Abstract
The Enterovirus genus of the Picornaviridae family comprises many important human pathogens, including polioviruses, rhinovirus, enterovirus A71, and enterovirus D68. They cause a wide variety of diseases, ranging from mild to severe life-threatening diseases. Currently, no effective vaccine is available against enteroviruses except for poliovirus. Enteroviruses subvert the autophagic machinery to benefit their assembly, maturation, and exit from host. Some enteroviruses spread between cells via a process described as autophagosome-mediated exit without lysis (AWOL). The early and late phases of autophagy are regulated through various lipids and their metabolizing enzymes. Some of these lipids and enzymes are specifically regulated by enteroviruses. In the present review, we summarize the current understanding of the regulation of autophagic machinery by enteroviruses, and provide updates on recent developments in this field.
Keywords:
picornavirus; enterovirus; replication; antiviral; autophagy; lipids; autophagosome maturation 1. Enteroviruses
Enteroviruses belong to the family of Picornaviridae, and are grouped into 12 species: enteroviruses A to H, enterovirus J, and rhinoviruses A to C. Within these 12 species, many serotypes exist as a result of mutations in the viral structural protein [1]. The serotypes that cause disease in human are formally grouped into five main groups: poliovirus, coxsackievirus, rhinovirus, enterovirus, and echovirus. The icosahedral capsid of enteroviruses consists of 60 copies of four functional subunits, VP1 to VP4. The P1 polyprotein is cleaved to VP0, VP1 and VP3. Subsequently, viral particles mature after cleavage of VP0 into VP2 and VP4. The P2 and P3 polyproteins include the non-structural proteins required for replication. Cleavage of 2A initially occurs, followed by cleavage of the precursor proteins 2BC, 3AB, and 3CD into 2B, 2C, 3A, 3B, 3C and 3D [2].
Enteroviruses cause a diverse range of human diseases, from mild illnesses including undifferentiated fever, conjunctivitis, and hand, foot, and mouth disease, to more severe illnesses such as pneumonia, meningitis, myocarditis, pericarditis, encephalitis, and paralysis [3]. Enterovirus A71 (EV-A71) has been increasingly reported to cause serious illness particularly in young children in Asia [4]. This virus causes hand, foot, and mouth disease, and in rare cases, severe and fatal neurological complications including brain stem encephalitis. In 2014, outbreaks of severe respiratory illness associated with enterovirus D68 occurred among children in the United States [3]. EV-A71 and enterovirus D68 infections are managed only with supportive and symptomatic care, as specific and effective antivirals are not available. Human rhinovirus (HRV) is the main cause of the common cold, and is a significant cause of infective exacerbation of underlying respiratory diseases such as asthma and chronic obstructive pulmonary disease [5]. Effective treatment for HRV is still limited. Coxsackievirus B3 (CV-B3) is an important cause of acute myocarditis, pancreatitis, and meningitis [3].
Poliovirus is the only enterovirus for which an effective vaccine is available. The Global Polio Eradication Initiative, established in 1988, aimed to eradicate polio worldwide by 2000 with the live-attenuated oral polio vaccine [3]. Unfortunately, more than a decade has passed since the targeted date, and the virus remains endemic in a few countries [6]. In view of this, there is also a pressing need to develop novel anti-poliovirus drugs with different mechanisms of action, to be used in the post-vaccine era [7].
The most common strategy in developing antivirals for enteroviruses is to target the viral proteins. However, the high mutation rates of enteroviruses often reduce the effectiveness of these compounds. A switch of target from viral proteins to host factors may be a promising approach, as this would reduce the risk of emergence of enterovirus resistance. The genomic RNA replication of enteroviruses has been shown to occur in membranous vesicles resembling coat protein complex (COPI and COPII)-coated transport vesicles derived from the host secretory pathway during early infection [8,9,10,11]. During late infection, there is a switch to membranes derived from autophagosomes, the degradative compartments of the autophagic machinery, for viral assembly, maturation, and exit from host cells [12,13,14,15]. Therefore, the autophagic machinery could potentially provide novel antiviral targets to treat enterovirus infections. In this review, we summarize the current understanding of the regulation of autophagic machinery by enteroviruses, and provide updates on recent developments in this field.
2. The Autophagic Machinery
Autophagy is divided into three distinct processes known as macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy (hereafter referred to as autophagy) is one of the most widely studied pathways in viruses [16]. Autophagy is a process of homeostasis, and can be activated in response to starvation [17]. Autophagy is also a component of the innate immune response against viral infections [18]. However, some viruses counteract autophagy by expressing proteins that can interfere with the machinery, while other viruses have developed unique mechanisms to thrive in the acidic lysosomal compartments to benefit their replication, spread and survival [19,20].
Autophagy begins with the generation of phagophores which elongate and self-fuse to form double-membrane vesicles, known as autophagosomes [21]. Autophagy-related genes (ATGs) mediate the formation of phagophores and autophagosomes. The mammalian homolog of yeast ATG8, also known as microtubule-associated protein light chain 3 (LC3), is one well-studied marker of the presence of autophagic membranes [22]. LC3 localizes in the cytoplasm during low autophagic activity, in the form of LC3-I. Upon upregulation of autophagy, LC3-I is conjugated with phosphatidylethanolamine (PE) to form lipidated LC3-PE, also known as LC3-II. The p62 protein interacts with LC3-II to target cargo to the autophagosomes for degradation. Autophagosomes fuse with endosomes to generate amphisomes, which obtain vacuolar-ATPase and become acidic. Subsequently, amphisomes fuse with incoming lysosomes to form autolysosomes. The cargo in the lumina of autolysosomes is then degraded by lysosomal proteases. The p62 protein can also be used to indicate the level of autophagic flux, as this protein is degraded along with LC3-II during complete autophagy (Figure 1) [22,23].
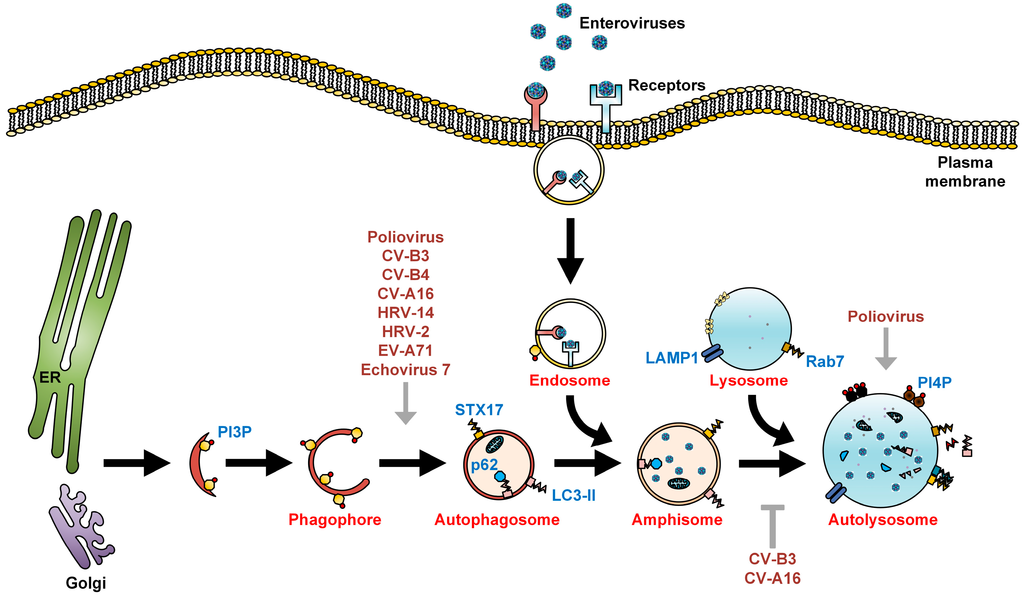
Figure 1.
The autophagic machinery and enteroviruses. Autophagy begins with the formation of phagophores that originate from either the endoplasmic reticulum (ER), Golgi, mitochondria, or plasma membrane, or are synthesized de novo, and undergo sealing to form autophagosomes. Autophagosomes then undergo a maturation process via fusion with endosomes to form acidic amphisomes. In EV-A71, HRV-A2, HRV-B14, and foot-and-mouth disease virus (FMDV) infections, upon internalization of the virus-receptor complex into endosomes, the acidified endosomes will then trigger viral uncoating [24,25,26,27]. In contrast, poliovirus, echovirus 1, and coxsackievirus A9 do not require endosomal acidification for viral entry [28,29]. During late autophagy, amphisomes fuse with lysosomes to form autolysosomes, where autophagic flux/degradation of sequestered organelles occurs. LC3-II and p62, both markers of autophagic membranes, are also degraded in autolysosomes by lysosomal proteases. Enteroviruses positively (grey arrows) and negatively (grey T arrow) regulate multiple steps of the autophagic machinery for their assembly, maturation, and exit from the host.
Figure 1.
The autophagic machinery and enteroviruses. Autophagy begins with the formation of phagophores that originate from either the endoplasmic reticulum (ER), Golgi, mitochondria, or plasma membrane, or are synthesized de novo, and undergo sealing to form autophagosomes. Autophagosomes then undergo a maturation process via fusion with endosomes to form acidic amphisomes. In EV-A71, HRV-A2, HRV-B14, and foot-and-mouth disease virus (FMDV) infections, upon internalization of the virus-receptor complex into endosomes, the acidified endosomes will then trigger viral uncoating [24,25,26,27]. In contrast, poliovirus, echovirus 1, and coxsackievirus A9 do not require endosomal acidification for viral entry [28,29]. During late autophagy, amphisomes fuse with lysosomes to form autolysosomes, where autophagic flux/degradation of sequestered organelles occurs. LC3-II and p62, both markers of autophagic membranes, are also degraded in autolysosomes by lysosomal proteases. Enteroviruses positively (grey arrows) and negatively (grey T arrow) regulate multiple steps of the autophagic machinery for their assembly, maturation, and exit from the host.
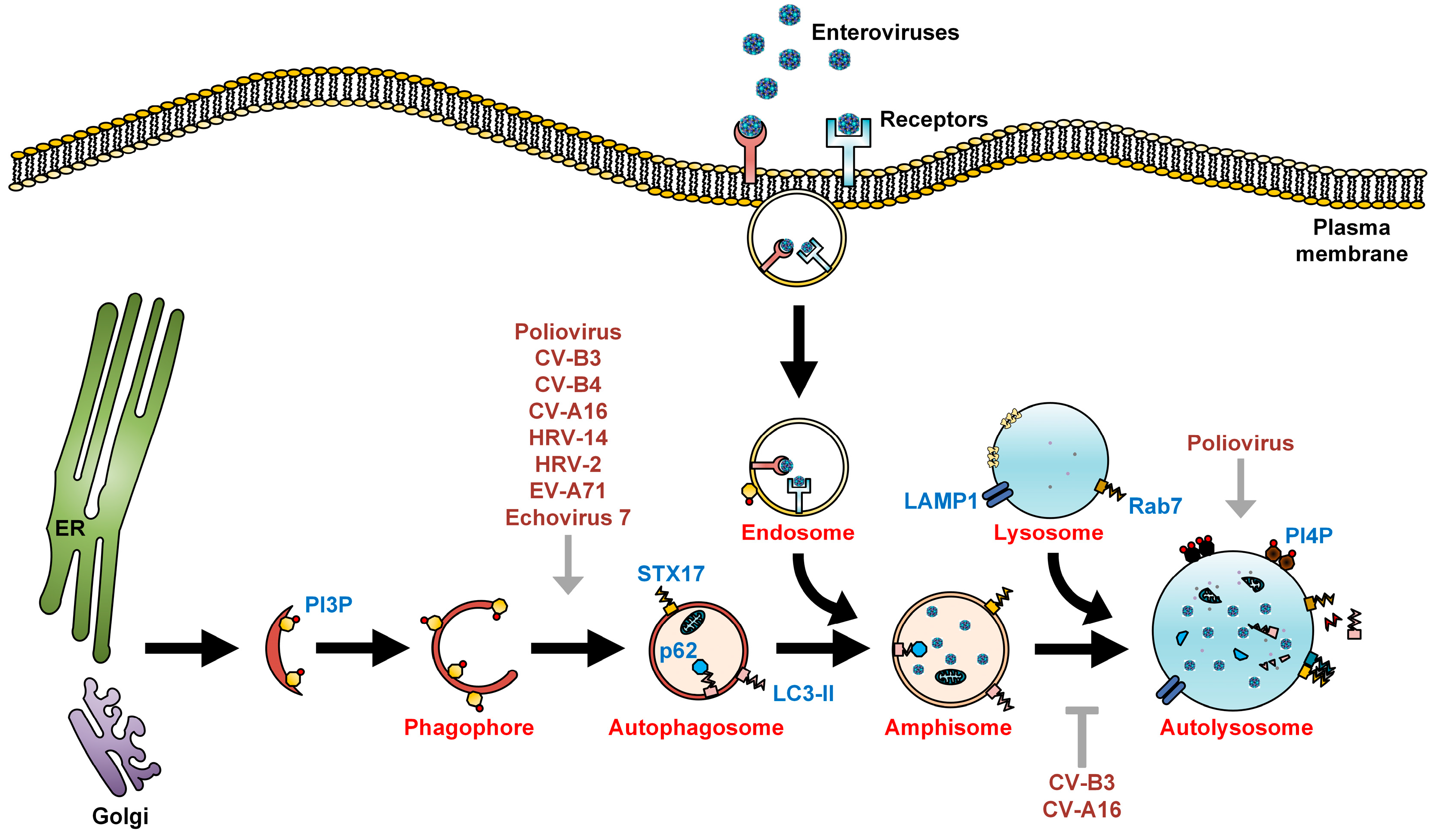
The late phase of autophagy involves fusion between autophagosomes and lysosomes. The precise mechanism that mediates this fusion event has been recently identified, and involves soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complexes, including STX17, SNAP29, and VAMP8 (Figure 2) [30]. Homotypic fusion and protein sorting (HOPS) tethering complex, containing proteins such as vacuolar protein sorting 33A (VPS33A) and VPS16, acts between autophagosomes and lysosomes. These factors are the interacting partners of STX17 [31]. Fusion between autophagosomes and lysosomes is regulated by the adaptor protein SNAP29 via two-way interactions, with SNAP29-STX17 located on the autophagosome membrane and SNAP29-VAMP8 located on the lysosome membrane (Figure 2) [30].
The involvement of Rab7 in autophagosome-lysosome fusion has been well characterized. Rab7 is a small GTPase belonging to the Ras-like GTPase superfamily [32]. In another study, Pleckstrin homology domain containing protein family member 1 (PLEKHM1), an adaptor protein that links LC3 to HOPS and Rab7, was found to mediate maturation of autophagosomes [33]. These findings allow the development of antivirals targeting the STX17-SNAP29-VAMP8 and LC3-II-PLEKHM1-Rab7 complexes.
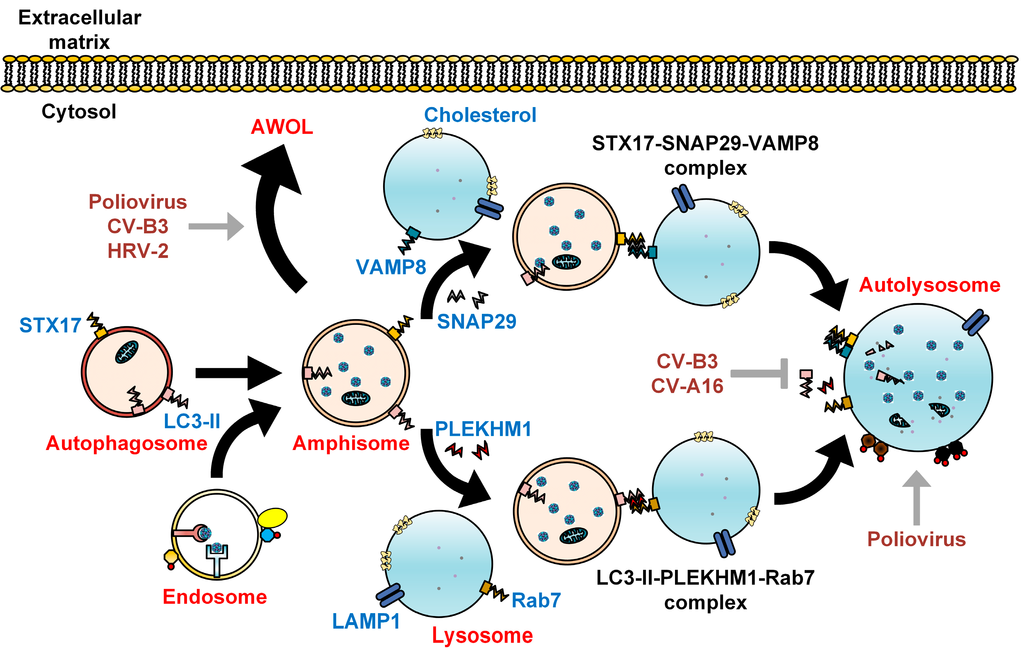
Figure 2.
Subversion of autophagosome maturation by enteroviruses. As recently described, autophagosomes fuse with lysosomes via the SNARE complex consisting of STX17, SNAP29, and VAMP8. Alternatively, autophagosome-lysosome fusion can be achieved via LC3-II, PLEKHM1, and Rab7. The engulfed cytoplasmic organelles are then degraded along with the inner membrane of the autolysosome, in a process termed autophagic flux. Cholesterol is commonly found on the membranes of autolysosome. Fusion between autophagosomes and endosomes forms amphisomes bearing viral particles, which can fuse with the plasma membrane to secrete enteroviruses via a proposed process known as autophagosome-mediated exit without lysis (AWOL). Enteroviruses positively (grey arrows) and negatively (grey T arrow) regulate autophagosome maturation to facilitate their own assembly, maturation, and exit from the host.
Figure 2.
Subversion of autophagosome maturation by enteroviruses. As recently described, autophagosomes fuse with lysosomes via the SNARE complex consisting of STX17, SNAP29, and VAMP8. Alternatively, autophagosome-lysosome fusion can be achieved via LC3-II, PLEKHM1, and Rab7. The engulfed cytoplasmic organelles are then degraded along with the inner membrane of the autolysosome, in a process termed autophagic flux. Cholesterol is commonly found on the membranes of autolysosome. Fusion between autophagosomes and endosomes forms amphisomes bearing viral particles, which can fuse with the plasma membrane to secrete enteroviruses via a proposed process known as autophagosome-mediated exit without lysis (AWOL). Enteroviruses positively (grey arrows) and negatively (grey T arrow) regulate autophagosome maturation to facilitate their own assembly, maturation, and exit from the host.
3. Enteroviruses and Autophagy
3.1. Poliovirus
During late infection, the double-membrane vesicles induced by poliovirus have a similar morphology to autophagosomes. These vesicles co-sediment with a variety of markers originating from the ER, Golgi, and lysosomes [34,35,36]. GFP-LC3 constructs have been used to study poliovirus-induced vesicles. These studies reveal co-localization of punctate GFP-LC3 signals with the 3A viral protein, a component of the viral RNA replication complex, and the double-stranded RNA of poliovirus [13,37]. The co-localization between punctate GFP-LC3-expressing cells and lysosomal-associated membrane protein 1 (LAMP1) was also observed, indicating that poliovirus induces autophagosome maturation during infection [37]. Expression of both 2BC and 3A viral proteins were required to induce autophagosome maturation in human embryonic kidney (HEK) 293T cells [37]. Production of intracellular infectious poliovirus in H1-HeLa cells was increased in the presence of autophagy inducers such as rapamycin and tamoxifen (Figure 3A). In contrast, treatment with inhibitors of autophagy, such as 3-methyladenine (3-MA) and RNA interference (RNAi) targeted against LC3 and ATG12, reduced the production of poliovirus (Figure 3B) [37]. Poliovirus also thrives in the mature acidic autophagosomes (Figure 2). Despite the fact that lysosomal proteases are not involved, intracellular vesicle acidification was found to promote the maturation of infectious poliovirus particles [19].
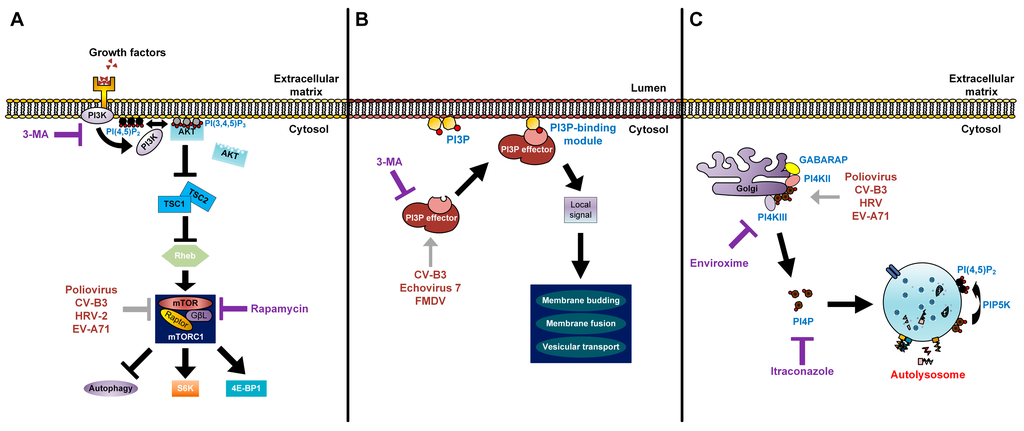
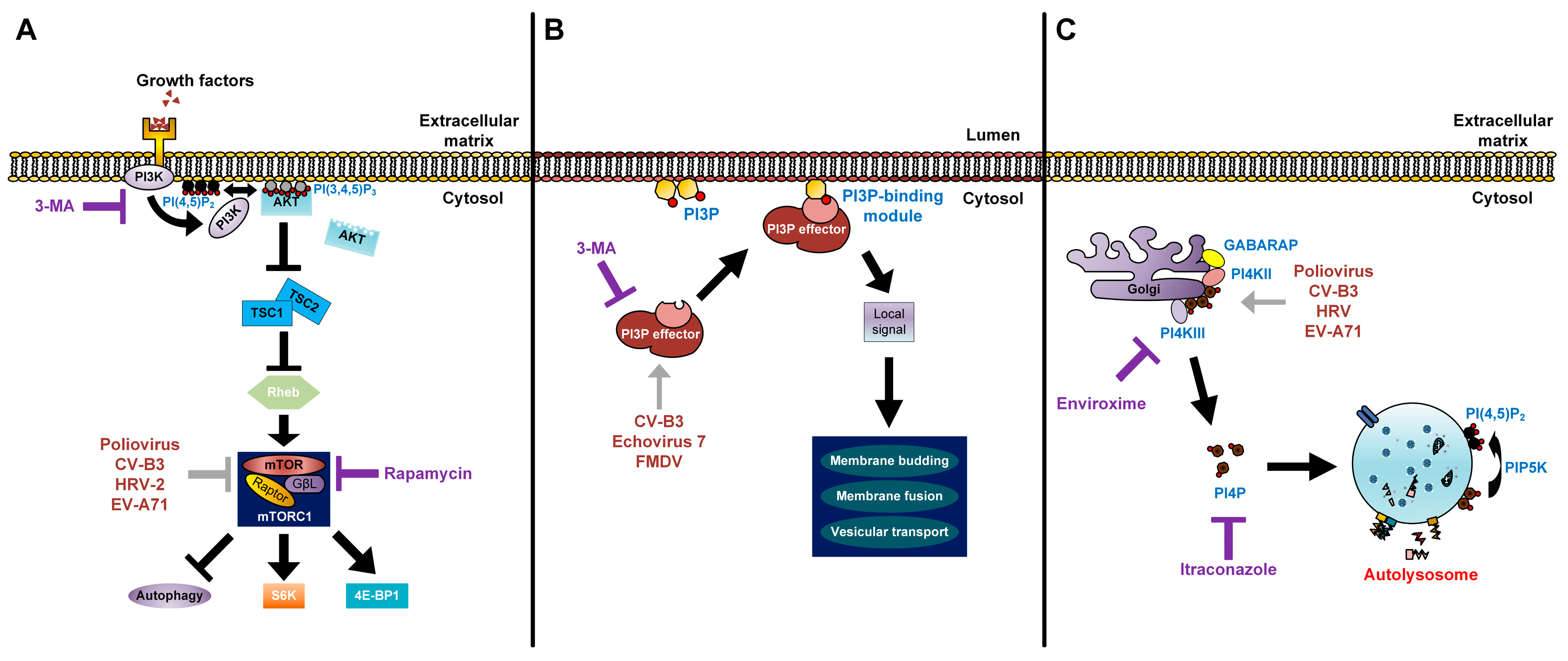
Figure 3.
Modes of action of lipids in enterovirus replication. (A) The processes of lipid signal transduction regulated by enteroviruses. Class I PI3Ks and their downstream product PI(3,4,5)P3 regulate mTORC1 signals. The phosphorylation of PI(4,5)P2 via PI3K activates the AKT signaling pathway. AKT blocks the inhibitory effect of TSC1-TSC2 complex on Rheb. Activated Rheb in turn promotes mTORC1 complex signaling. Rapamycin inhibits the mTORC1 complex (violet T arrow) and further enhances production of enteroviruses, which also have an inhibitory effect on mTORC1 (grey T arrow). 3-MA inhibits the class I PI3K and shuts off its downstream signaling (violet T arrow); (B) Picornaviruses recruit PI3P effectors (grey arrow) to the outer membranes of autophagosomes. PI3P lipids on autophagosome membranes directly interact with PI3P effectors to induce signals for membrane budding, membrane fusion, and vesicular transport of autophagosomes; 3-MA inhibits the recruitment of PI3P effectors to PI3P lipids (violet T arrow); (C) Enteroviruses recruit PI4KIIIβ (grey arrow) via Golgi-localized host factors, resulting in an increase of PI4P lipids. These lipids are commonly found to localize on autolysosome membranes and are converted to PI(4,5)P2 by PIP5K for autophagic lysosome reformation. Enviroxime inhibits PI4KIIIβ and itraconazole inhibits cholesterol and PI4P transfers (violet T arrow).
Figure 3.
Modes of action of lipids in enterovirus replication. (A) The processes of lipid signal transduction regulated by enteroviruses. Class I PI3Ks and their downstream product PI(3,4,5)P3 regulate mTORC1 signals. The phosphorylation of PI(4,5)P2 via PI3K activates the AKT signaling pathway. AKT blocks the inhibitory effect of TSC1-TSC2 complex on Rheb. Activated Rheb in turn promotes mTORC1 complex signaling. Rapamycin inhibits the mTORC1 complex (violet T arrow) and further enhances production of enteroviruses, which also have an inhibitory effect on mTORC1 (grey T arrow). 3-MA inhibits the class I PI3K and shuts off its downstream signaling (violet T arrow); (B) Picornaviruses recruit PI3P effectors (grey arrow) to the outer membranes of autophagosomes. PI3P lipids on autophagosome membranes directly interact with PI3P effectors to induce signals for membrane budding, membrane fusion, and vesicular transport of autophagosomes; 3-MA inhibits the recruitment of PI3P effectors to PI3P lipids (violet T arrow); (C) Enteroviruses recruit PI4KIIIβ (grey arrow) via Golgi-localized host factors, resulting in an increase of PI4P lipids. These lipids are commonly found to localize on autolysosome membranes and are converted to PI(4,5)P2 by PIP5K for autophagic lysosome reformation. Enviroxime inhibits PI4KIIIβ and itraconazole inhibits cholesterol and PI4P transfers (violet T arrow).
The spread of poliovirus from cell to cell is conventionally thought to require cellular lysis. As a fecal-oral pathogen, the continual release of viral particles into the intestinal lumen without cellular lysis would be beneficial. The escape of poliovirus from the intestine into the central nervous system can lead to destruction of motor neurons, causing acute flaccid paralysis. The finding of cell-to-cell spread of poliovirus in the spinal cords of Bonnet monkeys suggested the possibility of non-lytic spread in the central nervous system. During late infection, neurons in the spinal cord recovered without death of the infected cells [38]. Recently, it has been shown that poliovirus can spread between cells via an autophagy-dependent process known as autophagosome-mediated exit without lysis (AWOL) (Figure 2). The knockdown of LC3 by siRNA reduced the non-lytic spread of poliovirus, while the induction of autophagy with loperamide and nicardipine caused rapid spread of poliovirus in tissue culture and mice [14]. Another recent study demonstrated that clusters of poliovirus particles were sequestered within phosphatidylserine (PS) lipid-enriched autophagosome-like vesicles that are non-lytically released from cells. Interestingly, these vesicles bearing poliovirus had greater infection efficiency compared to free single viral particles [15]. Taken together, these findings indicate that poliovirus usurps the autophagic machinery to promote virus production.
3.2. Coxsackieviruses
Coxsackievirus B3 (CV-B3) exploits the autophagic machinery to facilitate its replication on the surface of autophagosomes [39]. The formation of double-membrane vesicles in CV-B3-infected HeLa and HEK293T cells was observed under transmission electron microscopy (EM). The punctate GFP-LC3 and LC3-II/LC3-I ratio were increased upon CV-B3 infection. In the presence of 3-MA and during transient knockdowns of Beclin1, ATG7, and VPS34, the expression of capsid VP1 and CV-B3 viral yield were decreased. In contrast, the treatment of rapamycin increased the expression of VP1 and viral titers (Figure 3A,B). Further investigations revealed that complete autophagy is blocked by CV-B3, as the level of p62 was not affected by viral infection. The inhibition of LAMP2 (the lysosomal transmembrane protein involved in autophagosome-lysosome fusion) increased both VP1 expression and viral production [40]. The results obtained from in vivo CV-B3-infected GFP-LC3 transgenic mice were also similar to in vitro findings [39,40]. Additionally, large autophagy-related structures bearing RNA polymerase-like components, termed megaphagosomes, were formed in mice infected with CVB3 [40]. Taken together, these findings suggest that CV-B3 subverts the autophagic machinery to facilitate viral replication.
Recently, the silencing of bactericidal/permeability-increasing protein fold-containing family B, member 3 (BPIFB3) was found to enhance the replication of CV-B3 by regulating the induction of autophagy [41]. Receptor interacting protein kinase-3 (RIP3), a regulator of programmed necrosis, increases autophagy flux during CV-B3 infection, which in turn promotes virus replication [42]. Another recent study also found that CV-B3 could replicate using both autophagy-dependent and autophagy-independent pathways in vivo [43]. By using three CV-B3 recombinants containing either intact or mutated autophagy-related genes (proLC3, proLC3G120A, and ATG4BC74A), the infection of CV-B3 was shown to be flexible in the usage of intracellular membranes. The proLC3 virus accumulated large quantities of LC3-II and replicated efficiently via the autophagic pathway. The proLC3G120A protein did not attach to PE to form LC3-II, but in return bound to the ER-resident protein SEL1L, to utilize membranes non-originated from autophagy for CV-B3 replication. The ATG4BC74A protein inhibited LC3-II accumulation, retained LC3-I, and accumulated immature phagophores that were crucial for viral replication [43].
In a recent study, a fluorescent timer protein marker was engineered within a CV-B3 clone to show that CV-B3 was associated with extensive intracellular membrane remodelling and was shed from cells in numerous extracellular microvesicles (EMVs). The detection of LC3-II in these EMVs suggests that the autophagy pathway, likely via the AWOL process, plays an important role in extracellular virus release within microvesicles (Figure 2) [44]. In another recent report, mature CV-B3 was observed engulfed in PS lipid-enriched autophagosome-like organelles and released non-lytically from cells [15].
CV-B4 infection of primary rat neurons has been reported to induce autophagy and accumulation of LC3-II, while 3-MA impaired both the formation of autophagosomes and viral production (Figure 3B) [45]. CV-A16 induces early autophagy, with the 2C and 3C non-structural proteins increasing the accumulation of autophagosomes without promoting fusion between autophagosomes and lysosomes. 2C-induced autophagosome formation was dependent on the human immunity-related GTPase family M protein (IRGM) activation (Figure 1) [46]. However, the role of autophagy in CV-A16 infection and pathogenesis remains unclear.
3.3. Human Rhinoviruses
The induction of autophagy has been shown to occur during infections with HRV-B14, a major receptor group virus, and HRV-A2, a minor receptor group virus. The major receptor group uses intracellular adhesion molecule-1 (ICAM-1), while the minor receptor group uses the low-density lipoprotein receptor (LDLR) family for entry. An earlier study demonstrated that HRV-A2 infection was not affected by the autophagy inducer, rapamycin or the autophagic inhibitor, 3-MA [47]. Later, another study showed the co-localization of punctate GFP-LC3 with LAMP1 was evident in both HRV-A2 and HRV-B14-infected cells [48]. Treatment with rapamycin increases HRV-A2 viral titer, while the presence of 3-MA inhibits viral yield, suggesting that autophagy has a role in HRV-A2 replication (Figure 3A,B) [48]. Similar to poliovirus and CV-B3, HRV-A2 is found to be engulfed in autophagosome-like vesicles enriched with PS lipids and released non-lytically into the extracellular matrix (Figure 2) [15].
3.4. Enterovirus A71
Autophagy is also activated during EV-A71 infection to facilitate viral replication [49]. Muscle and neuronal cell lines transfected with GFP-LC3 formed GFP+ punctate structures upon infection, and autophagosome-like vesicles were observed in EV-A71-infected cells. The increased accumulation of LC3-II was also evident in virus-infected cells. The viral titer of EV-A71 increased in the presence of rapamycin, but was impaired with 3-MA treatment (Figure 3A,B). Infection of mice with EV-A71 induced double-membrane vesicles in the neurons of the cervical spinal cord. Co-localization of LC3 and VP1 viral proteins was observed in these vesicles using immuno-EM analysis [49].
3.5. Echovirus 7
The core components of autophagy are required by echovirus 7 for its entry into host cells via endocytosis. Silencing of genes related to autophagy, including Beclin1, ATG12, ATG14, ATG16, and LC3, as well as 3-MA treatment impaired echovirus 7 infection (Figure 3B) [50]. Taken together, all the studies on poliovirus, coxsackieviruses, rhinoviruses, enterovirus A71, and echovirus 7 showed that utilization of the autophagic machinery is a crucial process for enterovirus infections.
4. Regulation of Autophagy by Enteroviruses via Lipids
The early and late phases of autophagy can be governed by lipids and lipid-metabolizing enzymes via three distinct routes. First, they govern the signaling machinery that links to mechanistic target of rapamycin (mTOR), which then regulates the activation of autophagy. This mechanism involves the class I phosphoinositide 3-kinases (PI3Ks) and their product, phosphatidylinositol-3,4,5-triphosphate (PI(3,4,5)P3). Secondly, lipids adhere to cytosolic protein mediators to facilitate deformation, expansion, and vesicle transport; an example is the modulation of PI3P that governs the generation and maturation of autophagosomes. Thirdly, the binding of PI4P lipids to autophagosome-lysosome membranes mediates the autophagic machinery [51,52].
4.1. The Regulation of Class I PI3K Signaling, PI(3,4,5)P3, and mTOR
Homeostasis between growth and starvation is regulated through a nutrient-sensing protein kinase, mTOR. In the presence of nutrients and growth signals, mTOR is activated while inhibiting autophagy (Figure 3A). The inactivation of mTOR during nutrient deprivation slows growth and anabolic processes, stimulating autophagy-mediated degradation of proteins and organelles as a recycling mechanism [53]. The release of growth factors such as insulin simulates the class I PI3K-dependent signaling cascade and activates mTOR complex 1 (mTORC1, Raptor-bound), which in turn suppresses autophagy. The PI(3,4,5)P3 converted from PI(4,5)P2 via the class I PI3Ks in response to insulin is the initial activator of signaling [53,54]. The phosphoinositide-dependent kinase 1 (PDK1) and AKT proteins are both involved downstream of PI(3,4,5)P3 [53,55].
Rapamycin, a potent activator of autophagy, impairs the activation of mTORC1 but not mTORC2 (Rictor-bound), and subsequently inhibits S6 kinase 1 (S6K1) and eIF4E-binding protein 1 (4E-BP1) phosphorylation [53,54]. As mentioned previously, treatment with rapamycin enhances the production of poliovirus, CV-B3, HRV-A2, and EV-A71 infectious progeny [37,39,48,49].
4.2. The Control of Class III PI3K Pathway and PI3P
The phosphorylation of PI at the 3’ position of the inositol ring is important for PI3P synthesis via the class III PI3K or VPS34 [55]. The addition of PI3P lipid alone suffices to stimulate the activation of autophagy (Figure 3B). There are two distinct complexes of VPS34 (PI3P effectors), each with its own unique functions [56]. In yeasts, complex I of VPS34, consisting of VPS34, Beclin1, VPS15, and ATG14, localizes at the pre-autophagosomal structure to initiate autophagy. Complex II of VPS34 is similar to complex I, except ATG14 is replaced by UV radiation resistance-associated gene protein (UVRAG), which targets Beclin1 to the endosomes, and functions in vacuolar protein sorting and autophagy induction in mammalian cells. In addition, UVRAG interacts with the class C VPS complex to facilitate fusion between autophagosomes and lysosomes. 3-MA non-specifically inhibits class III and I PI3Ks [57,58,59]. As mentioned previously, the transient knockdown of Beclin1 and VPS34 impairs the expression of VP1 viral protein and CV-B3 viral titers [39]. Echovirus 7 requires Beclin1 and other autophagic components to facilitate its genome release into cytoplasm via the endosomal system [50]. FMDV, a member of the Picornaviridae family which is closely related to enteroviruses, requires the interaction of Beclin1 with its 2C non-structural protein to facilitate virus survival (Figure 3B). This interaction is crucial to prevent Beclin1-mediated fusion between autophagosomes and lysosomes. However, the overexpression of Beclin1 in FMDV-infected cells reversed this effect, and facilitated the fusion process [60].
4.3. The Roles of PI4P in Autophagy
The yeast PI4-kinase (PI4K) PIK1 (and its vertebrate homolog, PI4KIIIβ) generates PI4P via its effector ATG26 [61]. Under normal physiological conditions, PIK1 and its product PI4P mediate the exit of secretory vesicles from the trans-Golgi (Figure 3C). Upon induction of autophagy, the recruitment of ATG proteins from the Golgi networks to phagophores is mediated by PIK1 [62]. The PI4P 5-kinase (PIP5K) facilitates the conversion of PI4P to PI(4,5)P2, which is important for autophagic lysosome reformation and recruitment of clathrin to the autolysosomes [63,64]. PI4P and PI(4,5)P2 are localized in distinct organelles, with PI4P uniformly distributed on the autolysosomal membrane, while PI(4,5)P2 is found on the autolysosomal tubules and buds [64]. Recently, it was reported that γ-aminobutyric acid receptor-associated protein (GABARAP), an autophagy-related protein, recruits PI4KIIα to generate PI4P, and this mediates the fusion of autophagosomes with lysosomes. The PI4P-GABARAP complex is localized along the perinuclear region of Golgi as well as on the autophagosome membrane [65].
In the life cycle of enteroviruses (poliovirus, CV-B3, HRV, and EV-A71), PI4KIIIβ and its product PI4P were identified as host mediators crucial in sustaining the replication of viruses (Figure 3C) [66]. PI4KIIIβ is a downstream effector of ARF1 that catalyzes the synthesis of PI4P lipids in the Golgi membranes [66]. During enterovirus infections, PI4KIIIβ is recruited to the sites of replication enriched with PI4P lipids, which is proposed to attract the viral 3D RNA-dependent RNA polymerase [67]. Apart from 3D polymerase, 2BC of poliovirus interacts directly with PI4KIIIβ. The production of PI4P via PI4KIIIβ and accumulation of unesterified cholesterol through osysterol-binding protein (OSBP) is mediated by 2BC, but is suppressed by 3A and 3AB viral proteins [68]. In contrast, the expression of 2BC precursor protein alone also increased the lipidation of LC3, a characteristic of autophagy induction [69]. Together these findings suggest the connection between PI4P lipids and autophagy as well as the importance of this interplay in the life cycles of enteroviruses.
5. Future Perspectives
It is of interest to determine if autophagic degradation of specific targets, including lipophagy (lipids), glycophagy (glycogens), mitophagy (mitochondria), pexophagy (peroxisomes), aggrephagy (protein aggregates), and ER-phagy (endoplasmic reticulum) are exploited by enteroviruses to facilitate their infections [70]. Additionally, the recent identification of specific regulators of autophagosome maturation, including STX17-SNAP29-VAMP8 and LC3-II-PLEKHM1-Rab7 complexes, may enable deeper understanding of mechanisms and roles of autophagy in enteroviral pathogenesis. The non-lytic spread of picornaviruses via exosomes was initially shown during hepatitis A infection, allowing hepatitis A virus to escape from neutralizing antibodies [71]. More recently, enteroviruses were discovered to transmit non-lytically via autophagosome-like vesicles, as populations of viral particles enwrapped in membranes of microvesicles [15]. Vesicular transmission could be beneficial for the overall fitness of enteroviruses, by allowing viral quasispecies to infect susceptible cells [72]. It could provide further insights into different tissue tropism of enteroviruses.
The absence of licensed antivirals against enteroviruses is partly due to high mutation rates of enteroviruses, with a resulting likelihood of emergence of resistance. Knowledge of the role of autophagy in enteroviral infection could lead to a promising area of therapeutics. However, targeting autophagic processes involved in infection may lead to side effects due to the importance of autophagy in cell homeostasis. Therefore, targeting the viral proteins which aid viral infection by interacting with autophagy proteins should provide more selective antiviral action [73]. In addition, lipids obtained from PI3P and PI4P are imperative for the formation of membranous vesicles involved in viral replication. Drugs targeting lipid signaling have shown potential as antivirals against enteroviruses. These include enviroxime, a potent and specific inhibitor of PI4KIIIβ, and itraconazole, an inhibitor of OSBP that shuts off cholesterol and PI4P transfers (Figure 3C) [74,75]. Recent advances in computational drug design have led to the identification of compounds that target the PI4KIIIβ lipid kinase and have potent antiviral activity against HRV, poliovirus, coxsackievirus, and EV-A71 [76]. However, the potential autophagy-modifying effects of these drugs have not yet been confirmed or correlated with their antiviral effects, which would be an area of considerable interest for future work.
6. Conclusions
In conclusion, the expanding knowledge of the relationship between enteroviruses and the autophagy pathway points to fascinating, diverse and complex mechanisms that can positively or negatively impact the survival of both viruses and host. Unraveling these mechanisms will provide insights into enterovirus life cycles and host immune responses, and a potential source of future drug development.
Acknowledgments
This study was supported by grants from the University of Malaya (High Impact Research Grant E000013-20001 as well as postgraduate research grant PG114-2012B and PG060-2015A), the Ministry of Education, Malaysia (Fundamental Research Grant Scheme FP015-2012A and FP022-2014A), L’Oréal-UNESCO for Women in Science Malaysia Fellowship 2014 (PV001-2015) and L’Oréal-UNESCO High Rising Talent Grant 2015 (IF006-2015).
Author Contributions
Jeffrey K. F. Lai, I-Ching Sam and Yoke Fun Chan wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest
References
- The Picornavirus Pages—Enterovirus. Available online: http://www.picornaviridae.com/enterovirus/enterovirus.htm (accessed on 2 December 2015).
- Lin, J.Y.; Chen, T.C.; Weng, K.F.; Chang, S.C.; Chen, L.L.; Shih, S.R. Viral and host proteins involved in picornavirus life cycle. J. Biomed. Sci. 2009, 16. [Google Scholar] [CrossRef] [PubMed]
- Symptoms—Non-Polio Enterovirus. Available online: http://www.cdc.gov/non-polio-enterovirus/about/symptoms.html (accessed on 3 December 2015).
- Yang, F.; Ren, L.; Xiong, Z.; Li, J.; Xiao, Y.; Zhao, R.; He, Y.; Bu, G.; Zhou, S.; Wang, J.; et al. Enterovirus 71 outbreak in the People’s Republic of China in 2008. J. Clin. Microbiol. 2009, 47, 2351–2352. [Google Scholar] [CrossRef] [PubMed]
- Mallia, P.; Message, S.D.; Gielen, V.; Contoli, M.; Gray, K.; Kebadze, T.; Aniscenko, J.; Laza-Stanca, V.; Edwards, M.R.; Slater, L.; et al. Experimental rhinovirus infection as a human model of chronic obstructive pulmonary disease exacerbation. Am. J. Crit. Care Med. 2011, 183, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Polio Global Eradication Initiative. Available online: http://www.polioeradication.org/casecount.asp (accessed on 2 December 2015).
- Couzin, J. Report concludes polio drugs are needed—After disease is eradicated. Science 2006, 311, 1539. [Google Scholar] [CrossRef] [PubMed]
- Rust, R.C.; Landmann, L.; Gosert, R.; Tang, B.L.; Hong, W.; Hauri, H.P.; Egger, D.; Bienz, K. Cellular COPII proteins are involved in production of the vesicles that form the poliovirus replication complex. J. Virol. 2001, 75, 9808–9818. [Google Scholar] [CrossRef] [PubMed]
- Egger, D.; Teterina, N.; Ehrenfeld, E.; Bienz, K. Formation of the poliovirus replication complex requires coupled viral translation, vesicle production, and viral RNA synthesis. J. Virol. 2000, 74, 6570–6580. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Altan-Bonnet, N.; Kovtunovych, G.; Jackson, C.L.; Lippincott-Schwartz, J.; Ehrenfeld, E. Hijacking components of the cellular secretory pathway for replication of poliovirus RNA. J. Virol. 2007, 81, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Van der Linden, L.; Wolthers, K.C.; van Kuppeveld, F.J. Replication and inhibitors of enteroviruses and parechoviruses. Viruses 2015, 7, 4529–4562. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, K.; Taylor, M.P.; Jackson, W.T. Cellular autophagy: Surrender, avoidance and subversion by microorganisms. Nat. Rev. Microbiol. 2004, 2, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Soares-Martins, J.A.; Riddell, G.T.; Jackson, W.T. Generation of unique poliovirus RNA replication organelles. MBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Bird, S.W.; Maynard, N.D.; Covert, M.W.; Kirkegaard, K. Nonlytic viral spread enhanced by autophagy components. Proc. Natl. Acad. Sci. USA 2014, 111, 13081–13086. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Du, W.; Hagemeijer, M.C.; Takvorian, P.M.; Pau, C.; Cali, A.; Brantner, C.A.; Stempinski, E.S.; Connelly, P.S.; Ma, H.C.; et al. Phosphatidylserine vesicles enable efficient en bloc transmission of enteroviruses. Cell 2015, 160, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of ATG proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Autophagy in the cellular energetic balance. Cell Metab. 2011, 13, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed]
- Richards, A.L.; Jackson, W.T. Intracellular vesicle acidification promotes maturation of infectious poliovirus particles. PLoS Pathog. 2012, 8, e1003046. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Zhang, G.; Yang, X.; Zhang, S.; Chen, L.; Yan, Q.; Xu, M.; Banerjee, A.K.; Chen, M. Phosphoprotein of human parainfluenza virus type 3 blocks autophagosome-lysosome fusion to increase virus production. Cell Host Microbe 2014, 15, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Eskelinen, E.L. Maturation of autophagic vacuoles in mammalian cells. Autophagy 2005, 1, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdalla, F.C.; Abeliovich, H.; Abraham, R.T.; Acevedo-Arozena, A.; Adeli, K.; Agholme, L.; Agnello, M.; Agostinis, P.; Aguirre-Ghiso, J.A.; et al. Guildelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012, 8, 445–544. [Google Scholar] [CrossRef] [PubMed]
- Yamayoshi, S.; Ohka, S.; Fujii, K.; Koike, S. Functional comparison of SCARB2 and PSGL1 as receptors for enterovirus 71. J. Virol. 2013, 87, 3335–3347. [Google Scholar] [CrossRef] [PubMed]
- Bayer, N.; Schober, D.; Prchla, E.; Murphy, R.F.; Blaas, D.; Fuchs, R. Effect of bafilomycin A1 and nocodazole on endocytic transport in HeLa cells: Implications for viral uncoating and infection. J. Virol. 1998, 72, 9645–9655. [Google Scholar] [PubMed]
- Grunert, H.P.; Wolf, K.U.; Langner, K.D.; Sawitzky, D.; Habermehl, K.O.; Zeichhardt, H. Internalization of human rhinovirus 14 into HeLa and ICAM-1-transfected BHK cells. Med. Microbiol. Immunol. 1997, 186, 1–9. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, V.; Larocco, M.; Baxt, B. Heparan sulfate-binding foot-and-mouth disease virus enters cells via caveola-mediated endocytosis. J. Virol. 2008, 82, 9075–9085. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.; Carrasco, L. Entry of poliovirus into cells does not require a low-pH step. J. Virol. 1993, 67, 4543–4548. [Google Scholar] [PubMed]
- Marjomaki, V.; Turkki, P.; Huttunen, M. Infectious entry pathway of enterovirus B species. Viruses 2015, 7, 6387–6399. [Google Scholar] [CrossRef] [PubMed]
- Itakura, E.; Kishi-Itakura, C.; Mizushima, N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012, 151, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Nishimura, T.; Sakamaki, Y.; Itakura, E.; Hatta, T.; Natsume, T.; Mizushima, N. The HOPS complex mediates autophagosome-lysosome fusion through interaction with syntaxin 17. Mol. Biol. Cell 2014, 25, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Alemu, E.A.; Brech, A.; Bruun, J.A.; Lamark, T.; Overvatn, A.; Bjorkoy, G.; Johansen, T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J. Cell Biol. 2010, 188, 253–269. [Google Scholar] [CrossRef] [PubMed]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; Miranda de Stegmann, D.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, A.; Giddings, T.H., Jr.; Ladinsky, M.S.; Kirkegaard, K. Cellular origin and ultrastructure of membranes induced poliovirus infection. J. Virol. 1996, 70, 6576–6588. [Google Scholar] [PubMed]
- Suhy, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Remodelling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: An autophagy-like origin for virus-induced vesicles. J. Virol. 2000, 75, 8953–8965. [Google Scholar] [CrossRef]
- Giachetti, C.; Hwang, S.S.; Semler, B.L. Cis-acting lesions targeted to the hydrophobic domain of a poliovirus membrane protein involved in RNA replication. J. Virol. 1992, 66, 6045–6057. [Google Scholar] [PubMed]
- Jackson, W.T.; Giddings, T.H., Jr.; Taylor, M.P.; Mulinyawe, S.; Rabinovitch, M.; Kopito, R.R.; Kirkegaard, K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005, 3, e156. [Google Scholar] [CrossRef] [PubMed]
- Ponnuraj, E.M.; John, T.J.; Levin, M.J.; Simoes, E.A. Cell-to-cell spread of poliovirus in the spinal cord of bonnet monkeys (Macada radiata). J. Gen. Virol. 1998, 79, 2393–2403. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Zhang, J.; Si, X.; Gao, G.; Mao, I.; McManus, B.M.; Luo, H. Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 2008, 82, 9143–9145. [Google Scholar] [CrossRef] [PubMed]
- Kemball, C.C.; Alirezaei, M.; Flynn, C.T.; Wood, M.R.; Harkins, S.; Kiosses, W.B.; Whitton, J.L. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J. Virol. 2010, 84, 12110–12124. [Google Scholar] [CrossRef] [PubMed]
- Delorme-Axford, E.; Morosky, S.; Bomberger, J.; Stolz, D.B.; Jackson, W.T.; Coyne, C.B. BPIFB3 regulates autophagy and coxsackievirus B replication through a noncanonical pathway independent of the core initiation machinery. MBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.G.; Morosky, S.A.; Drummond, C.G.; Patel, M.; Kim, C.; Stolz, D.B.; Bergelson, J.M.; Cherry, S.; Coyne, C.B. RIP3 regulates autophagy and promotes coxsackievirus B3 infection of intestinal epithelial cells. Cell Host Microbe 2015, 18, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Alirezaei, M.; Flynn, C.T.; Wood, M.R.; Harkins, S.; Whitton, J.L. Coxsackievirus can exploit LC3 in both autophagy-dependent and-independent manners in vivo. Autophagy 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M.; et al. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.Y.; Ha, Y.E.; Choi, J.E.; Ahn, J.; Lee, H.; Kweon, H.S.; Lee, J.Y.; Kim, D.H. Coxsackievirus B4 uses autophagy for replication after calpain activation in rat primary neurons. J. Virol. 2008, 82, 11976–11978. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; He, X.; Zhu, G.; Tu, H.; Liu, Z.; Li, W.; Han, S.; Yin, J.; Peng, B.; Liu, W. Coxsackievirus A16 elicits incomplete autophagy involving the mTOR and ERK pathways. PLoS ONE 2015, 10, e0122109. [Google Scholar] [CrossRef] [PubMed]
- Brabec-Zaruba, M.; Berka, U.; Blaas, D.; Fuchs, R. Induction of autophagy does not affect human rhinovirus type 2 production. J. Virol. 2007, 81, 10815–10817. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.A.; Jackson, W.T. Human rhinovirus 2 induces the autophagic pathway and replicates more efficiently in autophagic cells. J. Virol. 2011, 85, 9651–9654. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.; Chang, C.L.; Wang, P.S.; Tsai, Y.; Liu, H.S. Enterovirus 71-induced autophagy detected in vitro and in vivo promotes viral replication. J. Med. Virol. 2009, 81, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Bergelson, J.M. Echovirus 7 entry into polarized Caco-2 intestinal epithelial cells involves core components of the autophagic machinery. J. Virol. 2014, 88, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, A.; Tooze, S.A. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J. Cell Biol. 2009, 186, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Burman, C.; Ktistakis, N.T. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010, 584, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Verge, I.; Deretic, V. The role of PI3P phosphatases in the regulation of autophagy. FEBS Lett. 2010, 584, 1313–1318. [Google Scholar] [CrossRef] [PubMed]
- Toker, A. Phosphoinositide 3-kinases-a historical perspective. Subcell. Biochem. 2012, 58, 95–110. [Google Scholar] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Petiot, A.; Ogier-Denis, E.; Blommaart, E.F.; Meijer, A.J.; Codogno, P. Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J. Biol. Chem. 2000, 275, 992–998. [Google Scholar] [CrossRef] [PubMed]
- Obara, K.; Sekito, T.; Ohsumi, Y. Assortment of phosphatidylinositol 3-kinase complexes-ATG14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol. Biol. Cell 2006, 17, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Obara, K.; Ohsumi, Y. PtdIns 3-kinase orchestrates autophagosome formation in yeast. J. Lipids 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Gladue, D.P.; O’Donnell, V.; Baker-Branstetter, R.; Holinka, L.G.; Pacheco, J.M.; Fernandez-Sainz, I.; Lu, Z.; Baxt, B.; Piccone, M.E.; Rodriguez, L.; et al. Foot-and-mouth disease virus non-structural protein 2C interacts with Beclin1, modulating virus replication. J. Virol. 2012, 86, 12080–12090. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Oku, M.; Wasada, Y.; Ano, Y.; Sakai, Y. PI4P-signaling pathway for the synthesis of a nascent membrane structure in selective autophagy. J. Cell Biol. 2006, 173, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yang, Z.; Nair, U.; Mao, K.; Liu, X.; Klionsky, D.J. Phosphatidylinositol 4-kinases are required for autophagic membrane trafficking. J. Biol. Chem. 2012, 287, 37964–37972. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Rong, Y.; Liu, M.; Ma, L.; Du, W.; Zhang, H.; Tian, Y.; Cao, Z.; Li, Y.; Ren, H.; Zhang, C.; et al. Clathrin and phosphatidylinositol-4,5-biphosphate regulate autophagic lysosome reformation. Nat. Cell Biol. 2012, 14, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, H.Q.; Zhu, X.; Zhang, L.; Albanesi, J.; Levine, B.; Yin, H. GABARAPs regulate PI4P-dependent autophagosome: Lysosome fusion. Proc. Natl. Acad. Sci. USA 2015, 112, 7015–7020. [Google Scholar] [CrossRef] [PubMed]
- Van der Schaar, H.M.; van der Linden, L.; Lanke, K.H.; Strating, J.R.; Purstinger, G.; de Vries, E.; de Haan, C.A.; Neyts, J.; van Kuppeveld, F.J. Coxsackievirus mutants that can bypass host factor PI4KIIIβ and the need for high levels of PI4P lipids for replication. Cell Res. 2012, 22, 1576–1592. [Google Scholar] [CrossRef] [PubMed]
- Hsu, N.Y.; Iinytska, O.; Belov, G.; Santiana, M.; Chen, Y.H.; Takvorian, P.M.; Pau, C.; van der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Arita, M. Phosphatidylinositol-4 kinase III β and oxysterol-binding protein accumulate unesterified cholesterol on poliovirus-induced membrane structure. Microbiol. Immunol. 2014, 58, 239–256. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Kirkegaard, K. Modification of cellular autophagy protein LC3 by poliovirus. J. Virol. 2007, 81, 12543–12553. [Google Scholar] [CrossRef] [PubMed]
- Lennemann, N.J.; Coyne, C.B. Catch me if you can: The link between autophagy and viruses. PLoS Pathog. 2015, 11, e1004685. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hensley, L.; McKnight, K.L.; Hu, F.; Madden, V.; Ping, L.; Jeong, S.H.; Walker, C.; Lanford, R.E.; Lemon, S.M. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 2013, 496, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Altan-Bonnet, N.; Chen, Y.H. Intercellular transmission of viral populations with vesicles. J. Virol. 2015, 89, 12242–12244. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug. Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [PubMed]
- Strating, J.R.; van der Linden, L.; Albulescu, L.; Bigay, J.; Arita, M.; Delang, L.; Leyssen, P.; van der Schaar, H.M.; Lanken, K.H.; Thibaut, H.J.; et al. Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep. 2015, 10, 600–615. [Google Scholar] [CrossRef] [PubMed]
- Heinz, B.A.; Vance, L.M. The antiviral compound enviroxime targets the 3A coding region of rhinovirus and poliovirus. J. Virol. 1995, 69, 4189–4197. [Google Scholar] [PubMed]
- MacLeod, A.M.; Mitchell, D.R.; Palmer, N.J.; van de Poel, H.; Conrath, K.; Andrews, M.; Leyssen, P.; Neyts, J. Identification of a series of compounds with potent antiviral activity for the treatment of enterovirus infections. ACS Med. Chem. Lett. 2013, 4, 585–589. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).