Diverse Mechanisms Underlie Enhancement of Enteric Viruses by the Mammalian Intestinal Microbiota

{kind=link}
Abstract
1. Introduction
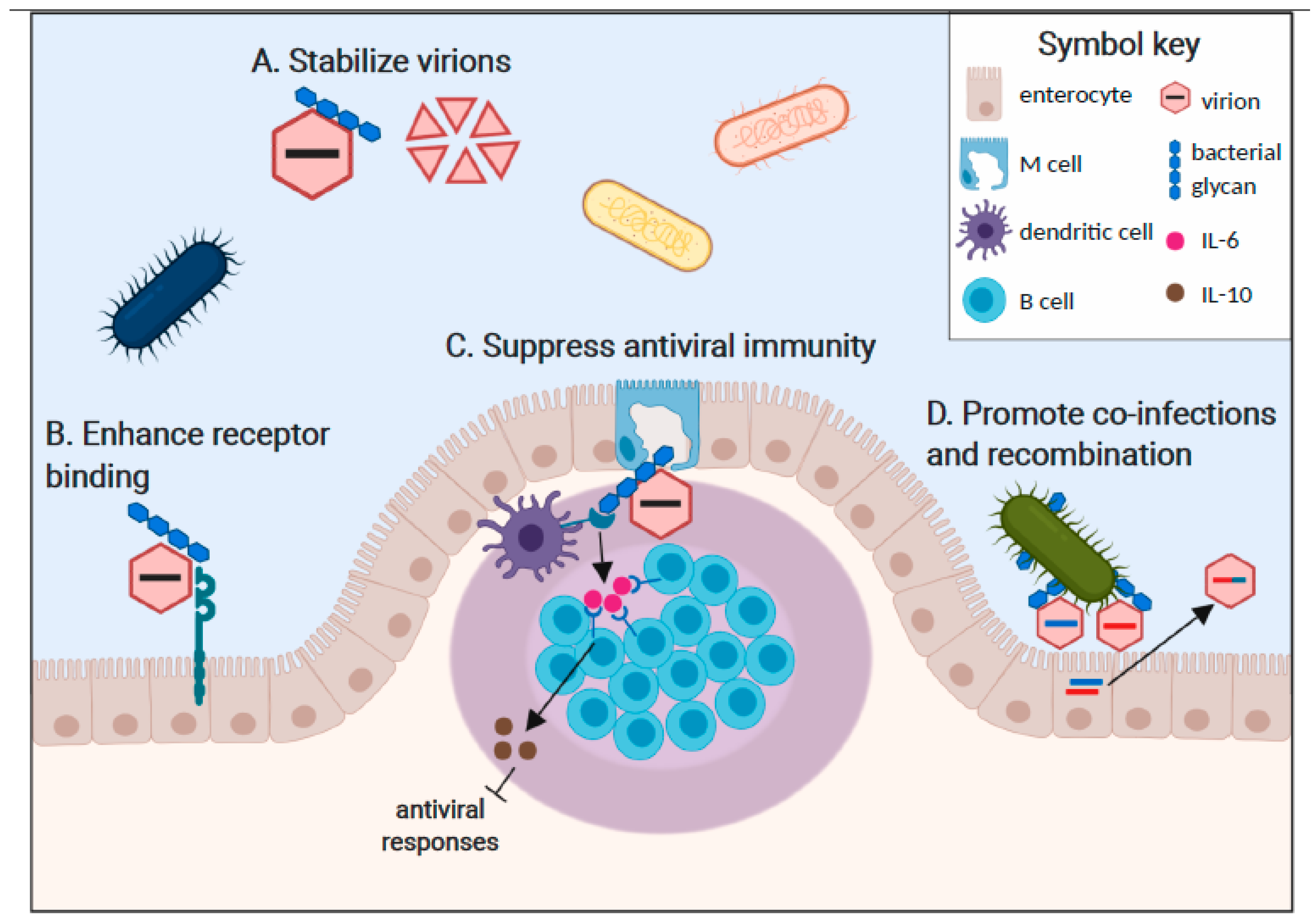
2. Mechanisms of Bacterial Enhancement of Enteric Virus Infections
2.1. Bacterial Stabilization of Virus Particles
2.2. Bacterial Enhancement of Virus Attachment to Target Cells
2.3. Bacterial Modulation of Host Immunity in a Proviral Manner
2.4. Bacterial Promotion of Viral Recombination
3. Discussion and Future Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Crawford, S.E.; Ramani, S.; Tate, J.E.; Parashar, U.D.; Svensson, L.; Hagbom, M.; Franco, M.A.; Greenberg, H.B.; O’Ryan, M.; Kang, G.; et al. Rotavirus infection. Nat. Rev. Dis. Primer 2017, 3, 17083. [Google Scholar] [CrossRef] [PubMed]
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Fields Virology; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; ISBN 978-0-7817-6060-7. [Google Scholar]
- GBD Diarrhoeal Diseases Collaborators. Estimates of global, regional, and national morbidity, mortality, and aetiologies of diarrhoeal diseases: A systematic analysis for the Global Burden of Disease Study. Lancet Infect. Dis. 2017, 17, 909–948. [Google Scholar] [CrossRef]
- Parashar, U.D.; Hummelman, E.G.; Bresee, J.S.; Miller, M.A.; Glass, R.I. Global illness and deaths caused by rotavirus disease in children. Emerg. Infect. Dis. 2003, 9, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Lanata, C.F.; Fischer-Walker, C.L.; Olascoaga, A.C.; Torres, C.X.; Aryee, M.J.; Black, R.E. Global causes of diarrheal disease mortality in children <5 years of age: A systematic review. PLoS ONE 2013, 8, e72788. [Google Scholar] [CrossRef]
- Payne, D.C.; Vinjé, J.; Szilagyi, P.G.; Edwards, K.M.; Staat, M.A.; Weinberg, G.A.; Hall, C.B.; Chappell, J.; Bernstein, D.I.; Curns, A.T.; et al. Norovirus and medically attended gastroenteritis in U.S. children. N. Engl. J. Med. 2013, 368, 1121–1130. [Google Scholar] [CrossRef]
- Koo, H.L.; Neill, F.H.; Estes, M.K.; Munoz, F.M.; Cameron, A.; DuPont, H.L.; Atmar, R.L. Noroviruses: The most common pediatric viral enteric pathogen at a large university hospital after introduction of rotavirus vaccination. J. Pediatr. Infect. Dis. Soc. 2013, 2, 57–60. [Google Scholar] [CrossRef]
- Koo, H.L.; Ajami, N.; Atmar, R.L.; DuPont, H.L. Noroviruses: The leading cause of gastroenteritis worldwide. Discov. Med. 2010, 10, 61–70. [Google Scholar]
- Wohlgemuth, N.; Honce, R.; Schultz-Cherry, S. Astrovirus evolution and emergence. Infect. Genet. Evol. 2019, 69, 30–37. [Google Scholar] [CrossRef]
- Mehndiratta, M.M.; Mehndiratta, P.; Pande, R. Poliomyelitis. Neurohospitalist 2014, 4, 223–229. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Virgin, H.W. Kingdom-agnostic metagenomics and the importance of complete characterization of enteric microbial communities. Gastroenterology 2014, 146, 1459–1469. [Google Scholar] [CrossRef]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar] [CrossRef]
- Round, J.L.; Mazmanian, S.K. The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Gordon, J.I. Commensal host-bacterial relationships in the gut. Science 2001, 292, 1115–1118. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Sekirov, I.; Russell, S.L.; Antunes, L.C.M.; Finlay, B.B. Gut microbiota in health and disease. Physiol. Rev. 2010, 90, 859–904. [Google Scholar] [CrossRef]
- Kamada, N.; Chen, G.Y.; Inohara, N.; Núñez, G. Control of pathogens and pathobionts by the gut microbiota. Nat. Immunol. 2013, 14, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Macpherson, A.J.; Harris, N.L. Interactions between commensal intestinal bacteria and the immune system. Nat. Rev. Immunol. 2004, 4, 478–485. [Google Scholar] [CrossRef]
- Falk, P.G.; Hooper, L.V.; Midtvedt, T.; Gordon, J.I. Creating and maintaining the gastrointestinal ecosystem: What we know and need to know from gnotobiology. Microbiol. Mol. Biol. Rev. MMBR 1998, 62, 1157–1170. [Google Scholar]
- Bouskra, D.; Brézillon, C.; Bérard, M.; Werts, C.; Varona, R.; Boneca, I.G.; Eberl, G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 2008, 456, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Garrett, W.S. The gut microbiota and mucosal T cells. Front. Microbiol. 2011, 2, 111. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Case, L.K.; Kopaskie, K.; Kozlova, A.; MacDearmid, C.; Chervonsky, A.V.; Golovkina, T.V. Successful transmission of a retrovirus depends on the commensal microbiota. Science 2011, 334, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Kuss, S.K.; Best, G.T.; Etheredge, C.A.; Pruijssers, A.J.; Frierson, J.M.; Hooper, L.V.; Dermody, T.S.; Pfeiffer, J.K. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 2011, 334, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Tsubura, A.; Inaba, M.; Imai, S.; Murakami, A.; Oyaizu, N.; Yasumizu, R.; Ohnishi, Y.; Tanaka, H.; Morii, S.; Ikehara, S. Intervention of T-cells in transportation of mouse mammary tumor virus (milk factor) to mammary gland cells in vivo. Cancer Res. 1988, 48, 6555–6559. [Google Scholar] [PubMed]
- Jones, M.K.; Watanabe, M.; Zhu, S.; Graves, C.L.; Keyes, L.R.; Grau, K.R.; Gonzalez-Hernandez, M.B.; Iovine, N.M.; Wobus, C.E.; Vinjé, J.; et al. Enteric bacteria promote human and murine norovirus infection of B cells. Science 2014, 346, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Baldridge, M.T.; Nice, T.J.; McCune, B.T.; Yokoyama, C.C.; Kambal, A.; Wheadon, M.; Diamond, M.S.; Ivanova, Y.; Artyomov, M.; Virgin, H.W. Commensal microbes and interferon-λ determine persistence of enteric murine norovirus infection. Science 2015, 347, 266–269. [Google Scholar] [CrossRef] [PubMed]
- Kernbauer, E.; Ding, Y.; Cadwell, K. An enteric virus can replace the beneficial function of commensal bacteria. Nature 2014, 516, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, R.; Chassaing, B.; Zhang, B.; Gewirtz, A.T. Antibiotic treatment suppresses rotavirus infection and enhances specific humoral immunity. J. Infect. Dis. 2014, 210, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.K.; Jesudhasan, P.R.; Mayer, M.J.; Narbad, A.; Winter, S.E.; Pfeiffer, J.K. Bacteria facilitate enteric virus co-infection of mammalian cells and promote genetic recombination. Cell Host Microbe 2018, 23, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.K.; Yi, H.; Kearns, D.B.; Mainou, B.A. Bacteria and bacterial envelope components enhance mammalian reovirus thermostability. PLoS Pathog. 2017, 13, e1006768. [Google Scholar] [CrossRef] [PubMed]
- Almand, E.A.; Moore, M.D.; Outlaw, J.; Jaykus, L.-A. Human norovirus binding to select bacteria representative of the human gut microbiota. PLoS ONE 2017, 12, e0173124. [Google Scholar] [CrossRef] [PubMed]
- Miura, T.; Sano, D.; Suenaga, A.; Yoshimura, T.; Fuzawa, M.; Nakagomi, T.; Nakagomi, O.; Okabe, S. Histo-blood group antigen-like substances of human enteric bacteria as specific adsorbents for human noroviruses. J. Virol. 2013, 87, 9441–9451. [Google Scholar] [CrossRef] [PubMed]
- Wilks, J.; Lien, E.; Jacobson, A.N.; Fischbach, M.A.; Qureshi, N.; Chervonsky, A.V.; Golovkina, T.V. Mammalian lipopolysaccharide receptors incorporated into the retroviral envelope augment virus transmission. Cell Host Microbe 2015, 18, 456–462. [Google Scholar] [CrossRef]
- Tan, M.; Hegde, R.S.; Jiang, X. The P domain of norovirus capsid protein forms dimer and binds to histo-blood group antigen receptors. J. Virol. 2004, 78, 6233–6242. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Jesudhasan, P.R.; Pfeiffer, J.K. Bacterial lipopolysaccharide binding enhances virion stability and promotes environmental fitness of an enteric virus. Cell Host Microbe 2014, 15, 36–46. [Google Scholar] [CrossRef]
- Li, D.; Breiman, A.; le Pendu, J.; Uyttendaele, M. Binding to histo-blood group antigen-expressing bacteria protects human norovirus from acute heat stress. Front. Microbiol. 2015, 6, 659. [Google Scholar] [CrossRef] [PubMed]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.-L.; Qu, L.; et al. Replication of human noroviruses in stem cell–derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef]
- Springer, G.F.; Williamson, P.; Brandes, W.C. Blood group activity of gram-negative bacteria. J. Exp. Med. 1961, 113, 1077–1093. [Google Scholar] [CrossRef]
- Rasko, D.A.; Wang, G.; Monteiro, M.A.; Palcic, M.M. Synthesis of mono- and di-fucosylated type I Lewis blood group antigens by Helicobacter pylori. Eur. J. Biochem. 2000, 267, 6059–6066. [Google Scholar] [CrossRef]
- Yi, W.; Shao, J.; Zhu, L.; Li, M.; Singh, M.; Lu, Y.; Lin, S.; Li, H.; Ryu, K.; Shen, J.; et al. Escherichia coli O86 O-antigen biosynthetic gene cluster and stepwise enzymatic synthesis of human blood group B antigen tetrasaccharide. J. Am. Chem. Soc. 2005, 127, 2040–2041. [Google Scholar] [CrossRef]
- Orchard, R.C.; Wilen, C.B.; Doench, J.G.; Baldridge, M.T.; McCune, B.T.; Lee, Y.-C.J.; Lee, S.; Pruett-Miller, S.M.; Nelson, C.A.; Fremont, D.H.; et al. Discovery of a proteinaceous cellular receptor for a norovirus. Science 2016, 353, 933–936. [Google Scholar] [CrossRef]
- Haga, K.; Fujimoto, A.; Takai-Todaka, R.; Miki, M.; Doan, Y.H.; Murakami, K.; Yokoyama, M.; Murata, K.; Nakanishi, A.; Katayama, K. Functional receptor molecules CD300lf and CD300ld within the CD300 family enable murine noroviruses to infect cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6248–E6255. [Google Scholar] [CrossRef]
- Wilen, C.B.; Lee, S.; Hsieh, L.L.; Orchard, R.C.; Desai, C.; Hykes, B.L.; McAllaster, M.R.; Balce, D.R.; Feehley, T.; Brestoff, J.R.; et al. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science 2018, 360, 204–208. [Google Scholar] [CrossRef]
- Grau, K.R.; Roth, A.N.; Zhu, S.; Hernandez, A.; Colliou, N.; DiVita, B.B.; Philip, D.T.; Riffe, C.; Giasson, B.; Wallet, S.M.; et al. The major targets of acute norovirus infection are immune cells in the gut-associated lymphoid tissue. Nat. Microbiol. 2017, 2, 1586–1591. [Google Scholar] [CrossRef]
- Nelson, C.A.; Wilen, C.B.; Dai, Y.-N.; Orchard, R.C.; Kim, A.S.; Stegeman, R.A.; Hsieh, L.L.; Smith, T.J.; Virgin, H.W.; Fremont, D.H. Structural basis for murine norovirus engagement of bile acids and the CD300lf receptor. Proc. Natl. Acad. Sci. USA 2018, 115, E9201–E9210. [Google Scholar] [CrossRef]
- Chang, K.-O.; Sosnovtsev, S.V.; Belliot, G.; Kim, Y.; Saif, L.J.; Green, K.Y. Bile acids are essential for porcine enteric calicivirus replication in association with down-regulation of signal transducer and activator of transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 8733–8738. [Google Scholar] [CrossRef]
- Jude, B.A.; Pobezinskaya, Y.; Bishop, J.; Parke, S.; Medzhitov, R.M.; Chervonsky, A.V.; Golovkina, T.V. Subversion of the innate immune system by a retrovirus. Nat. Immunol. 2003, 4, 573–578. [Google Scholar] [CrossRef]
- Basic, M.; Keubler, L.M.; Buettner, M.; Achard, M.; Breves, G.; Schroder, B.; Smoczek, A.; Jorns, A.; Wedekind, D.; Zschemisch, N.H.; et al. Norovirus triggered microbiota-driven mucosal inflammation in interleukin 10-deficient mice. Inflamm. Bowel Dis. 2014, 20, 431–443. [Google Scholar] [CrossRef]
- Turula, H.; Bragazzi Cunha, J.; Mainou, B.A.; Ramakrishnan, S.K.; Wilke, C.A.; Gonzalez-Hernandez, M.B.; Pry, A.; Fava, J.; Bassis, C.M.; Edelman, J.; et al. Natural secretory immunoglobulins promote enteric viral infections. J. Virol. 2018, 92, e00826-18. [Google Scholar] [CrossRef]
- Jones, M.K.; Karst, S.M. Enteric viruses hitch a ride on the evolutionary highway. Cell Host Microbe 2018, 23, 5–6. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roth, A.N.; Grau, K.R.; Karst, S.M. Diverse Mechanisms Underlie Enhancement of Enteric Viruses by the Mammalian Intestinal Microbiota. Viruses 2019, 11, 760. https://doi.org/10.3390/v11080760
Roth AN, Grau KR, Karst SM. Diverse Mechanisms Underlie Enhancement of Enteric Viruses by the Mammalian Intestinal Microbiota. Viruses. 2019; 11(8):760. https://doi.org/10.3390/v11080760
Chicago/Turabian StyleRoth, Alexa N., Katrina R. Grau, and Stephanie M. Karst. 2019. "Diverse Mechanisms Underlie Enhancement of Enteric Viruses by the Mammalian Intestinal Microbiota" Viruses 11, no. 8: 760. https://doi.org/10.3390/v11080760
APA StyleRoth, A. N., Grau, K. R., & Karst, S. M. (2019). Diverse Mechanisms Underlie Enhancement of Enteric Viruses by the Mammalian Intestinal Microbiota. Viruses, 11(8), 760. https://doi.org/10.3390/v11080760