
Multiple Lineages of Hantaviruses Harbored by the Iberian Mole (Talpa occidentalis) in Spain
 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Specimens
2.2. RNA Extraction and RT-PCR Analysis
2.3. Next-Generation Sequencing
2.4. Genetic and Phylogenetic Analyses
2.5. Host Identification and Phylogeny
3. Results
3.1. Hantavirus RNA Detection
3.2. Sequence Analysis
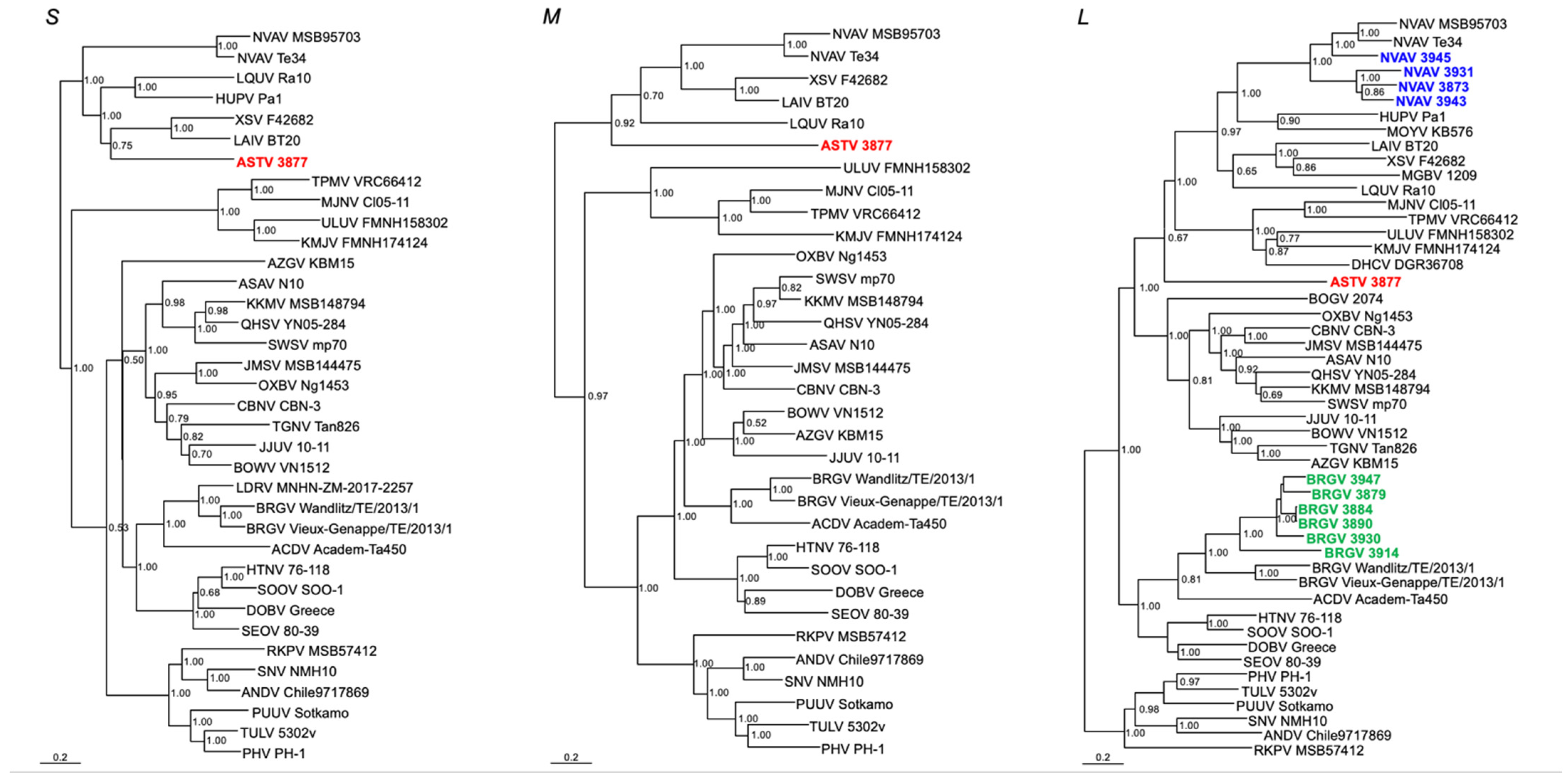
3.3. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, H.W.; Lee, P.W.; Johnson, K.M. Isolation of the etiologic agent of Korean hemorrhagic fever. J. Infect. Dis. 1978, 137, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Brummer-Korvenkontio, M.; Vaheri, A.; Hovi, T.; von Bonsdorff, C.H.; Vuorimies, J.; Manni, T.; Penttinen, K.; Oker-Blom, N.; Lähdevirta, J. Nephropathia epidemica: Detection of antigen in bank voles and serologic diagnosis of human infection. J. Infect. Dis. 1980, 141, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Baek, L.J.; Johnson, K.M. Isolation of Hantaan virus, the etiologic agent of Korean hemorrhagic fever, from wild urban rats. J. Infect. Dis. 1982, 146, 638–644. [Google Scholar] [CrossRef]
- Nichol, S.T.; Spiropoulou, C.F.; Morzunov, S.; Rollin, P.E.; Ksiazek, T.G.; Feldmann, H.; Sanchez, A.; Childs, J.; Zaki, S.; Peters, C.J. Genetic identification of a hantavirus associated with an outbreak of acute respiratory illness. Science 1993, 262, 914–917. [Google Scholar] [CrossRef]
- Duchin, J.S.; Koster, F.T.; Peters, C.J.; Simpson, G.L.; Tempest, B.; Zaki, S.R.; Ksiazek, T.G.; Rollin, P.E.; Nichol, S.; Umland, E.T.; et al. Hantavirus pulmonary syndrome: A clinical description of 17 patients with a newly recognized disease. N. Engl. J. Med. 1994, 330, 949–955. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Reported Cases of Hantavirus Pulmonary Syndrome in the United States. Available online: https://www.cdc.gov/hantavirus/surveillance/index.html#Infection (accessed on 17 April 2023).
- Alonso, D.O.; Iglesias, A.; Coelho, R.; Periolo, N.; Bruno, A.; Córdoba, M.T.; Filomarino, N.; Quipildor, M.; Biondo, E.; Fortunato, E.; et al. Epidemiological description, case-fatality rate, and trends of hantavirus pulmonary syndrome: 9 years of surveillance in Argentina. J. Med. Virol. 2019, 91, 1173–1181. [Google Scholar] [CrossRef]
- Plyusnin, A.; Vapalahti, O.; Vaheri, A. Hantaviruses: Genome structure, expression and evolution. J. Gen. Virol. 1996, 77, 2677–2687. [Google Scholar] [CrossRef] [PubMed]
- Khaiboullina, S.F.; Morzunov, S.P.; Jeor, S.C.S. Hantaviruses: Molecular biology, evolution and pathogenesis. Curr. Mol. Med. 2005, 5, 773–790. [Google Scholar] [CrossRef]
- Maes, P.; Adkins, S.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, E.; Blair, C.D.; Briese, T.; et al. Taxonomy of the order Bunyavirales: Second update 2018. Arch. Virol. 2019, 164, 927–941. [Google Scholar] [CrossRef]
- Laenen, L.; Vergote, V.; Calisher, C.H.; Klempa, B.; Klingström, J.; Kuhn, J.H.; Maes, P. Hantaviridae: Current classification and future perspectives. Viruses 2019, 11, 788. [Google Scholar] [CrossRef]
- Yanagihara, R.; Gu, S.H.; Arai, S.; Kang, H.J.; Song, J.-W. Hantaviruses: Rediscovery and new beginnings. Virus Res. 2014, 187, 6–14. [Google Scholar] [CrossRef]
- Arai, S.; Yanagihara, R. Genetic diversity and geographic distribution of bat-borne hantaviruses. Curr. Issues Mol. Biol. 2020, 39, 1–28. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Schmaljohn, C.S. A brief history of Bunyaviral family Hantaviridae. Diseases 2023, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Ohdachi, S.D.; Asakawa, M.; Kang, H.J.; Mocz, G.; Arikawa, J.; Okabe, N.; Yanagihara, R. Molecular phylogeny of a newfound hantavirus in the Japanese shrew mole (Urotrichus talpoides). Proc. Natl. Acad. Sci. USA 2008, 105, 16296–16301. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Bennett, S.N.; Dizney, L.; Sumibcay, L.; Arai, S.; Ruedas, L.A.; Song, J.W.; Yanagihara, R. Host switch during evolution of a genetically distinct hantavirus in the American shrew mole (Neurotrichus gibbsii). Virology 2009, 388, 8–14. [Google Scholar] [CrossRef]
- Kang, H.J.; Bennett, S.N.; Hope, A.G.; Cook, J.A.; Yanagihara, R. Shared ancestry between a newfound mole-borne hantavirus and hantaviruses harbored by cricetid rodents. J. Virol. 2011, 85, 7496–7503. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Bennett, S.N.; Sumibcay, L.; Arai, S.; Hope, A.G.; Mocz, G.; Song, J.-W.; Cook, J.A.; Yanagihara, R. Evolutionary insights from a genetically divergent hantavirus harbored by the European common mole (Talpa europaea). PLoS ONE 2009, 4, e6149. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.H.; Dormion, J.; Hugot, J.-P.; Yanagihara, R. High prevalence of Nova hantavirus infection in the European mole (Talpa europaea) in France. Epidemiol. Infect. 2014, 142, 1167–1171. [Google Scholar] [CrossRef]
- Gu, S.H.; Hejduk, J.; Markowski, J.; Kang, H.J.; Markowski, M.; Połatyńska, M.; Sikorska, B.; Liberski, P.P.; Yanagihara, R. Co-circulation of soricid- and talpid-borne hantaviruses in Poland. Infect. Genet. Evol. 2014, 28, 296–303. [Google Scholar] [CrossRef]
- Laenen, L.; Dellicour, S.; Vergote, V.; Nauwelaers, I.; De Coster, S.; Verbeeck, I.; Vanmechelen, B.; Lemey, P.; Maes, P. Spatio-temporal analysis of Nova virus, a divergent hantavirus circulating in the European mole in Belgium. Mol. Ecol. 2016, 25, 5994–6008. [Google Scholar] [CrossRef]
- Kang, H.J.; Gu, S.H.; Cook, J.A.; Yanagihara, R. Dahonggou Creek virus, a divergent lineage of hantavirus harbored by the long-tailed mole (Scaptonyx fusicaudus). Trop. Med. Health 2016, 44, 16. [Google Scholar] [CrossRef] [PubMed]
- Laenen, L.; Vergote, V.; Kafetzopoulou, L.E.; Wawina, T.B.; Vassou, D.; Cook, J.A.; Hugot, J.P.; Deboutte, W.; Kang, H.J.; Witkowski, P.T.; et al. A novel hantavirus of the European mole, Bruges virus, is involved in frequent Nova virus coinfections. Genome Biol. Evol. 2018, 10, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Yashina, L.N.; Panov, V.V.; Abramov, S.A.; Smetannikova, N.A.; Luchnikova, E.M.; Dupal, T.A.; Krivopalov, A.V.; Arai, S.; Yanagihara, R. Academ virus, a novel hantavirus in the Siberian mole (Talpa altaica) from Russia. Viruses 2022, 14, 309. [Google Scholar] [CrossRef]
- Hugot, J.-P.; Vanmechelen, B.; Maes, P. Landiras virus, a novel hantavirus hosted by Talpa aquitania n. sp., a recently discovered south European mole species. Bull. Acad. Vet. Fr. 2023, 176. [Google Scholar] [CrossRef]
- Miñarro, M.; Montiel, C.; Dapena, E. Vole pests in apple orchards: Use of presence signs to estimate the abundance of Arvicola terrestris cantabriae and Microtus lusitanicus. J. Pest Sci. 2014, 85, 477–488. [Google Scholar] [CrossRef]
- Klempa, B.; Fichet-Calvet, E.; Lecompte, E.; Auste, B.; Aniskin, V.; Meisel, H.; Denys, C.; Koivogui, L.; ter Meulen, J.; Krüger, D.H. Hantavirus in African wood mouse, Guinea. Emerg. Infect. Dis. 2006, 12, 838–840. [Google Scholar] [CrossRef] [PubMed]
- Vasilakis, N.; Forrester, N.L.; Palacios, G.; Nasar, F.; Savji, N.; Rossi, S.L.; Guzman, H.; Wood, T.G.; Popov, V.; Gorchakov, R.; et al. Negevirus: A proposed new taxon of insect-specific viruses with wide geographic distribution. J. Virol. 2013, 87, 2475–2488. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, D.; Marciel de Souza, W.; Silvas, J.A.; Deardorff, E.R.; Widen, S.G.; Estrada-Franco, J.G.; Weaver, S.C.; Nunes, M.; Aguilar, P.V. Barrita virus, a novel virus of the Patois serogroup (Genus Orthobunyavirus; Family Peribunyaviridae). Am. J. Trop. Med. Hyg. 2020, 103, 190–192. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web-servers. Syst. Biol. 2008, 75, 758–771. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. MODELTEST: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.M.; Kocher, T.D.; Wilson, A.C. Evolution of the cytochrome b gene of mammals. J. Mol. Evol. 1991, 32, 128–144. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Song, J.-W.; Gu, S.H.; Bennett, S.N.; Arai, S.; Puorger, M.; Hilbe, M.; Yanagihara, R. Seewis virus, a genetically distinct hantavirus in the Eurasian common shrew (Sorex araneus). Virol. J. 2007, 4, 114. [Google Scholar] [CrossRef]
- Schlegel, M.; Radosa, L.; Rosenfeld, U.M.; Schmidt, S.; Triebenbacher, C.; Löhr, P.W.; Fuchs, D.; Heroldová, M.; Jánová, E.; Stanko, M.; et al. Broad geographical distribution and high genetic diversity of shrew-borne Seewis hantavirus in Central Europe. Virus Genes 2012, 45, 48–55. [Google Scholar] [CrossRef]
- Yashina, L.N.; Abramov, S.A.; Gutorov, V.V.; Dupal, T.A.; Krivopalov, A.V.; Panov, V.V.; Danchinova, G.; Vinogradov, V.; Luchnikova, E.; Hay, J.; et al. Seewis virus: Phylogeography of a shrew-borne hantavirus in Siberia, Russia. Vector Borne Zoonotic Dis. 2010, 10, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Kang, H.J.; Gu, S.H.; Ohdachi, S.D.; Cook, J.A.; Yashina, L.N.; Tanaka-Taya, K.; Abramov, S.A.; Morikawa, S.; Okabe, N.; et al. Genetic diversity of Artybash virus in the Laxmann’s shrew (Sorex caecutiens). Vector Borne Zoonotic Dis. 2016, 16, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Yanagihara, R.; Gu, S.H.; Song, J.-W. Expanded host diversity and global distribution of hantaviruses: Implications for identifying and investigating previously unrecognized hantaviral diseases. In Global Virology—Identifying and Investigating Viral Diseases; Shapshak, P., Sinnott, J.T., Somboonwit, C., Kuhn, J., Eds.; Springer Publishing Company: New York, NY, USA, 2015; pp. 161–198. [Google Scholar]
- Palma, R.E.; Polop, J.J.; Owen, R.D.; Mills, J.N. Ecology of rodent-associated hantaviruses in the Southern Cone of South America: Argentina, Chile, Paraguay, and Uruguay. J. Wildl. Dis. 2012, 48, 267–281. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.J.; Gu, S.H.; Yashina, L.N.; Cook, J.A.; Yanagihara, R. Highly divergent genetic variants of soricid-borne Altai virus (Hantaviridae) in Eurasia suggest ancient host-switching events. Viruses 2019, 11, 857. [Google Scholar] [CrossRef] [PubMed]
- Yashina, L.N.; Abramov, S.A.; Zhigalin, A.V.; Smetannikova, N.A.; Dupal, T.A.; Krivopalov, A.V.; Kikuchi, F.; Senoo, K.; Arai, S.; Mizutani, T.; et al. Geographic distribution and phylogeny of soricine shrew-borne Seewis virus and Altai virus in Russia. Viruses 2021, 13, 1286. [Google Scholar] [CrossRef] [PubMed]
- Jalal, S.; Kim, C.M.; Kim, D.M.; Song, H.J.; Lee, J.C.; Shin, M.Y.; Lim, H.C. Geographical clustering of hantavirus isolates from Apodemus agrarius identified in the Republic of Korea indicate the emergence of a new hantavirus genotype. J. Clin. Virol. 2022, 146, 105030. [Google Scholar] [CrossRef] [PubMed]
- Nemirov, K.; Vapalahti, O.; Lundkvist, A.; Vasilen Golovljova, I.; Plyusnina, A.; Niemmimaa, J. Isolation and characterization of Dobrava hantavirus in the striped field mouse (Apodemus agrarius) in Estonia. J. Gen. Virol. 1999, 80, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Plyusnin, A.; Nemirov, K.; Apekina, N.; Plyusnina, A.; Lundkvist, A.; Vaheri, A. Dobrava hantavirus in Russia. Lancet 1999, 353, 207. [Google Scholar] [CrossRef]
- Scharninghausen, J.J.; Meyer, H.; Pfeffer, M.; Davis, D.S.; Honeycutt, R.L. Genetic evidence of Dobrava virus in Apodemus agrarius in Hungary. Emerg. Infect. Dis. 1999, 5, 468–470. [Google Scholar] [CrossRef]
- Chu, Y.K.; Owen, R.D.; Gonzalez, L.M.; Jonsson, C.B. The complex ecology of hantavirus in Paraguay. Am. J. Trop. Med. Hyg. 2003, 69, 263–268. [Google Scholar] [CrossRef]
- Chu, Y.K.; Goodin, D.; Owen, R.D.; Koch, D.; Jonsson, C.B. Sympatry of 2 hantavirus strains, Paraguay, 2003–2007. Emerg. Infect. Dis. 2009, 15, 1977–1980. [Google Scholar] [CrossRef]
- Chu, Y.K.; Owen, R.D.; Jonsson, C.B. Phylogenetic exploration of hantaviruses in Paraguay reveals reassortment and host switching in South America. Virol. J. 2011, 8, 399. [Google Scholar] [CrossRef]
- Klempa, B. Reassortment events in the evolution of hantaviruses. Virus Genes 2018, 54, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Liphardt, S.W.; Kang, H.J.; Arai, S.; Gu, S.H.; Cook, J.A.; Yanagihara, R. Reassortment between divergent strains of Camp Ripley virus (Hantaviridae) in the northern short-tailed shrew (Blarina brevicauda). Front. Cell Infect. Microbiol. 2020, 10, 460. [Google Scholar] [CrossRef] [PubMed]
- Feuda, R.; Bannikova, A.A.; Zemlemerova, E.D.; Di Febbraro, M.; Loy, A.; Hutterer, R.; Aloise, G.; Zykov, A.E.; Annesi, F.; Colangelo, P. Tracing the evolutionary history of the mole, Talpa europaea, through mitochondrial DNA phylogeography and species distribution modelling. Biol. J. Linn. Soc. 2015, 114, 495–512. [Google Scholar] [CrossRef]
- Nicolas, V.; Martinez-Vargas, J.; Hugot, J.-P. Talpa aquitania nov. sp. (Talpidae, Soricomorpha) a new mole species from southwest France and north Spain. Bull. Acad. Vet. Fr. 2015, 168, 329–334. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Bradfute, S.B.; Calisher, C.H.; Klempa, B.; Klingström, J.; Laenen, L.; Palacios, G.; Schmaljohn, C.S.; Tischler, N.D.; Maes, P. Pending reorganization of Hantaviridae to include only completely sequenced viruses: A call to action. Viruses 2023, 15, 660. [Google Scholar] [CrossRef] [PubMed]
- Quizon, K.; Holloway, K.; Iranpour, M.; Warner, B.M.; Deschambault, Y.; Soule, G.; Tierney, K.; Kobasa, D.; Sloan, A.; Safronetz, D. Experimental infection of Peromyscus species rodents with Sin Nombre virus. Emerg. Infect. Dis. 2022, 28, 1882–1885. [Google Scholar] [CrossRef]
- Goodfellow, S.M.; Nofchissey, R.A.; Schwalm, K.C.; Cook, J.A.; Dunnum, J.L.; Guo, Y.; Ye, C.; Mertz, G.J.; Chandran, K.; Harkins, M.; et al. Tracing transmission of Sin Nombre virus and discovery of infection in multiple rodent species. J. Virol. 2021, 95, e0153421. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Collection Site | Year | Talpa occidentalis | Crocidura russula | Sorex coronatus | Microtus lusitanicus | Arvicola scherman |
|---|---|---|---|---|---|---|
| Coceña | 2011 | 3/11 | 0/2 | 0/2 | ||
| 2012 | 0/1 | |||||
| Fresnadiellu | 2012 | 2/9 | ||||
| 2013 | 0/6 | |||||
| Fresnu | 2012 | 0/2 | 1/5 | 0/3 | 0/1 | |
| Oles | 2011 | 1/15 | 0/2 | |||
| 2012 | 1/3 | |||||
| 2013 | 1/2 | 0/1 | 0/4 | |||
| 2014 | 0/1 | |||||
| Priesca | 2011 | 3/13 | 0/1 | |||
| 2012 | 0/2 | 0/2 | ||||
| 2013 | 0/1 | 0/1 | ||||
| 2014 | 0/3 | |||||
| VEV (Villaviciosa) | 2011 | 0/2 | 1/4 |
| Virus | Collection Site | Nucleotides and GenBank Accession Numbers | ||
|---|---|---|---|---|
| S Segment | M Segment | L Segment | ||
| ASTV 3877 | Coceña | 1979 bp KY040518 | 1971 bp KY040519 | 1369 bp KY040520 |
| BRGV 3879 | Coceña | 1327 bp KY040512 | ||
| BRGV 3884 | Fresnadiellu | 1327 bp KY040513 | ||
| BRGV 3890 | Fresnadiellu | 553 bp KY040514 | ||
| BRGV 3914 | Oles | 553 bp KY040515 | ||
| BRGV 3930 | Oles | 553 bp KY040516 | ||
| BRGV 3947 | Priesca | 1327 bp KY040517 | ||
| NVAV 3873 | Coceña | 693 bp KY040508 | ||
| NVAV 3931 | Oles | 352 bp KY040509 | ||
| NVAV 3943 | Priesca | 352 bp KY040510 | ||
| NVAV 3945 | Priesca | 977 bp KY040511 | ||
| S Segment | M Segment | L Segment | |||||
|---|---|---|---|---|---|---|---|
| Host | Hantavirus | 1979 nt | 429 aa | 1971 nt | 657 aa | 1369 nt | 456 aa |
| Mole | ACDV Academ-Ta450 | 54.4 | 52.2 | 60.2 | 52.1 | 69.1 | 71.3 |
| ASAV N10 | 49.5 | 48.7 | 59.5 | 49.0 | 69.2 | 71.3 | |
| BRGV 3879 | - | - | - | - | 67.7 | 69.5 | |
| BRGV 3884 | - | - | - | - | 67.2 | 69.9 | |
| BRGV 3890 | - | - | - | - | 69.8 | 73.9 | |
| BRGV 3914 | - | - | - | - | 80.5 | 82.1 | |
| BRGV 3930 | - | - | - | - | 70.2 | 73.9 | |
| BRGV 3947 | - | - | - | - | 69.0 | 70.8 | |
| BRGV Vieux-Genappe/TE/2013/1 | 52.0 | 51.3 | 59.3 | 49.8 | 67.8 | 69.5 | |
| BRGV Wandlitz/TE/2013/1 | 52.6 | 51.0 | 59.6 | 49.8 | 66.3 | 68.9 | |
| DHCV DGR36708 | - | - | - | - | 66.0 | 66.5 | |
| LDRV MNHN-ZM-2017-2257 | 50.1 | 50.8 | - | - | - | - | |
| NVAV 3873 | - | - | - | - | 39.4 | 44.2 | |
| NVAV 3931 | - | - | - | - | 69.9 | 68.4 | |
| NVAV 3943 | - | - | - | - | 70.7 | 69.2 | |
| NVAV 3945 | - | - | - | - | 64.8 | 66.2 | |
| NVAV MSB95703 | 61.3 | 56.3 | 60.6 | 53.3 | 67.1 | 69.3 | |
| NVAV Te34 | 62.1 | 56.1 | 62.5 | 55.3 | 67.5 | 68.9 | |
| OXBV Ng1453 | 59.6 | 49.1 | 59.3 | 47.6 | 69.2 | 68.9 | |
| RKPV MSB57412 | 60.4 | 53.0 | 58.9 | 49.0 | 67.3 | 69.1 | |
| Shrew | MJNV Cl05-11 | 51.4 | 45.7 | 59.8 | 48.4 | 66.1 | 68.6 |
| TPMV VRC66412 | 47.9 | 48.0 | 59.2 | 49.2 | 67.2 | 69.3 | |
| ULUV FMNH158302 | 61.8 | 50.9 | 57.4 | 37.7 | 66.8 | 67.1 | |
| KMJV FMNH174124 | 59.6 | 51.9 | 57.7 | 40.2 | 67.8 | 69.3 | |
| BOGV 2074 | - | - | - | - | 68.5 | 71.9 | |
| JMSV MSB144475 | 49.6 | 47.6 | 58.6 | 50.4 | 67.9 | 69.3 | |
| KKMV MSB148794 | 50.7 | 50.8 | 53.1 | 46.1 | 67.6 | 71.3 | |
| SWSV mp70 | 51.0 | 50.3 | 61.4 | 57.8 | 68.0 | 70.0 | |
| SWSV 4050 | - | - | - | - | 67.1 | 73.0 | |
| SWSV 4056 | - | - | - | - | 67.9 | 73.0 | |
| QHSV YN05-284 | 49.3 | 50.6 | 60.5 | 49.2 | 60.0 | 62.4 | |
| CBNV CBN-3 | 50.6 | 50.2 | 61.1 | 50.5 | 68.2 | 70.6 | |
| JJUV 10-11 | 52.5 | 48.4 | 60.9 | 49.0 | 66.5 | 67.5 | |
| TGNV Tan826 | 30.4 | 46.3 | - | - | 59.6 | 58.4 | |
| AZGV KBM15 | 30.2 | 48.3 | 55.7 | 45.9 | 67.4 | 67.5 | |
| BOWV VN1512 | 60.1 | 48.7 | 59.4 | 47.5 | 67.9 | 68.2 | |
| Bat | XSV F42682 | 62.6 | 56.2 | 59.8 | 53.8 | 47.2 | 50.3 |
| LAIV BT20 | 63.5 | 58.1 | 62.2 | 56.0 | 68.3 | 72.6 | |
| LQUV Ra10 | 63.3 | 54.4 | 60.3 | 47.6 | 69.1 | 71.3 | |
| HUPV Pa1 | 62.5 | 56.1 | - | - | 66.5 | 74.6 | |
| MOYV KB576 | - | - | - | - | 67.6 | 71.1 | |
| MGBV 1209 | - | - | - | - | 62.6 | 61.3 | |
| Rodent | HTNV 76-118 | 53.5 | 53.1 | 57.7 | 51.8 | 67.0 | 67.5 |
| SEOV 80-39 | 49.5 | 51.5 | 59.7 | 51.6 | 67.1 | 70.6 | |
| SOOV SOO-1 | 54.2 | 53.4 | 59.3 | 51.0 | 67.6 | 68.2 | |
| DOBV Greece | 50.9 | 53.1 | 60.5 | 51.9 | 68.3 | 70.8 | |
| ANDV Chile9717869 | 59.1 | 53.3 | 60.1 | 49.9 | 66.0 | 66.0 | |
| SNV NMH10 | 58.0 | 53.5 | 59.5 | 49.6 | 66.4 | 67.8 | |
| PUUV Sotkamo | 61.5 | 52.2 | 59.8 | 50.8 | 67.6 | 68.0 | |
| TULV 5302v | 58.1 | 53.1 | 61.0 | 49.8 | 67.7 | 66.2 | |
| PHV PH-1 | 61.9 | 51.5 | 59.5 | 49.5 | 65.4 | 66.7 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gu, S.H.; Miñarro, M.; Feliu, C.; Hugot, J.-P.; Forrester, N.L.; Weaver, S.C.; Yanagihara, R. Multiple Lineages of Hantaviruses Harbored by the Iberian Mole (Talpa occidentalis) in Spain. Viruses 2023, 15, 1313. https://doi.org/10.3390/v15061313
Gu SH, Miñarro M, Feliu C, Hugot J-P, Forrester NL, Weaver SC, Yanagihara R. Multiple Lineages of Hantaviruses Harbored by the Iberian Mole (Talpa occidentalis) in Spain. Viruses. 2023; 15(6):1313. https://doi.org/10.3390/v15061313
Chicago/Turabian StyleGu, Se Hun, Marcos Miñarro, Carlos Feliu, Jean-Pierre Hugot, Naomi L. Forrester, Scott C. Weaver, and Richard Yanagihara. 2023. "Multiple Lineages of Hantaviruses Harbored by the Iberian Mole (Talpa occidentalis) in Spain" Viruses 15, no. 6: 1313. https://doi.org/10.3390/v15061313
APA StyleGu, S. H., Miñarro, M., Feliu, C., Hugot, J.-P., Forrester, N. L., Weaver, S. C., & Yanagihara, R. (2023). Multiple Lineages of Hantaviruses Harbored by the Iberian Mole (Talpa occidentalis) in Spain. Viruses, 15(6), 1313. https://doi.org/10.3390/v15061313