation and Study Sample Analysis. 2022. Available online: www.ema.europa.eu/contact (accessed on 23 June 2025).
- Food and Drug Administration. M10 Bioanalytical Method Validation and Study Sample Analysis. Guidance for Industry. 2022. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/m10-bioanalytical-method-validation-and-study-sample-analysis (accessed on 23 June 2025).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inclusions | Exclusions |
|---|---|
| Age 18 years or over | On regular medications (except contraception) |
| On established contraception if female and of childbearing age | History of medical or mental health illnesses |
| Contraindications to MRI scanning | |
| Pregnant or breastfeeding | |
| History of smoking or substance misuse | |
| Known allergy to CAR, B-ALA or HIS | |
| Known allergy to meat or fish products | |
| BMI of >30 | |
| Resting BP of <100/70 mmHg |
| All Participants (n = 16) (Mean ± SD) | Range | |
|---|---|---|
| Age (Yrs) | 29.4 (10) | 22–56 |
| Sex (%) | ||
| Male | 37.5 | |
| Female | 62.5 | |
| Weight (Kg) | 70.7 ± 13.8 | 49–95 |
| BMI (Kg/m2) | 24.4 ± 3.5 | 19.7–29.7 |
| Ethnicity (%) | ||
| White Caucasian | 75 | |
| Asian | 25 | |
| Diet (%) | ||
| Meat eater | 68.8 | |
| Pescetarian | 12.5 | |
| Vegetarian | 12.5 | |
| Vegan | 6.2 | |
| Baseline CrCl (mL/min) | 96.2 ± 11.4 | 72.1–105.6 |
| Bodyweight-adjusted (BWA) dosing (mg/Kg) | ||
| 4 g | 58.7 ± 12.1 | 41.9–81.0 |
| 6 g | 88.1 ± 18.2 | 62.9–121.0 |
| 10 g | 146.7 ± 30.6 | 104.8–202.4 |
| 15 g | 220.2 ± 45.6 | 157.2–303.6 |
| Adverse Events Experienced at Each Dose Escalation | ||
|---|---|---|
| Dose | N | % |
| 4 g | 0/16 | 0 |
| 6 g | 1/16 | 6.3 |
| 10 g | 3/15 | 20 |
| 15 g | 10/13 | 77 |
| Long-term dosing 5 g bd | 0/4 | 0 |
| Adverse events (AEs) and their prevalence | % AEs | |
| Headache | 43.5 | |
| Nausea/vomiting | 21.7 | |
| Paraesthesia | 21.7 | |
| Hypotension | 4.3 | |
| Flushing | 4.3 | |
| Carnosine Pharmacokinetic Parameters | ||||
|---|---|---|---|---|
| Carnosine Dose | AUC (μM * h) (±SD) | Cmax (μM) (±SD) | Tm (min) | T 1/2 (min) |
| 4 g | 39.6 ± 82.6 | 17.2 ± 45.4 | 15 | NA |
| 6 g | 113.0 ± 153.7 | 56.0 ± 78.1 | 60 | NA |
| 10 g | 450.6 ± 610.5 | 129.8 ± 195.5 | 60 | NA |
| 15 g | 762.1 ± 934.6 | 370.9 ± 428.2 | 60 | NA |
| L-Histidine pharmacokinetic parameters | ||||
| Carnosine Dose | AUC (μM * h) (±SD) | Cmax (μM) (±SD) | Tm (min) | T 1/2 (min) (±SD) |
| 4 g | 456.7 ± 563.8 | 194 ± 243.2 | 60 | 130.8 ± 152.4 |
| 6 g | 786.9 ± 921.9 | 283.6 ± 416.4 | 60 | 85.2 ± 98.3 |
| 10 g | 1256.0 ± 1482.5 | 371.0 ± 487.3 | 60 | 192.0 ± 213.7 |
| 15 g | 1791.0 ± 1993.5 | 465.3 ± 572.9 | 60 | 246.0 ± 298.6 |
| Systolic Blood Pressure Drop from Baseline at Nadir | ||
|---|---|---|
| Carnosine Dose | Blood Pressure Drop (mmHg) | Blood Pressure Drop (%) |
| 4 g | 8.7 | 7.5 |
| 6 g | 6 | 5.1 |
| 10 g | 8.2 | 7 |
| 15 g | 5.2 | 4.3 |
| Diastolic Blood Pressure Drop from Baseline at Nadir | ||
| Carnosine Dose | Blood Pressure Drop (mmHg) | Blood Pressure Drop (%) |
| 4 g | 10.7 | 14.4 |
| 6 g | 6 | 8 |
| 10 g | 6.1 | 8.2 |
| 15 g | 7 | 9.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, A.N.; Su, L.; Newton, J.; Grayson, A.K.; Taggart, D.; Bell, S.M.; Baig, S.; Gardner, I.; de Courten, B.; Majid, A. Dietary Carnosine Supplementation in Healthy Human Volunteers: A Safety, Tolerability, Plasma and Brain Concentration Study. Nutrients 2025, 17, 2130. https://doi.org/10.3390/nu17132130
Ali AN, Su L, Newton J, Grayson AK, Taggart D, Bell SM, Baig S, Gardner I, de Courten B, Majid A. Dietary Carnosine Supplementation in Healthy Human Volunteers: A Safety, Tolerability, Plasma and Brain Concentration Study. Nutrients. 2025; 17(13):2130. https://doi.org/10.3390/nu17132130
Chicago/Turabian StyleAli, Ali N., Li Su, Jillian Newton, Amy K. Grayson, David Taggart, Simon M. Bell, Sheharyar Baig, Iain Gardner, Barbora de Courten, and Arshad Majid. 2025. "Dietary Carnosine Supplementation in Healthy Human Volunteers: A Safety, Tolerability, Plasma and Brain Concentration Study" Nutrients 17, no. 13: 2130. https://doi.org/10.3390/nu17132130
APA StyleAli, A. N., Su, L., Newton, J., Grayson, A. K., Taggart, D., Bell, S. M., Baig, S., Gardner, I., de Courten, B., & Majid, A. (2025). Dietary Carnosine Supplementation in Healthy Human Volunteers: A Safety, Tolerability, Plasma and Brain Concentration Study. Nutrients, 17(13), 2130. https://doi.org/10.3390/nu17132130