Abstract
Chemical investigation of the marine sponge-derived Streptomyces xiamenensis 1310KO-148 afforded six polycyclic tetramate macrolactams (PTMs), including three known compounds (1–3) and three previously undescribed chlorohydrin-containing analogues, chlokamycins B–D (4–6). Their planar structures were elucidated by extensive analysis of 1D and 2D NMR spectra and HR-ESIMS data, while the relative configurations were assigned using NOESY correlations. The absolute configurations were further confirmed by electronic circular dichroism (ECD) calculations. Compounds 3–6 exhibited significant cytotoxic activity against 14 human cancer cell lines (GI50 = 2.68–24.92 μM) and antibacterial activity against Staphylococcus aureus (MIC = 16.00–32.00 μg/mL) and Micrococcus luteus (MIC = 4.00–32.00 μg/mL) among six tested bacterial strains.
1. Introduction
Tetramic acid-derived natural products constitute a prominent class of microbial secondary metabolites that are widely produced by bacteria and fungi, particularly members of the actinomycetes. These compounds are characterized by a 2,4-pyrrolidinedione scaffold and have attracted considerable interest because of their pronounced structural diversity and broad spectrum of biological activities, including antibacterial, antifungal, antiviral, and cytotoxic properties [1,2]. Structural diversification within this family commonly arises from various biosynthetic tailoring processes, generating multiple subclasses that exhibit distinct molecular frameworks and biological functions. Among these, polycyclic tetramate macrolactams (PTMs) constitute a structurally unique group of tetramic acid metabolites.
PTMs represent a distinct group of natural products characterized by a rigid three-dimensional framework comprising a large macrolactam ring, a tetramate core, and fused polycyclic rings. Ikarugamycin (1), the earliest identified compound in this class, was first isolated from Streptomyces phaeochromogenes in 1972 [3] and has been extensively studied for its strong antimicrobial and anticancer properties [4,5,6,7,8]. Subsequent studies have revealed that the structural and biological diversity of PTMs is expanded by diverse substituents and extensive modification processes, which collectively contribute to their pharmacological potential [8,9,10,11].
According to previous studies, capsimycin C from S. xiamenensis 318 bearing hydroxyl groups at the C-7 and C-8 positions (Figure S1) exhibited no cytotoxic activity at 10 μM against eight pancreatic cancer cell lines: HPAC, Patu8988, BxPC-3, PANC-1, AsPC-1, Capan-2, CFPAC-1, and MiaPaca-2 [12]. Similarly, hydroxyikarugamycin B from Streptomyces sp. SCSIO 40060 showed no cytotoxicity at 100 μM against SF268, NCI-H460, and HepG2 cell lines [13]. In particular, halogenated PTMs are extremely rare in nature. To date, only three chloride-containing PTMs have been reported, all of which were isolated from Streptomyces species: capsimycin D (3) [12] and chlokamycin [14], both possessing a 5/6/5 ring system, and pactamide F [8] with a 5/5/6 ring system (Figure S1). Cytotoxic activity has been reported for chlokamycin (IC50 = 24.7–33.5 μM against Jurkat and HCT116 cell lines) and pactamide F (IC50 = 2.65–2.85 μM against SF-268, MCF-7, NCI-H460, and Hep-G2 cell lines). The absolute configuration of chlokamycin remains unresolved, and pactamide F was produced from S. pactum SCSIO 02999 by promoter engineering and heterologous expression.
Given the structural diversity, limited occurrence, and incomplete biological evaluation of chloride-containing PTMs, further studies are required to clarify their chemical characteristics and pharmacological potential. Herein, we describe the isolation and identification of the chlorohydrin-containing 5/6/5 ring system of PTMs (3–6) from S. xiamenensis 1310KO-148 (Figure 1). In addition, compounds 3–6 were evaluated for their cytotoxicity and antibacterial activity, and a structure–activity relationship (SAR) was examined based on the positions of the chlorine and methoxy substitutions.
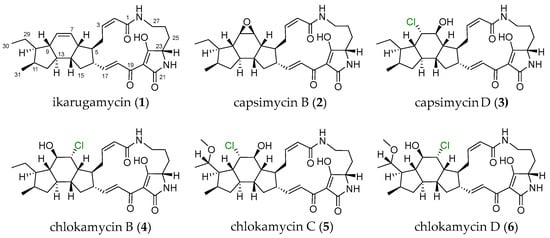
Figure 1.
Structures of PTMs (1–6) produced by S. xiamenensis 1310KO-148.
2. Results
2.1. Structure Elucidation of New Compounds
Extraction of the culture broth of the marine-derived actinomycete S. xiamenensis 1310KO-148 with ethyl acetate (EtOAc) and chemical investigation of the extract afforded three previously undescribed chlorohydrin-containing PTMs (4–6) along with three known analogues (1–3). Compounds 1–3 were identified as ikarugamycin (1), capsimycin B (2), and capsimycin D (3), respectively. Their structures were confirmed by comparing their 1H and 13C NMR, 2D NMR, and MS data with previously reported data (Figures S2–S20 and Tables S1–S3) [12,15].
Chlokamycin B (4) was obtained as a yellow amorphous powder. The molecular formula of 4 was deduced as C29H39N2O5Cl based on the HR-ESIMS peaks at m/z 531.2626 [M+H]+ (calcd for C29H40N2O5Cl, 531.2626) and 553.2446 [M+Na]+ (calcd for C29H39N2O5NaCl, 553.2445), indicating 11 degrees of unsaturation. Owing to the solubility issue of 4, its 2D NMR spectra for structural analysis were acquired in 90% CDCl3/CD3OD (Table 1). The 1H NMR spectrum of 4 exhibited two types methyl protons (δH 0.81, d, J = 6.5 Hz; δH 0.87, t, J = 7.3, Hz), four olefinic protons (δH 5.79, d, J = 11.4 Hz; δH 6.00, t, J = 11.4 Hz; δH 6.76, dd, J = 15.4, 10.1 Hz; δH 7.07, d, J = 15.4 Hz), three heteroatom-connected methine protons (δH 3.81, dd, J = 4.8, 3.0 Hz; δH 3.91, dd, J = 7.6, 4.1 Hz; δH 4.10, t, J = 4.1 Hz), eight sp3 methine protons (δH 1.02, td, J = 11.4, 7.6 Hz; δH 1.52, m; δH 1.77, td, J = 11.4, 7.4 Hz; δH 1.83, m; δH 2.16, m; δH 2.21, m; δH 2.43, m; δH 2.52, m), and seven sp3 methylene protons (δH 0.61, 2.11, m; δH 1.01, 1.50, m; δH 1.26, 1.93, m; δH 1.32, 1.71, m; δH 1.73, 1.99, m; δH 2.50, 3.35, m; δH 2.54, 3.58, m) (Table 1 and Figure S20). From the 13C NMR and HSQC spectra, two methyl carbons (δC 13.2, C-30; δC 17.9, C-31), four olefinic carbons (δC 122.8, C-18; δC 123.8, C-2; δC 141.8, C-3; δC 151.8, C-17), three heteroatom-connected methine carbons (δC 61.6, C-23; δC 68.5, C-7; δC 76.3, C-8), eight sp3 methine carbons (δC 32.4, C-11; δC 43.1, C-13; δC 43.7, C-6; δC 45.3, C-5; δC 46.1, C-14; δC 49.6, C-16; δC 50.2, C-10; δC 52.6, C-9), seven sp3 methylene carbons (δC 21.2, C-26; δC 21.7, C-29; δC 27.0, C-4; δC 27.4, C-25; δC 36.6, C-15; δC 39.0, C-27; δC 39.1, C-12), and five nonprotonated sp2 carbons (δC 101.0, C-20; δC 167.2, C-1; δC 173.6, C-19; δC 175.6, C-21; δC 197.3, C-24) were identified (Table 1). The 1H and 13C NMR spectra of 4 closely resembled those of capsimycin D (3), which belongs to the 5/6/5 type of chloride-containing PTMs (Table 1, Table 2 and Table S3). A comprehensive comparison of the NMR data for 4 and 3 indicates that 4 differs from 3 in the positions of a chlorine atom (δH 4.10, t, J = 4.1 Hz; δC 68.5, C-7) and a hydroxy group (δH 3.91, dd, J = 7.6, 4.1 Hz; δC 76.3, C-8). This conclusion was further supported by the 1H–1H COSY correlations of H-6 (δH 2.52)/H-7 (δH 4.10), H-6 (δH 2.52)/H-14 (δH 1.83), H-8 (δH 3.91)/H-9 (δH 1.02), and H-9 (δH 1.02)/H-10 (δH 1.52). In addition, key HMBC correlations from H-7 (δH 4.10) to C-5 (δC 45.3)/C-9 (δC 52.6) and from H-8 (δH 3.91) to C-6 (δC 43.7)/C-7 (δC 68.5)/C-10 (δC 50.2)/C-13 (δC 43.1) further confirmed the structural assignment (Figure 2). Thus, the planar structure of 4 was identified as a new positional isomer of 3 (Figure 1).

Table 1.
1H NMR (600 MHz) data of 3–6.
Table 2.
13C NMR (150 MHz) data of 3–6.
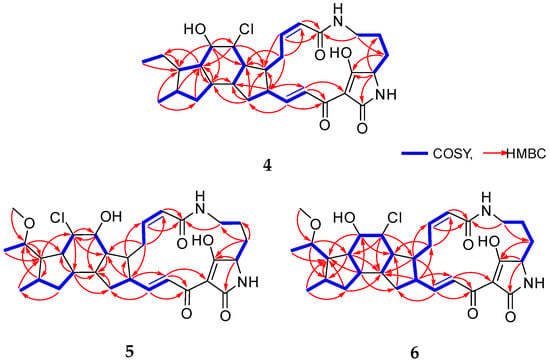
Figure 2.
Key 1H–1H COSY and HMBC correlations of 4–6.
The coupling constant between the olefinic protons H-2 (δH 5.79, J = 11.4 Hz) and H-3 (δH 6.00, J = 11.4 Hz) was indicative of a Z-configured double bond, whereas that between H-17 (δH 6.76, J = 15.4, 10.1 Hz) and H-18 (δH 7.07, J = 15.4 Hz) supported an E-configuration (Table 1). The relative configuration of the 5/6/5 tricyclic ring system in 4 was determined based on the NOESY spectrum. The NOE correlations of H-8 (δH 3.91) with H-10 (δH 1.52)/H-13 (δH 1.77), H-13 (δH 1.77) with H-8 (δH 3.91)/H-11 (δH 2.16), and H-5 (δH 2.21) with H-17 (δH 6.76) established that H-5, H-8, H-10, H-11, and H-13 were located on the same side of the 5/6/5 tricyclic unit (blue dashed curves for 4, Figure 3). In contrast, the correlations of H-9 (δH 1.02) with H-6 (δH 2.52)/H-7 (δH 4.10)/H-14 (δH 1.83)/H-29 (δH 1.32, 1.71)/H-31 (δH 0.81), H-6 (δH 2.52) with H-3 (δH 6.00)/H-9 (δH 1.02), and H-16 (δH 2.43) with H-14 (δH 1.83)/H-18 (δH 7.07) indicated that H-6, H-7, H-9, H-14, and H-16 were positioned on the opposite side (pink curves for 4, Figure 3). These spatial arrangements were further corroborated by coupling constant analysis. The large coupling constant between H-8 (dd, J = 7.6, 4.1 Hz) and H-9 (td, J = 11.4, 7.6 Hz), between H-9 (dd, J = 11.4, 7.6 Hz) and H-13 (td, J = 11.4, 7.4 Hz) was consistent with trans-diaxial orientation across the C-8/C-9 and C-9/C-13 bonds. Moreover, a key NOESY correlation of H-9 with H-7, along with the small coupling constant between H-7 (t, J = 4.1 Hz) and H-8 (dd, J = 7.6, 4.1 Hz), suggested a trans (axial–equatorial) orientation across the C-7/C-8 bond. These findings collectively implied trans–cis–trans A/B–B/C–C/D ring fusions [12,16]. Thus, the relative configuration of 4 was elucidated as 5S*,6R*,7R*,8R*,9S*,10R*,11R*,13R*,14R*,16S* for the 5/6/5 tricyclic unit of 4.
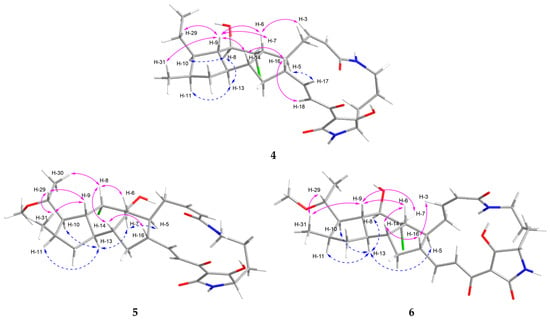
Figure 3.
NOESY correlations of 4–6.
To determine the absolute configuration of 4, additional ECD calculations were conducted for three possible configurations of (5S,6R,7R,8R,9S,10R,11R,13R,14R,16S,23S)-4A, (5R,6S,7S,8S,9R,10S,11S,13S,14S,16R,23R)-4B, and (5S,6R,7R,8R,9S,10R,11R,13R,14R,16S,23R)-4C using the TDDFT at the B3LYP/6-311G+(d,p) level of method (Figure 4B). The results of the 5/6/5 tricyclic ring system in 4 demonstrated good agreement between the experimental ECD spectrum and the calculated spectrum of isomer 4A with the (5S,6R,7R,8R,9S,10R,11R,13R,14R,16S,23S-4) configuration (Figure 4B). Furthermore, the absolute configuration at C-23 was assigned as 23S based on comparison with the calculated ECD spectra of conformers 4A and 4C (Figure 4B). In previously reported PTMs, the ornithine unit was derived from L-ornithine, as evidenced by the S-configuration at C-23 [9,15,17]. This fact supported the conclusion that the ornithine moiety in 4 also originates from L-ornithine. Thus, the structure of 4 was elucidated as a new isomer of 3, and 4 was named chlokamycin B, a previously undescribed chlorohydrin-containing PTMs metabolite.
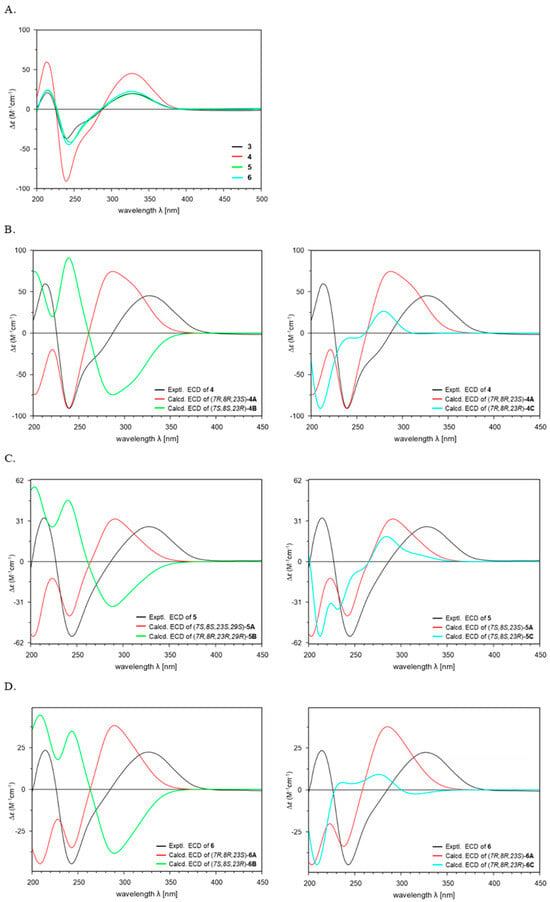
Figure 4.
Comparison of the experimental ECD spectra of 3–6 (A). Experimental and Calculated ECD spectra of 4–6 (B–D).
Chlokamycin C (5) was isolated as a yellow amorphous powder. The molecular formula of 5 was identified as C30H40N2O6Cl by the HR-ESIMS peaks at m/z 561.2733 [M+H]+ (calcd for C30H42N2O6Cl, 561.2731) and 583.2551 [M+Na]+ (calcd for C30H41N2O6NaCl, 583.2551), accounting for 11 degrees of unsaturation. Comparison of the NMR data for 5 with those of capsimycin D (3) demonstrated that their structures were highly similar. The key difference was the presence of a methoxy group (δH 3.30, s; δC 56.1; C-32) attached to C-29 (δC 78.7) in 5 (Table 1 and Table 2). The methylene carbon signal at C-29 (δC 22.3) in 3 was replaced by an oxygenated carbon (δC 78.7) in 5. The location of the methoxy substituent was assigned to the C-29 position based on the HMBC correlation from H-32 (δH 3.30) to C-29 (δC 78.7), and the COSY correlation between H-29 (δH 3.43, m) and H-30 (δH 1.21, d, J = 6.2 Hz) (Figure 2).
The relatively small vicinal coupling constant (J2,3 = 11.5 Hz) across the C-2/C-3 double bond supported the assignment of a Z-configuration, whereas the large vicinal coupling constant (J17,18 = 15.4 Hz) across the C-17/C-18 double bond indicated an E-configuration (Table 1). The NOE correlations of H-5 (δH 2.09) with H-7 (δH 4.11) and H-13 (δH 1.65) with H-7 (δH 4.11)/H-10 (δH 2.08)/H-11 (δH 2.24) indicated that H-5, H-7, H-10, H-11, and H-13 were orientated in the same direction, as shown by the blue dashed curves (Figure 3). Conversely, the cross-peaks of H-8 (δH 4.32) with H-6 (δH 2.13)/H-14 (δH 2.07)/H-30 (δH 1.21), H-9 (δH 2.10) with H-29 (δH 3.43)/H-31 (δH 1.04), H-16 (δH 2.38) with H-14 (δH 2.07), and H-29 (δH 3.43) with H-9 (δH 2.10)/H-31 (δH 1.04) supported that H-6, H-8, H-9, H-14, H-16, and H-29 were located on the other side, as represented by the pink curves for 5 in Figure 3. Thus, these results suggested that 5 possesses a relative configuration of 5S*,6R*,7S*,8S*,9R*,10R*,11R*,13R*,14R*,16S*,29S*.
To determine the absolute configuration, theoretical calculations of the ECD spectra of 5 were performed. The calculated ECD spectra of 5 were generated for three possible stereoisomers (5S,6R,7S,8S,9R,10R,11R,13R,14R,16S,23S,29S-5A; 5R,6S,7R,8R,9S,10S,11S,13S,14S,16R,23R,29R-5B; 5S,6R,7S,8S,9R,10R,11R,13R,14R,16S,23R,29S-5C) (Figure 4C). The absolute configuration of 5 (5S,6R,7S,8S,9R,10R,11R,13R,14R,16S,23S,29S) was established by comparison of the calculated and experimental ECD spectra (Figure 4C). Accordingly, the structure of 5 was determined as 29-methoxy capsimycin D, and 5 was named chlokamycin C.
Chlokamycin D (6) was obtained as a yellow amorphous powder. Its molecular formula was established as C30H41N2O6Cl based on the HR-ESIMS peaks at m/z 561.2730 [M+H]+ (calcd for C30H42N2O6Cl, 561.2731) and 583.2550 [M+Na]+ (calcd for C30H41N2O6NaCl, 583.2551), corresponding to 11 degrees of unsaturation. The 1H and 13C NMR data of 6 closely resembled those of 5, with notable differences observed at the positions corresponding to a chlorine atom (δH 4.23, t, J = 5.0 Hz; δC 68.6, C-7) and a hydroxy group (δH 3.90, dd, J = 8.0, 5.0 Hz; δC 77.3, C-8) (Table 1 and Table 2). This assignment was further supported by HMBC correlations from H-7 (δH 4.23) to C-5 (δC 46.8)/C-9 (δC 51.8)/C-14 (δC 46.7), from H-8 (δH 3.99) to C-6 (δC 45.8)/C-10 (δC 52.2), and from H-13 (δH 1.87) to C-8 (δC 77.3)/C-10 (δC 52.2) (Figure 2). In addition, the presence of an extra set of signals corresponding to a methoxy group (δH 3.36, s; δC 56.3, C-32) in 6, compared to those in 4, indicated that 6 is a 29-methoxylated derivative of 4 (Table 1 and Table 2).
The configurations of the C-2 (Z, ∆2,3) and C-17 (E, ∆17,18) double bonds in 6 were determined from the coupling constants J2,3 = 11.3 Hz and J17,18 = 15.4 Hz, respectively (Table 1). The NOE correlations showed that 6 has the same relative configuration as 4, featuring trans–cis–trans ring fusions (Figure 3). The relative configuration at C-29 was deduced as 29S* based on the NOE correlations of H-29 with H-31 and H-31 with H-9.
Theoretical ECD calculations were conducted on three possible stereoisomers of 6 (5S,6R,7R,8R,9R,10R,11R,13R,14R,16S,23S,29S-6A, 5R,6S,7S,8S,9S,10S,11S,13S,14S,16R,23R,29R-6B, and 5S,6R,7R,8R,9R,10R,11R,13R,14R,16S,23R,29S-6C) to assign the absolute configuration (Figure 4D). As shown in Figure 4D, the experimental ECD curve of 6 closely matched the calculated spectrum for the 5S,6R,7R,8R,9R,10R,11R,13R,14R,16S,23S,29S-6A isomer. Overall, these results indicated that 6 is a 29-methoxylated derivative of chlokamycin C (5), and 6 was designated chlokamycin D.
The biosynthetic pathway of PTMs has been broadly described as involving the assembly of three distinct fragments: one derived from L-ornithine and two C12 chains originating from six acetate units, consistent with what is now recognized as a hybrid polyketide synthase-nonribosomal peptide synthetase (PKS-NRPS) system [9,11,17,18,19]. As illustrated in Figure 5, compounds 1–6 are proposed to originate from a common PTM biosynthetic pathway in S. xiamenensis 1310KO-148. The characteristic 5/6/5 tricyclic core of ikarugamycin (1), the first identified PTM, is plausibly biosynthesized through IkaA-mediated assembly of two PKS-derived polyketide chains via consecutive condensation reactions. The resulting intermediates are subsequently processed by the oxidoreductase IkaB, followed by a cyclization step involving a Diels–Alder reaction, in which the alcohol dehydrogenase IkaC is thought to participate indirectly. Furthermore, IkaD introduces two modifications to ikarugamycin (1): epoxidation of the C-7/C-8 double bond to yield capsimycin B (2), and hydroxylation followed by methylation at the C-29 position, potentially leading to the formation of capsimycin [12,13,20]. The epoxide moiety present in the intermediate 2 may serve as a key precursor for the formation of the chlorohydrin functionality observed in compounds 3–6. In this scenario, nucleophilic attack by chloride on the oxirane ring would result in regioselective epoxide opening and subsequent formation of the corresponding chlorohydrin derivatives. Considering the conformational rigidity of the PTM framework, such nucleophilic attack is likely to occur from the axial direction of the epoxide. This stereochemical course is consistent with the Fürst–Plattner rule, which predicts preferential trans-diaxial ring opening of cyclic epoxides by nucleophiles [21]. Although the specific enzyme responsible for this transformation has not yet been identified in the genome of S. xiamenensis 1310KO-148, similar enzymatic processes leading to epoxide-to-halohydrin conversion have been suggested in the biosynthetic pathways of several halogenated natural products [22,23,24,25]. From the 70 L culture extract of strain 1310KO-148, compounds 3 (42.4 mg), 4 (8.0 mg), 5 (13.2 mg), and 6 (7.4 mg) were isolated. These results suggest that chlorohydrin formation at the C-7/C-8 double bond of the PTMs produced by this strain occurs regioselectively, with a preference for chlorination at C-8.
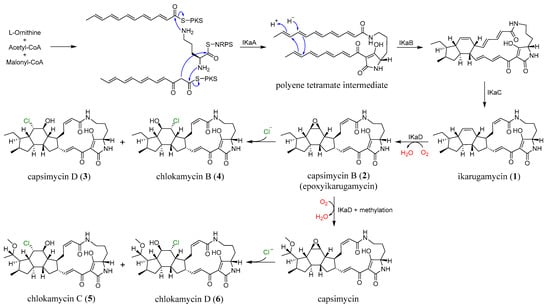
Figure 5.
The plausible biosynthetic pathway of 1–6.
2.2. Bioactivity Evaluation of Compounds
The biological activities of 3–6 were summarized in Table 3 and Table 4. Cytotoxicity was evaluated against six solid and eight blood cancer cell lines (Table 3 and Tables S16 and S17). Compounds 3–6 showed significant cytotoxicity across all 14 cancer cell lines. In the SAR analysis, 3 (GI50 = 2.68–17.82 μM) and 4 (GI50 = 3.78–19.58 μM), which lack a methoxy group at C-29, demonstrated greater cytotoxicity than 5 (GI50 = 4.95–13.90 μM) and 6. Furthermore, compounds 3 (GI50 = 2.68–17.82 μM) and 5 (GI50 = 4.95–13.90 μM), bearing a chlorine atom at the C-8 position, exerted superior anticancer activity compared to 4 (GI50 = 3.78–19.58 μM) and 6 (GI50 = 10.36–24.92 μM), which are chlorinated at C-7. These findings suggest that the presence and regioselective positioning of the chlorine substituent at C-7 and C-8 significantly influence the cytotoxic activity of the PTMs, indicating a possible SAR associated with the halogenation pattern.
Table 3.
Cytotoxicity of 3–6 against solid and blood cancer cell lines.
Table 4.
Antibacterial activities of 3–6.
The antibacterial activities of 3–6 were tested against six bacterial strains, comprising three Gram-positive and three Gram-negative species (Table 4). Compounds 3 and 4 exhibited antibacterial activity against Staphylococcus aureus, with minimum inhibitory concentrations (MICs) of 32.0 and 16.0 μg/mL, respectively. Compounds 3–6 exhibited notable activity against Micrococcus luteus, with MIC values ranging from 4.0 to 32.0 μg/mL. In the antibacterial assay against M. luteus, 3 and 4, which lack a methoxy group at C-29, exhibited significant activity (MIC = 4.00 μg/mL). Additionally, 4 and 6, bearing a chlorine atom at C-7, showed relatively enhanced antibacterial activity against both S. aureus (MIC = 16.00 μg/mL) and M. luteus (MIC = 16.00 μg/mL) compared to 3 (MIC = 32.00 μg/mL) and 5 (MIC = 32.00 μg/mL), which are chlorinated at C-8.
3. Materials and Methods
3.1. General Experimental Procedures and Reagents
Optical rotations were measured with a Rudolph analytical Autopol III S2 polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA). UV spectra were recorded with a Shimadzu UV-1650PC spectrophotometer (Shimadzu Corporation, Kyoto, Japan). IR spectra were obtained on an OPUS FT/IR-ALPHA II spectrophotometer (Bruker OPTIK GmbH & Co. KG, Ettlingen, Germany). LR-ESIMS data were obtained with an ISQ EM mass spectrometer. HR-ESIMS data were obtained with a Waters SYNPT G2 Q-TOF mass spectrometer (Waters Corporation, Milford, MA, USA) at Korea Basic Science Institute (KBSI) in Cheongju, Republic of Korea, with samples dissolved in MeOH prior to analysis. NMR spectra were acquired with a Bruker AVANCE III 600 spectrometer (Bruker Biospin GmbH, Rheinstetten, Germany) with a 3 mm probe operating at 600 MHz (1H) and 150 MHz (13C). Chemical shifts were expressed in ppm with reference to the solvent peaks (δH 3.31 and δC 49.15 ppm for CD3OD; δH 7.26 and δC 77.25 ppm for 90% CDCl3/CD3OD). ECD spectra were recorded with a JASCO J-1500 circular dichroism spectrometer (JASCO Corporation, Tokyo, Japan) at the Gyeongnam Bio and Anti-aging Core Facility Center for Changwon National University in Changwon, Republic of Korea. HPLC was performed using a BLS-Class pump (Teledyne SSI, Inc., State College, PA 16803, USA) with a Shodex RI-201H refractive index detector (Shoko Scientific Co. Ltd., Yokohama, Japan). Columns for HPLC were YMC-Triart C18 (250 mm × 10 mm, 5 μm), YMC-Triart C8 (250 mm × 10 mm, 5 μm), and YMC-CHIRAL PREP CD PM (250 mm × 4.6 mm, S-10 μm). RP silica gel (YMC-Gel ODS-A, 12 nm, S-75 μm) was used for open-column chromatography. Organic solvents were purchased as HPLC-grade, and ultrapure water was obtained from a Milipore Mili-Q Direct 8 system. The reagents used in the bioassay were purchased from Sigma-Aldrich (St. Louis, MO, USA), and Tokyo Chemical Industry (Tokyo, Japan).
3.2. Strain and Fermentation
The marine-derived strain 1310KO-148 was isolated from a sponge sample collected at Kosrae State, Federated States of Micronesia, in October 2013 (5°20′14.78″ N 162°56′41.32″ E). The strain was identified as Streptomyces xiamenensis based on 16S rRNA gene sequence analysis. The GenBank accession number is PP863885.
The strain 1310KO-148 was inoculated into modified Bennett’s broth medium (0.1% D-glucose, 0.02% tryptone, 0.01% yeast extract, 0.01% beef extract, 0.05% glycerol, sea salt 32 g/L, pH 7.0). The seed culture was incubated at 28 °C for 14 days at 120 rpm and aseptically transferred into a 100 L fermenter containing 70 L of sterilized culture medium (0.1% v/v). The culture was fermented at 28 °C with agitation at 40 rpm and an airflow rate of 10 L/min (LPM) for 16 days.
3.3. Extraction and Isolation of Compounds 3–6
The 70 L fermentation broth was extracted twice with ethyl acetate (EtOAc, 70 L each) and concentrated under reduced pressure to yield a crude extract (6.9 g). The extract was subjected to ODS column chromatography (YMC Gel ODS-A, 12 nm, S75 μm) followed by stepwise gradient elution with MeOH/H2O (v/v) (ranging from 20:80 to 100:0) as eluent to obtain 10 fractions (Fr.1–Fr.10). The Fr.10 (199.0 mg) was further fractionated on a column of silica gel (Merck silica gel 60, 6 nm, 40–63 μm) by eluting with n-hexane, CH2Cl2, and EtOAc to afford three subfractions (Fr.10.1–Fr.10.3), including pure 1 (Fr.10.3, 48.8 mg). The 90% MeOH fraction (Fr.8, 637 mg) was suspended in MeOH and extracted with n-hexane to yield MeOH- and n-hexane-soluble layers. The MeOH layer (412.5 mg) was separated into four subfractions (Fr.8.1–Fr.8.4) via an ODS column chromatography (MeOH/H2O, 80–100%). A semi-preparative reversed-phase HPLC (YMC-Triart C18 column, 250 mm × 10 mm i.d., 5 μm; 80% MeOH in H2O; flow rate: 2.0 mL/min; detector: RI) was used to divide the Fr.8.2 fraction (217.0 mg) into 21 subfractions (Fr.8.2.1–Fr.8.2.34), including pure 3 (42.4 mg, tR 41.0 min). Compound 2 was purified from the Fr.8.2.11 (4.2 mg) by a semi-preparative RP HPLC (YMC-ODS A C18 column, 250 mm × 10 mm i.d., 5 μm; 65% MeCN in H2O; flow rate: 1.5 mL/min; detector: RI) to yield 2 (1.3 mg, tR 26.0 min). The Fr.8.2.15 (20.6 mg) was purified by a semi-preparative RP HPLC (YMC-Pack C8 column, 250 mm × 10 mm i.d., 5 μm; 75% MeOH in H2O; flow rate: 2.0 mL/min; detector: RI) to yield 4 (8.0 mg, tR 44.0 min). The 80% MeOH fraction (Fr.7, 375.0 mg) was separated into four subfractions (Fr.7.1–Fr.7.5) via ODS column chromatography (MeOH/H2O, 70–100%). A semi-preparative reversed-phase HPLC (YMC-Pack C8 column, 250 mm ×10 mm i.d., 5 μm; 70% MeOH in H2O; flow rate: 2.0 mL/min; detector: RI) was used to divide the Fr.7.3 (255.0 mg) into 25 subfractions (Fr.7.3.1–Fr.7.3.25). The Fr.7.3.7 (33.4 mg) was purified using a semi-preparative reversed-phase HPLC (YMC-ODS A C18 column, 250 mm × 10 mm i.d., 5 μm; 65% MeCN in H2O; flow rate: 1.5 mL/min; detector: RI) to yield 5 (13.2 mg, tR 18.0 min). Compound 6 was purified from the Fr.7.3.9 (49.3 mg) by a semi-preparative reversed-phase HPLC (YMC-ODS A C18 column, 250 mm × 10 mm i.d., 5 μm; 65% MeCN in H2O; flow rate: 1.5 mL/min; detector: RI) to yield 6 (7.4 mg, tR 22.0 min).
Capsimycin D (3): Yellow amorphous powder; [α: +168.3 (c 0.2, MeOH); UV (MeOH) λmax (logԑ): 236 (3.85), 320 (3.83) nm; ECD (MeOH, λ [nm] (∆ԑ), c = 0.75 mM) 328 (+19.84), 240 (−36.91), 214 (+20.91); IR (MeOH) νmax 3307, 2940, 1647, 1579, 1438, 1244 cm−1; 1H and 13C NMR data see Table 1 and Table 2; HRESIMS m/z 531.2626 [M+H]+ (calcd for C29H40N2O5Cl, 531.2626) and 553.2444 [M+Na]+ (calcd for C29H39N2O5NaCl, 553.2445).
Chlokamycin B (4): Yellow amorphous powder; [α: +150.0 (c 0.2, MeOH); UV (MeOH) λmax (logԑ): 232 (2.95), 320 (2.74) nm; ECD (MeOH, λ [nm] (∆ԑ), c = 1.13 mM) 326 (+45.07), 240 (−90.75), 213 (+59.47); IR (MeOH) νmax 3311, 2933, 1650, 1605, 1459, 1236, 1028 cm−1; 1H and 13C NMR data see Table 1 and Table 2; HRESIMS m/z 531.2626 [M+H]+ (calcd for C29H40N2O5Cl, 531.2626) and 553.2446 [M+Na]+ (calcd for C29H39N2O5NaCl, 553.2445).
Chlokamycin C (5): Yellow amorphous powder; [α: +110.0 (c 0.2, MeOH); UV (MeOH) λmax (logԑ): 230 (3.92), 320 (3.77) nm; ECD (MeOH, λ [nm] (∆ԑ), c = 0.36 mM) 328 (+20.01), 242 (−44.84), 216 (+26.88); IR (MeOH) νmax 3307, 2929, 1647, 1579, 1448, 1025 cm−1; 1H and 13C NMR data see Table 1 and Table 2; HRESIMS m/z 561.2733 [M+H]+ (calcd for C30H42N2O6Cl, 561.2731) and 583.2551 [M+Na]+ (calcd for C30H41N2O6NaCl, 583.2551).
Chlokamycin D (6): Yellow amorphous powder; [α: +106.6 (c 0.2, MeOH); UV (MeOH) λmax (logԑ): 230 (4.04), 324 (3.91) nm; ECD (MeOH, λ [nm] (∆ԑ), c = 0.36 mM) 324 (+22.91), 242 (−48.57), 213 (+26.28); IR (MeOH) νmax 3303, 2933, 1647, 1583, 1448, 1244, 1028 cm−1; 1H and 13C NMR data see Table 1 and Table 2; HRESIMS m/z 561.2730 [M+H]+ (calcd for C30H42N2O6Cl, 561.2731) and 583.2550 [M+Na]+ (calcd for C30H41N2O6NaCl, 583.2551).
3.4. ECD Calculations
The preliminary geometry optimization and conformational search were performed using Conflex 8 [26]. The optimization and calculation for electronic circular dichroism (ECD) were conducted utilizing the Gaussian 16 programme (rev. B.01, Gaussian Inc., Wallingford, CT, USA). Conformational searches were carried out using MMFF94s force field calculations, with a search threshold of 5 kcal/mol. The conformers of 4–6 were optimized using the ground state DFT geometry optimization method at the B3LYP/6-311G+(d,p) level in MeOH with an IEFPCM model for ECD. The theoretical calculations of ECD spectra were performed using TD-SCF at the B3LYP/6-311G+(d,p) (NStates = 60). The calculated ECD spectra of the conformers were Boltzmann-weighted and averaged using SpecDis 1.71 software with a σ value of 0.28 eV.
3.5. Cytotoxic Assay
The cytotoxic activity of 3–6 was evaluated using the CellTiter-Glo® luminescent cell viability assay (Promega, Madison, WI, USA) and the sulforhodamine B (SRB) assay, following previously reported protocols [27]. Cancer cell lines were purchased from the Japanese Cancer Research Resources Bank (JCRB, Osaka, Japan). Luminescence signals were recorded with a GloMax® Multi Detection System (Promega, Madison, WI, USA), and GI50 values were calculated using the relative GI50 model implemented in GraphPad Prism 10 (GraphPad Software, San Diego, CA, USA). Doxorubicin served as the positive control.
3.6. Antibacterial Assay
The antibacterial activity was evaluated using the 96-well microplate method, as described in a published report [27], against three Gram-positive bacteria (Staphylococcus aureus KCTC1927, Bacillus subtilis KCTC1021, and Micrococcus luteus KCTC1915) and three Gram-negative bacteria (Escherichia coli KCTC2441, Salmonella typhimurium KCTC2515, and Klebsiella pneumoniae KCTC2690). The six bacterial species were cultured in Müller Hinton (MH) broth medium at 30 °C and 140 rpm for 24 h. The 0.5 McFarland bacterial suspension (1 × 108 CFU/mL) was diluted to 5 × 106 CFU/mL. Aliquots of 100 μL of the diluted suspension and 100 μL of Mueller–Hinton (MH) broth containing compounds 3–6 were dispensed into each well of a 96-well plate. Following incubation at 30 °C for 24 h, minimum inhibitory concentrations (MICs) were determined by measuring the optical density at 610 nm (OD610). Kanamycin was used as a positive control.
4. Conclusions
In summary, three previously unreported chlorohydrin-containing PTMs (4–6) were isolated and structurally characterized from S. xiamenensis 1310KO-148 obtained from a marine sponge. Comparison of the ECD spectra of 3–6 revealed a high degree of similarity in their Cotton effects (Figure 4A). In addition, all four compounds (3–6) exhibited positive optical rotation values, suggesting that variations in substitution at C-7, C-8, and C-29 have little influence on either the ECD spectra or optical rotation. Notably, potent anticancer activity was observed for capsimycin D (3), whose cytotoxicity has not been previously reported, as well as for the three newly isolated PTMs (4–6), with GI50 values ranging from 2.68 to 24.92 μM. SAR analysis revealed that both the position of the chlorine substitution at C-7/C-8 and the presence of a methoxy group at C-29 significantly influenced the cytotoxic activity, suggesting that structural differences can significantly influence their biological activity. Overall, these findings expand the chemical diversity and potential bioactivity of halogen-containing PTMs and provide new insights into the SAR of this class of compounds.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/md24030117/s1, Figure S1: PTMs; Figures S2–S6: 1H, 13C NMR, HSQC, COSY, HR-ESIMS data, and Structure of 1; Figures S7–S11: 1H, 13C NMR, HSQC, LR-ESIMS data, and Structure of 2; Figures S12–S20: 1H, 13C NMR, HSQC, COSY, HMBC, HR-ESIMS, IR, UV data, and Structure of 3; Figures S21–S29: 1H, 13C NMR, DEPT, HSQC, COSY, HMBC, NOESY, HR-ESIMS, IR, and UV data of 4; Figures S30–S38: 1H, 13C NMR, DEPT, HSQC, COSY, HMBC, NOESY, HR-ESIMS, IR, and UV data of 5; Figures S39–S46: 1H, 13C NMR, DEPT, HSQC, COSY, HMBC, NOESY, HR-ESIMS, IR, and UV data of 6; Tables S1–S3: 1H and 13C NMR data of 1–3; Tables S4–S15: TDSCF-ECD calculation data of 4–6; Tables S16 and S17: Cytotoxicity of 3–6.
Author Contributions
M.A.L.: Writing—Original draft, Resources, Investigation, Data curation, Visualization. J.S.K.: Data curation. J.-H.K.: Investigation. J.-W.Y.: Investigation. H.-S.L.: Resources. C.-S.H.: Resources. H.J.S.: Writing—Review and Editing, Validation, Supervision, Project Administration, and Funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Korea Institute of Marine Science & Technology Promotion (KIMST) grant funded by the Ministry of Oceans and Fisheries, Korea (Grant no. 20220027) and by the Korea Institute of Ocean Science and Technology (KB0012 and PEA0314). The authors express gratitude to Jung Hoon Choi, Korea Basic Science Institute, Ochang, Korea, for providing mass data.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
The data presented in this study are available upon request from the corresponding author.
Acknowledgments
The authors express gratitude to Jung Hoon Choi, Korea Basic Science Institute, Ochang, Korea, for providing mass data.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Mo, X.; Li, Q.; Ju, J. Naturally occurring tetramic acid products: Isolation, structure elucidation and biological activity. RSC Adv. 2014, 4, 50566–50593. [Google Scholar] [CrossRef]
- Jiang, M.; Chen, S.; Li, J.; Liu, L. The Biological and Chemical Diversity of Tetramic Acid Compounds from Marine-Derived Microorganisms. Mar. Drugs 2020, 18, 114. [Google Scholar] [CrossRef]
- Jomon, K.; Kuroda, Y.; Ajisaka, M.; Sakai, H. A new antibiotic, ikarugamycin. J. Antibiot. 1972, 5, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Zhang, S.-D.; Haidar, A.K.; Bajimaya, M.; Guo, Y.; Larsen, T.O.; Gram, L. Polycyclic Tetramate Macrolactams-A Group of Natural Bioactive Metallophores. Front. Chem. 2021, 9, 772858. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.H.; Dong, F.Y.; Da, L.T.; Yang, X.M.; Wang, X.X.; Weng, J.Y.; Feng, L.; Zhu, L.L.; Zhang, Y.L.; Zhang, Z.G.; et al. Ikarugamycin inhibits pancreatic cancer cell glycolysis by targeting hexokinase 2. FASEB J. 2020, 34, 3943–3955. [Google Scholar] [CrossRef] [PubMed]
- Lacret, R.; Oves-Costales, D.; Gómez, C.; Díaz, C.; De la Cruz, M.; Pérez-Victoria, I.; Vicente, F.; Genilloud, O.; Reyes, F. New Ikarugamycin Derivatives with Antifungal and Antibacterial Properties from Streptomyces zhaozhouensis. Mar. Drugs 2014, 13, 128–140. [Google Scholar] [CrossRef]
- Saeed, S.I.; Aklilu, E.; Mohammedsalih, K.M.; Adekola, A.A.; Mergani, A.E.; Mohamad, M.; Kamaruzzaman, N.F. Antibacterial Activity of Ikarugamycin against Intracellular Staphylococcus aureus in Bovine Mammary Epithelial Cells In Vitro Infection Model. Biology 2021, 10, 958. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Zhang, W.; Zhang, G.; Zhu, Y.; Chen, Y.; Liu, W.; Yuan, C.; Zhang, Q.; Zhang, H.; Zhang, L.; et al. Activation and characterization of a cryptic gene cluster reveals a cyclization cascade for polycyclic tetramate macrolactams. Chem. Sci. 2017, 8, 1607–1612. [Google Scholar] [CrossRef]
- Glöckle, A.; Schuler, S.; Einsiedler, M.; Gulder, T.A.M. A plug-and-play system for polycyclic tetramate macrolactam production and functionalization. Microb. Cell Fact. 2025, 24, 13. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.-J.; Liu, Y.; Wang, H.-X.; Zhu, D.-Y.; Shen, Y.-M.; Li, Y.-Y. Expression of the Clifednamide Biosynthetic Pathway in Streptomyces Generates 27,28-seco-Derivatives. J. Nat. Prod. 2020, 83, 2803–2808. [Google Scholar] [CrossRef]
- Qiu, J.; Qin, R.; Zhi, S.; Liu, L. Recent advance in macrolactams: Structure, bioactivity, and biosynthesis. Bioorg. Chem. 2025, 159, 108406. [Google Scholar] [CrossRef]
- Yu, H.-L.; Jiang, S.-H.; Bu, X.-L.; Wang, J.-H.; Weng, J.-Y.; Yang, X.-M.; He, K.-Y.; Zhang, Z.-G.; Ao, P.; Xu, J.; et al. Structural diversity of anti-pancreatic cancer capsimycins identified in mangrove-derived Streptomyces xiamenensis 318 and post-modification via a novel cytochrome P450 monooxygenase. Sci. Rep. 2017, 7, 40689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, G.; Zhang, L.; Liu, W.; Jiang, X.; Jin, H.; Liu, Z.; Zhang, H.; Zhou, A.; Zhang, C. New polycyclic tetramate macrolactams from marine-derived Streptomyces sp. SCSIO 40060. Tetrahedron 2018, 74, 6839–6845. [Google Scholar] [CrossRef]
- Fukuda, T.; Takahashi, M.; Kasai, H.; Nagai, K.; Tomoda, H. Chlokamycin, a New Chloride from the Marine-derived Streptomyces sp. MA2-12. Nat. Prod. Commun. 2017, 12, 1223–1226. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, W.; Zhang, Q.; Shi, T.; Ma, L.; Zhu, Y.; Li, S.; Zhang, H.; Zhao, Y.L.; Shi, R.; et al. Mechanistic Insights into Polycycle Formation by Reductive Cyclization in Ikarugamycin Biosynthesis. Angew. Chem. Int. Ed. Engl. 2014, 53, 4840–4844. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Blodgett, J.A.; Clardy, J. Targeted Discovery of Polycyclic Tetramate Macrolactams from an Environmental Streptomyces Strain. Org. Lett. 2010, 12, 4652–4653. [Google Scholar] [CrossRef] [PubMed]
- Schuler, S.; Einsiedler, M.; Evers, J.K.; Malay, M.; Uka, V.; Schneider, S.; Gulder, T.A.M. Expanding Polycyclic Tetramate Macrolactam (PoTeM) Core Structure Diversity by Chemo-Enzymatic Synthesis and Bioengineering. Angew. Chem. Int. Ed. Engl. 2025, 64, e202420335. [Google Scholar] [CrossRef]
- Harper, C.P.; Day, A.; Tsingos, M.; Ding, E.; Zeng, E.; Stumpf, S.D.; Qi, Y.; Robinson, A.; Greif, J.; Blodgett, J.A. Critical analysis of polycyclic tetramate macrolactam biosynthetic gene cluster phylogeny and functional diversity. Appl. Environ. Microbiol. 2024, 18, e0060024. [Google Scholar] [CrossRef] [PubMed]
- Montavon, A.; Marchán-Rivadeneira, M.R.; Han, Y. Polycyclic Tetramate Macrolactams and Their Potential as Anticancer Agents. Organics 2024, 5, 361–377. [Google Scholar] [CrossRef]
- Shigeno, S.; Kadowaki, M.; Nagai, K.; Hosoda, K.; Terahara, T.; Nishimura, T.; Hasegawa, N.; Tomoda, H.; Ohshiro, T. New polycyclic tetramate macrolactams with antimycobacterial activity produced by marine-derived Streptomyces sp. KKMA-0239. J. Antibiot 2024, 77, 265–271. [Google Scholar] [CrossRef]
- Horvath, A.; Bolla, K.; Wachtler, A.; Makso, L.; Papp, M.; Maho, S.; Dubrovay, Z.; Koti, J.; Skoda-Foldes, R. A Temperature-Controlled Switch between Furst-Plattner Rule and Anti-Furst-Plattner Rule Ring Opening of 2,3-Epoxy-steroids with Various Halide Sources in the Presence of Imidazolium Ionic Liquids. ACS Omega 2021, 6, 26846–26856. [Google Scholar] [CrossRef]
- Gribble, G.W. The diversity of naturally produced organohalogens. Chemosphere 2003, 52, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Vaillancourt, F.H.; Yeh, E.; Vosburg, D.A.; Garneau-Tsodikova, S.; Walsh, C.T. Nature’s Inventory of Halogenation Catalysts: Oxidative Strategies Predominate. Chem. Rev. 2006, 106, 3364–3378. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.S.; Fujimori, D.G.; Walsh, C.T. Halogenation strategies in natural product biosynthesis. Chem. Biol. 2008, 15, 99–109. [Google Scholar] [CrossRef]
- Schallmey, A.; Schallmey, M. Recent advances on halohydrin dehalogenases-from enzyme identification to novel biocatalytic applications. Appl. Microbiol. Biotechnol. 2016, 100, 7827–7839. [Google Scholar] [CrossRef]
- Lee, M.A.; Kang, J.S.; Yoon, Y.D.; Lee, H.S.; Heo, C.S.; Park, S.J.; Shin, H.J. Xiapyrroles A-F: N-Alkylpyrrole Alkaloids from the Marine-Derived Actinomycete Streptomyces xiamenensis 1310KO-148. J. Nat. Prod. 2025, 88, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.A.; Kang, J.S.; Yang, J.W.; Lee, H.S.; Heo, C.S.; Park, S.J.; Shin, H.J. Meirols A-C: Bioactive Catecholic Compounds from the Marine-Derived Fungus Meira sp. 1210CH-42. Mar. Drugs 2024, 22, 87. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.