
Discovery of Small Molecules Against Foot-and-Mouth Disease Virus Replication by Targeting 2C Helicase Activity

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
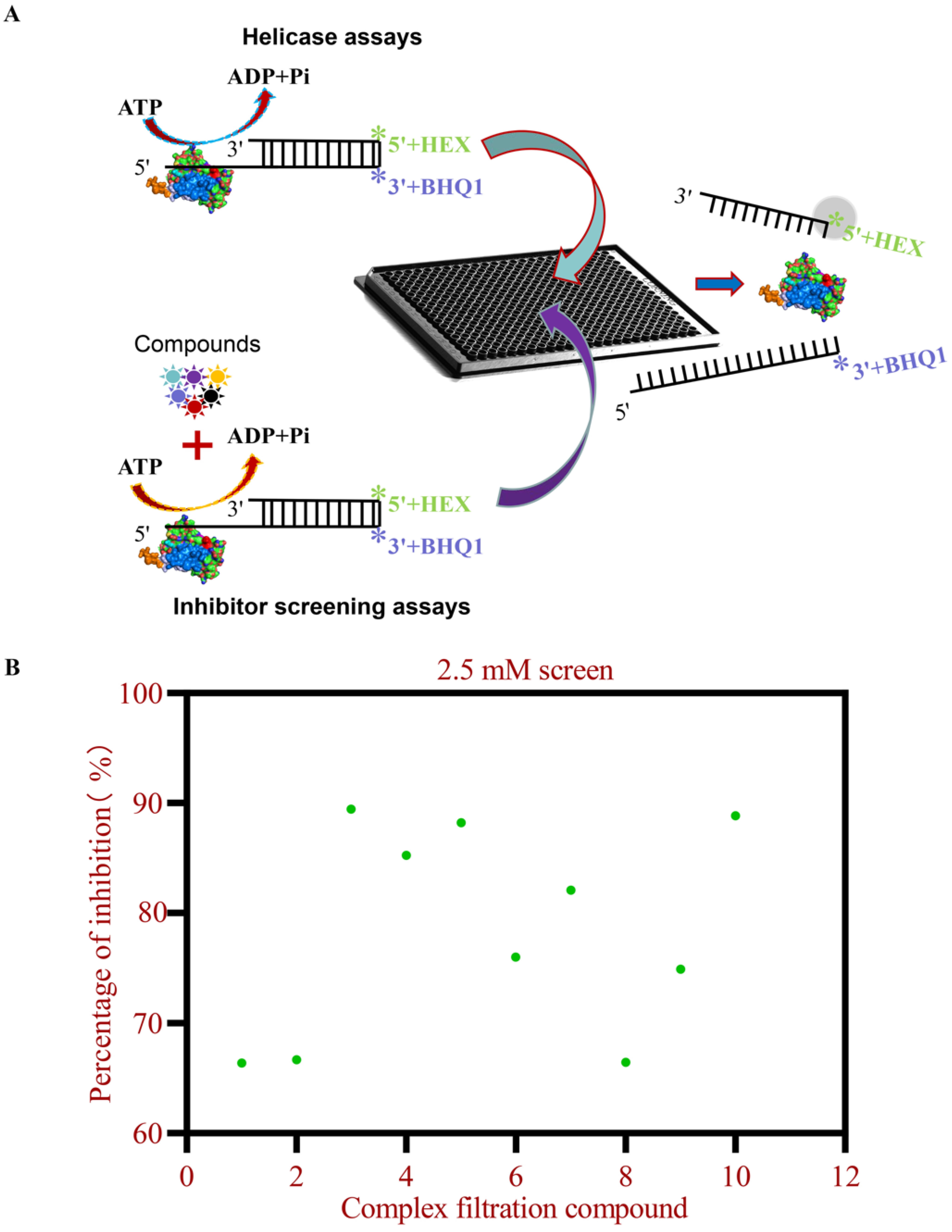
2.2. Establishment of Screening Methods for Inhibitors of 2C Protein Helicase Activity
2.3. HTS Assay of 2C Helicase Activity
2.4. Enzyme Inhibition Kinetic Assay
2.5. Gel-Based Helicase Inhibitor Assays
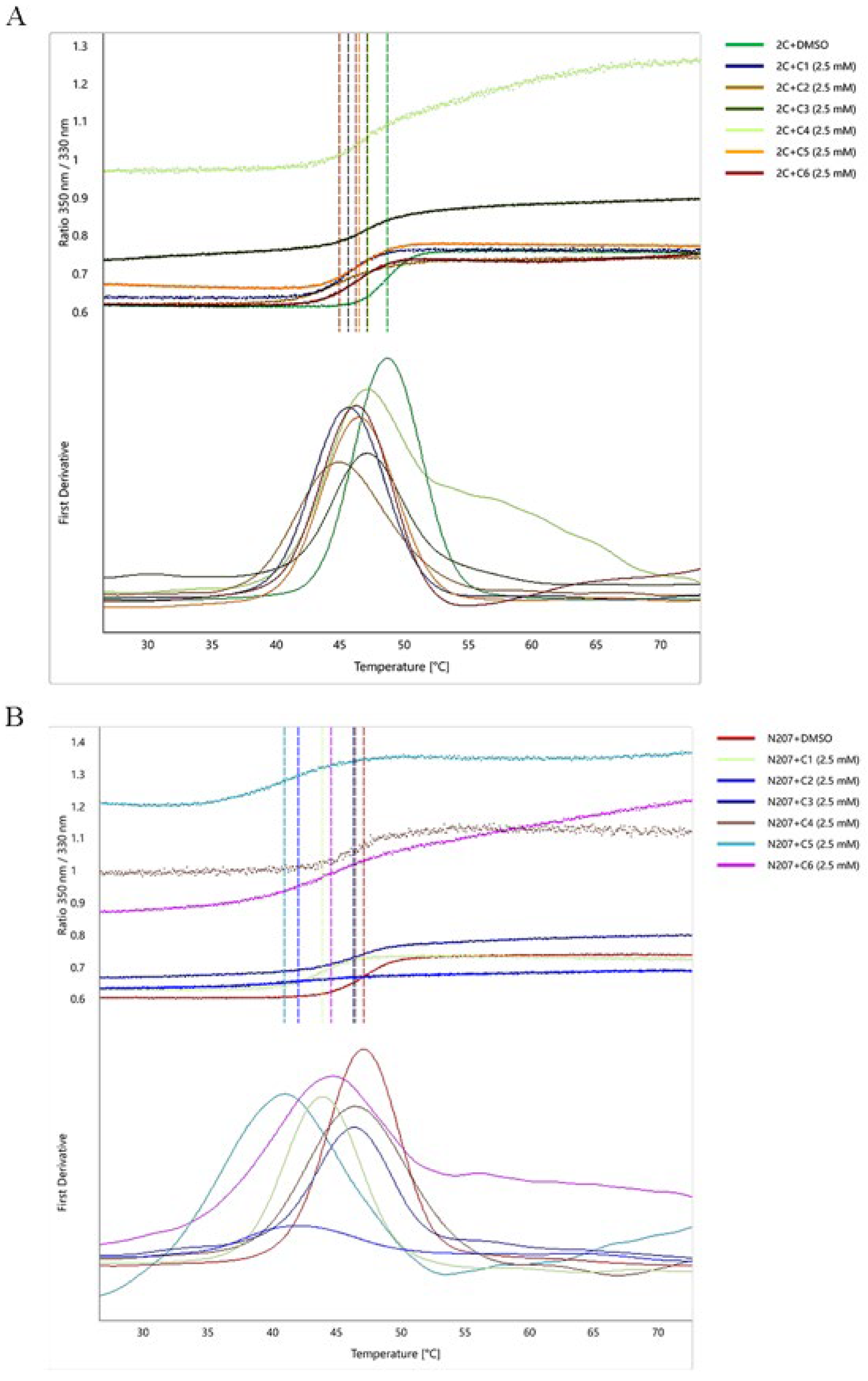
2.6. NanoDSF Analysis of FMDV 2C Protein–Ligand Interactions
2.7. Cytotoxicity Assay
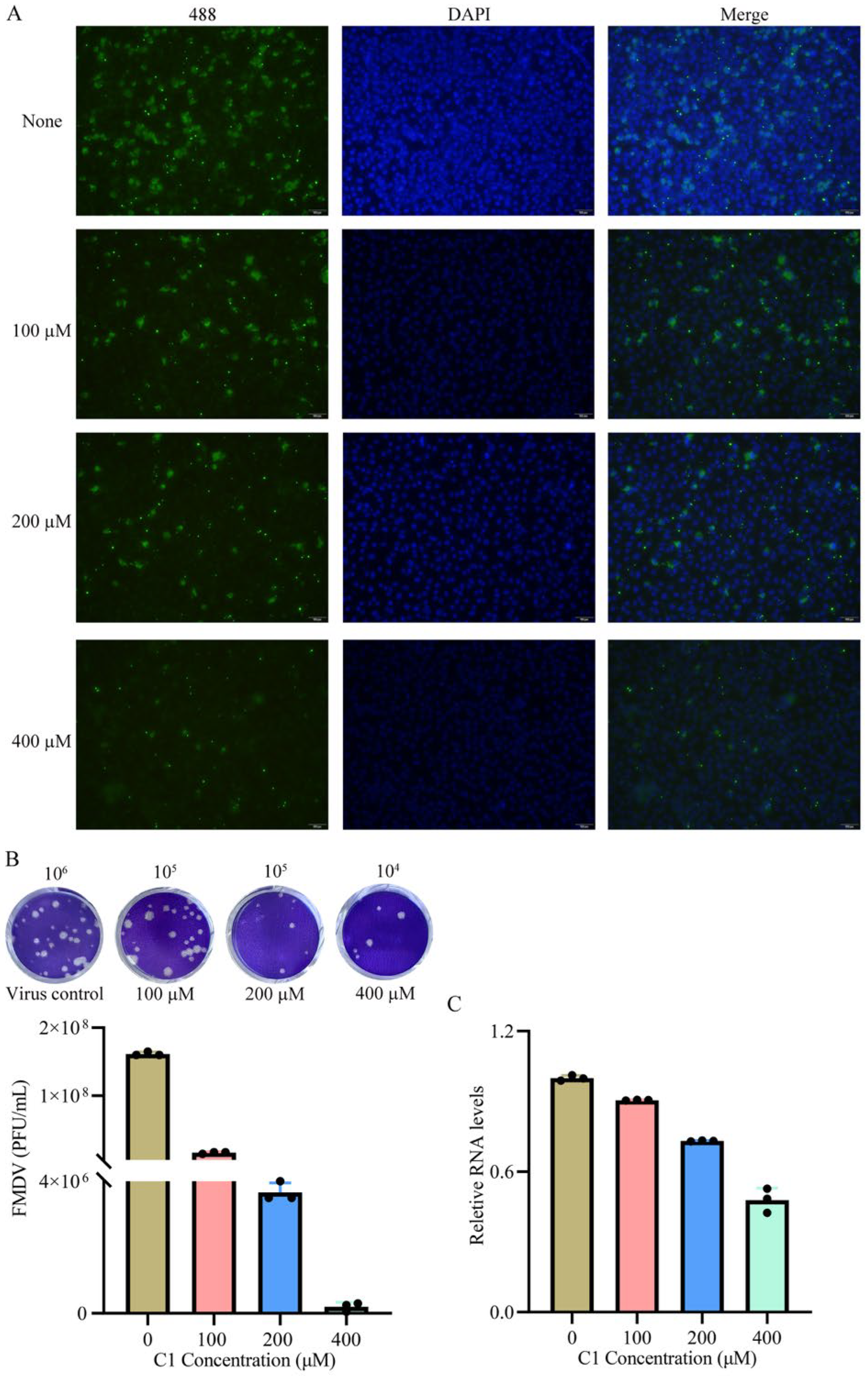
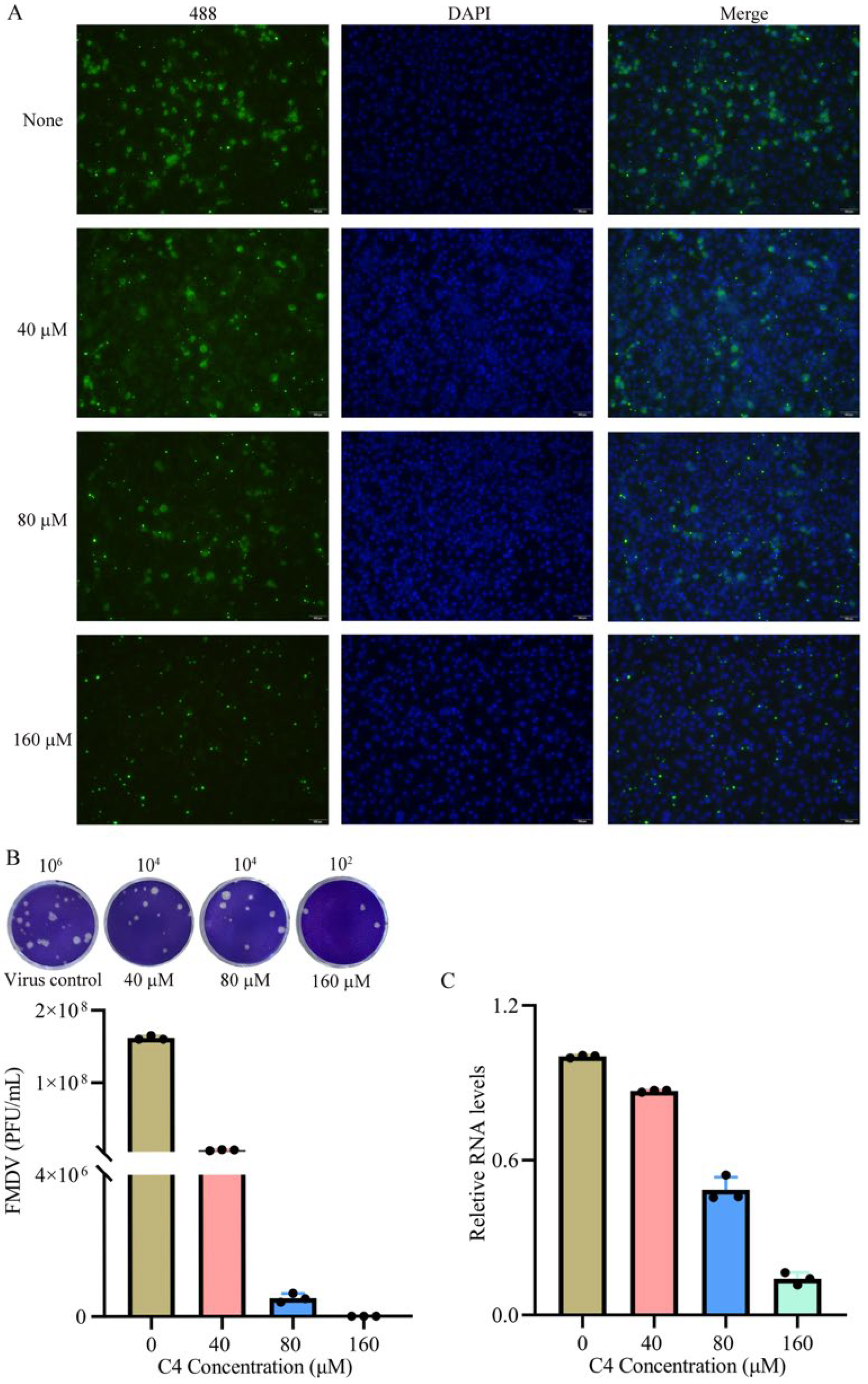
2.8. Viral Inhibition Assay
2.9. Indirect Immunofluorescence Assay
2.10. Plaque Assay
2.11. Real-Time qPCR
2.12. Statistical Analysis
3. Results
3.1. Optimization of 2C Helicase Enzymatic Activity
3.2. HTS for 2C Helicase Inhibitors
3.3. Analysis of 2C Protein-Ligand Interactions Based on NanoDSF
3.4. Enzyme Inhibition Kinetics Profiling
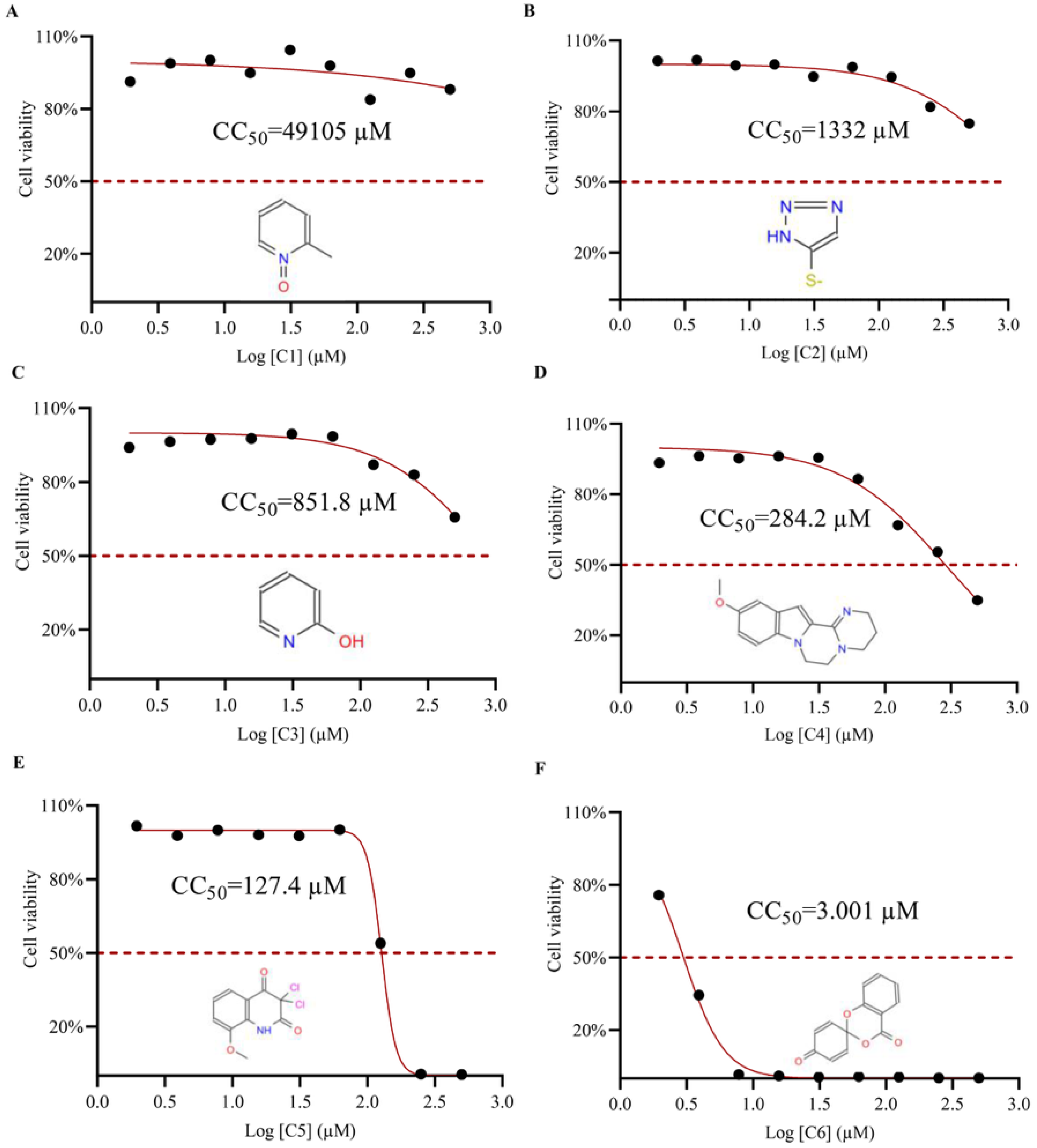
3.5. Determination of Half Cytotoxic Concentration (CC50) of Small Molecule Compounds
3.6. Antiviral Effect in Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| FMDV | Foot-and-mouth disease virus |
| FMD | Foot-and-mouth disease |
| FRET | Fluorescence resonance energy transfer |
| HTS | High-throughput screening |
| nanoDSF | Differential scanning fluorimetry |
| ORF | Open reading frame |
| NSP | Nonstructural protein |
| Arg-finger | Arginine finger |
| HAV | Hepatitis A virus |
| EV71 | Enterovirus 71 |
| CVB3/5 | Coxsackievirus B3/5 |
| PBL | Pocket-binding loop |
| PV | Poliovirus |
| EMCV | Encephalomyocarditis virus |
| IFA | Immunofluorescence |
| RT | Room temperature |
| DAPI | 4′,6-Diamidino-2-phenylindole |
| GuHCl | Guanidine hydrochloride |
References
- Arzt, J.; Sanderson, M.W.; Stenfeldt, C. Foot-and-Mouth Disease. Vet. Clin. North America. Food Anim. Pract. 2024, 40, 191–203. [Google Scholar] [CrossRef]
- Grubman, M.J.; Baxt, B. Foot-and-mouth disease. Clin. Microbiol. Rev. 2004, 17, 465–493. [Google Scholar] [CrossRef]
- Gao, Y.; Sun, S.Q.; Guo, H.C. Biological function of Foot-and-mouth disease virus non-structural proteins and non-coding elements. Virol. J. 2016, 13, 107. [Google Scholar] [CrossRef]
- Norder, H.; De Palma, A.M.; Selisko, B.; Costenaro, L.; Papageorgiou, N.; Arnan, C.; Coutard, B.; Lantez, V.; De Lamballerie, X.; Baronti, C.; et al. Picornavirus non-structural proteins as targets for new anti-virals with broad activity. Antivir. Res. 2011, 89, 204–218. [Google Scholar] [CrossRef]
- Sweeney, T.R.; Cisnetto, V.; Bose, D.; Bailey, M.; Wilson, J.R.; Zhang, X.; Belsham, G.J.; Curry, S. Foot-and-mouth disease virus 2C is a hexameric AAA+ protein with a coordinated ATP hydrolysis mechanism. J. Biol. Chem. 2010, 285, 24347–24359. [Google Scholar] [CrossRef]
- Hurdiss, D.L.; El Kazzi, P.; Bauer, L.; Papageorgiou, N.; Ferron, F.P.; Donselaar, T.; van Vliet, A.L.W.; Shamorkina, T.M.; Snijder, J.; Canard, B.; et al. Fluoxetine targets an allosteric site in the enterovirus 2C AAA+ ATPase and stabilizes a ring-shaped hexameric complex. Sci. Adv. 2022, 8, eabj7615. [Google Scholar] [CrossRef]
- Zhang, C.; Yang, F.; Wojdyla, J.A.; Qin, B.; Zhang, W.; Zheng, M.; Cao, W.; Wang, M.; Gao, X.; Zheng, H.; et al. An anti-picornaviral strategy based on the crystal structure of foot-and-mouth disease virus 2C protein. Cell Rep. 2022, 40, 111030. [Google Scholar] [CrossRef]
- Chen, P.; Wojdyla, J.A.; Colasanti, O.; Li, Z.; Qin, B.; Wang, M.; Lohmann, V.; Cui, S. Biochemical and structural characterization of hepatitis A virus 2C reveals an unusual ribonuclease activity on single-stranded RNA. Nucleic Acids Res. 2022, 50, 9470–9489. [Google Scholar] [CrossRef]
- Xia, H.; Wang, P.; Wang, G.C.; Yang, J.; Sun, X.; Wu, W.; Qiu, Y.; Shu, T.; Zhao, X.; Yin, L.; et al. Human Enterovirus Nonstructural Protein 2CATPase Functions as Both an RNA Helicase and ATP-Independent RNA Chaperone. PLoS Pathog. 2015, 11, e1005067. [Google Scholar] [CrossRef]
- Chen, Z.; Xiong, X.; Li, Y.; Huang, M.; Ren, Y.; Wu, D.; Qiu, Y.; Chen, M.; Shu, T.; Zhou, X. The nonstructural protein 2C of Coxsackie B virus has RNA helicase and chaperoning activities. Virol. Sin. 2022, 37, 656–663. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, N.; Tian, Y.; Pan, H.; Han, Y.; Li, Z.; Zhang, J.; Guan, S.; Chen, H.; Song, Y. Enzymatic characterization and dominant sites of foot-and-mouth disease virus 2C protein. Heliyon 2024, 10, e35449. [Google Scholar] [CrossRef]
- Guan, H.; Tian, J.; Qin, B.; Wojdyla, J.A.; Wang, B.; Zhao, Z.; Wang, M.; Cui, S. Crystal structure of 2C helicase from enterovirus 71. Sci. Adv. 2017, 3, e1602573. [Google Scholar] [CrossRef]
- Laufman, O.; Perrino, J.; Andino, R. Viral Generated Inter-Organelle Contacts Redirect Lipid Flux for Genome Replication. Cell 2019, 178, 275–289.e16. [Google Scholar] [CrossRef]
- Tang, J.; Abdullah, S.W.; Li, P.; Wu, J.; Pei, C.; Mu, S.; Wang, Y.; Sun, S.; Guo, H. Heat Shock Protein 60 Is Involved in Viral Replication Complex Formation and Facilitates Foot and Mouth Virus Replication by Stabilizing Viral Nonstructural Proteins 3A and 2C. mBio 2022, 13, e0143422. [Google Scholar] [CrossRef]
- Guan, H.; Tian, J.; Zhang, C.; Qin, B.; Cui, S. Crystal structure of a soluble fragment of poliovirus 2CATPase. PLoS Pathog. 2018, 14, e1007304. [Google Scholar] [CrossRef]
- He, Q.Y.; Zhao, H.F.; Meng, L.; Geng, Z.; Gao, Z.Q.; Qi, X.Y.; Dong, Y.H.; Zhang, H. A cardioviral 2C-ATP complex structure reveals the essential role of a conserved arginine in regulation of cardioviral 2C activity. J. Virol. 2024, 98, e0091124. [Google Scholar] [CrossRef]
- Lv, B.; Yuan, Y.; Yang, Z.; Wang, X.; Hu, J.; Sun, Y.; Du, H.; Liu, X.; Duan, H.; Ding, R.; et al. Stearoyl coenzyme A desaturase 1 (SCD1) regulates foot-and-mouth disease virus replication by modulating host cell lipid metabolism and viral protein 2C-mediated replication complex formation. J. Virol. 2024, 98, e0090224. [Google Scholar] [CrossRef]
- Cui, Z.; Liu, J.; Xie, C.; Wang, T.; Sun, P.; Wang, J.; Li, J.; Li, G.; Qiu, J.; Zhang, Y.; et al. High-throughput screening unveils nitazoxanide as a potent PRRSV inhibitor by targeting NMRAL1. Nat. Commun. 2024, 15, 4813. [Google Scholar] [CrossRef]
- Meyer, C.; Garzia, A.; Miller, M.W.; Huggins, D.J.; Myers, R.W.; Hoffmann, H.H.; Ashbrook, A.W.; Jannath, S.Y.; Liverton, N.; Kargman, S.; et al. Small-molecule inhibition of SARS-CoV-2 NSP14 RNA cap methyltransferase. Nature 2025, 637, 1178–1185. [Google Scholar] [CrossRef]
- Miao, J.; Yuan, H.; Rao, J.; Zou, J.; Yang, K.; Peng, G.; Cao, S.; Chen, H.; Song, Y. Identification of a small compound that specifically inhibits Zika virus in vitro and in vivo by targeting the NS2B-NS3 protease. Antivir. Res. 2022, 199, 105255. [Google Scholar] [CrossRef]
- Azzam, T.; Du, J.J.; Flowers, M.W.; Ali, A.V.; Hunn, J.C.; Vijayvargiya, N.; Knagaram, R.; Bogacz, M.; Maravillas, K.E.; Sastre, D.E.; et al. Combinatorially restricted computational design of protein-protein interfaces to produce IgG heterodimers. Sci. Adv. 2024, 10, eadk8157. [Google Scholar] [CrossRef] [PubMed]
- Bailly, M.; Mieczkowski, C.; Juan, V.; Metwally, E.; Tomazela, D.; Baker, J.; Uchida, M.; Kofman, E.; Raoufi, F.; Motlagh, S.; et al. Predicting Antibody Developability Profiles Through Early Stage Discovery Screening. mAbs 2020, 12, 1743053. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Zhang, Y.; Geng, B.; Tian, Y.; Tian, W.; Zou, Y.; Chen, H.; Chen, J. Fluorescence labeling-based differential scanning fluorimetry, an effective method for protein thermal stability and protein-compound binding analysis. Int. J. Biol. Macromol. 2024, 281 Pt 4, 136667. [Google Scholar] [CrossRef]
- Zeng, J.; Weissmann, F.; Bertolin, A.P.; Posse, V.; Canal, B.; Ulferts, R.; Wu, M.; Harvey, R.; Hussain, S.; Milligan, J.C.; et al. Identifying SARS-CoV-2 antiviral compounds by screening for small molecule inhibitors of nsp13 helicase. Biochem. J. 2021, 478, 2405–2423. [Google Scholar] [CrossRef] [PubMed]
- Jamal, S.M.; Belsham, G.J. Foot-and-mouth disease: Past, present and future. Vet. Res. 2013, 44, 116. [Google Scholar] [CrossRef]
- Ren, X.; Li, P.; Li, X.; Qian, P. Epidemiological and genetic characteristics of foot-and-mouth disease virus in China from 2010 to 2022. Virology 2024, 589, 109940. [Google Scholar] [CrossRef]
- Lu, Z.; Yu, S.; Wang, W.; Chen, W.; Wang, X.; Wu, K.; Li, X.; Fan, S.; Ding, H.; Yi, L.; et al. Development of Foot-and-Mouth Disease Vaccines in Recent Years. Vaccines 2022, 10, 1817. [Google Scholar] [CrossRef]
- Theerawatanasirikul, S.; Lueangaramkul, V.; Pantanam, A.; Mana, N.; Semkum, P.; Lekcharoensuk, P. Small Molecules Targeting 3C Protease Inhibit FMDV Replication and Exhibit Virucidal Effect in Cell-Based Assays. Viruses 2023, 15, 1887. [Google Scholar] [CrossRef]
- Kim, Y.; Pool, E.; Kim, E.; Dampalla, C.S.; Nguyen, H.N.; Johnson, D.K.; Lovell, S.; Groutas, W.C.; Chang, K.O. Potent small molecule inhibitors against the 3C protease of foot-and-mouth disease virus. Microbiol. Spectr. 2024, 12, e0337223. [Google Scholar] [CrossRef]
- Theerawatanasirikul, S.; Thangthamniyom, N.; Kuo, C.J.; Semkum, P.; Phecharat, N.; Chankeeree, P.; Lekcharoensuk, P. Natural Phytochemicals, Luteolin and Isoginkgetin, Inhibit 3C Protease and Infection of FMDV, In Silico and In Vitro. Viruses 2021, 13, 2118. [Google Scholar] [CrossRef]
- Mana, N.; Theerawatanasirikul, S.; Semkum, P.; Lekcharoensuk, P. Naturally Derived Terpenoids Targeting the 3D(pol) of Foot-and-Mouth Disease Virus: An Integrated In Silico and In Vitro Investigation. Viruses 2024, 16, 1128. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Paget, M.; Wang, C.; Zhu, Z.; Zheng, H. Innate immune evasion by picornaviruses. Eur. J. Immunol. 2020, 50, 1268–1282. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhou, X.; Wang, A.; Zheng, Y.; Gao, Y.; Zhou, J. Evolutions in fragment-based drug design: The deconstruction-reconstruction approach. Drug Discov. Today 2015, 20, 105–113. [Google Scholar] [CrossRef]
- Kirsch, P.; Hartman, A.M.; Hirsch, A.K.H.; Empting, M. Concepts and Core Principles of Fragment-Based Drug Design. Molecules 2019, 24, 4309. [Google Scholar] [CrossRef]
- Jhoti, H.; Williams, G.; Rees, D.C.; Murray, C.W. The ‘rule of three’ for fragment-based drug discovery: Where are we now? Nat. reviews. Drug Discov. 2013, 12, 644–645. [Google Scholar] [CrossRef]
- Pfister, T.; Wimmer, E. Characterization of the nucleoside triphosphatase activity of poliovirus protein 2C reveals a mechanism by which guanidine inhibits poliovirus replication. J. Biol. Chem. 1999, 274, 6992–7001. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Kitamura, N.; Musharrafieh, R.; Wang, J. Discovery of Potent and Broad-Spectrum Pyrazolopyridine-Containing Antivirals against Enteroviruses D68, A71, and Coxsackievirus B3 by Targeting the Viral 2C Protein. J. Med. Chem. 2021, 64, 8755–8774. [Google Scholar] [CrossRef]
- Xing, Y.; Zuo, J.; Krogstad, P.; Jung, M.E. Synthesis and Structure-Activity Relationship (SAR) Studies of Novel Pyrazolopyridine Derivatives as Inhibitors of Enterovirus Replication. J. Med. Chem. 2018, 61, 1688–1703. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound (Acronym) | Specs ID-Number | Name | Structure | InChIKeyTM | Molecular Formula |
|---|---|---|---|---|---|
| C1 (2-MPO) | AC-907/25014127 | 2-methylpyridine 1-oxide | 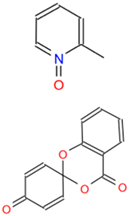 | CFZKDDTWZYUZKS-UHFFFAOYSA-N | C6H7NO |
| C2 (5-TzS−) | AO-090/25092001 | 1H-1,2,3-triazole-5-thiolate |  | LLCOQBODWBFTDD-UHFFFAOYSA-M | C2H2N3S− |
| C3 (2-PyOH) | AC-907/25014059 | pyridin-2-ol | 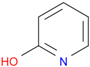 | UBQKCCHYAOITMY-UHFFFAOYSA-N | C5H5NO |
| C4 (MPPI) | AO-082/13829008 | 11-methoxy-3,4,6,7-tetrahydro-2H-pyrimido[2′,1′:3,4]pyrazino[1,2-a]indole |  | RPYFWWHSPMJDNM-UHFFFAOYSA-N | C15H17N3O |
| C5 (DCMQ) | AE-406/41056091 | 3,3-dichloro-8-methoxy-2,4(1H,3H)-quinolinedione | 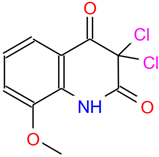 | QUECMMKDBHBZHZ-UHFFFAOYSA-N | C10H7Cl2NO3 |
| C6 (Spiro-BD-CHD-dione) | AN-970/40920757 | Spiro (4H-[1,3]benzodioxine-2,4′-[2,5]cyclohexadiene)-1′,4-dione |  | URIYZXDBDJZQMY-UHFFFAOYSA-N | C13H8O4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, S.; Mu, S.; Yu, S.; Tian, Y.; Lu, S.; Li, Z.; Wu, H.; Zhao, J.; Chen, H.; Sun, S.; et al. Discovery of Small Molecules Against Foot-and-Mouth Disease Virus Replication by Targeting 2C Helicase Activity. Viruses 2025, 17, 785. https://doi.org/10.3390/v17060785
Zhou S, Mu S, Yu S, Tian Y, Lu S, Li Z, Wu H, Zhao J, Chen H, Sun S, et al. Discovery of Small Molecules Against Foot-and-Mouth Disease Virus Replication by Targeting 2C Helicase Activity. Viruses. 2025; 17(6):785. https://doi.org/10.3390/v17060785
Chicago/Turabian StyleZhou, Saisai, Suyu Mu, Shuqi Yu, Yang Tian, Sijia Lu, Zhen Li, Hao Wu, Jiaying Zhao, Huanchun Chen, Shiqi Sun, and et al. 2025. "Discovery of Small Molecules Against Foot-and-Mouth Disease Virus Replication by Targeting 2C Helicase Activity" Viruses 17, no. 6: 785. https://doi.org/10.3390/v17060785
APA StyleZhou, S., Mu, S., Yu, S., Tian, Y., Lu, S., Li, Z., Wu, H., Zhao, J., Chen, H., Sun, S., & Song, Y. (2025). Discovery of Small Molecules Against Foot-and-Mouth Disease Virus Replication by Targeting 2C Helicase Activity. Viruses, 17(6), 785. https://doi.org/10.3390/v17060785