
Influence of Particle Size, Defect Density and Salts on the Dissolution and Early Hydration of a Model System (C3A + Quartz)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Dissolution and Early Hydration Kinetic Tracking
2.2. Characterization of Hydrated Samples
3. Results and Discussion
3.1. Role of Product Layer Formation on Early Reaction
3.2. Intrinsic Factors Affecting the Early Reaction
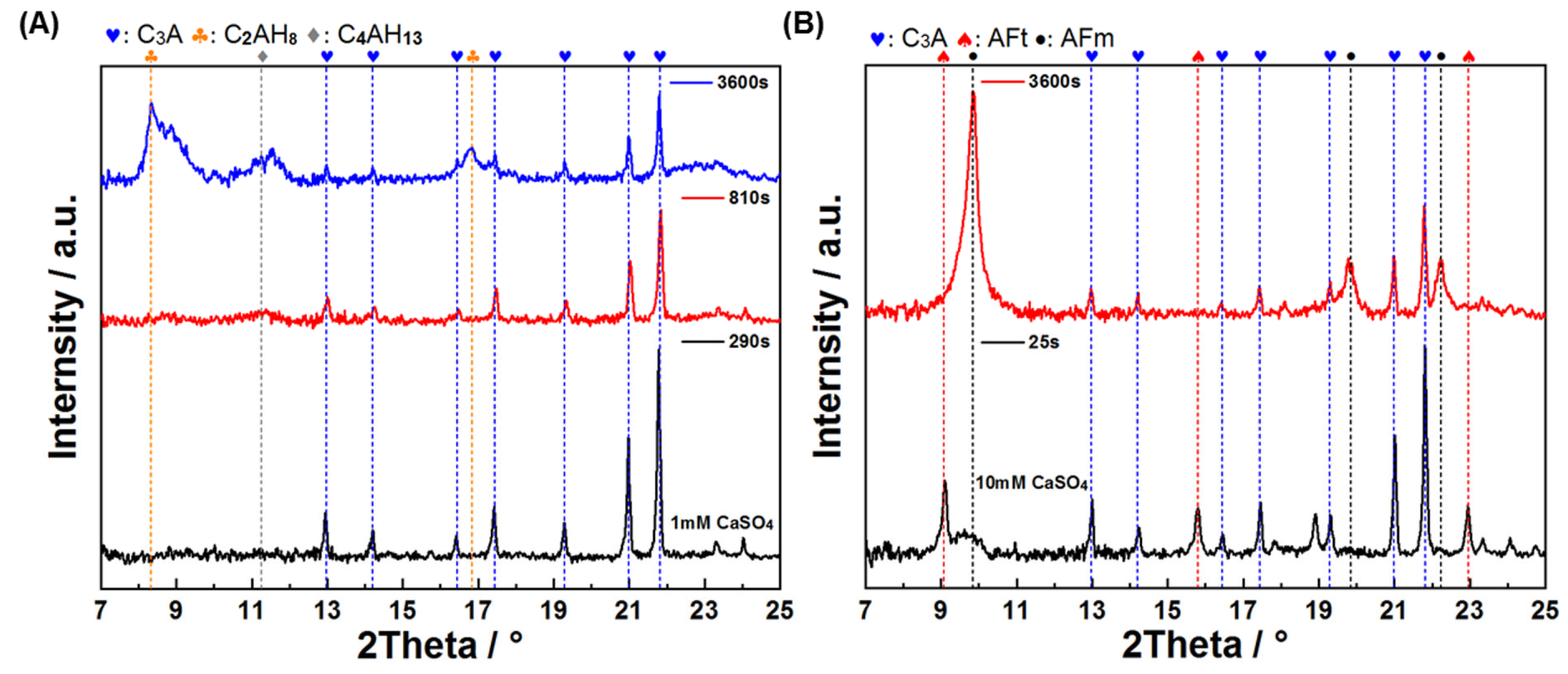
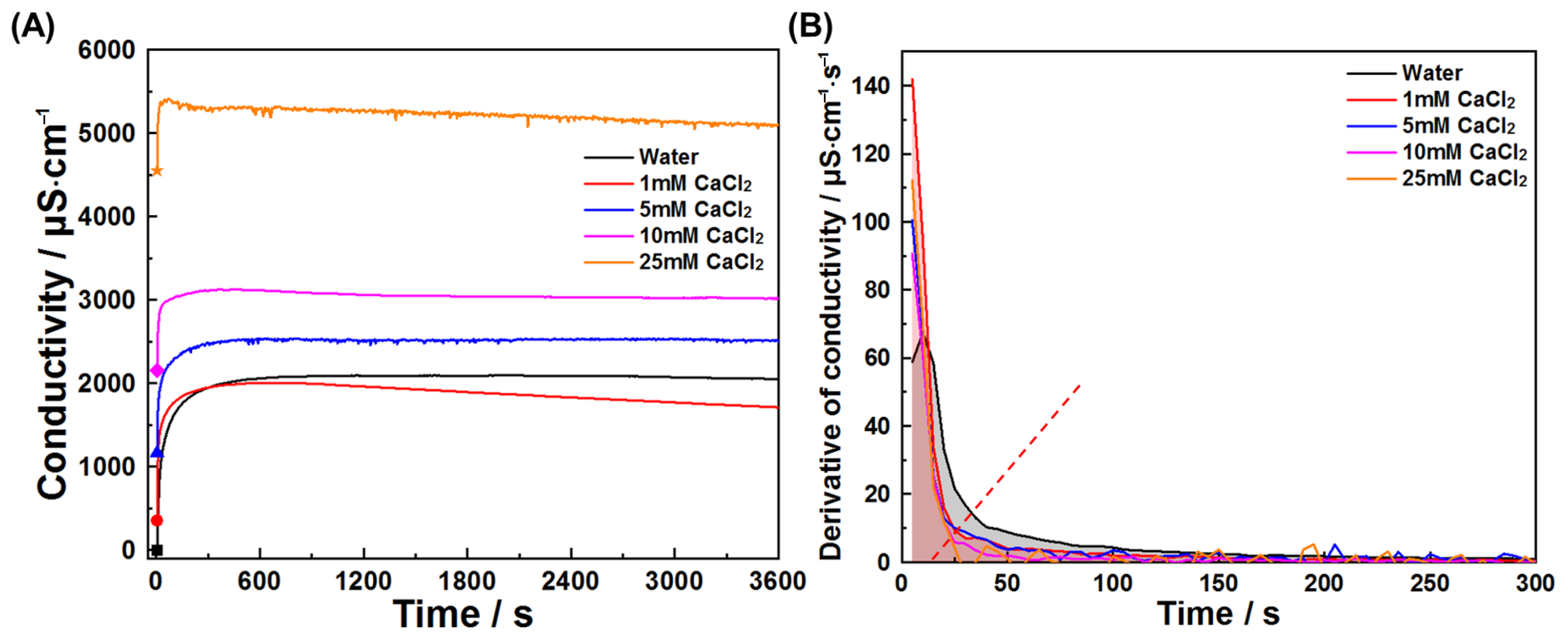
3.3. Sulfates and Chlorides Affected the Early Reaction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Taylor, H.F.W. Cement Chemistry, 2nd ed.; Thomas Telford: London, UK, 1997. [Google Scholar]
- Cuesta, A.; Morales-Cantero, A.; De la Torre, A.G.; Aranda, M.A.G. Recent Advances in C-S-H Nucleation Seeding for Improving Cement Performances. Materials 2023, 16, 1462. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Wang, J.; Cui, S.; Liu, H.; Wang, Y. Hydration, Microstructure and Properties of Cement-Based Materials in the Presence of Tertiary Alkanolamines: A Review. Constr. Build. Mater. 2024, 424, 135954. [Google Scholar] [CrossRef]
- Scrivener, K.; Ouzia, A.; Juilland, P.; Kunhi Mohamed, A. Advances in Understanding Cement Hydration Mechanisms. Cem. Concr. Res. 2019, 124, 105823. [Google Scholar] [CrossRef]
- Scrivener, K.L.; Matschei, T.; Georget, F.; Juilland, P.; Mohamed, A.K. Advances in Hydration and Thermodynamics of Cementitious Systems. Cem. Concr. Res. 2023, 174, 107332. [Google Scholar] [CrossRef]
- Nicoleau, L.; Nonat, A. A New View on the Kinetics of Tricalcium Silicate Hydration. Cem. Concr. Res. 2016, 86, 1–11. [Google Scholar] [CrossRef]
- Ye, S.; Feng, P.; Liu, Y.; Liu, J.; Bullard, J.W. In Situ Nano-Scale Observation of C3A Dissolution in Water. Cem. Concr. Res. 2020, 132, 106044. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, M.; Li, H.; Luo, G.; Tan, H.; Liu, Q. Hydration Kinetics of Portland Cement Shifting from Silicate to Aluminate Dominance Based on Multi-Mineral Reactions and Interactions. Mater. Des. 2023, 233, 112228. [Google Scholar] [CrossRef]
- Joseph, S.; Skibsted, J.; Cizer, Ö. A Quantitative Study of the C3A Hydration. Cem. Concr. Res. 2019, 115, 145–159. [Google Scholar] [CrossRef]
- Hirsch, T.; Matschei, T.; Stephan, D. The Hydration of Tricalcium Aluminate (Ca3Al2O6) in Portland Cement-Related Systems: A Review. Cem. Concr. Res. 2023, 168, 107150. [Google Scholar] [CrossRef]
- Ming, X.; Si, W.; Yu, Q.; Sun, Z.; Qiu, G.; Cao, M.; Li, Y.; Li, Z. Molecular Insight into the Initial Hydration of Tricalcium Aluminate. Nat. Commun. 2024, 15, 2929. [Google Scholar] [CrossRef]
- Mondal, P.; Jeffery, J.W. The Crystal Structure of Tricalcium Aluminate, Ca3Al2O6. Acta Crystallogr. Sect. B 1975, B31, 689–697. [Google Scholar] [CrossRef]
- Shin, G.Y.; Glasser, F.P. Interdependence of Sodium and Potassium Substitution in Tricalcium Aluminate. Cem. Concr. Res. 1983, 13, 135–140. [Google Scholar] [CrossRef]
- Myers, R.J.; Geng, G.; Rodriguez, E.D.; da Rosa, P.; Kirchheim, A.P.; Monteiro, P.J.M. Solution Chemistry of Cubic and Orthorhombic Tricalcium Aluminate Hydration. Cem. Concr. Res. 2017, 100, 176–185. [Google Scholar] [CrossRef]
- Pourchet, S.; Regnaud, L.; Perez, J.P.; Nonat, A. Early C3A Hydration in the Presence of Different Kinds of Calcium Sulfate. Cem. Concr. Res. 2009, 39, 989–996. [Google Scholar] [CrossRef]
- Ye, S.; Feng, P.; Liu, Y.; Liu, J.; Bullard, J.W. Dissolution and Early Hydration of Tricalcium Aluminate in Aqueous Sulfate Solutions. Cem. Concr. Res. 2020, 137, 106191. [Google Scholar] [CrossRef]
- Quennoz, A.; Scrivener, K.L. Hydration of C3A-Gypsum Systems. Cem. Concr. Res. 2012, 42, 1032–1041. [Google Scholar] [CrossRef]
- He, H.; Cao, J.; Duan, N. Defects and Their Behaviors in Mineral Dissolution under Water Environment: A Review. Sci. Total Environ. 2019, 651, 2208–2217. [Google Scholar] [CrossRef]
- Kurganskaya, I.; Luttge, A. Probability Distributions of Mineral Dissolution Rates: The Role of Lattice Defects. Front. Water 2023, 5, 1225837. [Google Scholar] [CrossRef]
- Kim, C.B.; Choi, S.C. Quantitative Interpretation of Cooling Rate of Clinker and It’s Effects on the Cement Strength Development. J. Korean Ceram. Soc. 2007, 44, 224–229. [Google Scholar] [CrossRef]
- Novosyolov, A.G.; Klassen, V.K.; Novoselova, I.N.; Dorokhova, E.S. Influence of Cooling on the Quality of Clinker with Different Content. Int. J. Appl. Eng. Res. 2015, 10, 42688–42691. [Google Scholar]
- Echigo, T.; Aruguete, D.M.; Murayama, M.; Hochella, M.F. Influence of Size, Morphology, Surface Structure, and Aggregation State on Reductive Dissolution of Hematite Nanoparticles with Ascorbic Acid. Geochim. Cosmochim. Acta 2012, 90, 149–162. [Google Scholar] [CrossRef]
- Pollet-Villard, M.; Daval, D.; Fritz, B.; Knauss, K.G.; Schäfer, G.; Ackerer, P. Influence of Etch Pit Development on the Surface Area and Dissolution Kinetics of the Orthoclase (001) Surface. Chem. Geol. 2016, 447, 79–92. [Google Scholar] [CrossRef]
- Lange, I.; Toro, M.; Arvidson, R.S.; Kurganskaya, I.; Luttge, A. The Role of Crystal Heterogeneity in Alkali Feldspar Dissolution Kinetics. Geochim. Cosmochim. Acta 2021, 309, 329–351. [Google Scholar] [CrossRef]
- Quennoz, A.; Scrivener, K.L. Interactions between Alite and C3A-Gypsum Hydrations in Model Cements. Cem. Concr. Res. 2013, 44, 46–54. [Google Scholar] [CrossRef]
- Zunino, F.; Scrivener, K. Factors Influencing the Sulfate Balance in Pure Phase C3S/C3A Systems. Cem. Concr. Res. 2020, 133, 106085. [Google Scholar] [CrossRef]
- Zunino, F.; Scrivener, K. The Influence of Sulfate Addition on Hydration Kinetics and C-S-H Morphology of C3S and C3S/C3A Systems. Cem. Concr. Res. 2022, 160, 106930. [Google Scholar] [CrossRef]
- Kapeluszna, E.; Kotwica, L. New Insights into the Role of Highly Reactive Pozzolans in the Early Hydration Process of C3S and C3A Monitored by Conductometry, Calorimetry, Phase Composition and Microstructure Analyses. Constr. Build. Mater. 2024, 452, 138950. [Google Scholar] [CrossRef]
- Andrade, S.; De Matos, P.R.; De, A.G.; Campos, C.E.M.; Torres, S.M.; Monteiro, P.J.M.; Paula, A. Hydration and Interactions between Pure and Doped C 3 S and C 3 A in the Presence of Different Calcium Sulfates. Cem. Concr. Res. 2022, 159, 106893. [Google Scholar] [CrossRef]
- Black, L.; Breen, C.; Yarwood, J.; Phipps, J.; Maitland, G. In Situ Raman Analysis of Hydrating C3A and C4AF Pastes in Presence and Absence of Sulphate. Adv. Appl. Ceram. 2006, 105, 209–216. [Google Scholar] [CrossRef]
- Zhang, G.; Ren, Q.; He, J.; Jiang, S.; Cheng, X.; Yu, Y.; Huang, S.; Zhang, C.; Zhou, M. New Understanding of Early Hydration of C4AF under Surface Vitrification. Powder Technol. 2021, 377, 372–378. [Google Scholar] [CrossRef]
- Petra, M.; Mazur, A.S. Long-Term Sulfate Resistance of Synthesized Cement Systems with Variable C3A/C4AF Ratio at Low Temperature or Ambient Conditions: Insights into the Crystalline and Amorphous Phase Assemblage. Cem. Concr. Res. 2022, 160, 106902. [Google Scholar]
- Axthammer, D.; Lange, T.; Dengler, J.; Gädt, T. Early Hydration and Viscoelastic Properties of Tricalcium Aluminate Pastes Influenced by Soluble Sodium Salts. Cem. Concr. Res. 2025, 190, 107788. [Google Scholar] [CrossRef]
- Maier, M.; Scherb, S.; Neißer-Deiters, A.; Beuntner, N.; Thienel, K.-C. Hydration of Cubic Tricalcium Aluminate in the Presence of Calcined Clays. J. Am. Ceram. Soc. 2021, 104, 3619–3631. [Google Scholar] [CrossRef]
- Wesselsky, A.; Jensen, O.M. Synthesis of Pure Portland Cement Phases. Cem. Concr. Res. 2009, 39, 973–980. [Google Scholar] [CrossRef]
- Ye, S.; Feng, P.; Liu, J. Dissolution and Early Hydration of Tetracalcium Aluminoferrite (C4AF) in Water and in Aqueous Sulfate Solutions. Cem. Concr. Res. 2024, 186, 107676. [Google Scholar] [CrossRef]
- Ouyang, X.; Xu, S.; Ma, Y.; Ye, G. Effect of Aggregate Chemical Properties on Nucleation and Growth of Calciun SilicateHydrate. J. Chin. Ceram. Soc. 2021, 49, 972–979. [Google Scholar]
- Ye, S.; Feng, P.; Lu, J.; Zhao, L.; Liu, Q.; Zhang, Q.; Liu, J.; Bullard, J.W. Solubility of Tricalcium Aluminate from 10 °C to 40 °C. Cem. Concr. Res. 2022, 162, 106989. [Google Scholar] [CrossRef]
- Nicoleau, L.; Nonat, A.; Perrey, D. The Di- and Tricalcium Silicate Dissolutions. Cem. Concr. Res. 2013, 47, 14–30. [Google Scholar] [CrossRef]
- Juilland, P.; Gallucci, E.; Flatt, R.; Scrivener, K. Dissolution Theory Applied to the Induction Period in Alite Hydration. Cem. Concr. Res. 2010, 40, 831–844. [Google Scholar] [CrossRef]
- Scrivener, K.; Snellings, R.; Lothenbach, B. A Practical Guide to Microstructural Analysis of Cementitious Materials; CRC Press: Boca Raton, FL, USA, 2016; ISBN 9781498738675. [Google Scholar]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ye, S.; Feng, P. Influence of Particle Size, Defect Density and Salts on the Dissolution and Early Hydration of a Model System (C3A + Quartz). Materials 2025, 18, 3560. https://doi.org/10.3390/ma18153560
Ye S, Feng P. Influence of Particle Size, Defect Density and Salts on the Dissolution and Early Hydration of a Model System (C3A + Quartz). Materials. 2025; 18(15):3560. https://doi.org/10.3390/ma18153560
Chicago/Turabian StyleYe, Shaoxiong, and Pan Feng. 2025. "Influence of Particle Size, Defect Density and Salts on the Dissolution and Early Hydration of a Model System (C3A + Quartz)" Materials 18, no. 15: 3560. https://doi.org/10.3390/ma18153560
APA StyleYe, S., & Feng, P. (2025). Influence of Particle Size, Defect Density and Salts on the Dissolution and Early Hydration of a Model System (C3A + Quartz). Materials, 18(15), 3560. https://doi.org/10.3390/ma18153560