
Hallmarks of Metabolic Reprogramming and Their Role in Viral Pathogenesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
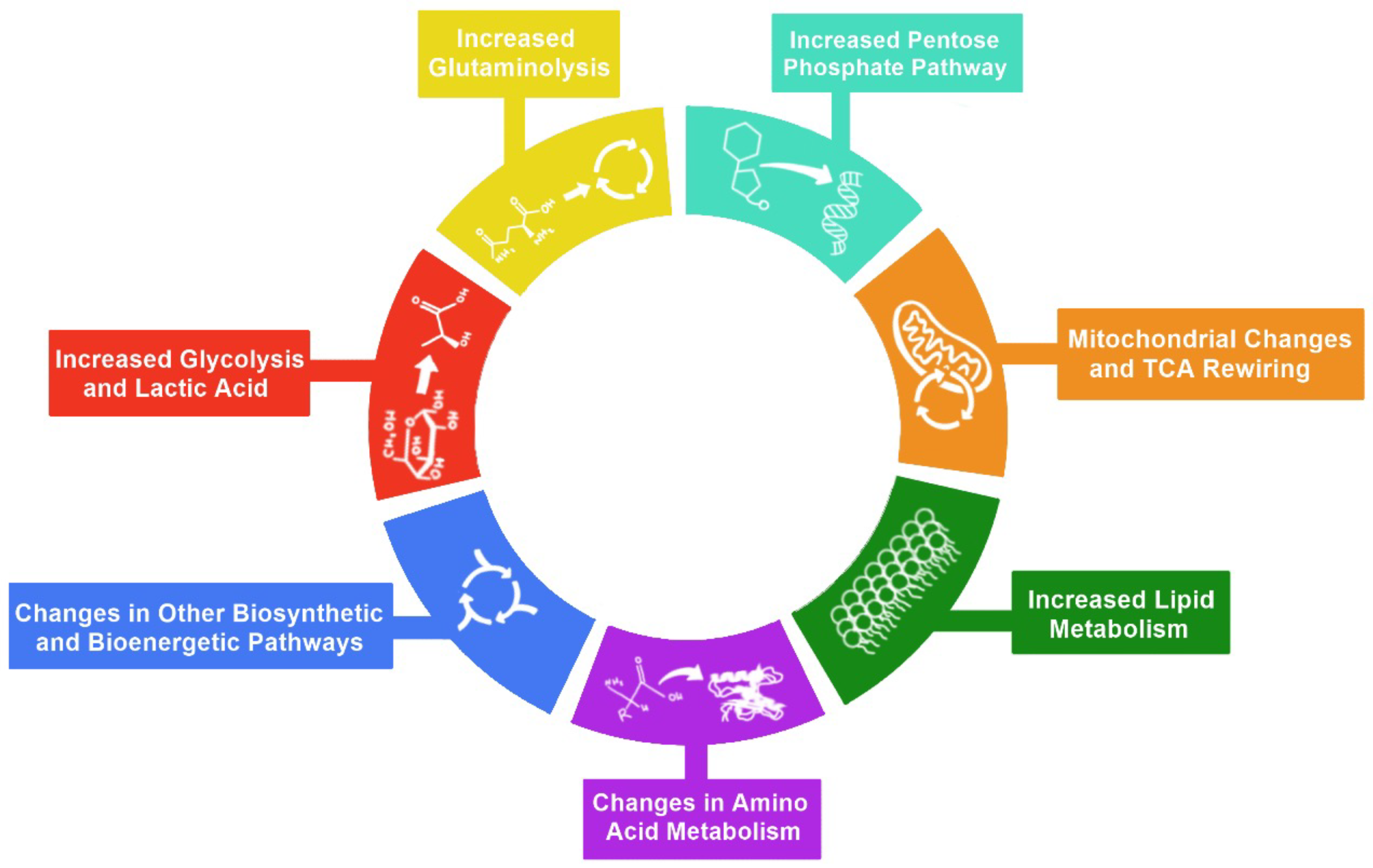
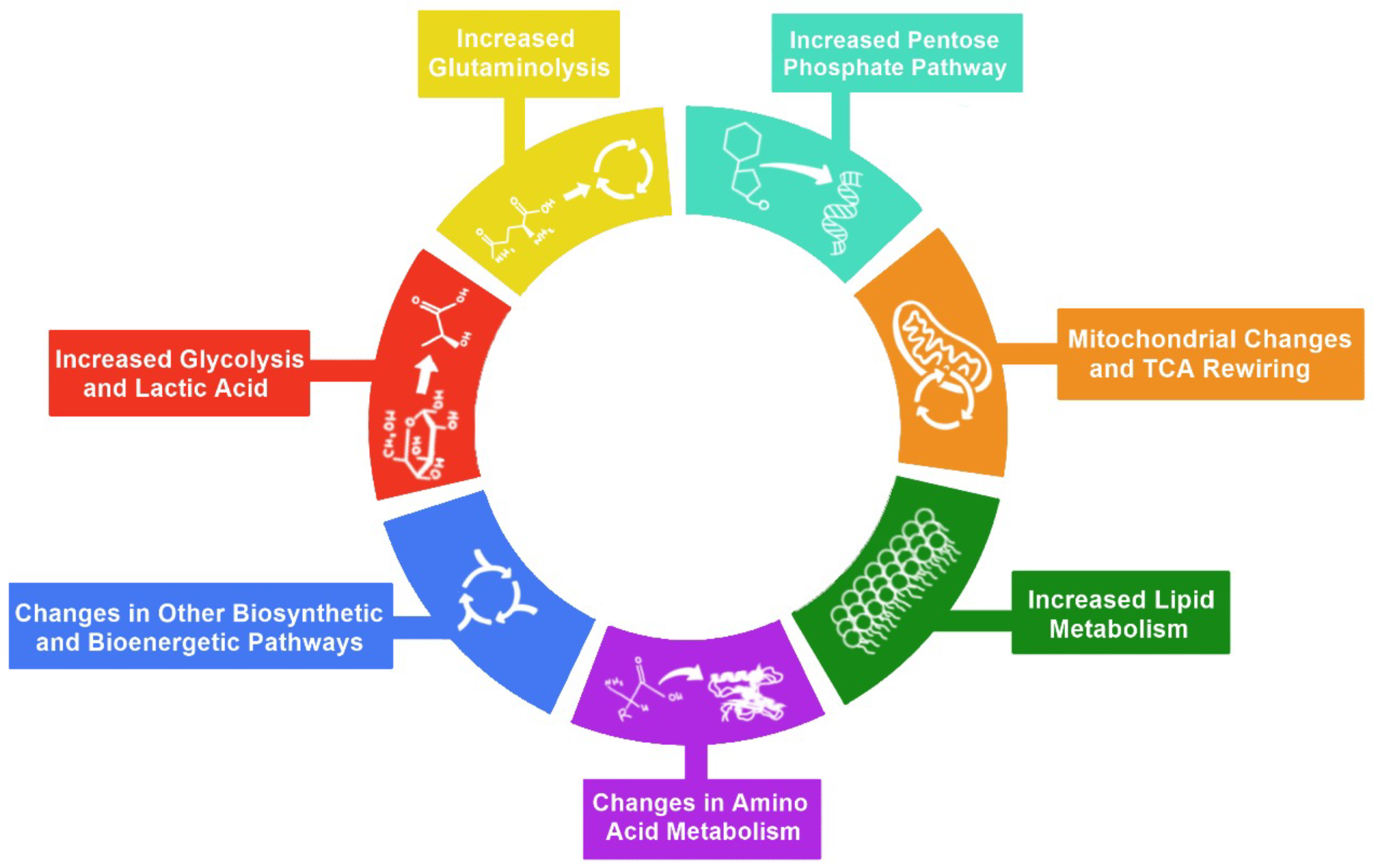
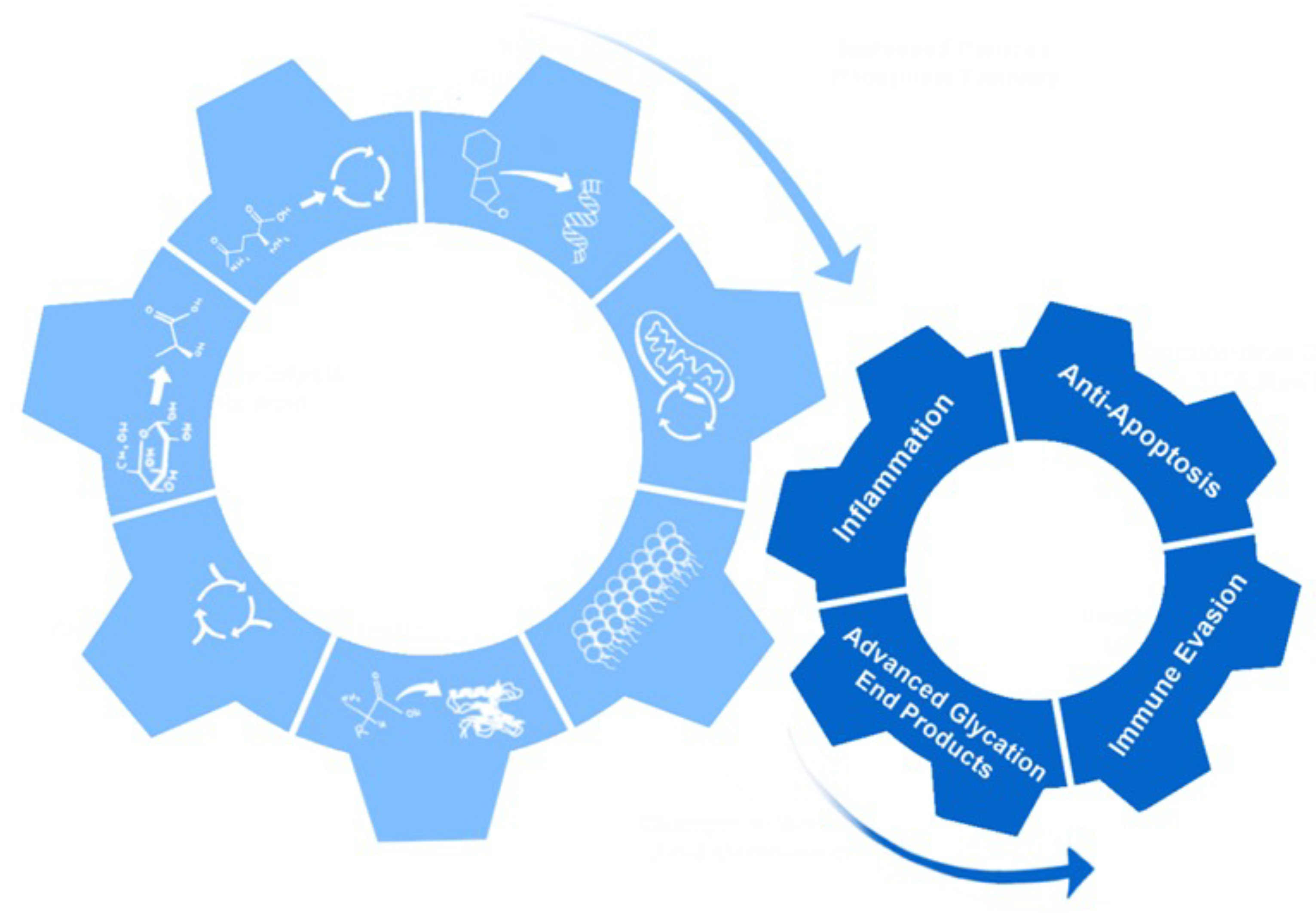
2. Hallmarks of Metabolic Reprogramming
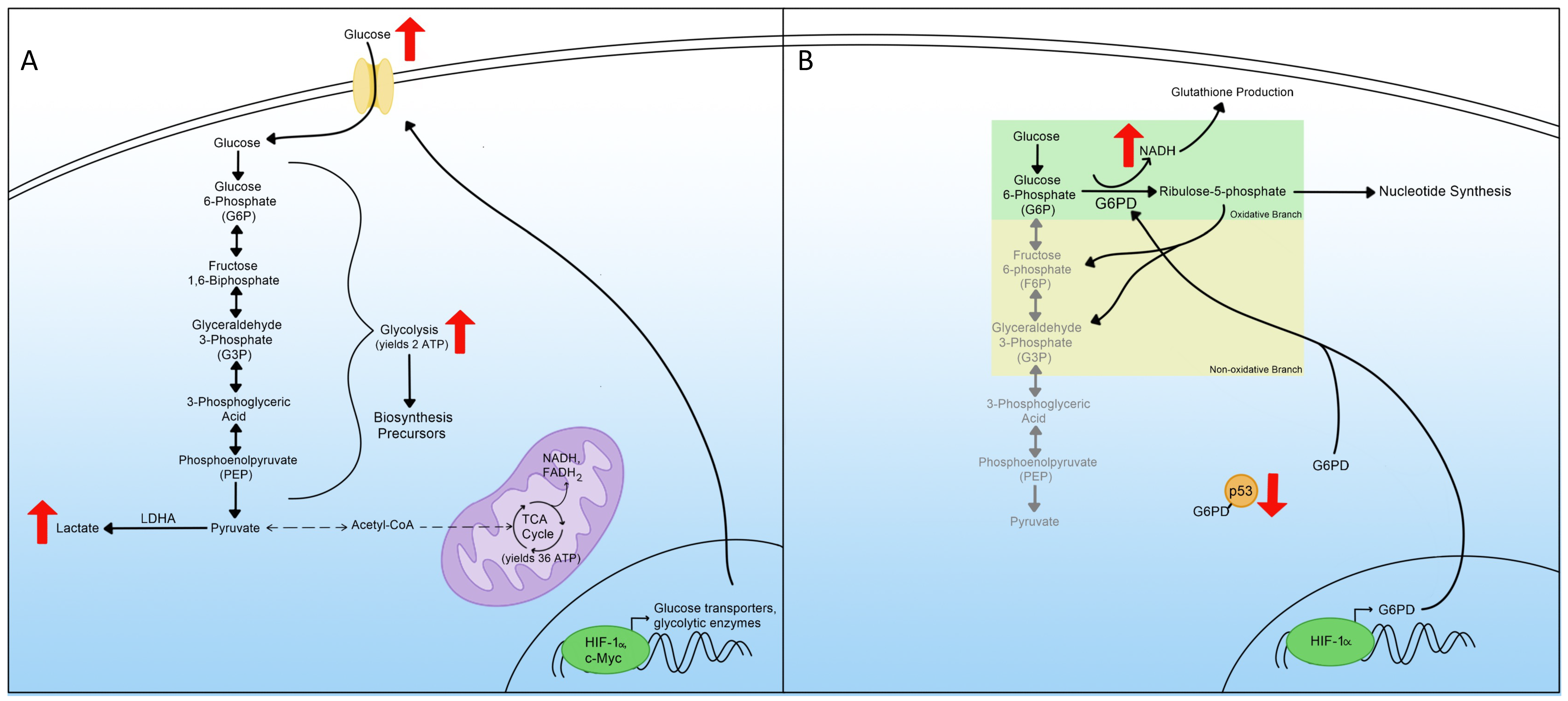
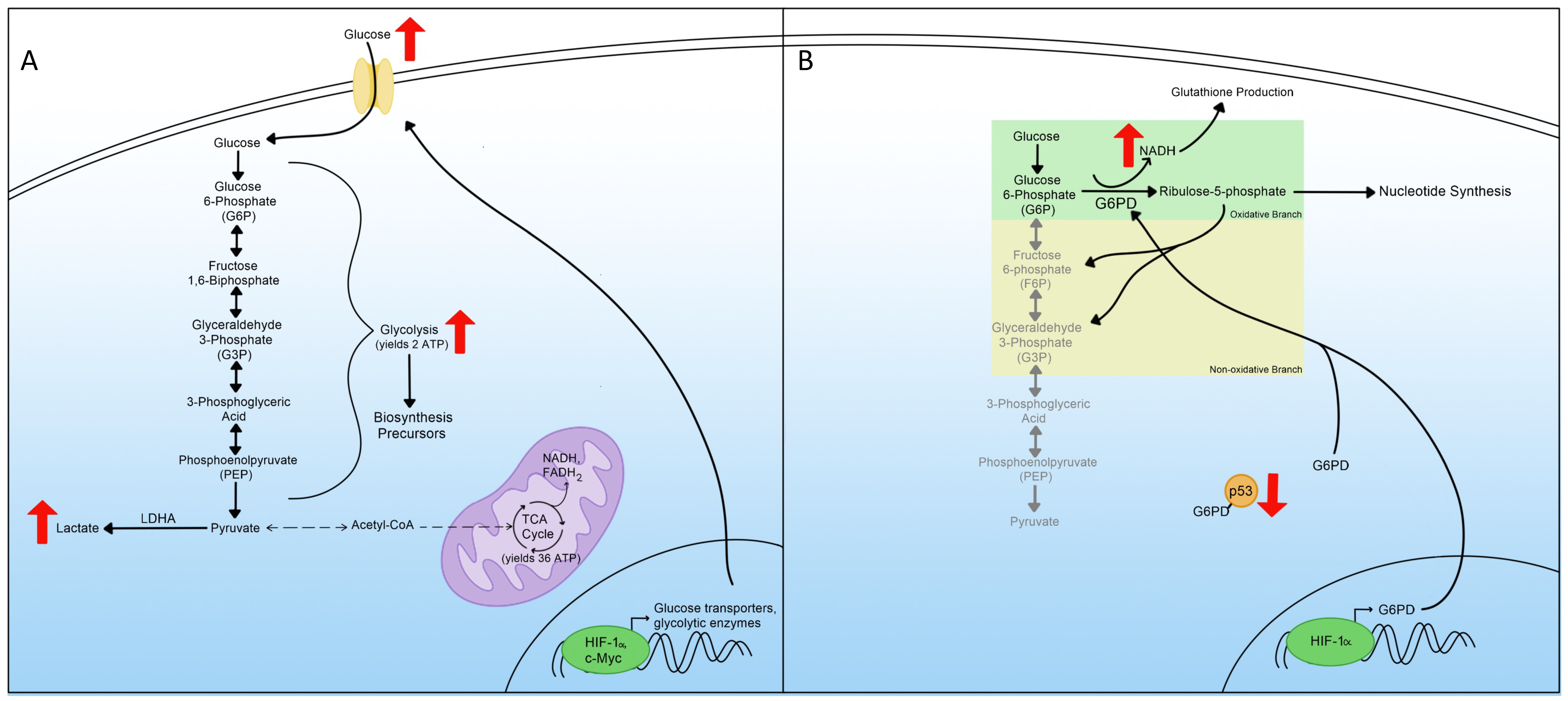
3. Glycolysis
4. Pentose Phosphate Pathway
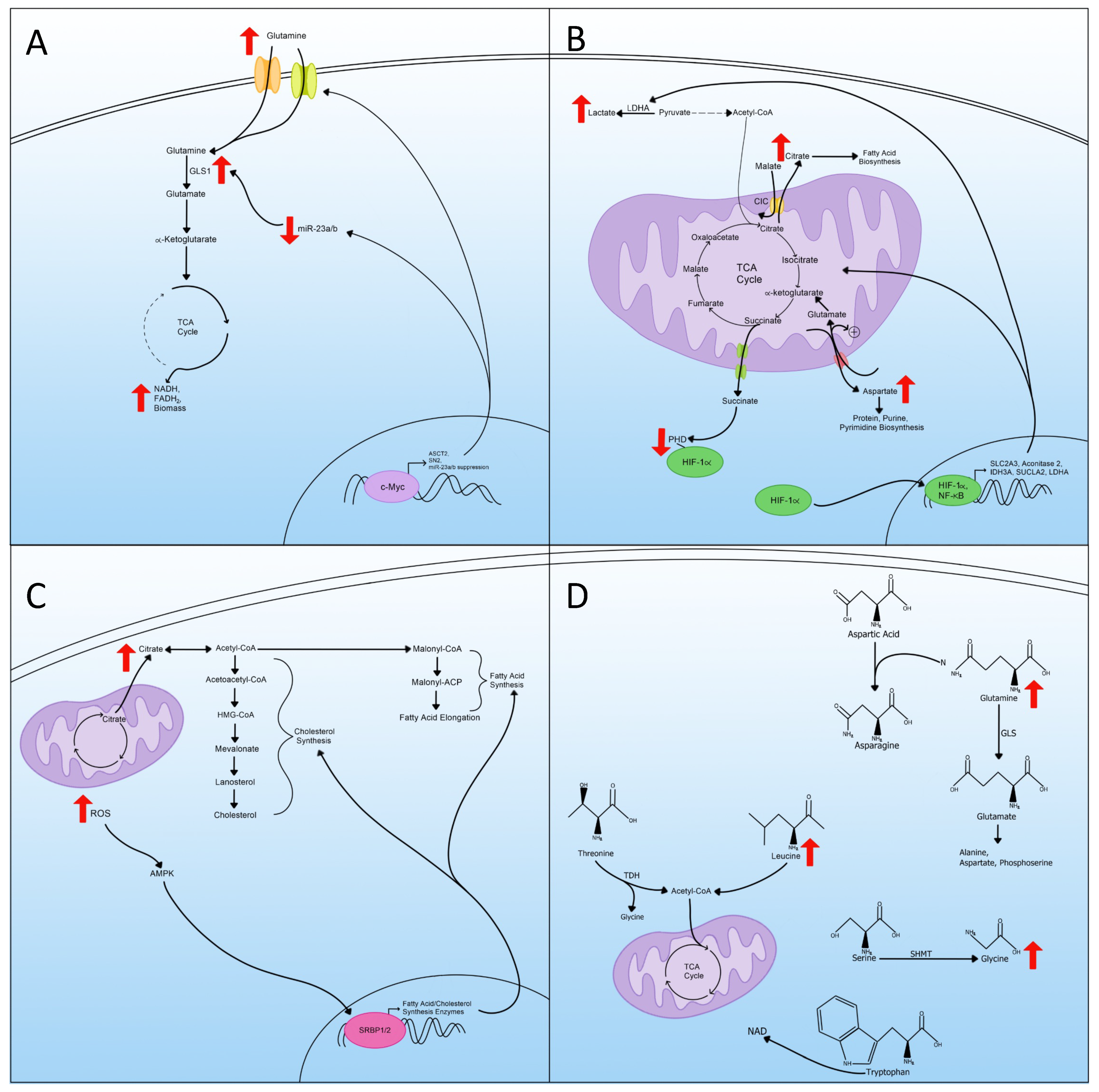
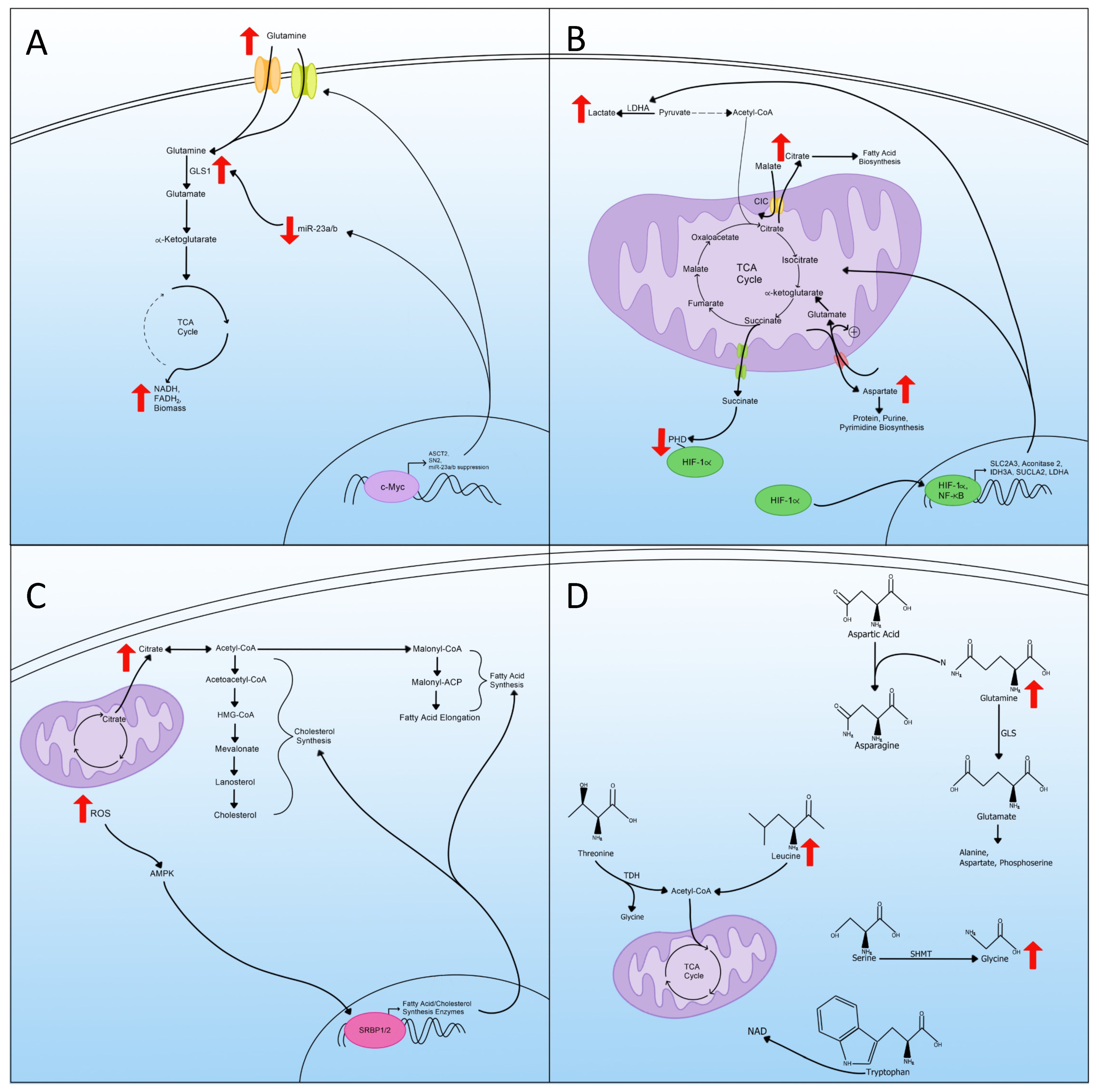
5. Glutaminolysis
6. Mitochondrial Changes and TCA Cycle Rewiring
7. Lipid Metabolism
8. Amino Acid Metabolism
9. Other Biosynthetic and Bioenergetic Pathways
10. Metabolic Reprogramming and Viruses
11. Viruses and Glycolysis
12. Viruses and Pentose Phosphate Pathway
13. Viruses and Glutaminolysis
14. Viruses and Mitochondrial Changes and TCA Cycle Rewiring
15. Viruses and Lipid Metabolism
16. Viruses and Amino Acids
17. Viruses and Other Biosynthetic and Bioenergetic Pathways
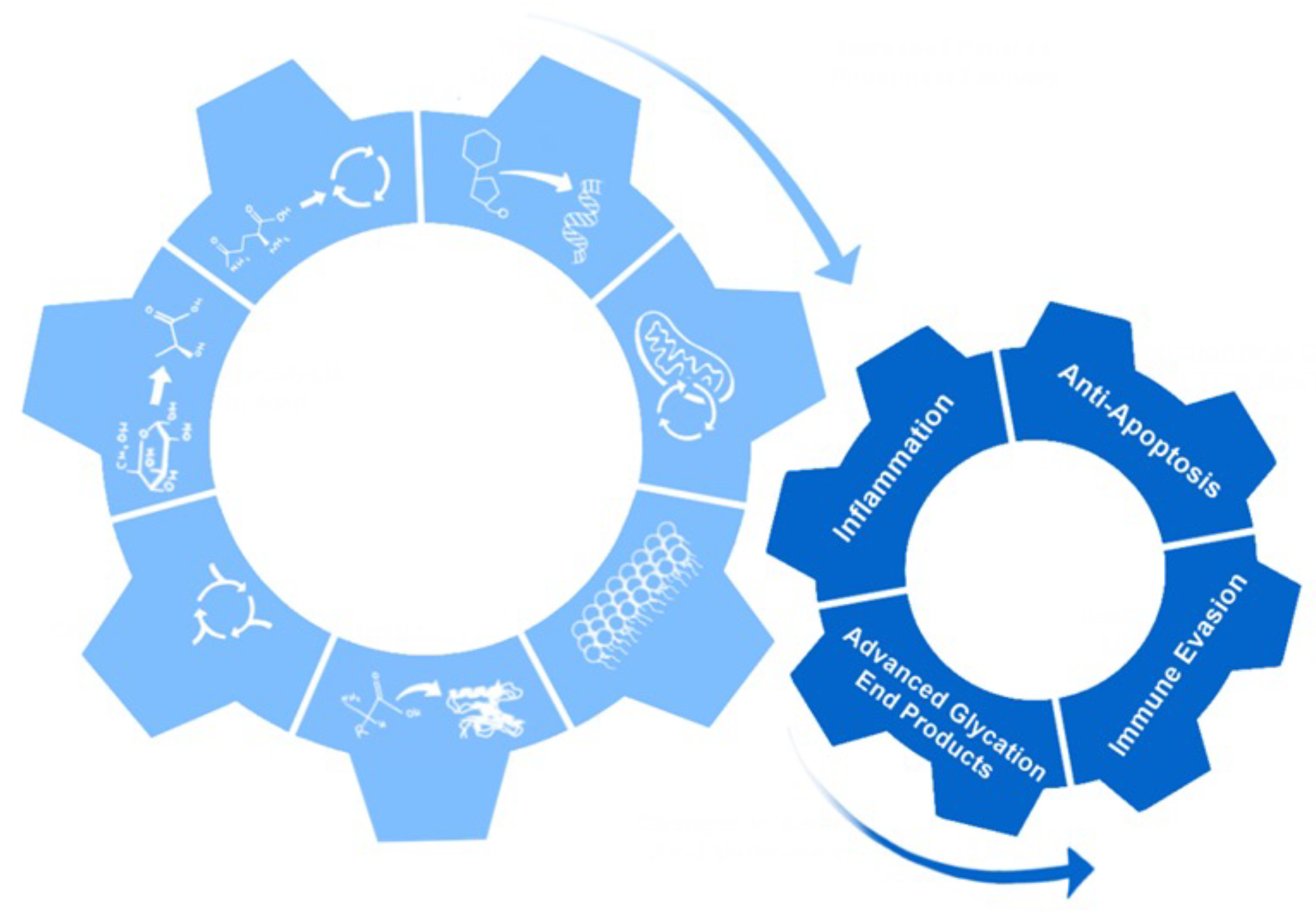
18. Non-Metabolic Effects of Metabolic Reprogramming
19. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Medina, M.Á. Metabolic Reprogramming is a Hallmark of Metabolism Itself. Bioessays 2020, 42, e2000058. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Posener, K.; Negelein, E. Ueber den Stoffwechsel der Tumoren. Biochem. Z. 1924, 152, 319–344. (In German). Reprinted in English in the book On metabolism of tumors by O. Warburg, Publisher: Constable, London, 1930 [Google Scholar]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaupel, P.; Schmidberger, H.; Mayer, A. The Warburg effect: Essential part of metabolic reprogramming and central contributor to cancer progression. Int. J. Radiat. Biol. 2019, 95, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.M.; Xu, S.; Munger, J. Stealing the Keys to the Kitchen: Viral Manipulation of the Host Cell Metabolic Network. Trends Microbiol. 2015, 23, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Purdy, J.G.; Luftig, M.A. Reprogramming of cellular metabolic pathways by human oncogenic viruses. Curr. Opin. Virol. 2019, 39, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Thyrsted, J.; Holm, C.K. Virus-induced metabolic reprogramming and innate sensing hereof by the infected host. Curr. Opin. Biotechnol. 2021, 68, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Polcicova, K.; Badurova, L.; Tomaskova, J. Metabolic reprogramming as a feast for virus replication. Acta Virol. 2020, 64, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Schiliro, C.; Firestein, B.L. Mechanisms of Metabolic Reprogramming in Cancer Cells Supporting Enhanced Growth and Proliferation. Cells 2021, 10, 1056. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Li, X.B.; Gu, J.D.; Zhou, Q.H. Review of aerobic glycolysis and its key enzymes-new targets for lung cancer therapy. Thorac. Cancer 2015, 6, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Williams, N.C.; O’Neill, L.A.J. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, D.G.; O’Neill, L.A.J. Krebs cycle rewired for macrophage and dendritic cell effector functions. FEBS Lett. 2017, 591, 2992–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeBerardinis, R.J.; Lum, J.J.; Hatzivassiliou, G.; Thompson, C.B. The biology of cancer: Metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008, 7, 11–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Li, J.; Wu, L.; Yu, Q.; Ji, J.; Wu, J.; Dai, W.; Guo, C. Emerging roles and the regulation of aerobic glycolysis in hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 2020, 39, 126. [Google Scholar] [CrossRef] [PubMed]
- Marelli-Berg, F.M.; Fu, H.; Mauro, C. Molecular mechanisms of metabolic reprogramming in proliferating cells: Implications for T-cell-mediated immunity. Immunology 2012, 136, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Jassim, A.H.; Coughlin, L.; Harun-Or-Rashid, M.; Kang, P.T.; Chen, Y.R.; Inman, D.M. Higher Reliance on Glycolysis Limits Glycolytic Responsiveness in Degenerating Glaucomatous Optic Nerve. Mol. Neurobiol. 2019, 56, 7097–7112. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Reckzeh, E.S.; Karageorgis, G.; Schwalfenberg, M.; Ceballos, J.; Nowacki, J.; Stroet, M.C.M.; Binici, A.; Knauer, L.; Brand, S.; Choidas, A.; et al. Inhibition of Glucose Transporters and Glutaminase Synergistically Impairs Tumor Cell Growth. Cell Chem. Biol. 2019, 26, 1214–1228.e25. [Google Scholar] [CrossRef]
- Faas, M.; Ipseiz, N.; Ackermann, J.; Culemann, S.; Grüneboom, A.; Schröder, F.; Rothe, T.; Scholtysek, C.; Eberhardt, M.; Böttcher, M.; et al. IL-33-induced metabolic reprogramming controls the differentiation of alternatively activated macrophages and the resolution of inflammation. Immunity 2021, 54, 2531–2546.e5. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.N.; Luo, Y.; Yang, Y.H.; Fu, J.T.; Geng, X.M.; Shi, J.P.; Yang, J. Lactylation, a Novel Metabolic Reprogramming Code: Current Status and Prospects. Front. Immunol. 2021, 12, 688910. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Ordoñez, A.; Seoane, S.; Avila, L.; Eiro, N.; Macía, M.; Arias, E.; Pereira, F.; García-Caballero, T.; Gómez-Lado, N.; Aguiar, P.; et al. POU1F1 transcription factor induces metabolic reprogramming and breast cancer progression via LDHA regulation. Oncogene 2021, 40, 2725–2740. [Google Scholar] [CrossRef] [PubMed]
- Phypers, B.; Pierce, T. Lactate physiology in health and disease. Contin. Educ. Anaesth. Crit. Care Pain 2006, 6, 128–132. [Google Scholar] [CrossRef]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef] [PubMed]
- De Vitto, H.; Arachchige, D.B.; Richardson, B.C.; French, J.B. The Intersection of Purine and Mitochondrial Metabolism in Cancer. Cells 2021, 10, 2603. [Google Scholar] [CrossRef] [PubMed]
- Hay, N. Reprogramming glucose metabolism in cancer: Can it be exploited for cancer therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, R.J. Glucose metabolism and cancer. Curr. Opin. Cell Biol. 2006, 18, 598–608. [Google Scholar] [CrossRef]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The pentose phosphate pathway: An antioxidant defense and a crossroad in tumor cell fate. Free Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Sánchez, R.; Gallardo-Pérez, J.C.; Rodríguez-Enríquez, S.; Saavedra, E.; Marín-Hernández, Á. Control of the NADPH supply for oxidative stress handling in cancer cells. Free Radic. Biol. Med. 2017, 112, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Chen, T.; Liu, L.; Cao, C.; Lv, F.; Rabinowitz, J.D.; Huang, Y.; Chen, X. Live-cell imaging of NADPH production from specific pathways. CCS Chem. 2020, 3, 1642–1648. [Google Scholar] [CrossRef]
- Polat, I.H.; Tarrado-Castellarnau, M.; Benito, A.; Hernandez-Carro, C.; Centelles, J.; Marin, S.; Cascante, M. Glutamine Modulates Expression and Function of Glucose 6-Phosphate Dehydrogenase via NRF2 in Colon Cancer Cells. Antioxidants 2021, 10, 1349. [Google Scholar] [CrossRef] [PubMed]
- Vizán, P.; Alcarraz-Vizán, G.; Díaz-Moralli, S.; Solovjeva, O.N.; Frederiks, W.M.; Cascante, M. Modulation of pentose phosphate pathway during cell cycle progression in human colon adenocarcinoma cell line HT29. Int. J. Cancer 2009, 124, 2789–2796. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Mejías, R.; Echevarría, M.; López-Barneo, J. Induction of the glucose-6-phosphate dehydrogenase gene expression by chronic hypoxia in PC12 cells. FEBS Lett. 2004, 569, 256–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayar, İ.; Bildik, A. Investigation of glucose catabolism in hypoxic Mcf 7 breast cancer culture. Cytotechnology 2021, 73, 217–232. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, S.E.; O’Neill, L.A. HIF1α and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef]
- Yang, L.; Venneti, S.; Nagrath, D. Glutaminolysis: A Hallmark of Cancer Metabolism. Annu. Rev. Biomed. Eng. 2017, 19, 163–194. [Google Scholar] [CrossRef]
- Mayers, J.R.; Vander Heiden, M.G. Famine versus feast: Understanding the metabolism of tumors in vivo. Trends Biochem. Sci. 2015, 40, 130–140. [Google Scholar] [CrossRef]
- Gaglio, D.; Metallo, C.M.; Gameiro, P.A.; Hiller, K.; Danna, L.S.; Balestrieri, C.; Alberghina, L.; Stephanopoulos, G.; Chiaradonna, F. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol. Syst. Biol. 2011, 7, 523. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, H.C.; Yu, Y.C.; Sung, Y.; Han, J.M. Glutamine reliance in cell metabolism. Exp. Mol. Med. 2020, 52, 1496–1516. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Mancuso, A.; Daikhin, E.; Nissim, I.; Yudkoff, M.; Wehrli, S.; Thompson, C.B. Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 19345–19350. [Google Scholar] [CrossRef] [Green Version]
- Bott, A.J.; Peng, I.C.; Fan, Y.; Faubert, B.; Zhao, L.; Li, J.; Neidler, S.; Sun, Y.; Jaber, N.; Krokowski, D.; et al. Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab. 2015, 22, 1068–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [Green Version]
- Ryan, D.G.; Yang, M.; Prag, H.A.; Blanco, G.R.; Nikitopoulou, E.; Segarra-Mondejar, M.; Powell, C.A.; Young, T.; Burger, N.; Miljkovic, J.L.; et al. Disruption of the TCA cycle reveals an ATF4-dependent integration of redox and amino acid metabolism. Elife 2021, 10, e72593. [Google Scholar] [CrossRef]
- Todisco, S.; Convertini, P.; Iacobazzi, V.; Infantino, V. TCA Cycle Rewiring as Emerging Metabolic Signature of Hepatocellular Carcinoma. Cancers 2019, 12, 68. [Google Scholar] [CrossRef] [Green Version]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petillo, A.; Abruzzese, V.; Koshal, P.; Ostuni, A.; Bisaccia, F. Extracellular Citrate Is a Trojan Horse for Cancer Cells. Front. Mol. Biosci. 2020, 7, 593866. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, T.; Chen, X.; Zhang, B.; Wang, Y.; Xie, M.; Ji, X.; Sun, M.; Huang, W.; Xia, L. ONECUT2 facilitates hepatocellular carcinoma metastasis by transcriptionally upregulating FGF2 and ACLY. Cell Death Dis. 2021, 12, 1113. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, F. The mitochondrial transporter family (SLC25): Physiological and pathological implications. Pflug. Arch. 2004, 447, 689–709. [Google Scholar] [CrossRef]
- Broeks, M.H.; van Karnebeek, C.D.M.; Wanders, R.J.A.; Jans, J.J.M.; Verhoeven-Duif, N.M. Inborn disorders of the malate aspartate shuttle. J. Inherit. Metab. Dis. 2021, 44, 792–808. [Google Scholar] [CrossRef]
- Alkan, H.F.; Bogner-Strauss, J.G. Maintaining cytosolic aspartate levels is a major function of the TCA cycle in proliferating cells. Mol. Cell Oncol. 2019, 6, e1536843. [Google Scholar] [CrossRef] [Green Version]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Luengo, A.; Danai, L.V.; Bush, L.N.; Diehl, F.F.; Hosios, A.M.; Lau, A.N.; Elmiligy, S.; Malstrom, S.; Lewis, C.A.; et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 2018, 20, 782–788. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-McDermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Robinson, A.J.; Overy, C.; Kunji, E.R. The mechanism of transport by mitochondrial carriers based on analysis of symmetry. Proc. Natl. Acad. Sci. USA 2008, 105, 17766–17771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhunussova, A.; Sen, B.; Friedman, L.; Tuleukhanov, S.; Brooks, A.D.; Sensenig, R.; Orynbayeva, Z. Tumor microenvironment promotes dicarboxylic acid carrier-mediated transport of succinate to fuel prostate cancer mitochondria. Am. J. Cancer Res. 2015, 5, 1665–1679. [Google Scholar] [PubMed]
- Lukyanova, L.D.; Kirova, Y.I.; Germanova, E.L. The Role of Succinate in Regulation of Immediate HIF-1α Expression in Hypoxia. Bull. Exp. Biol. Med. 2018, 164, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.F.; Perkins, N.D. Nuclear factor-κB, p53, and mitochondria: Regulation of cellular metabolism and the Warburg effect. Trends Biochem. Sci. 2012, 37, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Londhe, P.; Yu, P.Y.; Ijiri, Y.; Ladner, K.J.; Fenger, J.M.; London, C.; Houghton, P.J.; Guttridge, D.C. Classical NF-κB Metabolically Reprograms Sarcoma Cells Through Regulation of Hexokinase 2. Front. Oncol. 2018, 8, 104. [Google Scholar] [CrossRef]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Xu, X.; Wu, J.; Wang, D.; Wang, J. NF-κB controls four genes encoding core enzymes of tricarboxylic acid cycle. Gene 2017, 621, 12–20. [Google Scholar] [CrossRef]
- Gordan, J.D.; Simon, M.C. Hypoxia-inducible factors: Central regulators of the tumor phenotype. Curr. Opin. Genet. Dev. 2007, 17, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Liang, S.; Cheng, Z.; Zhang, X.; Luo, L.; Li, L.; Zhang, W.; Li, S.; Xu, Q.; Zhong, M.; et al. ROS/PI3K/Akt and Wnt/β-catenin signalings activate HIF-1α-induced metabolic reprogramming to impart 5-fluorouracil resistance in colorectal cancer. J. Exp. Clin. Cancer Res. 2022, 41, 15. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.G.; Han, Z.T.; He, S.H.; Wu, X.D.; Chen, T.R.; Shao, C.H.; Chen, D.L.; Su, N.; Chen, Y.M.; Wang, T.; et al. HIF1/2α mediates hypoxia-induced LDHA expression in human pancreatic cancer cells. Oncotarget 2017, 8, 24840–24852. [Google Scholar] [CrossRef] [PubMed]
- McFate, T.; Mohyeldin, A.; Lu, H.; Thakar, J.; Henriques, J.; Halim, N.D.; Wu, H.; Schell, M.J.; Tsang, T.M.; Teahan, O.; et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J. Biol. Chem. 2008, 283, 22700–22708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Cho, M.; Kim, B.S.; Han, J.H.; Park, S.; Lee, I.K.; Ryu, D.; Kim, J.H.; Bae, S.J.; Ha, K.T. Drug evaluation based on phosphomimetic PDHA1 reveals the complexity of activity-related cell death in A549 non-small cell lung cancer cells. BMB Rep. 2021, 54, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Randall, G. Multifaceted roles for lipids in viral infection. Trends MicroBiol. 2011, 19, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the Wheels of the Cancer Machine: The Role of Lipid Metabolism in Cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Seimiya, H.; Tsuruo, T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br. J. Cancer 2009, 100, 1369–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagiota, A.; Chachami, G.; Paraskeva, E. Lipid Metabolism in Cancer: The Role of Acylglycerolphosphate Acyltransferases (AGPATs). Cancers 2022, 14, 228. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, I.; Gianfanti, F.; Desbats, M.A.; Orso, G.; Berretta, M.; Prayer-Galetti, T.; Ragazzi, E.; Cocetta, V. Cholesterol Metabolic Reprogramming in Cancer and Its Pharmacological Modulation as Therapeutic Strategy. Front. Oncol. 2021, 11, 682911. [Google Scholar] [CrossRef]
- Ačimovič, J.; Goyal, S.; Košir, R.; Goličnik, M.; Perše, M.; Belič, A.; Urlep, Ž.; Guengerich, F.P.; Rozman, D. Cytochrome P450 metabolism of the post-lanosterol intermediates explains enigmas of cholesterol synthesis. Sci. Rep. 2016, 6, 28462. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Yang, H.; Song, B.L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef] [PubMed]
- Vona, R.; Iessi, E.; Matarrese, P. Role of Cholesterol and Lipid Rafts in Cancer Signaling: A Promising Therapeutic Opportunity? Front. Cell Dev. Biol. 2021, 9, 622908. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Shen, H.; Huang, H.; Yang, Z.; Zhou, Y.; Zhao, G. Cholesterol-lowering drug pitavastatin targets lung cancer and angiogenesis via suppressing prenylation-dependent Ras/Raf/MEK and PI3K/Akt/mTOR signaling. Anticancer Drugs 2020, 31, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Karamouzis, M.V.; Papavassiliou, A.G. Post-translational modifications and regulation of the RAS superfamily of GTPases as anticancer targets. Nat. Rev. Drug Discov. 2007, 6, 541–555. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.D. Sterol regulatory element-binding proteins: Transcriptional activators of lipid synthesis. Biochem. Soc. Trans. 2002, 30, 1091–1095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, L.; Qi, H.; Zhang, H.; Ding, L.; Huang, Q.; Zhao, D.; Wu, B.J.; Li, X. Targeting SREBP-2-Regulated Mevalonate Metabolism for Cancer Therapy. Front. Oncol. 2020, 10, 1510. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. AMP-activated/SNF1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [Green Version]
- Lieu, E.L.; Nguyen, T.; Rhyne, S.; Kim, J. Amino acids in cancer. Exp. Mol. Med. 2020, 52, 15–30. [Google Scholar] [CrossRef]
- Keenan, M.M.; Chi, J.T. Alternative fuels for cancer cells. Cancer J. 2015, 21, 49–55. [Google Scholar] [CrossRef] [Green Version]
- Mayers, J.R.; Wu, C.; Clish, C.B.; Kraft, P.; Torrence, M.E.; Fiske, B.P.; Yuan, C.; Bao, Y.; Townsend, M.K.; Tworoger, S.S.; et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med. 2014, 20, 1193–1198. [Google Scholar] [CrossRef] [PubMed]
- Sheen, J.H.; Zoncu, R.; Kim, D.; Sabatini, D.M. Defective regulation of autophagy upon leucine deprivation reveals a targetable liability of human melanoma cells in vitro and in vivo. Cancer Cell 2011, 19, 613–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, S.; Feng, J.; Cao, Y.; Shen, S.; Cai, Y.; Yang, D.; Yan, R.; Wang, L.; Zhang, H.; Zhong, X.; et al. Glycine cleavage system determines the fate of pluripotent stem cells via the regulation of senescence and epigenetic modifications. Life Sci. Alliance 2019, 2, e201900413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Alexander, P.; Wu, L.; Hammer, R.; Cleaver, O.; McKnight, S.L. Dependence of mouse embryonic stem cells on threonine catabolism. Science 2009, 325, 435–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, M.; Taurino, G.; Bianchi, M.G.; Kilberg, M.S.; Bussolati, O. Asparagine Synthetase in Cancer: Beyond Acute Lymphoblastic Leukemia. Front. Oncol. 2020, 9, 1480. [Google Scholar] [CrossRef] [PubMed]
- Curthoys, N.P.; Watford, M. Regulation of glutaminase activity and glutamine metabolism. Annu. Rev. Nutr. 1995, 15, 133–159. [Google Scholar] [CrossRef] [PubMed]
- Burke, L.; Guterman, I.; Palacios Gallego, R.; Britton, R.G.; Burschowsky, D.; Tufarelli, C.; Rufini, A. The Janus-like role of proline metabolism in cancer. Cell Death Discov. 2020, 6, 104. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bai, C.; Ruan, Y.; Liu, M.; Chu, Q.; Qiu, L.; Yang, C.; Li, B. Coordinative metabolism of glutamine carbon and nitrogen in proliferating cancer cells under hypoxia. Nat. Commun. 2019, 10, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, N.; Sampei, G.; Kawai, G. Free-Energy Profile Analysis of the Catalytic Reaction of Glycinamide Ribonucleotide Synthetase. Life 2022, 12, 281. [Google Scholar] [CrossRef]
- Zhang, Y.; Morar, M.; Ealick, S.E. Structural biology of the purine biosynthetic pathway. Cell Mol. Life Sci. 2008, 65, 3699–3724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navas, L.E.; Carnero, A. NAD+ metabolism, stemness, the immune response, and cancer. Signal Transduct. Target. Ther. 2021, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Stein, L.R.; Imai, S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012, 23, 420–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carracedo, A.; Cantley, L.C.; Pandolfi, P.P. Cancer metabolism: Fatty acid oxidation in the limelight. Nat. Rev. Cancer 2013, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012, 2012, 282041. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.C.; Tajika, M.; Shimura, M.; Carey, K.T.; Stroud, D.A.; Murayama, K.; Ohtake, A.; McKenzie, M. Loss of the Mitochondrial Fatty Acid β-Oxidation Protein Medium-Chain Acyl-Coenzyme A Dehydrogenase Disrupts Oxidative Phosphorylation Protein Complex Stability and Function. Sci. Rep. 2018, 8, 153. [Google Scholar] [CrossRef] [Green Version]
- Dalla Pozza, E.; Dando, I.; Pacchiana, R.; Liboi, E.; Scupoli, M.T.; Donadelli, M.; Palmieri, M. Regulation of succinate dehydrogenase and role of succinate in cancer. Semin. Cell Dev. Biol. 2020, 98, 4–14. [Google Scholar] [CrossRef]
- Liu, Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006, 9, 230–234. [Google Scholar] [CrossRef] [Green Version]
- Schönfeld, P.; Wieckowski, M.R.; Lebiedzińska, M.; Wojtczak, L. Mitochondrial fatty acid oxidation and oxidative stress: Lack of reverse electron transfer-associated production of reactive oxygen species. Biochim. Biophys. Acta 2010, 1797, 929–938. [Google Scholar] [CrossRef] [Green Version]
- Umemoto, T.; Johansson, A.; Ahmad, S.A.I.; Hashimoto, M.; Kubota, S.; Kikuchi, K.; Odaka, H.; Era, T.; Kurotaki, D.; Sashida, G.; et al. ATP citrate lyase controls hematopoietic stem cell fate and supports bone marrow regeneration. EMBO J. 2022, e109463. [Google Scholar] [CrossRef]
- Oeggl, R.; Neumann, T.; Gätgens, J.; Romano, D.; Noack, S.; Rother, D. Citrate as Cost-Efficient NADPH Regenerating Agent. Front. Bioeng. Biotechnol. 2018, 6, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiarugi, A.; Dölle, C.; Felici, R.; Ziegler, M. The NAD metabolome--a key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, L.; Qin, B.; Jiang, M.; Xie, L.; Luo, Z.; Guo, X.; Zhang, J.; Li, X.; Zhu, C.; Du, Y.; et al. Regulating immune memory and reversing tumor thermotolerance through a step-by-step starving-photothermal therapy. J. Nanobiotechnol. 2021, 19, 297. [Google Scholar] [CrossRef] [PubMed]
- Lyssiotis, C.A.; Cantley, L.C. Acetate fuels the cancer engine. Cell 2014, 159, 1492–1494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, J.M.; Kim, J.H.; Oh, H.J.; Park, H.E.; Lee, T.H.; Cho, N.Y.; Kang, G.H. Downregulation of acetyl-CoA synthetase 2 is a metabolic hallmark of tumor progression and aggressiveness in colorectal carcinoma. Mod. Pathol. 2017, 30, 267–277. [Google Scholar] [CrossRef]
- Schug, Z.T.; Voorde, J.V.; Gottlieb, E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer 2016, 16, 708–717. [Google Scholar] [CrossRef]
- Lakhter, A.J.; Hamilton, J.; Konger, R.L.; Brustovetsky, N.; Broxmeyer, H.E.; Naidu, S.R. Glucose-independent Acetate Metabolism Promotes Melanoma Cell Survival and Tumor Growth. J. Biol. Chem. 2016, 291, 21869–21879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaker, S.K.; Ch’ng, J.; Christofk, H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019, 17, 59. [Google Scholar] [CrossRef]
- Harrison, M.A.A.; Hochreiner, E.M.; Benjamin, B.P.; Lawler, S.E.; Zwezdaryk, K.J. Metabolic Reprogramming of Glioblastoma Cells during HCMV Infection Induces Secretome-Mediated Paracrine Effects in the Microenvironment. Viruses 2022, 14, 103. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.; Bajad, S.U.; Coller, H.A.; Shenk, T.; Rabinowitz, J.D. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006, 2, e132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munger, J.; Bennett, B.D.; Parikh, A.; Feng, X.J.; McArdle, J.; Rabitz, H.A.; Shenk, T.; Rabinowitz, J.D. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 2008, 26, 1179–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thai, M.; Graham, N.A.; Braas, D.; Nehil, M.; Komisopoulou, E.; Kurdistani, S.K.; McCormick, F.; Graeber, T.G.; Christofk, H.R. Adenovirus E4ORF1-induced MYC activation promotes host cell anabolic glucose metabolism and virus replication. Cell Metab. 2014, 19, 694–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripoli, M.; D’Aprile, A.; Quarato, G.; Sarasin-Filipowicz, M.; Gouttenoire, J.; Scrima, R.; Cela, O.; Boffoli, D.; Heim, M.H.; Moradpour, D.; et al. Hepatitis C virus-linked mitochondrial dysfunction promotes hypoxia-inducible factor 1 alpha-mediated glycolytic adaptation. J. Virol. 2010, 84, 647–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef] [Green Version]
- Chan, E.Y.; Sutton, J.N.; Jacobs, J.M.; Bondarenko, A.; Smith, R.D.; Katze, M.G. Dynamic host energetics and cytoskeletal proteomes in human immunodeficiency virus type 1-infected human primary CD4 cells: Analysis by multiplexed label-free mass spectrometry. J. Virol. 2009, 83, 9283–9295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallenberger, S.; Bosch, V.; Angliker, H.; Shaw, E.; Klenk, H.D.; Garten, W. Inhibition of furin-mediated cleavage activation of HIV-1 glycoprotein gp160. Nature 1992, 360, 358–361. [Google Scholar] [CrossRef]
- Ritter, J.B.; Wahl, A.S.; Freund, S.; Genzel, Y.; Reichl, U. Metabolic effects of influenza virus infection in cultured animal cells: Intra- and extracellular metabolite profiling. BMC Syst. Biol. 2010, 4, 61. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, K.A.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Dengue virus induces and requires glycolysis for optimal replication. J. Virol. 2015, 9, 2358–2366. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.; Tsang, J.O.; Yan, B.; Chik, K.K.; Chan, C.C.; Cao, J.; Liang, R.; Tang, K.; Yin, F.; Ye, Z.W.; et al. Metabolic Profiling Reveals Significant Perturbations of Intracellular Glucose Homeostasis in Enterovirus-Infected Cells. Metabolites 2020, 10, 302. [Google Scholar] [CrossRef] [PubMed]
- Delgado, T.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Global metabolic profiling of infection by an oncogenic virus: KSHV induces and requires lipogenesis for survival of latent infection. PLoS Pathog. 2012, 8, e1002866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado, T.; Carroll, P.A.; Punjabi, A.S.; Margineantu, D.; Hockenbery, D.M.; Lagunoff, M. Induction of the Warburg effect by Kaposi’s sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 10696–10701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yogev, O.; Lagos, D.; Enver, T.; Boshoff, C. Kaposi’s sarcoma herpesvirus microRNAs induce metabolic transformation of infected cells. PLoS Pathog. 2014, 10, e1004400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulse, M.; Caruso, L.B.; Madzo, J.; Tan, Y.; Johnson, S.; Tempera, I. Poly(ADP-ribose) polymerase 1 is necessary for coactivating hypoxia-inducible factor-1-dependent gene expression by Epstein-Barr virus latent membrane protein 1. PLoS Pathog. 2018, 14, e1007394. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Hu, Z.Y.; Dong, X.; Tan, Z.; Li, W.; Tang, M.; Chen, L.; Yang, L.; Tao, Y.; Jiang, Y.; et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene 2014, 33, 4568–4578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashida, Z.; Laxman, S. The pentose phosphate pathway and organization of Metabolic Networks Enabling Growth Programs. Curr. Opin. Syst. Biol. 2021, 28, 100390. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Grüning, N.M.; Krüger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef] [Green Version]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Münch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef]
- Keshavarz, M.; Solaymani-Mohammadi, F.; Namdari, H.; Arjeini, Y.; Mousavi, M.J.; Rezaei, F. Metabolic host response and therapeutic approaches to influenza infection. Cell Mol. Biol. Lett. 2020, 25, 15. [Google Scholar] [CrossRef]
- Prusinkiewicz, M.A.; Tu, J.; Dodge, M.J.; MacNeil, K.M.; Radko-Juettner, S.; Fonseca, G.J.; Pelka, P.; Mymryk, J.S. Differential Effects of Human Adenovirus E1A Protein Isoforms on Aerobic Glycolysis in A549 Human Lung Epithelial Cells. Viruses 2020, 12, 610. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, K.; Ebinuma, H.; Nakamoto, N.; Sakasegawa, N.; Murakami, Y.; Chu, P.S.; Usui, S.; Ishibashi, Y.; Wakayama, Y.; Taniki, N.; et al. Prominent steatosis with hypermetabolism of the cell line permissive for years of infection with hepatitis C virus. PLoS ONE 2014, 9, e94460. [Google Scholar] [CrossRef] [PubMed]
- Shytaj, I.L.; Procopio, F.A.; Tarek, M.; Carlon-Andres, I.; Tang, H.Y.; Goldman, A.R.; Munshi, M.; Kumar Pal, V.; Forcato, M.; Sreeram, S.; et al. Glycolysis downregulation is a hallmark of HIV-1 latency and sensitizes infected cells to oxidative stress. EMBO Mol. Med. 2021, 13, e13901. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.W.; Maguire, T.G.; Alwine, J.C. Glutamine metabolism is essential for human cytomegalovirus infection. J. Virol. 2010, 84, 1867–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vastag, L.; Koyuncu, E.; Grady, S.L.; Shenk, T.E.; Rabinowitz, J.D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011, 7, e1002124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thai, M.; Thaker, S.K.; Feng, J.; Du, Y.; Hu, H.; Ting Wu, T.; Graeber, T.G.; Braas, D.; Christofk, H.R. MYC-induced reprogramming of glutamine catabolism supports optimal virus replication. Nat. Commun. 2015, 6, 8873. [Google Scholar] [CrossRef] [PubMed]
- Gualdoni, G.A.; Mayer, K.A.; Kapsch, A.M.; Kreuzberg, K.; Puck, A.; Kienzl, P.; Oberndorfer, F.; Frühwirth, K.; Winkler, S.; Blaas, D.; et al. Rhinovirus induces an anabolic reprogramming in host cell metabolism essential for viral replication. Proc. Natl. Acad. Sci. USA 2018, 115, E7158–E7165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lévy, P.L.; Duponchel, S.; Eischeid, H.; Molle, J.; Michelet, M.; Diserens, G.; Vermathen, M.; Vermathen, P.; Dufour, J.F.; Dienes, H.P.; et al. Hepatitis C virus infection triggers a tumor-like glutamine metabolism. Hepatology 2017, 65, 789–803. [Google Scholar] [CrossRef] [PubMed]
- Clerc, I.; Moussa, D.A.; Vahlas, Z.; Tardito, S.; Oburoglu, L.; Hope, T.J.; Sitbon, M.; Dardalhon, V.; Mongellaz, C.; Taylor, N. Entry of glucose- and glutamine-derived carbons into the citric acid cycle supports early steps of HIV-1 infection in CD4 T cells. Nat. Metab. 2019, 1, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-García, F.J.; Pérez-Hernández, C.A.; Rodríguez-Murillo, M.; Moreno-Altamirano, M.M.B. The Role of Tricarboxylic Acid Cycle Metabolites in Viral Infections. Front. Cell. Infect. Microbiol. 2021, 11, 725043. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Clippinger, A.J.; Alwine, J.C. Viral effects on metabolism: Changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends MicroBiol. 2011, 19, 360–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapadia, S.B.; Chisari, F.V. Hepatitis C virus RNA replication is regulated by host geranylgeranylation and fatty acids. Proc. Natl. Acad. Sci. USA 2005, 102, 2561–2566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vescovo, T.; Refolo, G.; Manuelli, M.C.; Tisone, G.; Piacentini, M.; Fimia, G.M. The Impact of Mevastatin on HCV Replication and Autophagy of Non-Transformed HCV Replicon Hepatocytes Is Influenced by the Extracellular Lipid Uptake. Front. Pharmacol. 2019, 10, 718. [Google Scholar] [CrossRef] [Green Version]
- Dickens, A.M.; Anthony, D.C.; Deutsch, R.; Mielke, M.M.; Claridge, T.D.; Grant, I.; Franklin, D.; Rosario, D.; Marcotte, T.; Letendre, S.; et al. Cerebrospinal fluid metabolomics implicate bioenergetic adaptation as a neural mechanism regulating shifts in cognitive states of HIV-infected patients. AIDS 2015, 29, 559–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hileman, C.O.; Kalayjian, R.C.; Azzam, S.; Schlatzer, D.; Wu, K.; Tassiopoulos, K.; Bedimo, R.; Ellis, R.J.; Erlandson, K.M.; Kallianpur, A.; et al. Plasma Citrate and Succinate Are Associated with Neurocognitive Impairment in Older People With HIV. Clin. Infect. Dis. 2021, 73, e765–e772. [Google Scholar] [CrossRef] [PubMed]
- Cassol, E.; Misra, V.; Dutta, A.; Morgello, S.; Gabuzda, D. Cerebrospinal fluid metabolomics reveals altered waste clearance and accelerated aging in HIV patients with neurocognitive impairment. AIDS 2014, 28, 1579–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Griffiths, S.; Digard, P.; Pham, N.; Auer, M.; Haas, J.; Grey, F. Asparagine Deprivation Causes a Reversible Inhibition of Human Cytomegalovirus Acute Virus Replication. mBio 2019, 10, e01651-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Wang, N.; Zhou, Z.; Liang, H.; Pan, C.; Zhu, D.; Liu, F.; Zhang, C.Y.; Zhang, Y.; Zen, K. Circulating human cytomegalovirus-encoded HCMV-miR-US4-1 as an indicator for predicting the efficacy of IFNα treatment in chronic hepatitis B patients. Sci. Rep. 2016, 6, 23007. [Google Scholar] [CrossRef] [PubMed]
- Brault, C.; Levy, P.; Bartosch, B. Hepatitis C virus-induced mitochondrial dysfunctions. Viruses 2013, 5, 954–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santerre, M.; Arjona, S.P.; Allen, C.N.; Callen, S.; Buch, S.; Sawaya, B.E. HIV-1 VPR protein impairs lysosome clearance causing SNCA/alpha-synuclein accumulation in neurons. Autophagy 2021, 17, 1768–1782. [Google Scholar] [CrossRef]
- Gibellini, L.; De Biasi, S.; Paolini, A.; Borella, R.; Boraldi, F.; Mattioli, M.; Lo Tartaro, D.; Fidanza, L.; Caro-Maldonado, A.; Meschiari, M.; et al. Altered bioenergetics and mitochondrial dysfunction of monocytes in patients with Covid-19 pneumonia. EMBO Mol. Med. 2020, 12, e13001. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.L.; Weng, S.F.; Kuo, C.H.; Ho, H.Y. Enterovirus 71 induces mitochondrial reactive oxygen species generation that is required for efficient replication. PLoS ONE 2014, 9, e113234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foo, J.; Bellot, G.; Pervaiz, S.; Alonso, S. Mitochondria-mediated oxidative stress during viral infection. Trends MicroBiol. 2022, S0966-842X, 00318-8. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Cho, Y.Y.; Lee, G.Y.; You, D.G.; Yoo, Y.D.; Kim, Y.J. A direct role for hepatitis B virus X protein in inducing mitochondrial membrane permeabilization. J. Viral Hepat. 2018, 25, 412–420. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, C.; Scrima, R.; Quarato, G.; D’Aprile, A.; Ripoli, M.; Lecce, L.; Boffoli, D.; Moradpour, D.; Capitanio, N. Hepatitis C virus protein expression causes calcium-mediated mitochondrial bioenergetic dysfunction and nitro-oxidative stress. Hepatology 2007, 46, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Couret, J.; Chang, T.L. Reactive Oxygen Species in HIV Infection. EC Microbiol. 2016, 3, 597–604. [Google Scholar] [PubMed]
- Mukerjee, R.; Chang, J.R.; Del Valle, L.; Bagashev, A.; Gayed, M.M.; Lyde, R.B.; Hawkins, B.J.; Brailoiu, E.; Cohen, E.; Power, C.; et al. Deregulation of microRNAs by HIV-1 Vpr protein leads to the development of neurocognitive disorders. J. Biol. Chem. 2011, 286, 34976–34985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, C.M.; Schafer, X.L.; Moorman, N.J.; Munger, J. Human cytomegalovirus induces the activity and expression of acetyl-coenzyme A carboxylase, a fatty acid biosynthetic enzyme whose inhibition attenuates viral replication. J. Virol. 2011, 85, 5814–5824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xi, Y.; Harwood, S.; Wise, L.M.; Purdy, J.G. Human Cytomegalovirus pUL37x1 Is Important for Remodeling of Host Lipid Metabolism. J. Virol. 2019, 93, e00843-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokry, R.L.; Schumacher, M.L.; Hogg, N.; Terhune, S.S. Nitric Oxide Circumvents Virus-Mediated Metabolic Regulation during Human Cytomegalovirus Infection. mBio 2020, 11, e02630-20. [Google Scholar] [CrossRef]
- Yu, Y.; Maguire, T.G.; Alwine, J.C. ChREBP, a glucose-responsive transcriptional factor, enhances glucose metabolism to support biosynthesis in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. USA. 2014, 111, 1951–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Webster-Cyriaque, J.; Tomlinson, C.C.; Yohe, M.; Kenney, S. Fatty acid synthase expression is induced by the Epstein-Barr virus immediate-early protein BRLF1 and is required for lytic viral gene expression. J. Virol. 2004, 78, 4197–4206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, A.K.; Lung, R.W.; Dawson, C.W.; Young, L.S.; Ko, C.W.; Yeung, W.W.; Kang, W.; To, K.F.; Lo, K.W. Activation of sterol regulatory element-binding protein 1 (SREBP1)-mediated lipogenesis by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) promotes cell proliferation and progression of nasopharyngeal carcinoma. J. Pathol. 2018, 246, 180–190. [Google Scholar] [CrossRef] [Green Version]
- Hulse, M.; Johnson, S.M.; Boyle, S.; Caruso, L.B.; Tempera, I. Epstein-Barr Virus-Encoded Latent Membrane Protein 1 and B-Cell Growth Transformation Induce Lipogenesis through Fatty Acid Synthase. J. Virol. 2021, 95, e01857-20. [Google Scholar] [CrossRef] [PubMed]
- Namazue, J.; Kato, T.; Okuno, T.; Shiraki, K.; Yamanishi, K. Evidence for attachment of fatty acid to varicella-zoster virus glycoproteins and effect of cerulenin on the maturation of varicella-zoster virus glycoproteins. Intervirology 1989, 30, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Chukkapalli, V.; Heaton, N.S.; Randall, G. Lipids at the interface of virus-host interactions. Curr. Opin. MicroBiol. 2012, 15, 512–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; He, G.; Han, G.S.; Zhang, J.; Catanzaro, N.; Diaz, A.; Wu, Z.; Carman, G.M.; Xie, L.; Wang, X. Host Pah1p phosphatidate phosphatase limits viral replication by regulating phospholipid synthesis. PLoS Pathog. 2018, 14, e1006988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, E.A.; Chung, R.T. HCV and host lipids: An intimate connection. Semin. Liver Dis. 2013, 33, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xie, Q. Metabolic Dysfunction-associated Fatty Liver Disease (MAFLD) and Viral Hepatitis. J. Clin. Transl. Hepatol. 2022, 10, 128–133. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hood, B.L.; Chadwick, S.L.; Liu, S.; Watkins, S.C.; Luo, G.; Conrads, T.P.; Wang, T. Fatty acid synthase is up-regulated during hepatitis C virus infection and regulates hepatitis C virus entry and production. Hepatology 2008, 48, 1396–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Z.; Liu, Q.; Sun, F.; Qiao, L. Hepatitis C virus nonstructural protein 5A perturbs lipid metabolism by modulating AMPK/SREBP-1c signaling. Lipids Health Dis. 2019, 18, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; Lacount, D.J.; Kuhn, R.J.; Randall, G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 17345–17350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tongluan, N.; Ramphan, S.; Wintachai, P.; Jaresitthikunchai, J.; Khongwichit, S.; Wikan, N.; Rajakam, S.; Yoksan, S.; Wongsiriroj, N.; Roytrakul, S.; et al. Involvement of fatty acid synthase in dengue virus infection. Virol. J. 2017, 14, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasheed, S.; Yan, J.S.; Lau, A.; Chan, A.S. HIV replication enhances production of free fatty acids, low density lipoproteins and many key proteins involved in lipid metabolism: A proteomics study. PLoS ONE 2008, 3, e3003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, M.M.; Ratcliff, A.N.; Bhat, M.; Alwarawrah, Y.; Hughes, P.; Arcos, J.; Loiselle, D.; Torrelles, J.B.; Funderburg, N.T.; Haystead, T.A.; et al. Cellular fatty acid synthase is required for late stages of HIV-1 replication. Retrovirology 2017, 14, 45. [Google Scholar] [CrossRef] [Green Version]
- Ligat, G.; Da Re, S.; Alain, S.; Hantz, S. Identification of Amino Acids Essential for Viral Replication in the HCMV Helicase-Primase Complex. Front. Microbiol. 2018, 9, 2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, D.; Mohr, I. Viral subversion of the host protein synthesis machinery. Nat. Rev. MicroBiol. 2011, 9, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Juszkiewicz, S.; Blears, D.; Bajpe, P.K.; Han, Z.; Faull, P.; Mitter, R.; Stewart, A.; Snijders, A.P.; Hegde, R.S.; et al. Translation stress and collided ribosomes are co-activators of cGAS. Mol. Cell 2021, 81, 2808–2822.e10. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Sánchez, I.; Schafer, X.L.; Monaghan, M.; Munger, J. The Human Cytomegalovirus UL38 protein drives mTOR-independent metabolic flux reprogramming by inhibiting TSC2. PLoS Pathog. 2019, 15, e1007569. [Google Scholar] [CrossRef] [PubMed]
- Choi, U.Y.; Lee, J.J.; Park, A.; Zhu, W.; Lee, H.R.; Choi, Y.J.; Yoo, J.S.; Yu, C.; Feng, P.; Gao, S.J.; et al. Oncogenic human herpesvirus hijacks proline metabolism for tumorigenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 8083–8093. [Google Scholar] [CrossRef]
- Cheng, M.L.; Chien, K.Y.; Lai, C.H.; Li, G.J.; Lin, J.F.; Ho, H.Y. Metabolic Reprogramming of Host Cells in Response to Enteroviral Infection. Cells 2020, 9, 473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pant, A.; Cao, S.; Yang, Z. Asparagine Is a Critical Limiting Metabolite for Vaccinia Virus Protein Synthesis during Glutamine Deprivation. J. Virol. 2019, 93, e01834-18. [Google Scholar] [CrossRef] [Green Version]
- Grady, S.L.; Purdy, J.G.; Rabinowitz, J.D.; Shenk, T. Argininosuccinate synthetase 1 depletion produces a metabolic state conducive to herpes simplex virus 1 infection. Proc. Natl. Acad. Sci. USA 2013, 110, E5006-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ariav, Y.; Ch’ng, J.H.; Christofk, H.R.; Ron-Harel, N.; Erez, A. Targeting nucleotide metabolism as the nexus of viral infections, cancer, and the immune response. Sci. Adv. 2021, 7, eabg6165. [Google Scholar] [CrossRef] [PubMed]
- Gostner, J.M.; Becker, K.; Kurz, K.; Fuchs, D. Disturbed Amino Acid Metabolism in HIV: Association with Neuropsychiatric Symptoms. Front. Psychiatry 2015, 6, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, M.F. Tryptophan depletion and HIV infection: A metabolic link to pathogenesis. Lancet Infect. Dis. 2003, 3, 644–652. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Sun, L.; Chen, J.; Sun, W.W.; Ma, L.; Han, Y.; Jin, X.; Zhao, Q.X.; Li, T.; Lu, H.; et al. Tryptophan Metabolism Activates Aryl Hydrocarbon Receptor-Mediated Pathway to Promote HIV-1 Infection and Reactivation. mBio 2019, 10, e02591-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurichesse, H.; Tauveron, I.; Gourdon, F.; Cormerais, L.; Champredon, C.; Charrier, S.; Rochon, C.; Lamain, S.; Bayle, G.; Laveran, H.; et al. Threonine and methionine are limiting amino acids for protein synthesis in patients with AIDS. J. Nutr. 1998, 128, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Sitole, L.J.; Tugizimana, F.; Meyer, D. Multi-platform metabonomics unravel amino acids as markers of HIV/combination antiretroviral therapy-induced oxidative stress. J. Pharm. Biomed. Anal. 2019, 176, 112796. [Google Scholar] [CrossRef]
- Rasmussen, A.L.; Diamond, D.L.; McDermott, J.E.; Gao, X.; Metz, T.O.; Matzke, M.M.; Carter, V.S.; Belisle, S.E.; Korth, M.J.; Waters, K.M.; et al. Systems virology identifies a mitochondrial fatty acid oxidation enzyme, dodecenoyl coenzyme A delta isomerase, required for hepatitis C virus replication and likely pathogenesis. J. Virol. 2011, 85, 11646–11654. [Google Scholar] [CrossRef] [Green Version]
- Fernandes-Siqueira, L.O.; Zeidler, J.D.; Sousa, B.G.; Ferreira, T.; Da Poian, A.T. Anaplerotic Role of Glucose in the Oxidation of Endogenous Fatty Acids during Dengue Virus Infection. mSphere 2018, 3, e00458-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nanda, P.; Ghosh, A. Genome Scale-Differential Flux Analysis reveals deregulation of lung cell metabolism on SARS-CoV-2 infection. PLoS Comput. Biol. 2021, 17, e1008860. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, D.; Roy, D.; Cassol, E. Examining Relationships between Metabolism and Persistent Inflammation in HIV Patients on Antiretroviral Therapy. Mediat. Inflamm. 2018, 2018, 6238978. [Google Scholar] [CrossRef] [PubMed]
- Martín-Vicente, M.; González-Riaño, C.; Barbas, C.; Jiménez-Sousa, M.Á.; Brochado-Kith, O.; Resino, S.; Martínez, I. Metabolic changes during respiratory syncytial virus infection of epithelial cells. PLoS ONE 2020, 15, e0230844. [Google Scholar] [CrossRef] [PubMed]
- Páez-Franco, J.C.; Torres-Ruiz, J.; Sosa-Hernández, V.A.; Cervantes-Díaz, R.; Romero-Ramírez, S.; Pérez-Fragoso, A.; Meza-Sánchez, D.E.; Germán-Acacio, J.M.; Maravillas-Montero, J.L.; Mejía-Domínguez, N.R.; et al. Metabolomics analysis reveals a modified amino acid metabolism that correlates with altered oxygen homeostasis in COVID-19 patients. Sci. Rep. 2021, 11, 6350. [Google Scholar] [CrossRef] [PubMed]
- Gelpi, M.; Mikaeloff, F.; Knudsen, A.D.; Benfeitas, R.; Krishnan, S.; Svenssson Akusjärvi, S.; Høgh, J.; Murray, D.D.; Ullum, H.; Neogi, U.; et al. The central role of the glutamate metabolism in long-term antiretroviral treated HIV-infected individuals with metabolic syndrome. Aging 2021, 13, 22732–22751. [Google Scholar] [CrossRef]
- Tretter, L.; Adam-Vizi, V. Alpha-ketoglutarate dehydrogenase: A target and generator of oxidative stress. Philos. Trans. R Soc. B Biol. Sci. 2005, 360, 2335–2345. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Li, K.; Niu, Y.; Lin, Q.; Liang, H.; Luo, X.; Liu, L.; Li, N. The mTOR/PGC-1α/SIRT3 Pathway Drives Reductive Glutamine Metabolism to Reduce Oxidative Stress Caused by ISKNV in CPB Cells. Microbiol. Spectr. 2022, 10, e0231021. [Google Scholar] [CrossRef]
- Ju, H.Q.; Lin, J.F.; Tian, T.; Xie, D.; Xu, R.H. NADPH homeostasis in cancer: Functions, mechanisms and therapeutic implications. Signal Transduct. Target. Ther. 2020, 5, 231. [Google Scholar] [CrossRef]
- Panday, A.; Sahoo, M.K.; Osorio, D.; Batra, S. NADPH oxidases: An overview from structure to innate immunity-associated pathologies. Cell. Mol. Immunol. 2015, 12, 5–23. [Google Scholar] [CrossRef] [Green Version]
- Amatore, D.; Sgarbanti, R.; Aquilano, K.; Baldelli, S.; Limongi, D.; Civitelli, L.; Nencioni, L.; Garaci, E.; Ciriolo, M.R.; Palamara, A.T. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4-derived ROS. Cell. Microbiol. 2015, 17, 131–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudreau, H.E.; Emerson, S.U.; Korzeniowska, A.; Jendrysik, M.A.; Leto, T.L. Hepatitis C virus (HCV) proteins induce NADPH oxidase 4 expression in a transforming growth factor beta-dependent manner: A new contributor to HCV-induced oxidative stress. J. Virol. 2009, 83, 12934–12946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Violi, F.; Oliva, A.; Cangemi, R.; Ceccarelli, G.; Pignatelli, P.; Carnevale, R.; Cammisotto, V.; Lichtner, M.; Alessandri, F.; De Angelis, M.; et al. Nox2 activation in COVID-19. Redox Biol. 2020, 36, 101655. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Kumar, S.; Simon, S.D.; Singh, D.P.; Kumar, A. HIV gp120- and methamphetamine-mediated oxidative stress induces astrocyte apoptosis via cytochrome P450 2E1. Cell Death Dis. 2013, 4, e850. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amelio, I.; Cutruzzolá, F.; Antonov, A.; Agostini, M.; Melino, G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014, 39, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Serefidou, M.; Venkatasubramani, A.V.; Imhof, A. The Impact of One Carbon Metabolism on Histone Methylation. Front. Genet. 2019, 10, 764. [Google Scholar] [CrossRef] [PubMed]
- Ayari, A.; Rosa-Calatrava, M.; Lancel, S.; Barthelemy, J.; Pizzorno, A.; Mayeuf-Louchart, A.; Baron, M.; Hot, D.; Deruyter, L.; Soulard, D.; et al. Influenza infection rewires energy metabolism and induces browning features in adipose cells and tissues. Commun. Biol. 2020, 3, 237. [Google Scholar] [CrossRef]
- Francesconi, V.; Giovannini, L.; Santucci, M.; Cichero, E.; Costi, M.P.; Naesens, L.; Giordanetto, F.; Tonelli, M. Synthesis, biological evaluation and molecular modeling of novel azaspiro dihydrotriazines as influenza virus inhibitors targeting the host factor dihydrofolate reductase (DHFR). Eur. J. Med.Chem. 2018, 155, 229–243. [Google Scholar] [CrossRef]
- Tonelli, M.; Naesens, L.; Gazzarrini, S.; Santucci, M.; Cichero, E.; Tasso, B.; Moroni, A.; Costi, M.P.; Loddo, R. Host dihydrofolate reductase (DHFR)-directed cycloguanil analogues endowed with activity against influenza virus and respiratory syncytial virus. Eur. J. Med. Chem. 2017, 135, 467–478. [Google Scholar] [CrossRef]
- Zhang, Y.; Guo, R.; Kim, S.H.; Shah, H.; Zhang, S.; Liang, J.H.; Fang, Y.; Gentili, M.; Leary, C.N.O.; Elledge, S.J.; et al. SARS-CoV-2 hijacks folate and one-carbon metabolism for viral replication. Nat. Commun. 2021, 12, 1676. [Google Scholar] [CrossRef] [PubMed]
- Zgheib, R.; Battaglia-Hsu, S.F.; Hergalant, S.; Quéré, M.; Alberto, J.M.; Chéry, C.; Rouyer, P.; Gauchotte, G.; Guéant, J.L.; Namour, F. Folate can promote the methionine-dependent reprogramming of glioblastoma cells towards pluripotency. Cell Death Dis. 2019, 10, 596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhang, Z.; Hoshino, A.; Zheng, H.D.; Morley, M.; Arany, Z.; Rabinowitz, J.D. NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat. Metab. 2019, 1, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Vousden, K.H. Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Teoh, S.T.; Ensink, E.; Ogrodzinski, M.P.; Yang, C.; Vazquez, A.I.; Lunt, S.Y. Cysteine catabolism and the serine biosynthesis pathway support pyruvate production during pyruvate kinase knockdown in pancreatic cancer cells. Cancer Metab. 2019, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Suo, C.; Li, S.T.; Zhang, H.; Gao, P. Metabolic reprogramming for cancer cells and their microenvironment: Beyond the Warburg Effect. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Jacquet, P.; Stéphanou, A. Metabolic Reprogramming, Questioning, and Implications for Cancer. Biology 2021, 10, 129. [Google Scholar] [CrossRef] [PubMed]
- Adam, C.; Paolini, L.; Gueguen, N.; Mabilleau, G.; Preisser, L.; Blanchard, S.; Pignon, P.; Manero, F.; Le Mao, M.; Morel, A.; et al. Acetoacetate protects macrophages from lactic acidosis-induced mitochondrial dysfunction by metabolic reprograming. Nat. Commun. 2021, 12, 7115. [Google Scholar] [CrossRef]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [Green Version]
- Jing, C.; Castro-Dopico, T.; Richoz, N.; Tuong, Z.K.; Ferdinand, J.R.; Lok, L.S.C.; Loudon, K.W.; Banham, G.D.; Mathews, R.J.; Cader, Z.; et al. Macrophage metabolic reprogramming presents a therapeutic target in lupus nephritis. Proc. Natl. Acad. Sci. USA 2020, 117, 15160–15171. [Google Scholar] [CrossRef]
- Kim, J.; Kim, J.; Bae, J.S. ROS homeostasis and metabolism: A critical liaison for cancer therapy. Exp. Mol. Med. 2016, 48, e269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palsson-McDermott, E.M.; O’Neill, L.A. The Warburg effect then and now: From cancer to inflammatory diseases. Bioessays 2013, 35, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, S.; Jeon, R.; Vuckovic, I.; Jiang, X.; Lerman, A.; Folmes, C.D.; Dzeja, P.D.; Herrmann, J. Interferon Gamma Induces Reversible Metabolic Reprogramming of M1 Macrophages to Sustain Cell Viability and Pro-Inflammatory Activity. EBioMedicine 2018, 30, 303–316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Cazares, D.; Chavez-Dominguez, R.; Carlos-Reyes, A.; Lopez-Camarillo, C.; Hernadez de la Cruz, O.N.; Lopez-Gonzalez, J.S. Contribution of Angiogenesis to Inflammation and Cancer. Front. Oncol. 2019, 9, 1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Ignazio, L.; Batie, M.; Rocha, S. Hypoxia and Inflammation in Cancer, Focus on HIF and NF-κB. Biomedicines 2017, 5, 21. [Google Scholar] [CrossRef] [Green Version]
- Schoppmann, S.F.; Birner, P.; Stöckl, J.; Kalt, R.; Ullrich, R.; Caucig, C.; Kriehuber, E.; Nagy, K.; Alitalo, K.; Kerjaschki, D. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am. J. Pathol. 2002, 161, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.X.; Talebian, F.; Liu, J.Q.; Khattabi, M.; Yu, L.; Bai, X.F. IL-10 contributes to the suppressive function of tumour-associated myeloid cells and enhances myeloid cell accumulation in tumours. Scand. J. Immunol. 2012, 75, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Tarasenko, T.N.; Jestin, M.; Matsumoto, S.; Saito, K.; Hwang, S.; Gavrilova, O.; Trivedi, N.; Zerfas, P.M.; Barca, E.; DiMauro, S.; et al. Macrophage derived TNFα promotes hepatic reprogramming to Warburg-like metabolism. J. Mol. Med. 2019, 97, 1231–1243. [Google Scholar] [CrossRef]
- Kilic, M.; Kasperczyk, H.; Fulda, S.; Debatin, K.M. Role of hypoxia inducible factor-1 alpha in modulation of apoptosis resistance. Oncogene 2007, 26, 2027–2038. [Google Scholar] [CrossRef] [PubMed]
- Del Rey, M.J.; Valín, Á.; Usategui, A.; García-Herrero, C.M.; Sánchez-Aragó, M.; Cuezva, J.M.; Galindo, M.; Bravo, B.; Cañete, J.D.; Blanco, F.J.; et al. Hif-1α Knockdown Reduces Glycolytic Metabolism and Induces Cell Death of Human Synovial Fibroblasts Under Normoxic Conditions. Sci. Rep. 2017, 7, 3644. [Google Scholar] [CrossRef] [Green Version]
- Fulda, S.; Debatin, K.M. HIF-1-regulated glucose metabolism: A key to apoptosis resistance? Cell Cycle 2007, 6, 790–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Z.; Guo, M.; Yu, Y.; Zhang, F.F.; Ge, M.K.; Chen, G.Q.; Shen, S.M. Downregulation of AIF by HIF-1 contributes to hypoxia-induced epithelial-mesenchymal transition of colon cancer. Carcinogenesis 2016, 37, 1079–1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuura, K.; Canfield, K.; Feng, W.; Kurokawa, M. Metabolic Regulation of Apoptosis in Cancer. Int. Rev. Cell Mol. Biol. 2016, 327, 43–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughn, A.E.; Deshmukh, M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat. Cell Biol. 2008, 10, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Fu, A.; Wu, R.; Wei, N.; Song, K.; Lim, S.; Luo, K.Q. High Expression of G6PD Increases Doxorubicin Resistance in Triple Negative Breast Cancer Cells by Maintaining GSH Level. Int. J. Biol. Sci. 2022, 18, 1120–1133. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, S.; Koyama, S.; Itahashi, K.; Tanegashima, T.; Lin, Y.T.; Togashi, Y.; Kamada, T.; Irie, T.; Okumura, G.; Kono, H.; et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell 2022, 40, 201–218.e9. [Google Scholar] [CrossRef] [PubMed]
- Caronni, N.; Simoncello, F.; Stafetta, F.; Guarnaccia, C.; Ruiz-Moreno, J.S.; Opitz, B.; Galli, T.; Proux-Gillardeaux, V.; Benvenuti, F. Downregulation of Membrane Trafficking Proteins and Lactate Conditioning Determine Loss of Dendritic Cell Function in Lung Cancer. Cancer Res. 2018, 78, 1685–1699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetze, K.; Walenta, S.; Ksiazkiewicz, M.; Kunz-Schughart, L.A.; Mueller-Klieser, W. Lactate enhances motility of tumor cells and inhibits monocyte migration and cytokine release. Int. J. Oncol. 2011, 39, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef]
- Ratter, J.M.; Rooijackers, H.M.M.; Hooiveld, G.J.; Hijmans, A.G.M.; de Galan, B.E.; Tack, C.J.; Stienstra, R. In vitro and in vivo Effects of Lactate on Metabolism and Cytokine Production of Human Primary PBMCs and Monocytes. Front. Immunol. 2018, 9, 2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Li, S.; Tang, Q.; Zhu, G. Metabolic Reprogramming and Immune Evasion in Nasopharyngeal Carcinoma. Front. Immunol. 2021, 12, 680955. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.; Fendt, S.M.; Ho, P.C. Navigating metabolic pathways to enhance antitumour immunity and immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef] [PubMed]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Investig. 2007, 117, 1147–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.M.; Sage, L.K.; Huang, L.; Barber, J.; Klonowski, K.D.; Mellor, A.L.; Tompkins, S.M.; Tripp, R.A. Inhibition of indoleamine 2,3-dioxygenase enhances the T-cell response to influenza virus infection. J. Gen. Virol. 2013, 94, 1451–1461. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, F.C.; Coimbra, J.S.; de Oliveira, E.B.; Zuñiga, A.D.; Rojas, E.E. Food Protein-polysaccharide Conjugates Obtained via the Maillard Reaction: A Review. Crit. Rev. Food Sci. Nutr. 2016, 56, 1108–1125. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, S.R.; Baynes, J.W. Maillard reaction products in tissue proteins: New products and new perspectives. Amino Acids 2003, 25, 275–281. [Google Scholar] [CrossRef]
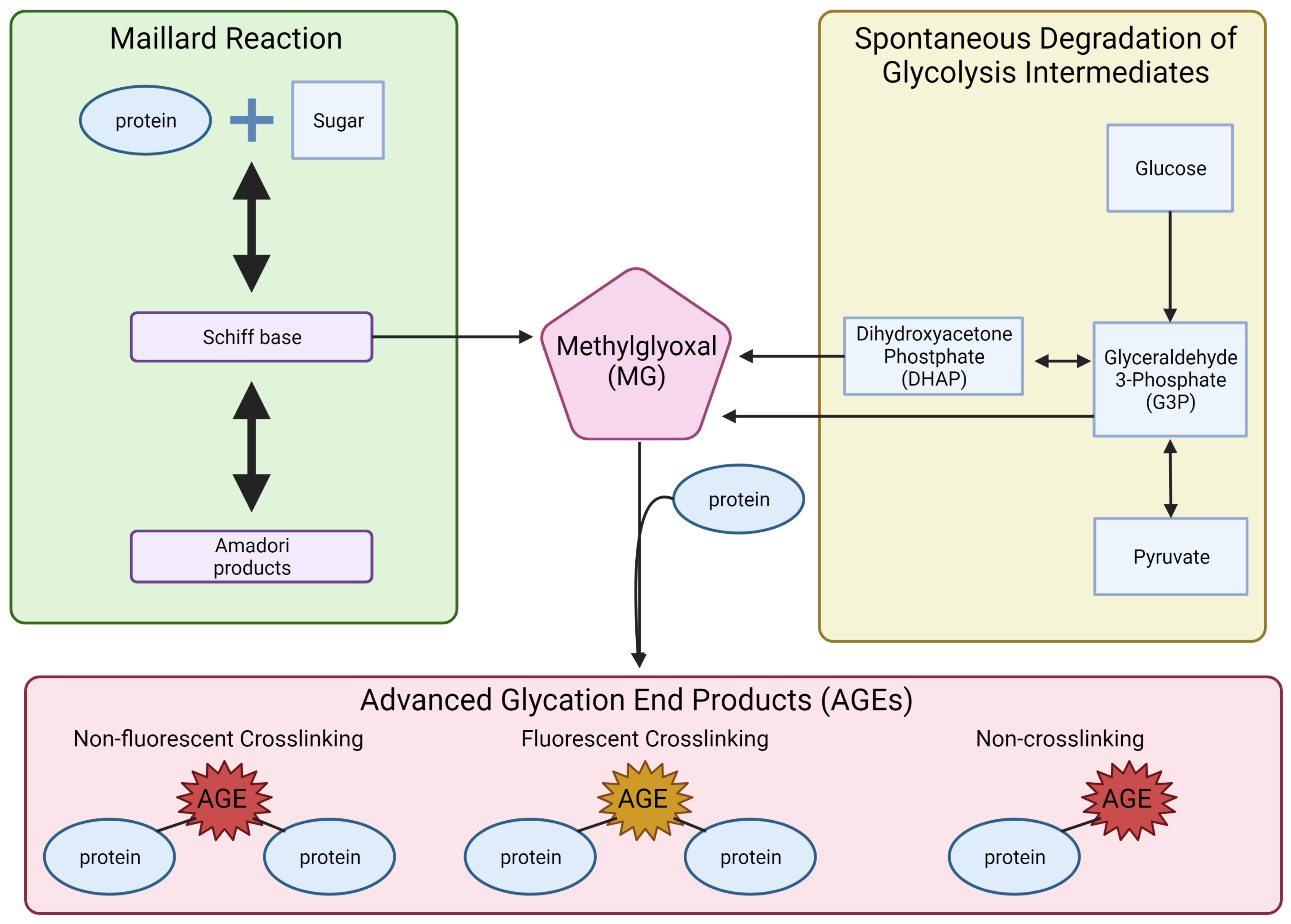
- Allaman, I.; Bélanger, M.; Magistretti, P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015, 9, 23. [Google Scholar] [CrossRef] [Green Version]
- Kold-Christensen, R.; Johannsen, M. Methylglyoxal Metabolism and Aging-Related Disease: Moving from Correlation toward Causation. Trends Endocrinol. Metab. 2020, 31, 81–92. [Google Scholar] [CrossRef]
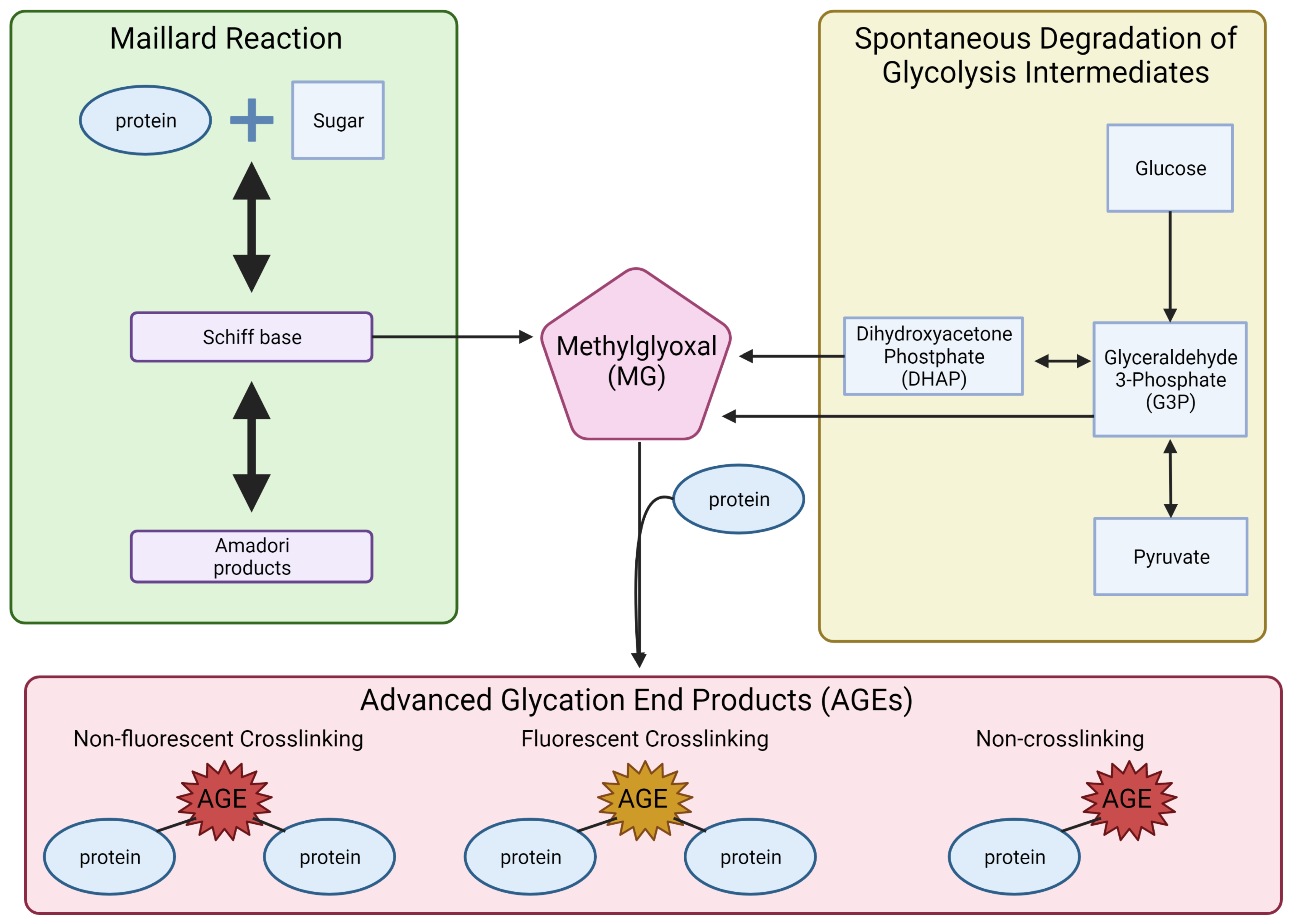
- Ahmed, N. Advanced glycation endproducts-role in pathology of diabetic complications. Diabetes Res. Clin. Pract. 2005, 67, 3–21. [Google Scholar] [CrossRef]
- Angeloni, C.; Zambonin, L.; Hrelia, S. Role of methylglyoxal in Alzheimer’s disease. BioMed Res. Int. 2014, 2014, 238485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vistoli, G.; De Maddis, D.; Cipak, A.; Zarkovic, N.; Carini, M.; Aldini, G. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): An overview of their mechanisms of formation. Free Radic. Res. 2013, 47 (Suppl S1), 3–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nandi, S.K.; Nahomi, R.B.; Rankenberg, J.; Glomb, M.A.; Nagaraj, R.H. Glycation-mediated inter-protein cross-linking is promoted by chaperone-client complexes of α-crystallin: Implications for lens aging and presbyopia. J. Biol. Chem. 2020, 295, 5701–5716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulrich, P.; Cerami, A. Protein glycation, diabetes, and aging. Recent Prog. Horm. Res. 2001, 56, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facchiano, F.; Lentini, A.; Fogliano, V.; Mancarella, S.; Rossi, C.; Facchiano, A.; Capogrossi, M.C. Sugar-induced modification of fibroblast growth factor 2 reduces its angiogenic activity in vivo. Am. J. Pathol. 2002, 161, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Iannuzzi, C.; Irace, G.; Sirangelo, I. Differential effects of glycation on protein aggregation and amyloid formation. Front. Mol. Biosci. 2014, 1, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kass, D.A. Getting better without AGE: New insights into the diabetic heart. Circ. Res. 2003, 92, 704–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirangelo, I.; Iannuzzi, C. Understanding the Role of Protein Glycation in the Amyloid Aggregation Process. Int. J. Mol. Sci. 2021, 22, 6609. [Google Scholar] [CrossRef] [PubMed]
- Bouma, B.; Kroon-Batenburg, L.M.; Wu, Y.P.; Brünjes, B.; Posthuma, G.; Kranenburg, O.; de Groot, P.G.; Voest, E.E.; Gebbink, M.F. Glycation induces formation of amyloid cross-beta structure in albumin. J. Biol. Chem. 2003, 278, 41810–41819. [Google Scholar] [CrossRef] [Green Version]
- Kichev, A.; Ilieva, E.V.; Piñol-Ripoll, G.; Podlesniy, P.; Ferrer, I.; Portero-Otín, M.; Pamplona, R.; Espinet, C. Cell death and learning impairment in mice caused by in vitro modified pro-NGF can be related to its increased oxidative modifications in Alzheimer disease. Am. J. Pathol. 2009, 175, 2574–2585. [Google Scholar] [CrossRef] [Green Version]
- Mendez, D.L.; Jensen, R.A.; McElroy, L.A.; Pena, J.M.; Esquerra, R.M. The effect of non-enzymatic glycation on the unfolding of human serum albumin. Arch. Biochem. Biophys. 2005, 444, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Groener, J.B.; Oikonomou, D.; Cheko, R.; Kender, Z.; Zemva, J.; Kihm, L.; Muckenthaler, M.; Peters, V.; Fleming, T.; Kopf, S.; et al. Methylglyoxal and Advanced Glycation End Products in Patients with Diabetes-What We Know so Far and the Missing Links. Exp. Clin. Endocrinol. Diabetes 2019, 127, 497–504. [Google Scholar] [CrossRef]
- Hanssen, N.M.; Beulens, J.W.; van Dieren, S.; Scheijen, J.L.; van der A, D.L.; Spijkerman, A.M.; van der Schouw, Y.T.; Stehouwer, C.D.; Schalkwijk, C.G. Plasma advanced glycation end products are associated with incident cardiovascular events in individuals with type 2 diabetes: A case-cohort study with a median follow-up of 10 years (EPIC-NL). Diabetes 2015, 64, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saulnier, P.J.; Wheelock, K.M.; Howell, S.; Weil, E.J.; Tanamas, S.K.; Knowler, W.C.; Lemley, K.V.; Mauer, M.; Yee, B.; Nelson, R.G.; et al. Advanced Glycation End Products Predict Loss of Renal Function and Correlate With Lesions of Diabetic Kidney Disease in American Indians With Type 2 Diabetes. Diabetes 2016, 65, 3744–3753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beisswenger, P.J.; Howell, S.K.; Russell, G.B.; Miller, M.E.; Rich, S.S.; Mauer, M. Early progression of diabetic nephropathy correlates with methylglyoxal-derived advanced glycation end products. Diabetes Care 2013, 36, 3234–3239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fishman, S.L.; Sonmez, H.; Basman, C.; Singh, V.; Poretsky, L. The role of advanced glycation end-products in the development of coronary artery disease in patients with and without diabetes mellitus: A review. Mol. Med. 2018, 24, 59. [Google Scholar] [CrossRef] [PubMed]
- Bellier, J.; Nokin, M.J.; Lardé, E.; Karoyan, P.; Peulen, O.; Castronovo, V.; Bellahcène, A. Methylglyoxal, a potent inducer of AGEs, connects between diabetes and cancer. Diabetes Res. Clin. Pract. 2019, 148, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Azizian-Farsani, F.; Abedpoor, N.; Hasan Sheikhha, M.; Gure, A.O.; Nasr-Esfahani, M.H.; Ghaedi, K. Receptor for Advanced Glycation End Products Acts as a Fuel to Colorectal Cancer Development. Front. Oncol. 2020, 10, 552283. [Google Scholar] [CrossRef] [PubMed]
- van Zoelen, M.A.; van der Sluijs, K.F.; Achouiti, A.; Florquin, S.; Braun-Pater, J.M.; Yang, H.; Nawroth, P.P.; Tracey, K.J.; Bierhaus, A.; van der Poll, T. Receptor for advanced glycation end products is detrimental during influenza A virus pneumonia. Virology 2009, 391, 265–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passos, F.R.S.; Heimfarth, L.; Monteiro, B.S.; Corrêa, C.B.; Moura, T.R.; Araújo, A.A.S.; Martins-Filho, P.R.; Quintans-Júnior, L.J.; Quintans, J.S.S. Oxidative stress and inflammatory markers in patients with COVID-19: Potential role of RAGE, HMGB1, GFAP and COX-2 in disease severity. Int. Immunopharmacol. 2022, 104, 108502. [Google Scholar] [CrossRef]
- Takeuchi, M.; Kikuchi, S.; Sasaki, N.; Suzuki, T.; Watai, T.; Iwaki, M.; Bucala, R.; Yamagishi, S. Involvement of advanced glycation end-products (AGEs) in Alzheimer’s disease. Curr. Alzheimer Res. 2004, 1, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Batkulwar, K.; Godbole, R.; Banarjee, R.; Kassaar, O.; Williams, R.J.; Kulkarni, M.J. Advanced Glycation End Products Modulate Amyloidogenic APP Processing and Tau Phosphorylation: A Mechanistic Link between Glycation and the Development of Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 988–1000. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.N.; Arjona, S.P.; Santerre, M.; De Lucia, C.; Koch, W.J.; Sawaya, B.E. Metabolic Reprogramming in HIV-Associated Neurocognitive Disorders (HAND). Front. Cell Neurosci. 2022. [Google Scholar] [CrossRef]
- Kong, Y.; Liu, C.; Zhou, Y.; Qi, J.; Zhang, C.; Sun, B.; Wang, J.; Guan, Y. Progress of RAGE Molecular Imaging in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 227. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Yamagishi, S. Possible involvement of advanced glycation end-products (AGEs) in the pathogenesis of Alzheimer’s disease. Curr. Pharm. Des. 2008, 14, 973–978. [Google Scholar] [CrossRef]
- Vitek, M.P.; Bhattacharya, K.; Glendening, J.M.; Stopa, E.; Vlassara, H.; Bucala, R.; Manogue, K.; Cerami, A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 4766–4770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alghamdi, A.; Forbes, S.; Birch, D.J.S.; Vyshemirsky, V.; Rolinski, O.J. Detecting beta-amyloid glycation by intrinsic fluorescence-Understanding the link between diabetes and Alzheimer’s disease. Arch. Biochem. Biophys. 2021, 704, 108886. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Allen, C.N.S.; Arjona, S.P.; Santerre, M.; Sawaya, B.E. Hallmarks of Metabolic Reprogramming and Their Role in Viral Pathogenesis. Viruses 2022, 14, 602. https://doi.org/10.3390/v14030602
Allen CNS, Arjona SP, Santerre M, Sawaya BE. Hallmarks of Metabolic Reprogramming and Their Role in Viral Pathogenesis. Viruses. 2022; 14(3):602. https://doi.org/10.3390/v14030602
Chicago/Turabian StyleAllen, Charles N. S., Sterling P. Arjona, Maryline Santerre, and Bassel E. Sawaya. 2022. "Hallmarks of Metabolic Reprogramming and Their Role in Viral Pathogenesis" Viruses 14, no. 3: 602. https://doi.org/10.3390/v14030602
APA StyleAllen, C. N. S., Arjona, S. P., Santerre, M., & Sawaya, B. E. (2022). Hallmarks of Metabolic Reprogramming and Their Role in Viral Pathogenesis. Viruses, 14(3), 602. https://doi.org/10.3390/v14030602