Abstract
Cyanobacteria are considered to be one of the most promising sources of new, natural products. Apart from non-ribosomal peptides and polyketides, ribosomally synthesized and post-translationally modified peptides (RiPPs) are one of the leading groups of bioactive compounds produced by cyanobacteria. Among these, cyanobactins have sparked attention due to their interesting bioactivities and for their potential to be prospective candidates in the development of drugs. It is assumed that the primary source of cyanobactins is cyanobacteria, although these compounds have also been isolated from marine animals such as ascidians, sponges and mollusks. The aim of this review is to update the current knowledge of cyanobactins, recognized as being produced by cyanobacteria, and to emphasize their genetic clusters and chemical structures as well as their bioactivities, ecological roles and biotechnological potential.
1. Introduction
Cyanobactins belong to the class of ribosomally synthetized peptides with post-translational modifications (RiPPs) that can be generally defined as cyclic peptides containing modifications, which include azole/azoline rings, d-sterocenters and in some cases, prenyl groups [1].
These compounds can be produced by distinct cyanobacteria strains through a pathway recently assigned as post-ribosomal peptide synthesis (PRPS) [2]. In PRPS, an unmodified precursor peptide produced by translation directly encodes the sequence that will form the mature cyanobactin [2,3]. Subsequent cleavage of the precursor peptide is followed by several modifications leading to the formation of the end product [2,4]. The denomination, cyanobactins, was introduced for the first time in 2008, based on the related features and biosynthetic pathways of these cyanobacterial compounds [5]. The first cyanobactins to be described were ulicyclamide and ulithiacyclamide, in the tunicate Lissoclinum patella by Ireland and Scheuer in 1980 [6]. Twenty-five years later, two separate studies demonstrated that the cyanobacterium, Prochloron, symbiont of the tunicate, was in fact responsible for the production of cyanobactins through a PRPS pathway [7,8]. The cyanobactins are found in different marine animals, such as ascidians, sponges and mollusks. At present, however, it is believed that cyanobactins are derived exclusively from cyanobacteria [9,10]. Interestingly, not all of the currently known cyanobactins can be associated with a cyanobacterial symbiont. This review aims to present the current state of knowledge of cyanobactins, which are produced solely from cyanobacteria, with special emphasis given to their chemical structures, genetic biosynthetic pathways, bioactivities, ecological roles and biotechnological potential.
2. Producing Cyanobacterial Strains
It is estimated that 10% to 30% of all cyanobacteria can produce cyanobactins [9,11,12]. These peptides seem to be widespread among symbiotic, as well as free-living cyanobacteria, from terrestrial to freshwater and marine environments [12,13,14]. A recent study analyzed the genome of 126 cyanobacteria strains, which revealed that 31 cyanobactin gene clusters were present in 24% of the strains. Although the cyanobactin genetic clusters appear to be sporadically distributed among cyanobacteria, they are more frequent in Arthrospira, Oscillatoria and Microcystis genera. In contrast, the Prochlorococcus or Synechococcus genomes seem to lack these cyanobactin gene clusters [11].
Thus far, cyanobactin biosynthetic gene clusters and their respective associated metabolites have been described in cyanobacteria belonging to the unicellular genera Prochloron (patellamide, lissoclinamides, ulithiacyclamides, patellin and trunkamide) [10], Microcystis (microcyclamide, piricyclamide and aerucyclamides) [10,15] and Cyanothece (cyanothecamides) [16,17]. Filamentous non-heterocystous genera such as Trichodesmium (trichamide) [10], Planktothrix (prenylagaramide) [16,18], Lyngbya (aesturamide) and Arthrospira (arthrospiramide) [5,16,19] and filamentous heterocystous genera such as Anabaena (anacyclamide) [20] and Nostoc (tenuecyclamide) [10] are also described as cyanobactin producers. Figure 1 shows cyanobactins time line evolution since their discovery to present.
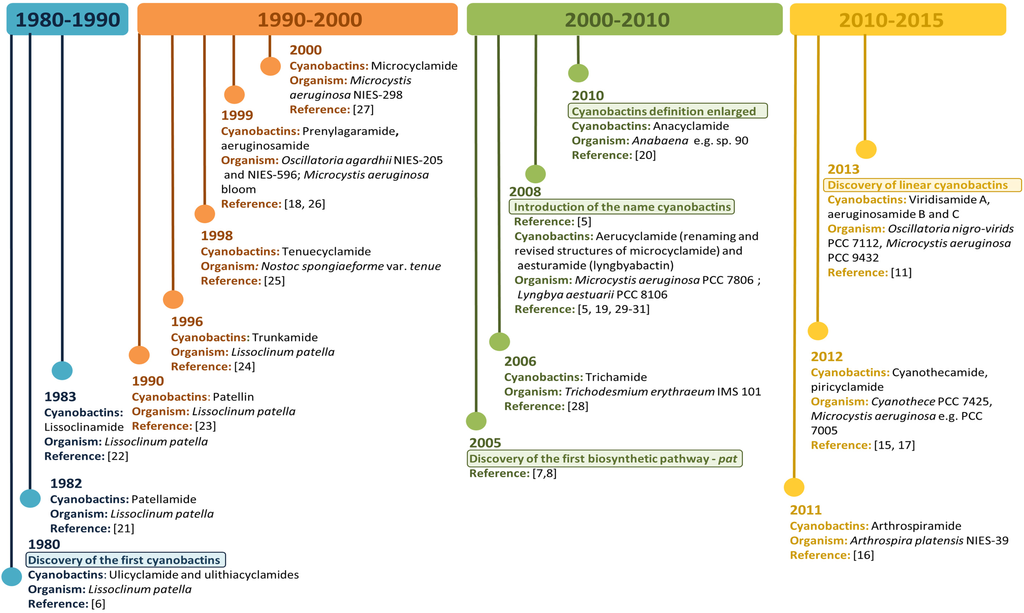
Figure 1.
Cyanobactins Time Line. Evolution of cyanobactins since their discovery in 1980 to present.
Figure 1.
Cyanobactins Time Line. Evolution of cyanobactins since their discovery in 1980 to present.
Additionally, cyanobactin genes have been found in distinct cyanobacteria orders [12,13,14] and several cyanobactin genetic clusters with unknown products have been described [11,16]. Therefore, it is important to highlight that the genetic and chemical diversity of the cyanobactins still remains to be explored in greater depth.
3. Chemical Structures
Cyanobactins are generally defined as ribosomally synthesized, N-C macrocyclic peptides produced by cyanobacteria [5,9]. Prenylation, or heterocyclization of amino acids, present a second feature of cyanobactins that altogether share a number of characteristics [9]. These compounds are 6–20 amino acids in length. l-amino acids are commonly found in these peptide structures, although more rarely d-amino acids may be present [4,32]. The epimerization to the non-proteinogenic D-form is thought to be spontaneous and not due to enzymatic activity [4,32]. Initially it was assumed that cyanobactins were only composed of cyclic peptides; however, in 2013, the structural diversity of cyanobactins was expanded to encompass highly modified linear peptides with rare post-translational modifications [11].
Cyanobacteria are able to produce cyanobactins that contain heterocyclized amino acids (see Section 5) as well as non-heterocyclized amino acids, which may be sporadically prenylated or geranylated (see Section 6). Cyanobactins that contain heterocycles (see Section 5) present 6–11 amino acids in length and both thiazole (described in 36 cyanobactins) and oxazoline (described in 27 cyanobactins) are frequent in their structures. Thiazoline (described in 16 cyanobactins) and oxazole (described in six cyanobactins) are less frequent. Disulfide bridges are only present in the ulithiacyclamide group—where two of the four encoded cysteines form a disulfide bridge and the other two are heterocyclized to thiazole. In addition, the formation of the disulfide bridge is considered to be spontaneous, owing to the fact that the cyanobactin genetic clusters do not encode an enzyme to carry out this reaction [4,9]. Some cyanobactins may present prenylation of threonine, serine and tyrosine amino acids and, more rarely, N-methylation, as is the case for microcyclamide. The linear cyanobactins, such as aeruginosamides and viridisamide A, range in length from 3 to 5 amino acids and present prenylated N-termini and methylated C-termini bound to thiazoles.
Cyanobactins lacking heterocyclized amino acids (see Section 6) range in length from 7 to 20 amino acids and have a conserved proline residue. The prenyl attachments are present in all cyanobactin families, whereas geranylation may occur in anacyclamide and piricyclamide families. The latter may contain disulfide bridges.
The enormous cyanobactin diversity results from the distinctive genetic features of this class of compounds. The presence of one or more precursor peptide genes with a hypervariable core region certainly contributes to the chemical diversity within this class of compounds [2,9,10].
4. Biosynthetic Genetic Clusters
Cyanobactin genetic clusters (approximately from 8 to 19 kb in length) have only been identified in cyanobacteria [11]. The active cyanobactin gene clusters always encode: (1) two protease genes, A (N-terminal protease) and G (C-terminal protease), that are related to patA and patG genes from the patellamide biosynthetic pathway, also known as pat (both A and G genes comprehend a domain of unknown function (DUF) and a macrocyclase domain which is only present in the G-protease [33]);(2) a precursor peptide gene E, homolog to patE, which directly encodes the cyanobactin structure that acts as a substrate for post-translational modifications and, (3) two genes B and C, which are related to patB and patC, that encode short conserved proteins of an unidentified function [9,10]. Although B and C genes are conserved among nearly all cyanobactin genetic clusters, studies have demonstrated that these genes are non-essential [34]. In addition, the cyanobactin gene clusters may also encode homologs of PatD and/or PatF, denoted as D-protein (cyclodehydratase) and F-protein (prenyltransferase), as well as thiazoline/oxazoline dehydrogenases (responsible for the aromatization of the heterocycles to thiazoles and oxazoles), methyltransferases and other non-characterized proteins (Figure 2) [10]. The D gene is only present in cyanobactin biosynthetic pathways that produce compounds with heterocyclized amino acids (see Section 5). In contrast, the F gene seems to be present in all cyanobactin pathways, with the exception of the non-prenylated trichamide (see Section 5 and Section 6). Nevertheless, this gene was proven to be essential for the synthesis of the non-prenylated patellamides, suggesting that it may have a distinct function in this biosynthetic pathway [34].
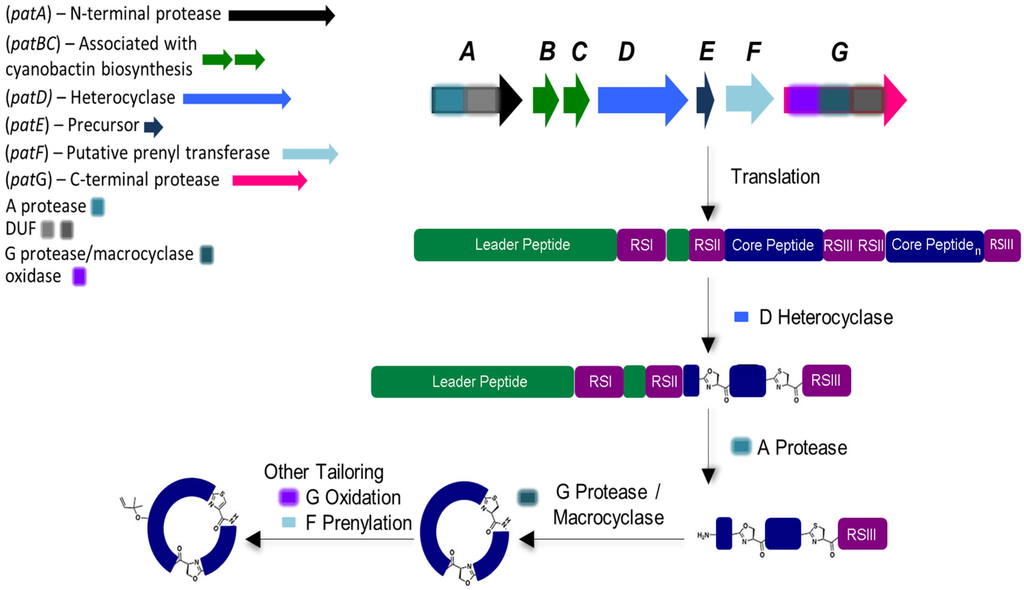
Figure 2.
General biosynthesis of cyanobactins that contain azoline heterocycles. Heterocyclization and proteolytic tailoring to produce macrocycles succeed translation of the core peptide. Multiple copies of the core peptide and recognition sequences may exist. Additional tailoring such as prenylation and oxidation/dehydrogenation to azoles may occur. Additional modifications are occasionally present. Adapted with permission from [2] (http://dx.doi.org/10.1039/c2np20085f). Copyright © The Royal Society of Chemistry, 2015.
Figure 2.
General biosynthesis of cyanobactins that contain azoline heterocycles. Heterocyclization and proteolytic tailoring to produce macrocycles succeed translation of the core peptide. Multiple copies of the core peptide and recognition sequences may exist. Additional tailoring such as prenylation and oxidation/dehydrogenation to azoles may occur. Additional modifications are occasionally present. Adapted with permission from [2] (http://dx.doi.org/10.1039/c2np20085f). Copyright © The Royal Society of Chemistry, 2015.
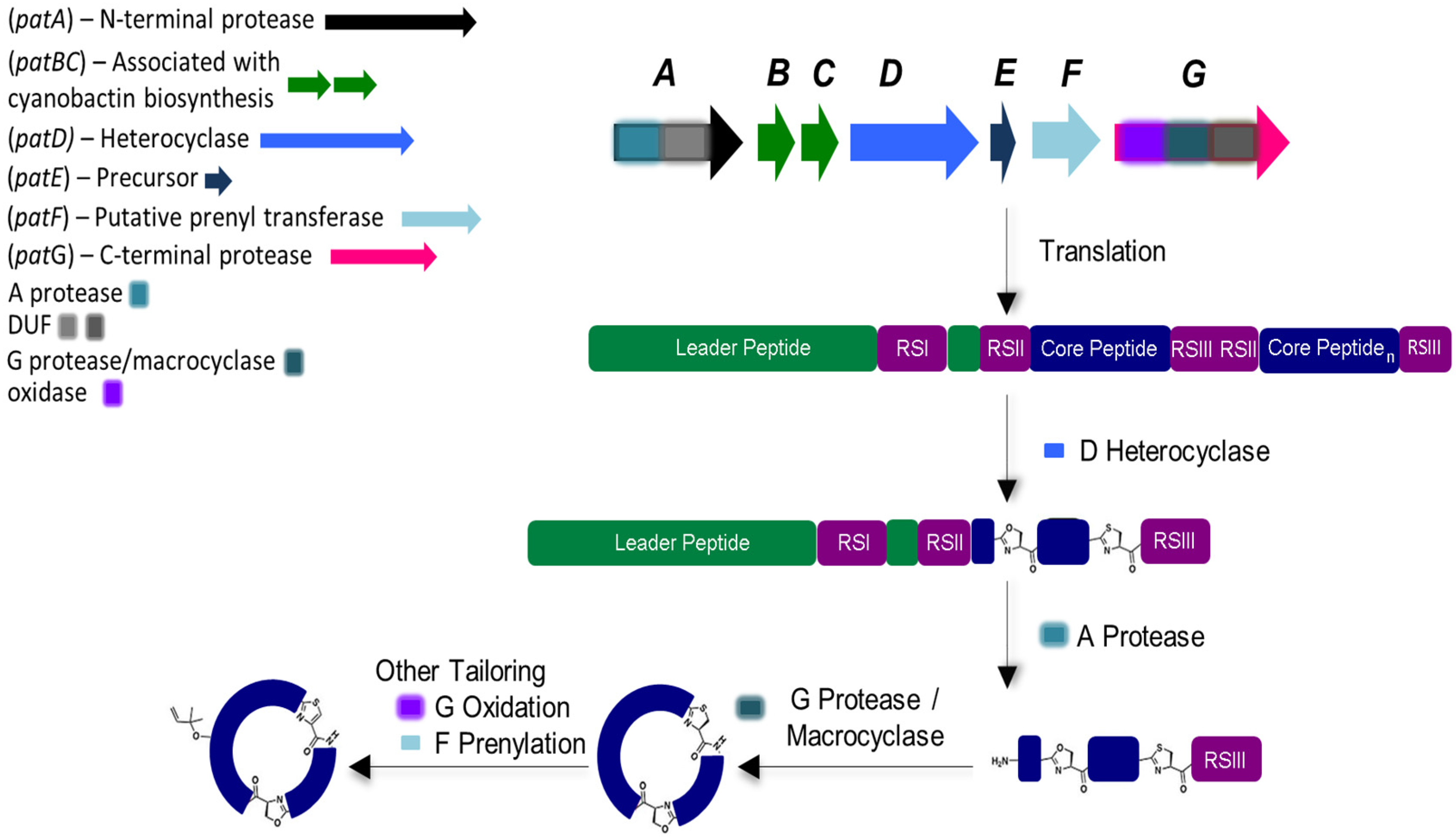
Cyanobactin biosynthesis begins with the precursor E-peptide, which is composebd of an N-terminal conserved leader sequence that is recognized by some of the modifying and cleaving enzymes [35]. However, reports indicate that cyanobactin genetic clusters may also employ more than one precursor peptide. In fact, it has been described that a genetic cluster may contain up to 10 precursor peptide genes [11,16]. Within the E-peptide that contains the enzymes recognition sequences (RSI, RSII and RSIII), 1–4 hypervariable core sequences may be present and dictate the amino acid backbone of the cyanobactin [2,3,9,10,36]. In the presence of more than one core peptide, each one is flanked by N-terminal (RSII) and C-terminal (RSIII) recognition sequences which may constitute four or five amino acids. In the presence of D-protein, cyclodehydratase, (and in order to form oxazoline and thiazole) heterocyclization of cysteines, serines and/or threonines will occur, directed by the recognition sequence RSI [37,38]. A-protease cleaves the precursor peptide RSII, leaving a free amine available for macrocyclization. Lastly, the G-protease splits the precursor peptide RSIII and catalyzes C-N macrocyclization. Subsequently, other transformations may occur such as: (1) the prenylation of serines/threonines or tyrosines/tryptophans residues, catalyzed by the PatF class of prenyltransferases (Figure 2) [16,39]; (2) the oxidation of heterocycles to oxazoles and thiazoles when the oxidase domain ispresent within the G gene or separate [11,33,40] and more uncommonly; (3) geranylation [9].
Cyanobactins have been classified into different groups based on the correspondence between genotypes and chemotypes [16]. Another classification scheme has also been proposed based solely on cyanobactin structures [41].
4.1. Heterocyclization
The heterocyclase (cyclodehydratase) accomplishes heterocyclization of cysteine, serine and threonine residues to thiazolines or oxazolines and eliminates water [2,42]. The cyanobactin heterocyclases (D) were studied in detail in the patellamide and trunkamide pathways [37] and reviewed in the broader context of RiPPs [4,42]. The activity of the heterocyclases (D), from both the above-mentioned pathways, is ATP-dependent and occurs in a defined order of reactions [4,37]. An adenylase mechanism has been proposed for TruD, from the trunkamide pathway, whose crystal structure presents as a three-domain protein [43]. The processivity of the enzyme requires the presence of the leader peptide to be attached to the core, indicating that heterocyclization occurs before cleavage and macrocyclization of the precursor peptide [37,42]. A short sequence element in the leader peptide is an indication for heterocycle formation [2,36]. Cyanobactin pathways encoding a heterocyclase may also codify an oxidase domain responsible for oxidation of thiazolines and oxazolines to thiazoles and oxazoles [9,11].
4.2. Cleavage and Macrocyclization
Two subtilisin-like proteins, N-terminal protease (A) and C-terminal protease (G), are encoded in cyanobactin genetic clusters. It was demonstrated that PatA protease (from the patellamide gene cluster) catalyzes the cleavage of the N-terminal protease site of the precursor peptide, removing the leader sequence, whereas PatG protease catalyzes the formation of the macrocyclic peptide while removing the C-terminal protease signature [8,44]. PatG protease from Prochloron was shown to macrocyclize a broad range of synthetic substrates with non-proteinogenic and d-amino acids at a lower rate [38].
The structure of the protease-macrocyclase domains of PatG and PagG (the latter from the prenylagaramide genetic cluster) was determined and the catalytic triad was presented [33,40]. The crystal structure of the macrocyclase domain of PatG shows that a subtilisin fold is present (like in PatA); however, it contains two helices designated by the “macrocyclization” insert, without conservation in terms of length and sequence. The macrocyclase domain is insensitive to the identity of the residues within the core peptide, since PatG acts on RSIII residues and catalyzes C-N macrocyclization [42]. During macrocyclization, the PatA protease removes the amino terminal linked to the core, yielding a free amino terminal, and PatG protease removes the carboxy terminal sequence flanking the core. The cleavage site is protected by the PatG protease preventing access to water and hydrolysis until the transamidation reaction is complete [33,40].
4.3. Prenylation, Oxidation and DUF
The presence of the gene encoding the prenyltransferase has been described in both prenylated and non-prenylated pathways [11]. For instance, the prenyltransferase gene is present in the patellamide (pat) pathway that generates the non-prenylated patellamides A and C [8]. In contrast, e.g., prenylagaramide (pag), trunkamide (tru), and aesturamide (lyn) pathways, which also encode the prenyltransferase gene, synthesize the prenylated compounds [5,16]. Prenylagaramide contains O-prenylated tyrosine [16], whereas trunkamide contains O-prenylated threonine and serine [34]. The prenyltransferases from lyn (LynF) and tru (TruF) pathways have been characterized biochemically [39,45]. LynF prenylates the oxygen atom of a tyrosine residue using dimethylallyl pyrophosphate (DMAPP). The reverse O-prenylated tyrosine undergoes spontaneous Claisen rearrangement yielding the ortho-substituted phenol [45]. In contrast, TruF prenylates serine and threonine residues on the hydroxyl side chain [39,45]. The structure of PatF from the patellamide pathway has been determined, revealing that it embraces, as the other prenyltransferases, the classic TIM barrel fold [46]. However, no enzymatic activity was detected, thus remaining consistent with the absence of prenylation in patellamides A and C. A careful examination revealed that it is in the positions usually occupied by conserved and catalytic active Asp and Lys residues that the residues His125 and Met136 are located, thereby justifying the inactivity of PatF [42,46]. It has already been demonstrated that PatF is essential for patellamide production in vivo [34] and consequently it must be responsible for another function in this pathway. Further studies will be required to understand the essential role of this protein in the production of the patellamides [46].
The oxidase domain is conserved among PatG homologs and studies reveal that it is FMN dependent [42]. The related thiazoline oxidase, in the microcin pathway, was the subject of a biochemical study [47] and although this enzyme is related in sequence to the patellamide enzymes, the basis of the substrate recognition remains unclear, as microcins are linear and patellamides are macrocycles [42]. Notwithstanding, it has been demonstrated that one homolog of the oxidase domain of PatG is able to oxidize both linear and macrocycle thiazoline-containing compounds [48] while another homolog only has the ability to perform oxidation on a macrocyclic substrate [4,6].
Both PatA and PatG proteases contain a domain of unknown function (DUF) sharing 56% sequence similarity [8,47]. The DUF domains are found in PatA and PatG from the patellamide-like biosynthetic clusters [49]. It is assumed that epimerization follows heterocyclization and precedes oxidation. Although epimerization has been proposed as a possible role for the DUF domain, it has been suggested that this phenomenon is chemically spontaneous, assigning no roles to DUF domains in the patellamide biosynthesis [32,42]. The crystal structure of PatG-DUF has been characterized as a novel fold dimer with two zinc ions. The practical importance of the dimer remains unclear since the residues involved in Zn2+ binding (necessary for dimerization) are not conserved among DUF domains of the other homologs; it is subsequently not clear if the other domains are dimers [49]. The DUF domain does not bind to linear substrates (simple peptides or peptides with heterocycles) [47] and additional experiments will be necessary to determine if the DUF domain binds to the macrocycle, or the core peptide alone [42].
5. Cyanobactins Encoding Heterocyclization Enzymes
5.1. Ulicyclamide, Ulithiacyclamides, Patellamides and Lissoclinamides
At the beginning of the 1980s, interest in compounds produced by didemnid tunicates started increased as a part of some programs to isolate antineoplastic natural products. These marine invertebrates were proven to be good candidates since they harbor unicellular prokaryotic algae. Although nitrogen fixation was not demonstrated in these symbionts at the time, this possibility prompted research to look for novel nitrogenous metabolites in tropical didemnid species [21]. Additionally, cytotoxic compounds were already documented from tunicate extracts [21]. A good example is didemnin B, the first marine natural product evaluated in anticancer clinical trials [50].
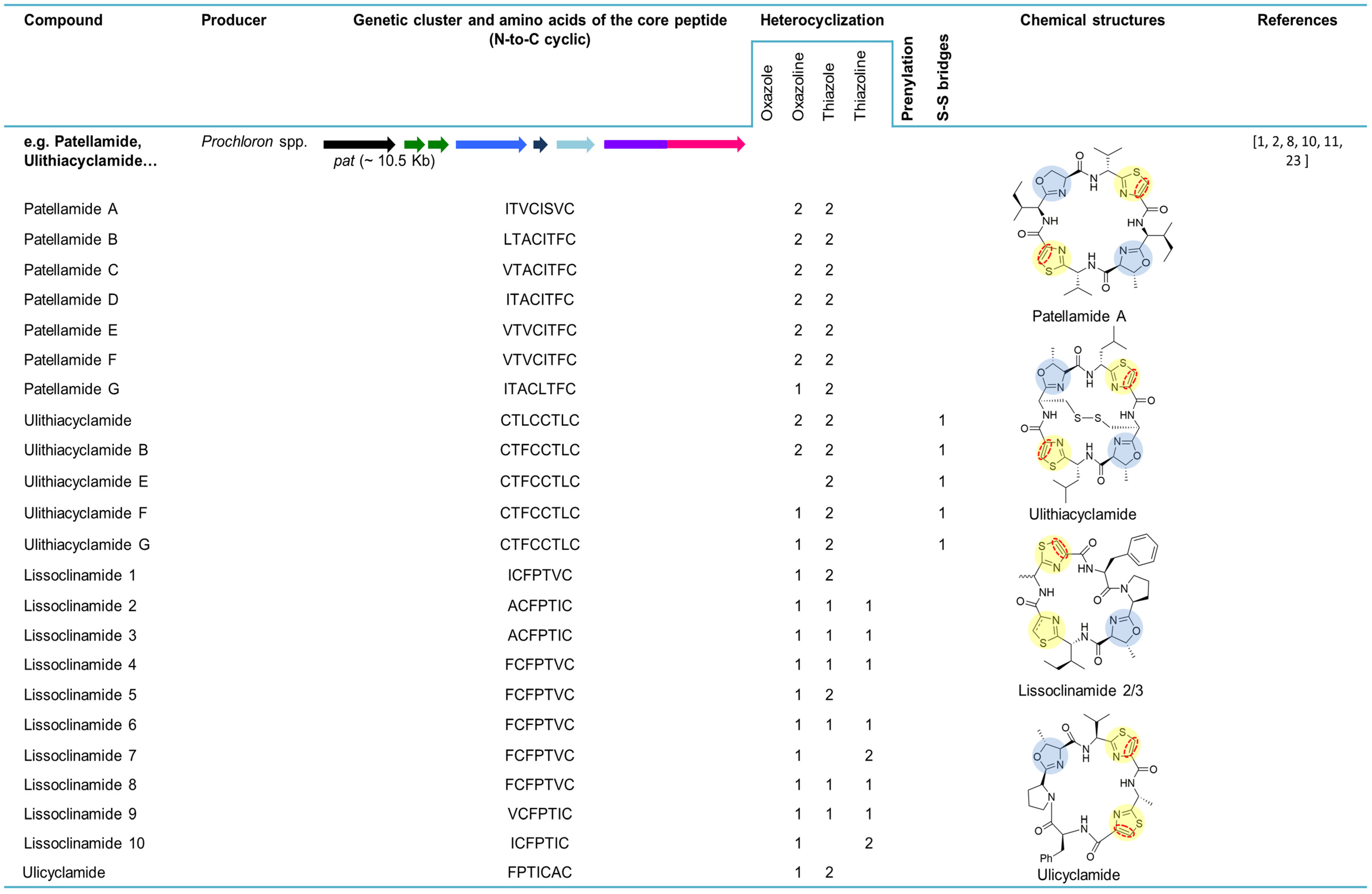
Ulicyclamide and ulithiacyclamide were the first cyanobactins to be discovered by Ireland and Scheuer in 1980 (Figure 3) [6]. These small cyclic peptides isolated from the ascidian L. patella from Palau, Western Caroline Islands, unveiled a combination of chemical features, including N-to-C macrocyclization and heterocyclization to form thiazol(in)e and oxazoline motifs that were unique [2,6]. It was unclear at the time if the ascidian hosts or their partners produced these compounds, since information about chemical switches between the ascidians and their symbiotic algae was scarce [6]. Two years later, the octapeptides patellamides A, B and C were reported and isolated from L. patella collected at Eil Malk Island, Palau Islands [21]. In 1983, three new cyclic heptapeptides (1, 2 and 3) recently named lissoclinamides from L. patella were described as the first thiazoline-containing peptides isolated from this organism [22]. This work developed by Wasylyk and co-workers also presented a revised structure for ulicyclamide. In fact, in 1983, Hamamoto reported the structure of patellamide A (ascidiacyclamide) with a different skeleton [51] and later studies assigned the correct structures of ulicyclamide and patellamide A, as described by Wasylyk and Hamamoto [1,52,53]. In 1989, four novel cyclic peptides, including patellamide D and lissoclinamides 4, 5 and 6 were isolated from the same tunicate species [54,55]. The known peptides derived from L. patella were categorized in two separate groups, one constituted by ulithiacyclamide (structure determined by Biskupiak and Ireland [56]) and patellamides A, B and C and the other represented by ulicyclamide and lissoclinamides 1–3. Both groups present uncommon amino acids containing thiazole moieties, but differ in the nature of their macrocyclic ring skeleton [55]. At the same time, Williams and Moore [57] reported on the isolation and structure determination of ulithiacyclamide B from L. patella from Pohnpei, Federated States of Micronesia. The difference between ulithiacyclamide and ulithiacyclamide B is that the latter lacks the symmetry associated with ulithiacyclamide and possesses a phenyl group [57]. Patellamide E was isolated from L. patella collected in Pulau Salu, Singapore. The tunicate extract also contained patellamides A and B and ulithiacyclamide [58]. Patellamide F was characterized from the extract of L. patella sampled from northwestern Australia. The extract also contained patellamide B, ulithiacyclamide and lissoclinamide 3 [59]. In 1998, during a screening for multidrug resistance (MDR) reversing agents from marine organisms, four new cyclic peptides, assigned patellamide G and ulithiacyclamides E−G, were isolated from the tunicate L. patella collected in Pohnpei, along with the known patellamides A−C and ulithiacyclamide B. A common aspect of the novel metabolites was that at least one of the threonine units was not cyclized to an oxazoline ring [60].
In 2005, the first cyanobactin gene cluster (assigned as pat, originated the patellamide group) (Figure 3) was described in Prochloron didemni, the cyanobacterial symbiont of L. patella [8]. This discovery allowed the attribution of patellamides to the cyanobacterium Prochloron and confirmed that these compounds are synthetized by a ribosomal pathway. The function of the genes was also confirmed by heterologous expression in Escherichia coli [8]. At the same time, heterologous production of patellamides D and A (ascidiacyclamide) was verified by shotgun-cloning techniques by cloning Prochloron gDNA into an E. coli host, affirming that the Prochloron sp. is the primary biosynthetic source of the patellamides isolated from L. patella [7]. The presence of patellamides A and C was previously reported in the L. patella reef sample, but failed to detect any other patellamide variants as a major product of the crude extract. The pat gene cluster (~10.5 kb) contains a single precursor peptide gene, patE, that encodes for patellamides A (ITVCISVC) and C (VTACITFC). PatA and patG genes border this genetic cluster while patD and patF genes surround patE. PatB and patC genes, with an unknown function, are also present in this biosynthetic pathway. At that time, the function of patF was not known. Since the pat cluster was only found in a patellamide-producing L. patella strain, it was suggested that pat was probably acquired through horizontal gene transfer [8]. In 2006, 46 Prochloron-harboring ascidians were analyzed for the presence of pat genes. It was demonstrated that each symbiotic strain contains a unique pathway and that an extended library of patellamides is produced due to small hypervariable cassettes present in a conserved genetic background. Six patE gene variants (E1–E6) encoding seven different cyclic products, including patellamides A, B and C, ulithiacyclamide, ulicyclamide and lissoclinamides 2/3 and 4/5, were found among the analyzed samples. For instance, patellamide C is encoded by E1 (position I) and E2 (position II). Additionally, 23 other patE variants were found in 96-well plate E gene clone libraries [34].
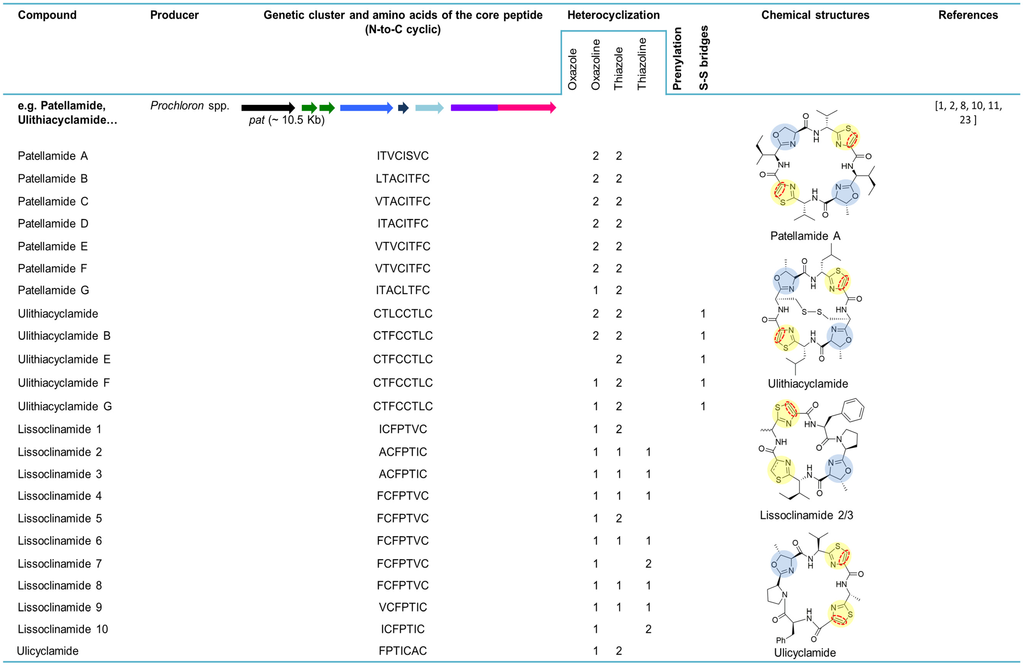
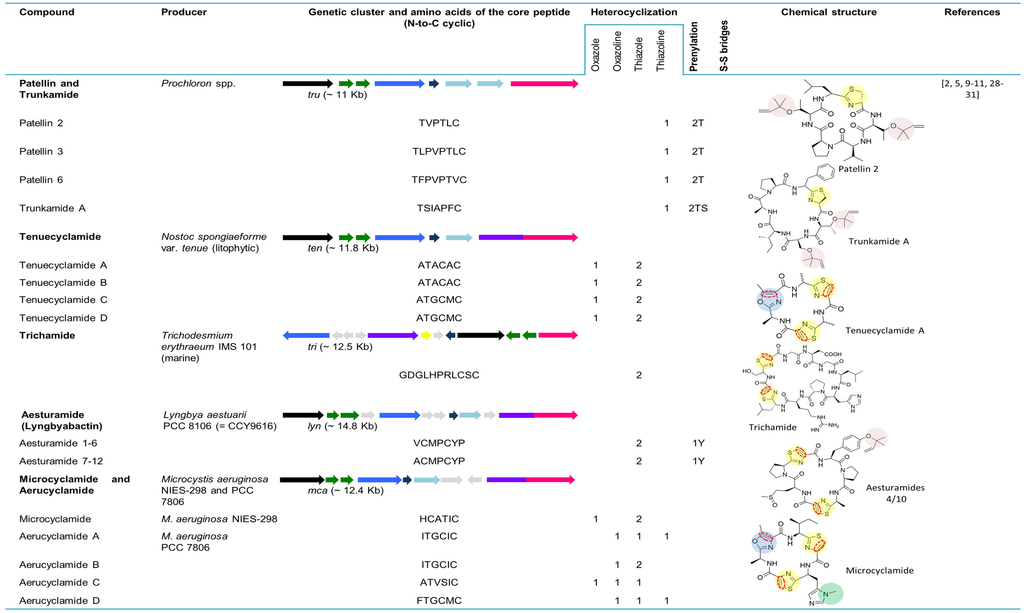
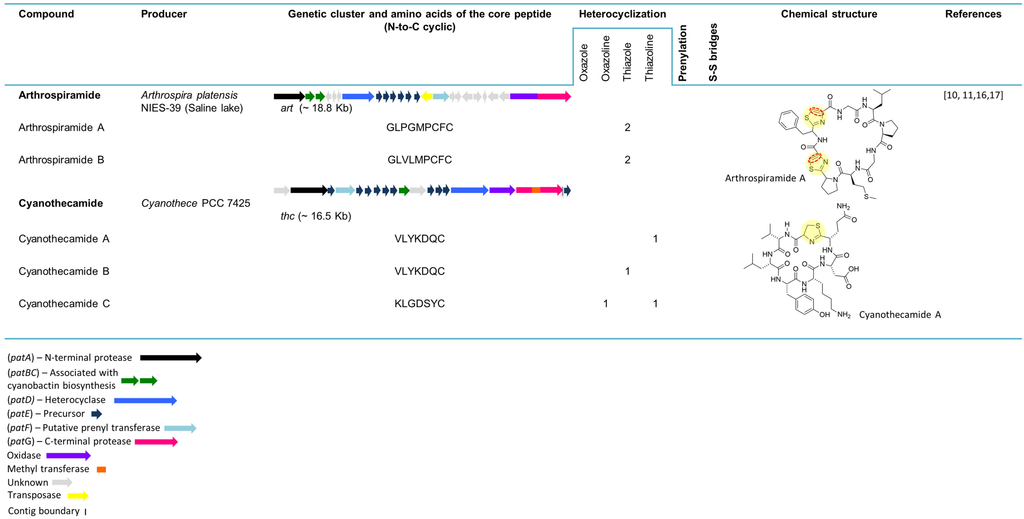
Figure 3.
Cyanobactins that encode heterocyclization enzymes. The peptides are organized in chronological order and the sequence of the core peptide is presented in the linear form. The location of the prenyl-group is indicated by one letter amino acid abbreviation in the correspondent column. The genes are identified by different colors. Heterocyclization of Cys (pale yellow) or Ser/Thr (pale blue), oxidation to azole (red dashed line), prenylation (pale red) and N-methylation (pale green) are indicated in the chemical structures.
Figure 3.
Cyanobactins that encode heterocyclization enzymes. The peptides are organized in chronological order and the sequence of the core peptide is presented in the linear form. The location of the prenyl-group is indicated by one letter amino acid abbreviation in the correspondent column. The genes are identified by different colors. Heterocyclization of Cys (pale yellow) or Ser/Thr (pale blue), oxidation to azole (red dashed line), prenylation (pale red) and N-methylation (pale green) are indicated in the chemical structures.
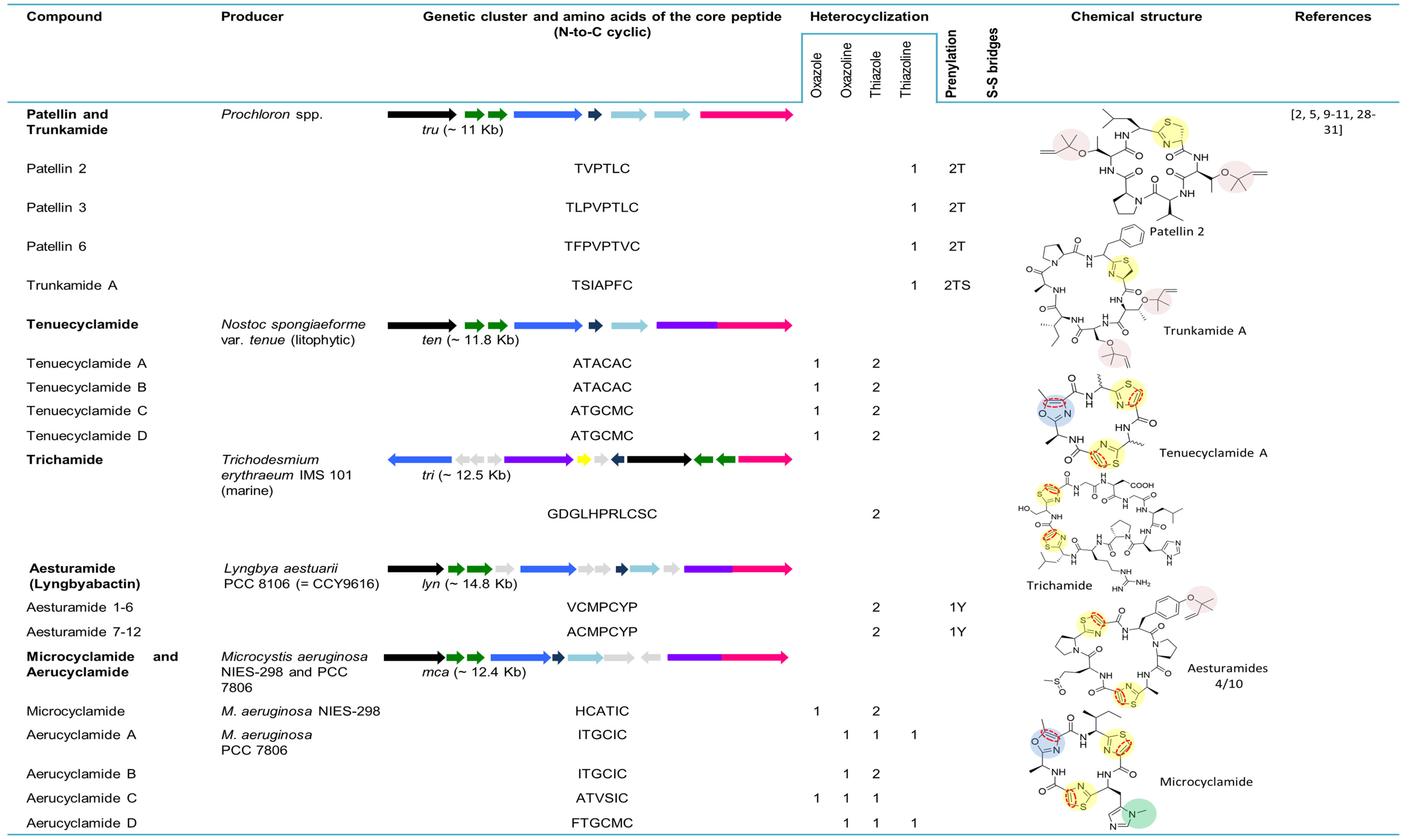

5.2. Patellins and Trunkamide
Patellin 2 (Figure 3), a modified cyclic peptide, was characterized from the Fijian marine tunicate L. patella. Contrary to the previously identified Lissoclinum peptides, this cyanobactin lacks the distinguishing thiazole and oxazoline amino acids and contains a thiazoline and two threonine residues modified as dimethylallyl ethers [23]. The cyclic compounds patellins 1–6 and trunkamide A were isolated in 1996 from Lissoclinum spp. collected in Viti Levu, Fiji (patellins 1–5) and in the Great Barrier Reef, Australia (patellins 3, 5 and 6 (G) and trunkamide). While patellins are hexa (patellins 1–2) or octa (patellins 3–5 and 6 (G)) peptides, trunkamide is a heptapeptide [24]. The presence of two dimethylallyl threonines (or one threonine and one serine) side chains and one thiazoline ring in the backbone of the patellins is the most important feature of these compounds [24].
Trunkamide A (Figure 3) was synthesized and its stereochemistry was revised in 2000 by Wipf and Uto [61]. The interesting activities of trunkamide A (see Section 7) further led to large-scale production of trunkamide A and enabled the completion of its biological activity as well as conformational studies. To that end, a total solid-phase synthesis and preliminary characterization of trunkamide A was performed and the configuration proposed by Wipf was confirmed [62]. In 2001, a solution was published by Mckeever and Pattenden [63] for the synthesis of trunkamide A. In order to obtain structure-activity relationships, the three-dimensional structure (solution structure) of this peptide was presented [64].
The patellin pathway tru (~11 kb) (Figure 3) directly encodes on the tru gene patellins 2 and 6 and was discovered in 2008 by Donia and co-workers [5]. The tru genetic cluster was cloned in E. coli, confirming that the same one was responsible for the biosynthesis of the prenylated compounds. As was seen in the pat gene cluster, hypervariability in small cassettes within the E gene led to the diversity of natural cyclic peptides. Variants of the E gene from the tru pathway were cloned from the ascidian samples containing patellins and trunkamide, revealing two new genes truE2 and truE3. While TruE2 encodes patellin 6 and trunkamide, TruE3 encodes three copies of trunkamide. The tru genetic cluster is syntenic to pat; however, two major differences are perceived: (1) truG does not contain an oxidase domain, which is congruent with the fact that patellins have thiazolines, whereas patellamides are oxidized to thiazoles; and, (2) two copies of truF located in the tru gene cluster are contiguous to each other. It was suggested that F or D genes were responsible for the heterocyclization of serine and threonine in the patellamides and/or prenylation in patellins, since the leading differences between pat and tru genetic clusters were detected in these genes [5].
5.3. Tenuecyclamides
In 1998, Banker and Carmeli announced the discovery of tenuecyclamides (A−D). These four hexapeptides, harboring two thiazoles and one oxazole, were isolated from the litophytic cyanobacterium strain Nostoc spongiaeforme var. tenue (TAU strain IL-184-6) [25]. The cyanobactin genetic cluster for tenuecyclamides, ten (~11.8 kb) was described from the same strain. The number and organization of the genes in this biosynthetic pathway is equal to that in the pat pathway. A unique feature of this gene cluster is its precursor peptide (TenE) that encodes four copies of the amino acid sequences of the core peptide, two for each of the hexapeptides A and C (Figure 3) [5]. Tenuecyclamide B differs from A only in stereochemistry [25].
5.4. Trichamide
Trichamide was the first natural product reported from the marine bloom-forming cyanobacterium Trichodesmium erythraeum ISM101 in 2006. The previous discovery of the patellamide gene cluster [8] contributed to the identification of a related cluster in this cyanobacterium strain and enabled the prediction of the tri biosynthetic pathway through the whole genome sequencing. This peptide, cyclized by an N-C terminal amide bond, comprises 11 amino acids, including two cysteine-derived thiazole groups (Figure 3). Contrary to pat, the tri gene cluster has a bidirectional gene order and the oxidase domain encodes separately from the protease gene. This gene cluster (~12.5 kb) consists of a unique precursor peptide gene encoding a single copy of the trichamide core peptide, a cyclodehydratase, an oxidase, two proteases and hypothetical genes. The presence of tRNA synthetase genes bordering the tri gene cluster proposed the possible procurement of this gene cluster through the horizontal gene transfer [28].
5.5. Aesturamides (Lyngbyabactins)
The genome mining of Lyngbya aestuarii CCY9616 (PCC 8106) by Donia and colleagues [5] allowed for the identification of a new cyanobactin genetic cluster dubbed lyn (~14.8 kb). This contains all of the homologs genes present in the patellamide genetic cluster (A–G), with the exception of the occurrence of four domains of unknown function. The possible new cyclic peptides structures were predicted based on the single lynE sequence that contains one copy of the Ala-containing sequence and two copies of the Val-containing sequence [5]. Twelve cyclic peptides assigned to aesturamides were described as a result of the single precursor peptide present in the lyn genetic cluster, representing the highest number of natural products produced by a ribosomal pathway. Aesturamides are synthetized enzymatically as reverse O-prenylated tyrosine ethers that subsequently undergo a Claisen rearrangement to produce forward C-prenylated tyrosine [19]. Two different groups of aesturamides arise from the two different sequences of the core peptide. Aesturamides 1 to 3 are the result of post-translational modifications of the lynE sequence, VCMPCYP (Figure 3). These compounds are reverse O-prenylated, non-prenylated and forward C-prenylated. Aesturamides 4 to 6 belong to this first group and are sulfoxide derivatives of 1–3. Aesturamides 7 to 12, the products of the lynE sequence ACMPCYP, are in the same manner as the above-mentioned aesturamides, reverse O-prenylated, non-prenylated and forward C-prenylated compounds, with or without Met oxidation. Aesturamides are N-C cyclic compounds, containing thiazole, which is congruent with the presence of lynA, lynG and lynD genes and an oxidase domain [19].
5.6. Microcyclamides and Aerucyclamides
Microcyclamide was the first cyclic hexapeptide composed of five-membered heterocycles (Figure 3) reported from the bloom-forming cyanobacterium strain Microcystis aeruginosa NIES-298, by Ishida and co-workers in 2000 [27]. Later, it was demonstrated that microcyclamide is a product of a ribosomal pathway, through the activity of a set of enzymes that are related to the ones involved in patellamide biosynthesis [31]. Genomic data analysis of M. aeruginosa PCC 7806 allowed for the identification of a gene cluster encoding proteins with more that 90% similarity to the nine proteins encoded in NIES-298 (~12.4 kb) and resulted in the discovery and isolation of two new microcyclamide family peptides: microcyclamide 7806A and microcyclamide 7806B. The genes from the cyanobactin genetic clusters (mca) in both strains have the same order as the ones from the patellamide gene cluster, with the exception of two open-reading frames with no assigned functions. The E gene in NIES-298 contains two-peptide coding regions (HCATIC; (Figure 3)), which is consistent with the amino acid composition and order in the microcyclamide molecule, whereas the corresponding gene in PCC 7806 encodes four (ITGCIC, ITGCIC, ATVSIC and FTGCMC; (Figure 3)). Microcyclamide 7806A and 7806B (later renamed aerucyclamide C and D) structures are assigned to the precursor sequences ATVSIC and FTGCMC (Figure 3) [31]. Subsequently, microcyclamide structures were revised and renamed. Aerucyclamides A−D were isolated from M. aeruginosa PCC 7806 [29,30]. It was proposed that they were the actual ribosomal-encoded metabolites, which is consistent with the genetic data provided by Ziemert [31]. More recently, microcyclamide-like genetic clusters were also detected in different Microcystis strains [11].
5.7. Arthrospiramide
The art gene cluster (~18.8 kb) (Figure 3) was uncovered from the genome of the economically important cyanobacterium Arthrospira platensis NIES-39 [16]. The organization of art (from A to G gene) is similar to that of the most common cyanobactin genetic clusters, with two exceptions: (1) it contains six precursor peptides that encode different products and (2) a transposase is present between artE and artF genes. Closely related art gene clusters were also found in the genome of A. platensis strain Paraca and an edible Arthrospira strain. In the first case, all the cyanobactin genes are present with the exception of the E genes, since the genome was highly fragmented on small contigs. The genome of the second Arthrospira strain contains three additional precursor peptides encoding for different products. In an attempt to isolate peptides from this genera, commercial preparations of dried A. platensis were acquired, and the compounds derived from the ArtE3 and ArtE5 precursor peptides were partially purified and named arthrospiramide A and B, respectively [16]. Recently, several arthrospiramide-like genetic clusters from different Arthrospira strains were uncovered through genome-mining techniques [11].
5.8. Cyanothecamides
Three new cyanobactins, (cyanothecamides (A−C)), were discovered from a culture of Cyanothece sp. PCC 7425 exposed to a heat shock [17]. The cyanothecamide gene cluster, thc (~16.5 kb) (Figure 3), was previously reported in [16], but no cyanobactins were detected in cultures growing in normal conditions over a two-year period. The organization of this genetic cluster is unique (representing a separate cyanobactin genotype), including a methyltransferase and an ABC transporter. Furthermore, nine copies of the thcE gene are present, each one encoding a different product. Among three plasmids present in the Cyanothece sp. PCC 7425, the tenth thcE gene was detected in the plasmid pP742501 [16]. The thcE2 gene, from the cyanothecamide gene cluster, was identified as the ortholog of patE. The ThcE2 cassette, VLYKDQC, is responsible for the synthesis of cyanothecamides A and B. Cyanothecamide C, detected by mass spectrometry (MS), was assigned to thcE3 gene with the coding sequence KLGDSYC and revealed a new cassette-flanking sequence (SNCIG), which has not yet been described in the canonical patA recognition sequence. Cyanothecamides are macrocycle heptapeptides with a single sulfur heterocycle that gives this group of compounds a novel backbone arrangement [17].
5.9. Aeruginosamides and Viridisamide—Linear Cyanobactins
In 2013, the genome mining of 126 cyanobacterial strains, led to the discovery of linear cyanobactins by Leikoski and colleagues [11]. The cyanobactin genetic clusters of M. aeruginosa PCC 9432 (~12.4 kb), Oscillatoria nigro-viridis PCC 7112 (~13 kb) and Leptolyngbya sp. PCC 7376 (~11.3 kb) (Figure 4), encode an unusual bimodular protein with homology to SAM-dependent methyltransferases and prenyltransferases, thereby indicating that a new cyanobactin variety could arise as an end product. However, in Leptolyngbya sp. PCC 7376, the cyanobactins could not be predicted since the identified precursors lacked the recognizable cleavage sites. The bioinformatics study predicted the presence of the linear cyanobactins in M. aeruginosa PCC 9432 and O. nigro-viridis PCC 7112 due to the presence of conserved cleavage sites. The tetrapeptide aeruginosamide B and the pentapeptide aeruginosamide C were detected in M. aeruginosa PCC 9432. A similar cytotoxic compound, named aeruginosamide, was previously reported from a M. aeruginosa bloom [26]. Both aeruginosamide B and C present a prenylated N-termini and a methylated C-termini bound to thiazole. In O. nigro-viridis PCC 7112, the linear tripeptide viridisamide A was identified. As in aeruginosamides B and C, viridisamide A contains a prenylated N-termini and a C-termini bound to thiazole [11].

Figure 4.
Linear cyanobactins. The genes are identified by different colors. Prenylated N-termini (pale red) and methylated C-termini bound to thiazoles are highlighted in the chemical structures.
Figure 4.
Linear cyanobactins. The genes are identified by different colors. Prenylated N-termini (pale red) and methylated C-termini bound to thiazoles are highlighted in the chemical structures.

6. Cyanobactins Non-Encoding Heterocyclization or Oxidation Enzymes
6.1. Anacyclamides
The whole genome sequence from the strain Anabaena sp. 90 allowed for the discovery of new cyanobactins, designated anacyclamides by Leikoski and colleagues [20]. Using bioinformatics tools coupled with stable isotope labeling and MS, these novel cyclic peptides were found in a total of 27 Anabaena strains. Anacyclamides can differ from 7 to 20 amino acids in length, enclosing only proteinogenic amino acids that can be prenylated or geranylated. The acy gene cluster (~10.7 kb) is composed of 11 genes organized bidirectionally, such as in the trichamide gene cluster. Only one precursor peptide gene is present in the acy genetic cluster. Nevertheless, 15 distinct amino acid sequences from the core peptide were detected from 27 strains yielding 18 unique anacyclamides. The acy gene cluster lacks acyD as well as an oxidase domain, which is consistent with the chemical structure of these metabolites. In contrast to the other reported cyanobactins, anacyclamides exhibit a pronounced amino acid variation with only one proline at the conserved C-terminus [20]. With the discovery of anacyclamides, the definition of cyanobactins was extended to include unmodified peptides that can have prenylated or geranylated amino acids (Figure 5).
6.2. Prenylagaramides
The isolation and structural explanation of the Tyr O-prenylated cyclic peptides prenylagaramides A and B were reported in 1999 [18]. The nonapeptide, prenylagaramide A was isolated from Oscillatoria agardhii NIES-205, whereas the heptapeptide prenylagaramide B was obtained from a culture of O. agardhii NIES-596. The prenylagaramide genetic cluster pag (~13.4 kb) in Planktothrix agardhii NIES-596 (foremost identified as O. agardhii NIES-596) is highly similar to acy, as it lacks the heterocyclase domain. Seven precursor peptide genes are present in the pag genetic cluster. The E6 gene (INPYLYP) directly encodes prenylagaramide B, while the E7 gene (QAYLGIPLP) encodes the new prenylagaramide C [16]. The difference between prenylagaramides and anacyclamides is that the latter are unadorned N-C cycles, without Tyr O-prenylation (Figure 5) [16].
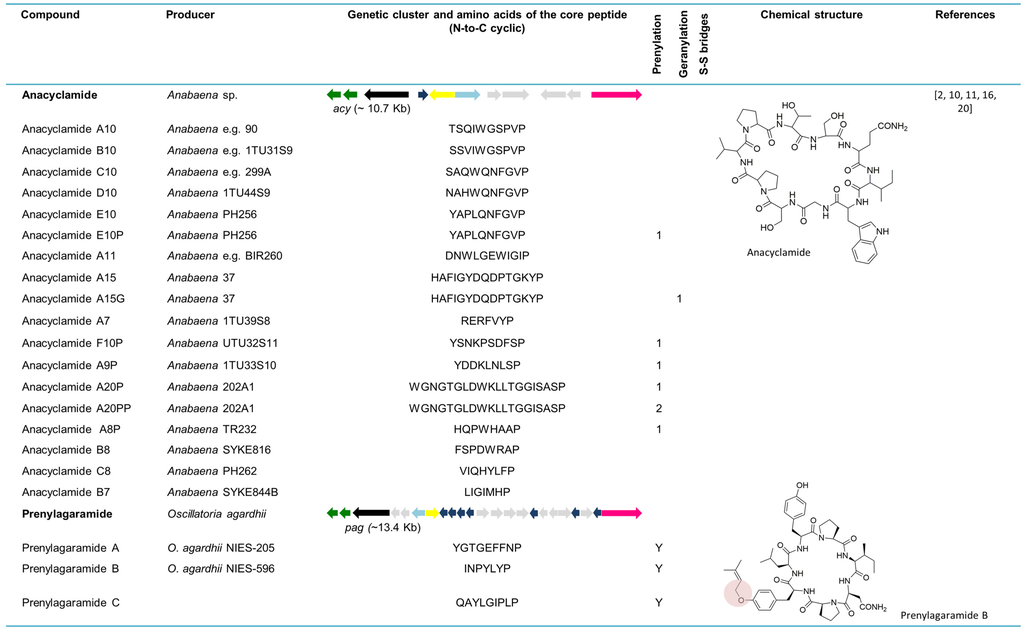

Figure 5.
Cyanobactins that do not encode heterocyclization or oxidationenzymes. Thepeptides are organized in chronological order and the sequence of the core peptide is presented in linear form. The location of the prenyl group is indicated by one-letter amino acid abbreviation in the corresponding column. The genes are identified by different colors. Prenylation (pale red) is indicated in the chemical structure.
Figure 5.
Cyanobactins that do not encode heterocyclization or oxidationenzymes. Thepeptides are organized in chronological order and the sequence of the core peptide is presented in linear form. The location of the prenyl group is indicated by one-letter amino acid abbreviation in the corresponding column. The genes are identified by different colors. Prenylation (pale red) is indicated in the chemical structure.
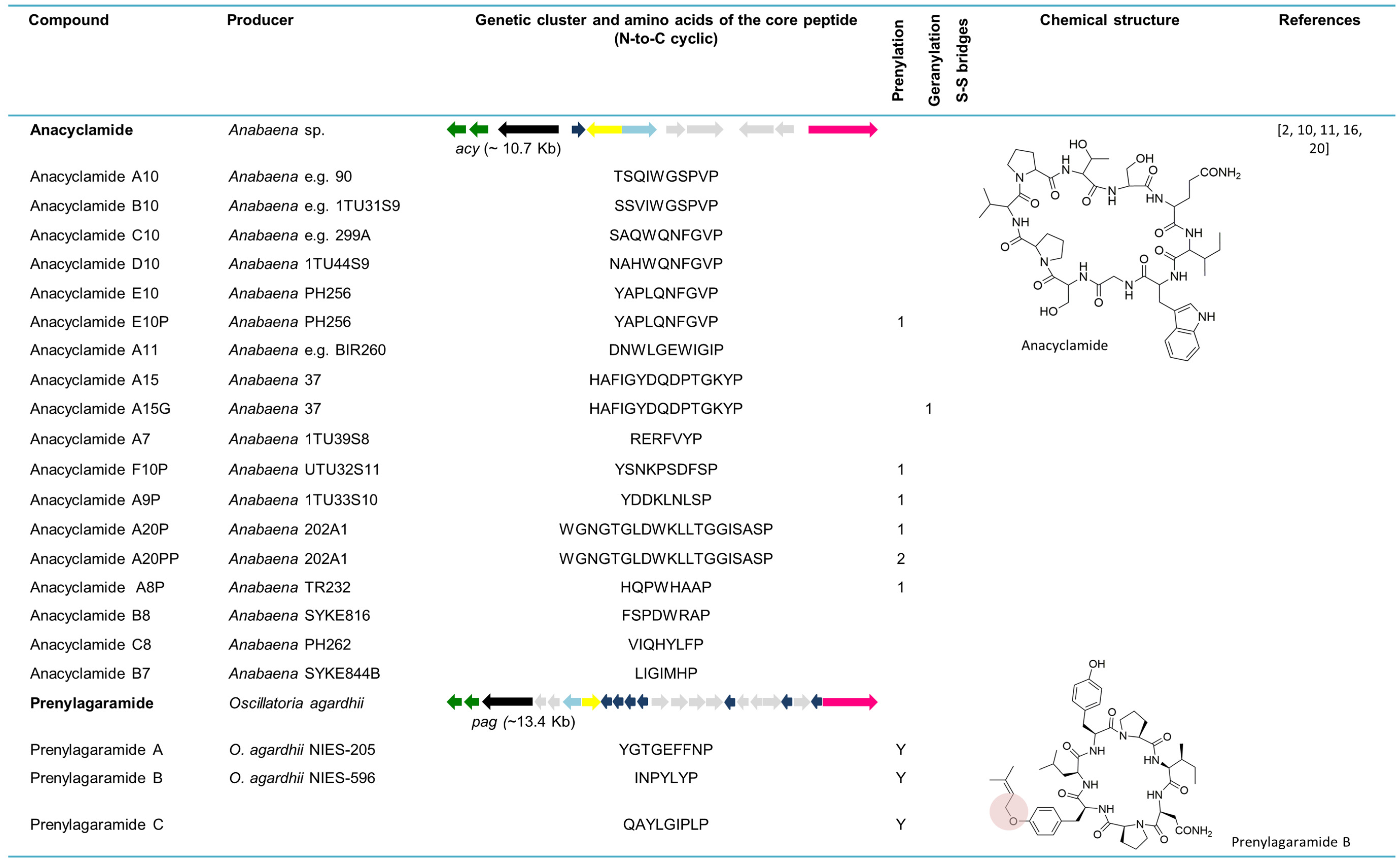

6.3. Piricyclamides
The investigation of the inactive cyanobactin gene cluster, mae, in M. aeruginosa NIES-843 (~19.7 kb) [16] resulted in the discovery of the piricyclamides in M. aeruginosa PCC 7005 (Figure 5) [15]. This new cyanobactin family is structurally diverse since some piricyclamides consist exclusively of proteinogenic amino acids, whereas others contain disulfide bridges, such as ulithiacyclamides, and some are prenylated or geranylated. Piricyclamides can differ from 7 to 17 amino acids in length. The pir gene cluster in M. aeruginosa PCC 7005 (~9.4 kb) is similar to the previously described gene cluster in NIES-843 [16], although the re-examination of the latter has revealed the presence of two extra precursor genes. The inactivation of the gene cluster in NIES-843 is due to a frameshift mutation in pirG gene and the presence of two insertion elements in the pirA gene. The pir genetic clusters constitute one to four precursor peptide genes. Additionally, the analysis of a bloom sample composed of Aphanizomenon (97% of the 16S rRNA genes sequenced) and Microcystis (3% of the 16S rRNA genes sequenced) revealed 19 different precursor peptide genes disclosing the vast genetic and concomitant chemical diversity of these compounds in a single environmental sample. In this study, 13 different piricyclamide core gene sequences were uncovered in seven Microcystis strains, whereas nine piricyclamides were detected by LC-MS in six strains. In strain PCC 7005, all the essential genes for cyanobactin production are present in the pir gene cluster and, similar to acy, the D gene and G gene oxidase domains are lacking. Four piricyclamides were found in PCC 7005, two of which are geranylated and one being composed of a disulfide bridge. The PCC 7005 pir gene cluster was cloned into E. coli from the beginning of pirE3 and only one piricyclamide TFCDLATKQCYP (pirE4) was detected. This was due to the lack of promoters and some precursor genes in the amplified part of the genetic cluster. Phylogenetic inferences using concatenated proteases A and G clearly differentiate piricyclamides from the other M. aeruginosa cyanobactins, microcyclamides (Figure 5) [15]. Furthermore, several piricyclamide-like genetic clusters were uncovered for different Microcystis strains through genome mining [11].
7. Bioactivities
Cyanobactins have various activities derived from their different structures (Table 1). The search for anticancer compounds resulted in the discovery of several cyanobactins, although cytotoxicity has been the most pronounced activity attributed to these peptides (see Table 1) [9,10].
The in vitro antitumor activity against L1210 murine leukemia cell cultures was evaluated for the cyanobactins; ulithiacyclamide, ulicyclamide, patellamides A−C and lissoclinamides 1–3 [21,22,51]. The most effective cyanobactin was found to be ulithiacyclamide, with an IC50 value of 0.35 µg/mL, followed by patellamides B, C and A, with IC50 values of 2.0, 3.2 and 3.9 µg/mL, respectively. Ulicyclamide was the least active with an IC50 value of 7.2 µg/mL [21]. Lissoclinamides 1, 2 and 3 exhibited borderline cytotoxicity, with IC50 values higher than 10 µg/mL [22,51]. Similarly, patellamide D and lissoclinamides 4–6 displayed slight cytotoxicity against PS lymphocytic leukemia cells with ED50 values (µg/mL) of 11, 10, 12 and 6.9, respectively [22]. The intriguing structures and cytotoxic activities reported for cyanobactins led to the discovery of the importance of the oxazoline function that was believed to play an essential role in cytotoxicity. The confirmation of the efficacy of ulithiacyclamide against L1210 murine leukemia cells, with cytotoxic values comparable to the ones from the clinical anticancer drugs, suggested that the potency of this cyanobactin was related to the unique disulfide bridge that supports the conformation of this compound [65]. The mechanistic characteristics involved in the cytotoxicity of ulithiacyclamide were studied by Shioiri and co-workers [66], and it was found that ulithiacyclamide inhibited cell growth in a self-destructive manner by inhibiting RNA and protein synthesis. Additionally, cytotoxicity of this compound against L1210 cells was synergistically enhanced when tested together with bleomycin, an anticancer drug with a different cytotoxic mechanism [66]. The confirmation of ulicyclamide cytotoxicity against L1210 cells (IC50 = 13 µg/mL) and its synthetic intermediate lacking an oxazoline function in its structure (IC50 = 35 µg/mL), contradicted the earlier idea that oxazoline was essential in cytotoxicity and related the same to its inhibitory effects on both DNA and RNA syntheses [67]. The cyanobactins ulithiacyclamide and ulithiacyclamide B, as well as patellamides A−C, were also tested against the KB cell line. Ulithiacyclamide (IC50 = 35 ng/mL) and ulithiacyclamide B (IC50 = 17 ng/mL) were found to be the most cytotoxic against the KB cell line, whereas patellamides A (IC50 = 3000 ng/mL), B (IC50 > 4000 ng/mL) and C (IC50 = 6000 ng/mL) were at least two orders of magnitude less active. Nevertheless, all the aforementioned cyanobactins were less toxic than other tested compounds. Ulithiacyclamide B and patellamides A−C were also tested against distinct tumor cell lines such as leukemia (murine L1210), solid tumor (murine colon adenocarcinoma C38 and human colon adenocarcinoma H116) and low malignancy (murine) cell lines. None of the compounds showed selective cytotoxicity against solid tumor cell lines. Ulithiacyclamide B presented similar cytotoxicity against all the tested cell lines, suggesting that this compound is a general cytotoxin and probably too toxic to be active against solid tumors in vivo. The patellamides presented very weak activity against the L210 cell line. Ulithiacyclamide B and patellamides B and C did not show selective activity against either L1210 or an acute myelogenous leukemia (C1498) compared with normal myeloid-committed stem cell (CFU-GM). This suggests that none of these compounds would reduce myelo suppression or have a distinct advantage over any clinically used agent if found to be active in vivo [57]. In fact, studies on ulithiacyclamide were interrupted due to its extreme cytotoxicity in animal models [68]. The capability of patellamide E as a possible cytotoxic agent was also determined against human colon tumor cells in vitro, but the results suggested weak cytotoxicity (IC50 = 125 µg/mL) [58]. When tested in the NCI 60 human tumor cell line panel, patellamides B and F and ulithiacyclamide exhibited modest to general cytotoxicity (average LC50 values of 48 µM, 13 µM and 3 µM, respectively) without a pattern of differential cytotoxicity of sufficient significance to warrant further studies based on the NCI screen [59].

Table 1.
Bioactivities of cyanobactins produced from cyanobacteria.
| Source Organism | Compound | Bioactivity | References |
|---|---|---|---|
| Lissoclinum patella (Prochloron spp.) | Lissoclinamides 1–3 | Borderline cytotoxicity against L1210 murine leukemia cells (IC50 > 10 µg/mL) | [22] |
| Lissoclinamides 4–6 | Slight cytotoxicity against PS lymphocytic leukemia cells (ID50 = 10, 12 and 6.9 µg/mL for lissoclinamides 4, 5 and 6, respectively) | [22] | |
| Patellamide A | Mild cytotoxicity against L1210 murine leukemia cells (IC50 = 3.9 µg/mL) | [21,57] | |
| Poor cytotoxicity against KB cell line (IC50 = 3000 ng/mL) | |||
| Patellamide B | Mild cytotoxicity against L1210 murine leukemia cells (IC50 = 2.0 µg/mL) | [21,57,59,60] | |
| Poor cytotoxicity against KB cell line (IC50 > 4000 ng/mL) | |||
| General cytotoxicity in NCI’s 60 human tumor cell line panel (Average LC50 = 48 µM) | |||
| Multidrug reversing activity | |||
| Patellamide C | Mild cytotoxicity against L1210 murine leukemia cells (IC50 = 3.2 µg/mL) | [21,57,60] | |
| Poor cytotoxicity against KB cell line (IC50 = 6000 ng/mL) | |||
| Multidrug reversing activity | |||
| Patellamide D | Slight cytotoxicity against PS lymphocytic leukemia cells (ID50 = 11 µg/mL) | [22] | |
| Multidrug reversing activity | |||
| Patellamide E | Weak cytotoxicity against human colon tumor cells (IC50 = 125 µg/mL) | [58] | |
| Patellamide F | General cytotoxicity in NCI’s 60 human tumor cell line panel (Average LC50 = 13 µM) | [59] | |
| Patellin 6 | Moderate cytotoxic against P388, A549, HT29 and CVI cells (Average IC50 = 2 µg/mL) and inhibition of topoisomerase II activity (IC50 = 2.5µg/mL) | [24] | |
| Trunkamide A | Active against P-388 mouse lymphoma, A-549 human lung carcinoma, HT-29 human colon carcinoma (IC50 = 0.5 µg/mL) and MEL-28 human melanoma (IC50 = 1.0 µg/mL) cell lines. | [69] | |
| Ulicyclamide | Poor cytotoxicity against L1210 murine leukemia cells (IC50 = 7.2 µg/mL) | [21] | |
| Ulithiacyclamide | Cytotoxicity against L1210 murine leukemia (IC50 = 0.35 µg/mL) and KB (IC50 = 35 ng/mL) cell lines | [21,57,59] | |
| General cytotoxicity in NCI’s 60 human tumor cell line panel (Average LC50 = 3 µM) | |||
| Ulithiacyclamide B | Cytotoxicity against KB cell line (IC50 = 17 ng/mL) | [57] | |
| Microcystis aeruginosa | Aerucyclamides | Toxic to freshwater crustacean Thamnocephalus platyurus (LC50 = 30.5 µM for aerucyclamide A and LC50 = 33.8 µM for aerucyclamide B) | [29,30,31] |
| Antimalarial (aerucyclamide B presented IC50 = 0.7 µM, aerucyclamide C presented IC50 = 2.3 µM and aerucyclamide D presented IC50 = 6.3 µM) | |||
| Aerucyclamide C—moderate activity against Trypanosoma brucei rhodesiense (IC50 = 9.2 µM) | |||
| No inhibitory activity against HeLa cells and standard antiproliferative, antibacterial and antifungal assays | |||
| Microcystis aeruginosa NIES-298 (freshwater) | Microcyclamide | Slight cytotoxicity against P388 murine leukemia cells (IC50 = 1.2 µg/mL) | [27] |
| Microcystis sp. | Microcyclamide | Microcyclamide MZ602—mild cytotoxicity against Molt4 leukemia cell line (20% cell grow inhibition) and mild inhibition of chymotrypsin (IC50 = 75 µM) | [70] |
| Microcyclamide MZ568—strong cytotoxicity against Molt4 leukemia cell line (36% cell grow inhibition) and no inhibition of serine proteases | |||
| Nostoc spongiaeforme var. tenue (litophytic) | Tenuecyclamide A, C and D | Inhibited division of sea urchin embryos Paracentrotus lividus (ED100 = 108 µM, for tenuecyclamide A, ED100 = 9.0 µM for C and ED100 = 19.1 µM for D). B not tested. | [25] |
| Trichodesmium erythraeum IMS 101 (marine) | Trichamide | No effects found (tested for cytotoxicity, antifungal, antibacterial and antiviral activities) | [28] |
Since an MDR phenotype is the problem associated with many chemotherapeutic administrations, cyanobactins have been evaluated for their potential as MDR inhibitors [9]. Patellamide D was the first compound to demonstrate this activity, improving the effectiveness of certain drugs in human cell lines (CEM/VLB 100) as a selective antagonist to MDR [9]. Anti-MDR activity against vinblastine-resistant CCRF-CEM human leukemic lymphoblasts was evaluated for patellamides A−C and G and ulithiacyclamides E−G. Patellamides B and C were considered significantly active in reducing the drug resistance by about ten-fold (the IC50 value for vinblastine in the presence of 2.5 µg/mL of patellamides B and C was 12 nM) [60]. Antimalarial activity of several cyanobactins has been also determined. Some patellamides present antimalarial activity even at doses ten-fold greater than the cytotoxic dose [9].
Patellins 1–6 and trunkamide A were also tested for their potential bioactivity in several cytotoxicity assays. While patellins 1–5 and trunkamide were inactive, patellin 6 displayed moderate cytotoxicity against P388, A549, HT29 and CV1 cells (IC50 = 2 µg/mL) and inhibition of topoisomerase II activity (IC50 of 2.5 µg/mL) [24]. In contrast to the initial findings, trunkamide A appeared to have quite promising antitumor activity [61], exhibiting the antitumor activity against cell lines derived from human tumors. Trunkamide was active against the P-388 mouse lymphoma (IC50 = 0.5 µg/mL), A-549 human lung carcinoma (IC50 = 0.5 µg/mL), HT-29 human colon carcinoma (IC50 = 0.5 µg/mL) and MEL-28 human melanoma (IC50 = 1.0 µg/mL) cell lines. Trunkamide A, as well as an acceptable pharmaceutical composition and carrier of this compound, were applied for patent by Bowden and Gravalos [69]. Trunkamide A was initially selected for further testing by the National Cancer Institute (NCI) due to a good COMPARE correlation analysis and specificity against the UO-31 renal cell line (an MDR line). This could suggest a potential novel mechanism of action and a possible non-MDR activity, as well [62,64]. Trunkamide was classified as a pre-clinical candidate by the marine natural products pharmaceutical company, PharmaMar, but no additional information has been accessible in the past twelve years [9].
The effect of tenuecyclamides A, C and D isolated from N. spongiaeforme var. tenue was determined by the division of the sea urchin embryos Paracentrotus lividus. Tenuecyclamide A inhibited the division of sea urchin embryos with ED100 of 108 µM, tenuecyclamide C with ED100 of 9.0 µM and tenuecyclamide D with ED100 of 19.1 µM [25].
Crude methanol extracts from T. erythraeum, tested for general cytotoxicity (HTC-116, CEM-TART) and antihuman immunodeficiency virus, antifungal (Candida albicans) and antimicrobial (Staphylococcus aureus and Enterococcus faecium) had no significant activity in these assays. The extracts from T. erythraeum IMS101 caused neurotoxicity in a mouse assay but the purified trichamide was not the active compound. Trichamide is not released in significant amounts to the cyanobacterial growth medium, suggesting that this cyanobactin may have an antipredation function [28].
Microcyclamide isolated from M. aeruginosa NIES-298 exhibited slight cytotoxic activity against P388 murine leukemia cells (IC50 = 1.2 µg/mL) [27]. Aerucyclamide 7806A and 7806B (previously designated as microcyclamides) from M. aeruginosa PCC 7806 were screened against HeLa cells and standard antiproliferative, antibacterial and antifungal assays, but no inhibitory activity was detected [31]. Aerucyclamides A and B were also tested in a 24-h acute toxicity test assay using the sensitive freshwater crustacean Thamnocephalus platyurus. Both peptides were toxic with LC50 values of 30.5 and 33.8 µM, respectively. However, it is unclear if this toxicity is ecologically relevant [29]. In search of cyanobacterial compounds against tropical diseases, aerucyclamides A−D were screened against Plasmodium falciparum K1, Trypanosoma brucei rhodesiense STIB 900, and rat myoblast L6 cells. Aerucyclamide B was the most active compound against the chloroquine-resistant strain K1 of P. falciparum (IC50 = 0.7 µM), presenting a large selectivity for the parasite in relation to the L6 rat myoblast cell line (IC50 = 120 µM). Additionally, an interesting fact was that the antiplasmodial activity decreased by one order of magnitude by one structural modification from aerucyclamide B to A (IC50 = 5.0 µM), i.e., reduction of thiazole to a thiazoline residue. Aerucyclamides C and D also presented low micromolar activity (IC50 values of 2.3 and 6.3 µM, respectively). All compounds displayed very weak to no toxicity for the L6 cell line (IC50 values ranging from 106 to >168 µM). Aerucyclamide C was the most active compound against T. brucei rhodesiense, with moderate activity (IC50 = 9.2 µM). Aerucyclamides, particularly aerucyclamide B, displayed preferably potent and selective antiplasmodial activity against P. falciparum K1 and a large selectivity over the mammalian L6 cell line. Aerucyclamide C exhibited low cytotoxicity against the crustacean grazer T. platyurus (LC50 = 70.5 µM) [30]. The biological activities of microcyclamides MZ602 and MZ568 were tested against the serine proteases trypsin, thrombin, chymotrypsin and elastase, and against the Molt4 (leukemia) cell line. Microcyclamide MZ602 presented very mild cytotoxicity against the Molt4 cell line (20% cell grow inhibition at 83 µM) and mild inhibition of chymotrypsin (IC50 = 75 µM). In contrast, microcyclamide MZ568 displayed a stronger cytotoxicity against the Molt4 cell line (36% cell grow inhibition at 1.8 µM) and no inhibition in the tested serine proteases [70].
Not all the cyanobactins have been evaluated for their bioactivity. Lack of information, especially concerning the cyanobactins discovered in the last several years, is due to the fact that bioactivity assessment is greatly dependent on the available amounts of cyanobactins. Integration of biosynthetic pathways with heterologous expression systems for cyanobactins may lead to the production of sufficient amounts providing new insights for its bioactivity.
8. Ecological Roles
Cyanobactins’ ecological functions have not been clearly studied, although they may be related to some of the bioactivities previously described, such as antibacterial activity and allelopathy.
Cyanobactin-like peptides, such as aeruginazoles [71,72] and kawaguchipeptin B [73], isolated from Microcystis sp. strain IL-323 and a Microcystis bloom showed antibacterial activities. The putative cyanobactins, nostocyclamide and nostocyclamide M isolated from the free-living cyanobacterium Nostoc sp. 31 showed allelopathy against competing cyanobacteria and small grazing organisms [74,75]. The allelopathic activity of nostocyclamides suggested that these molecules were essential for competition among cyanobacteria. For this reason, allelopathy emerged as a possible ecological role for cyanobactins. Nevertheless, more studies using other strains and species need to be done before extrapolations are made.
The metal-binding proprieties of some cyanobactins have been pointed out as a possible role for these peptides in nature. This subject has been well documented in several reviews [9,68,76,77,78,79] and therefore we will only shortly summarize some of the most significant work that has been done to date. Most of the cyanobactin metal-binding studies have focused on cyclic hepta- and octapeptides of the lissoclinamide and patellamide groups. Cyanobactins, such as ascidiacyclamide isolated from the ascidian L. patella [53], lissoclinamide 9 and 10, patellamides A−E and ulithiacyclamides are described as metal-binding compounds. Ascidiacyclamide was the first cyanobactin to be described as a metal binder. The characterized bis-copper (II) complex of ascidiacyclamide revealed two copper ions separated by a bridging carbonate anion fixed in the “saddle-shaped” molecule [80]. Likewise, studies regarding the binding of Cu(II) to patellamide D, which presents a “figure of eight” conformation in the solid state, determined a similar associated bis-copper complex [81]. Patellamides A−C were able to selectively bind to Cu(II) and Zn(II), but not to Ca(II) or Mg(II). These cyanobactins have the ability to bind two metals per molecule, and patellamide B and E experience a conformational change from the “figure of eight” to the “square” upon copper complexation [82]. In another study, Morris and colleagues [83] demonstrated that patellamide C presented extreme selectivity for Cu(II), even in the presence of excess Zn(II). In contrast, the same cyanobactin was not able to significantly bind to Co(II), Ni(II) and Hg(II). The higher affinity of patellamide C to Cu(II) over Zn(II) seems to be linked to the availability of the exposed nitrogen-binding sites for these ions. Ulithiacyclamide shows identical selectivity for Cu(II) and immediately forms Cu(II) complexes without any change in its conformation. Patellamide A, which mostly presents the “square” form in solution, was less selective for copper and the binding resulted in small conformational changes. Interestingly, patellamide C binding of Cu(II) led to a conformational change from the “figure of eight” to the “square,” allowing the favorable Cu(II) accommodation [83]. It seems clear that the metal-binding activity is dependent on the molecular shape. Cu(II) prefers the “square planar” or “square pyramidal” environments provided when the patellamides assume the “square” conformation. On the other hand, Zn(II) prefers a “tetrahedral” binding environment, that is not available in patellamide C. Nevertheless, the binding of Zn(II) does take place but it is probably less ideal than the binding of Cu(II) [83]. These studies revealed that copper is the biologically relevant metal for these cyanobactins. In addition to molecular shape, other factors seem to affect the interaction of cyanobactins with metals. The nature of the anions, the presence/absence of a base to deprotonate the cyclic peptide, the reaction solvent and the nature of the heterocycles that constitute the compounds are important factors that influence metal-binding complex formation [68]. Similar studies have been performed on lissoclinamides 9 and 10, revealing that lissoclinamide 10 is more selective for Cu(II) than lissoclinamide 9, even in the presence of excess Zn(II). Additionally, it was discovered that both cyanobactins present specific Cu(II)-binding motifs that are very close to the geometrical ideal [84].
It is possible that the natural role of ascidiacyclamide and patellamides includes Cu(II) coordination. However, cyanobactin metal-binding constants are usually low, when compared to physiologically relevant molecules. Furthermore, metal-binding constants were determined in vitro and hence determination of this in natural environments would be important.
Several biological functions have been proposed for the Cu(II) complexes of cyclic peptides. Those include: Cu(II) transport and storage, copper detoxification, CO2 hydration (carbonic anhydrase reactivity), oxygen activation and phosphoester hydrolysis (phosphatase reactivity). Additionally, Cu(II) cyclic peptide complexes may also act as enzyme co-factors [85].
The recent studies by Comba [85] suggest that the possible biological function of the patellamides produced by an organism is the carbonic anhydrase activity of their dicopper complexes. The dicopper(II)-patellamide complexes seem to be the first structural, spectroscopic and competent functional models for a carbonic anhydrase based on copper. However, it is important to point out that to date, there is no substantial evidence that Cu(II) complexation of patellamides is relevant for the ascidians [85].
Therefore, the essential question regarding the ecological roles of cyanobactins remains to be answered. It seems possible that cyanobactins may have several roles in nature and continuous efforts will be necessary to provide new insights concerning this intriguing subject.
9. Biotechnological Importance
Cyclic peptides offer several advantages over linear peptides for their pharmaceutical potential due to versatility, higher permeability across membrane barriers, greater resistance to enzymatic degradation, enhanced bioavailability [86,87] and higher affinity in receptor binding [87]. Mid-sized macrocycles and cyclic peptides can interact with extended binding sites that are not acquiescent to small molecules and have several conveniences compared to the native proteins and antibodies [1]. The biotechnological capability of cyanobactins’ biosynthetic pathways relies essentially on the guided macrocyclases and precursor peptide biosynthesis, and mobility of recognition sequences (RSs) [33,36,40,88]. Cyanobactins are directly encoded in the precursor peptide sequence(s), which are being defined by a set of adaptable tailoring enzymes. These proprieties make them very promising for bioengineering the compounds of interest, since the precursor peptide sequence(s) can be easily manipulated in order to produce the recombinant compounds.
The transportable nature of the leader peptide has already been demonstrated through the heterologous production of cyanobactins. For example, patellamides were heterologously produced by a microcin-like pathway in P. didemni [8] and by shotgun cloning [7]. Modification of the precursor peptide variable sequence was successfully achieved leading to the production of eptidemnamide in E. coli, a new cyclic peptide similar to an anticoagulant used clinically [34]. Likewise, the heptapeptide trunkamide was also produced in E. coli by co-expressing the mechanisms involved in the production of the hexapeptide patellin 2 and the octapeptide patellin 3, with the precursor peptide for trunkamide [5]. The cyanobactin biosynthetic pathway tru was engineered in order to integrate multiple tandem mutation and non-proteinogenic amino acids, using eight heterologous components simultaneously expressed in E. coli. Ptru-SD1 vector, encoding for the tru biosynthetic pathway, was cloned in E. coli together with vector ptruE, encoding for new cyanobactins. As a result, 16 out of the 22 recombinant compounds were expressed. In addition, patellamide precursor peptides responsible for ulithiacyclamide and patellamide C synthesis were cloned into the tru pathway in order to combine tru and pat biosynthetic pathways. As a result, one of the derivatives containing two thiazoline residues was successfully synthesized, thus presenting the first report of a tru derivate containing this modification [88]. Later, in order to determine the acceptable mutations in the trunkamide pathway, several random double and quadruple mutants were synthesized and expressed in E. coli. More than 300 new compounds were obtained as a result of the successful incorporation of 323 amino acids in 159 sequences, demonstrating the potential of the tru pathway in drug discovery, synthetic biology and biotechnology [89]. Conclusively, the use of the leader peptide and its ability to tolerate a broad range of biosynthetic enzymes allows the production of engineered compounds in heterologous hosts, thereby emphasizing its potential [3].
The cyanobactin pathways, pat and tru, appear to be unbiased by mutations. Enzymes are directed by short recognition sequences enclosed by substrates that can be substituted with the intention of effectively guiding the inclusion of specific functional groups to the mature cyanobactin. In order to test the mobility of post-translational modifications, hybrid precursor peptides comprising sections of tru, lyn and pag genetic clusters were constructed, leading to the production of the natural products in E. coli. The swapping of recognition sequences raises an interesting approach for synthetic biology concerning the tailoring of peptide products in vitro and in vivo [36].
A number of studies have been focusing on PatG activity with the aim to synthesize cyclic peptides [33,40,90]. Small libraries were also synthesized in vitro using distinct substrates for the prenyltransferase enzymes [39]. The discovery of the PatG macrocyclase domain [40], as well as the PatG macrocyclase crystal structure [33], allowed for the elucidation of the key features involved in substrate recognition and reactivities of this enzyme. Nevertheless, one of the major limitations of PatG is its slow enzyme activity. In that sense, studies exploring the PatG biotechnological potential started to arise and have focused on attempts to increase the catalytic efficiency of this enzyme. The engineering of the PatG macrocyclase domain from the patellamide pathway has enabled the macrocyclization of two peptides containing a mutated macrocyclase signature (AYRG) that are not synthesized by the wild-type enzyme. The reaction rate of this enzyme was also an order of magnitude faster than that in the wild-type PatG macrocyclase with a non-mutated macrocyclase signature (AYDG) [40]. In another study, an engineered PatG variant with mutations in the macrocyclase domain, named PatGsubtiligase, was generated. In the presence of Ala-Gly, the PatGsubtiligase was more efficient in catalyzing the cyclization via aminolysis than the wild-type enzyme. This reaction resulted in the production of the cyclic peptides without adducts. In contrast, the wild-type enzyme reaction displayed linear and cyclic peptides as well as linear adducts [33].
Recent progress in the chemistry of heterocyclization using PatD and homologs [36,43,91] has allowed the synthesis of short heterocycle-containing peptides, providing a simple method to generate native-like substrates for subsequent enzymes. An in vitro biosynthesis system for azoline-containing peptides, designated as FIT-PAD (flexible in vitro translation), was conceived through the integration of a cell-free translation system (composed by RNA polymerase, translation factors and ribosome) with the cyclodehydratase, PatD. In addition, extensive mutagenesis and deletion analysis were performed on PatE. This system interestingly enabled a more efficient synthesis of an extensive variety of azoline-containing peptides expressed from synthetic DNA templates. In addition, the substrate recognition determinants for PatD catalysis were identified, disclosing the biotechnological potential of the FIT-PAD system [91]. Recently, engineering studies on LynD, from the aesturamide pathway allowed the construction of a variant of this enzyme that efficiently processes substrates without a leader peptide, representing a great potential in biotechnology. The engineered LynD can act on peptides without the leader sequence and only three C-terminal residues (AYD) are necessary for those products where macrocyclization is required [92].
As previously mentioned, a more desirable approach for the synthesis of cyclic peptides is the successful introduction of tailored reactions that can significantly modify the potentially pharmaceutical peptides. The synthesis of azol(in)e-based cyclic peptides has already been reported. In this study, simple derivatives of the patellamide core sequence were synthesized in vitro through a new universal method [48]. More recently, cyanobactin-tailoring enzymes have been investigated in vitro in order to produce new natural compounds with tailored post-translational modifications. This was achieved using hybrid biosynthetic structures encompassing a mixture of enzymes, precursors and RS elements from trunkamide, prenylagaramide, cyanothecamide, patellamide, and aesturamide pathways. The synthesis of highly unnatural derivatives such as the N-C peptide macrocycle (22 amino acids in length) was accomplished by manipulating the order of addition and individuality of the enzymes. This study also demonstrates for the first time the reconstitution of a native, multistep RiPP pathway with multiple enzymes in one pot [93].
The continuous discovery of new genetic clusters [11] as well as the promiscuity of the tailoring enzymes involved in cyanobactin production suggest that investigations of their biotechnological potential should be continued in order to meet the pharmaceutical requirements and further contribution to the development of new compounds.
10. Conclusions
Cyanobactins constitute one of the largest classes of metabolites, with more than 100 cyanobactins known from free-living as well as symbiotic cyanobacteria, including symbionts of sponges and ascidians. Genome-mining techniques empower the continuous finding of these small peptides, and reveal that uncovered cyanobactins warrant further investigation. Several genetic clusters and their associated compounds have been described from a diverse range of cyanobacteria species, but the bioactivities or ecological roles of most of these compounds are still unknown. Since the first report of the patellamide pathway, pat, cyanobactin gene clusters have revealed an unexpected arrangement, especially in terms of the presence of several precursor peptides, encoding for different compounds, as well as the absence of heterocyclase and oxidase enzymes. Cyanobactins are known for their attractive bioactivities as disclosed thus far. Still, several issues remain concerning their relevant use in the development of new clinical drugs. Their mechanism of action needs to be understood in order to prioritize their application for the appropriate target. Moreover, the amount of cyanobactin obtained from cyanobacterial cultures is strain dependent and, for this reason, efforts in biotechnological approaches need to be pursued, particularly in terms of the promiscuity of tailoring enzymes. Furthermore, unraveling the ecological roles of cyanobactins, especially metal-binding ones, will be important for biotechnology development, since it may enable the design of synthetic analogs with a high affinity for reagents.
Acknowledgments
This research was partially funded by the FCT project UID/Multi/04423/2013 and by the projects MARBIOTECH (reference NORTE-07-0124-FEDER-000047) within the Scientific Research and Technological Development (SR&TD) Integrated Program MARVALOR—Building research and innovation capacity for improved management and valorization of marine resources, supported by the Programa Operacional Regional do Norte (ON.2-O Novo Norte) and NOVOMAR (reference 0687-NOVOMAR-1-P), supported by the European Regional Development Fund and ptdc/marbio/2818/2013. Joana Martins acknowledges FCT, Fundo Social Europeu (FSE) and Ministério da Ciência, Tecnologia e Ensino Superior (MCTES) for co-funding the PhD scholarship SFRH/80426/2011. The authors acknowledge Vítor Ramos and João Morais for providing the picture used in the graphical abstract and Pedro Lemos for the graphic design of the same.
Author Contributions
Joana Martins and Vitor Vasconcelos conceived the idea and wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jaspars, M. The origins of cyanobactin chemistry and biology. Chem. Commun. 2014, 50, 10174–10176. [Google Scholar] [CrossRef] [PubMed]
- Arnison, P.G.; Bibb, M.J.; Bierbaum, G.; Bowers, A.A.; Bugni, T.S.; Bulaj, G.; Camarero, J.A.; Campopiano, D.J.; Challis, G.L.; Clardy, J.; et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep. 2013, 30, 108–160. [Google Scholar] [CrossRef] [PubMed]
- Oman, T.J.; van der Donk, W.A. Follow the leader: The use of leader peptides to guide natural product biosynthesis. Nat. Chem. Biol. 2010, 6, 9–18. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Donia, M.S.; Schmidt, E.W. Ribosomal peptide natural products : Bridging the ribosomal and nonribosomal worlds. Nat. Prod. Rep. 2009, 26, 537–559. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.S.; Ravel, J.; Schmidt, E.W. A global assembly line for cyanobactins. Nat. Chem. Biol. 2008, 4, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Ireland, C.; Scheuer, P.J. Ulicyclamide and Ulithiacyclamide, Two New Small Peptides from a Marine Tunicate. J. Am. Chem. Soc. 1980, 102, 5688–5691. [Google Scholar] [CrossRef]
- Long, P.F.; Dunlap, W.C.; Battershill, C.N.; Jaspars, M. Shotgun Cloning and Heterologous Expression of the Patellamide Gene Cluster as a Strategy to Achieving Sustained Metabolite Production. ChemBioChem 2005, 6, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.W.; Nelson, J.T.; Rasko, D.A.; Sudek, S.; Eisen, J.A.; Haygood, M.G.; Ravel, J. Patellamide A and C biosynthesis by a microcin-like pathway in Prochloron didemni, the cyanobacterial symbiont of Lissoclinum patella. PNAS 2005, 102, 7315–7320. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.S.; Schmidt, E.W. Cyanobactins—Ubiquitous Cyanobacterial Ribosomal Peptide Metabolites. In Comprehensive Natural Products II. Chemistry and Biology; Mander, L., Lui, H.-W., Eds.; Elsevier: Oxford, UK, 2010; Volume 2, pp. 539–558. [Google Scholar]
- Sivonen, K.; Leikoski, N.; Fewer, D.P.; Jokela, J. Cyanobactins—Ribosomal cyclic peptides produced by cyanobacteria. Appl. Microbiol. Biotechnol. 2010, 86, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- Leikoski, N.; Liu, L.; Jokela, J.; Wahlsten, M.; Gugger, M.; Calteau, A.; Permi, P.; Kerfeld, C.A.; Sivonen, K.; Fewer, D.P. Genome Mining Expands the Chemical Diversity of the Cyanobactin Family to Include Highly Modified Linear Peptides. Chem. Biol. 2013, 20, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Shih, P.M.; Wu, D.; Latifi, A.; Axen, S.D.; Fewer, D.P.; Talla, E.; Calteau, A.; Cai, F.; Tandeau de Marsac, N.; Rippka, R.; et al. Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. PNAS 2013, 110, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Leikoski, N.; Fewer, D.P.; Sivonen, K. Widespread Occurrence and Lateral Transfer of the Cyanobactin Biosynthesis Gene Cluster in Cyanobacteria. Appl. Environ. Microbiol. 2009, 75, 853–857. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.; Leão, P.N.; Ramos, V.; Vasconcelos, V. N-terminal Protease Gene Phylogeny Reveals the Potential for Novel Cyanobactin Diversity in Cyanobacteria. Mar. Drugs 2013, 11, 4902–4916. [Google Scholar] [CrossRef] [PubMed]
- Leikoski, N.; Fewer, D.P.; Jokela, J.; Alakoski, P.; Wahlsten, M.; Sivonen, K. Analysis of an Inactive Cyanobactin Biosynthetic Gene Cluster Leads to Discovery of New Natural Products from Strains of the Genus Microcystis. PLoS ONE 2012, 7, e43002. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.S.; Schmidt, E. Linking chemistry and genetics in the growing cyanobactin natural products family. Chem. Biol. 2012, 18, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Houssen, W.E.; Koehnke, J.; Zollman, D.; Vendome, J.; Raab, A.; Smith, M.C.M.; Naismith, J.H.; Jaspars, M. The Discovery of New Cyanobactins from Cyanothece PCC 7425 Defines a New Signature for Processing of Patellamides. ChemBioChem 2012, 13, 2683–2689. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Itou, Y.; Ishida, K.; Shin, H.J. Prenylagaramides A and B, new cyclic peptides from two strains of Oscillatoria agardhii. J. Nat. Prod. 1999, 62, 752–755. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Lin, Z.; Tianero, M.D.B.; Schmidt, E.W. Aestuaramides, a Natural Library of Cyanobactin Cyclic Peptides Resulting from Isoprene-Derived Claisen Rearrangements. ACS Chem. Biol. 2013, 8, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Leikoski, N.; Fewer, D.P.; Jokela, J.; Wahlsten, M.; Rouhiainen, L.; Sivonen, K. Highly Diverse Cyanobactins in Strains of the Genus Anabaena. Appl. Environ. Microbiol. 2010, 76, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Ireland, C.M.; Durso, A.R.; Newman, R.A.; Hacker, M.P. Antineoplastic Cyclic Peptides from the Marine Tunicate Lissoclinum patella. J. Org. Chem. 1982, 47, 1807–1811. [Google Scholar] [CrossRef]
- Wasylyk, J.M.; Biskupiak, J.E.; Costello, C.E.; Ireland, C.M. Cyclic peptide structures from the tunicate Lissoclinum patella by FAB mass spectrometry. J. Org. Chem. 1983, 48, 4445–4449. [Google Scholar] [CrossRef]
- Zabriskie, T.M.; Foster, M.P.; Stout, T.J.; Clardy, J.; Ireland, C.M. Studies on the Solution- and Solid-state Structure of Patellin 2. J. Am. Chem. Soc. 1990, 112, 8080–8084. [Google Scholar] [CrossRef]
- Carroll, A.R.; Coll, J.C.; Bourne, D.J.; MacLeod, J.K.; Mark Zabriskie, T.; Ireland, C.M.; Bowden, B.F. Patellins 1-6 and Trunkamide A: Novel Cyclic Hexa-, Hepta- and Octa-peptides from Colonial Ascidians, Lissoclinurn sp. Aust. J. Chem. 1996, 49, 659–667. [Google Scholar]
- Banker, R.; Carmeli, S. Tenuecyclamides A-D, Cyclic Hexapeptides from the Cyanobacterium Nostoc spongiaeforme var. tenue. J. Nat. Prod. 1998, 61, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Lawton, L.; Morris, L.; Jaspars, M. A bioactive modified peptide, aeruginosamide, isolated from the cyanobacterium Microcystis aeruginosa. J. Am. Chem. Soc. 1999, 64, 5329–5332. [Google Scholar]
- Ishida, K.; Nakagawa, H.; Murakami, M. Microcyclamide, a Cytotoxic Cyclic Hexapeptide from the Cyanobacterium Microcystis aeruginosa. J. Nat. Prod. 2000, 63, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Sudek, S.; Haygood, M.G.; Youssef, D.T.A.; Schmidt, E.W. Structure of Trichamide, a Cyclic Peptide from the Bloom-Forming Cyanobacterium Trichodesmium erythraeum, Predicted from the Genome Sequence. Appl. Environ. Microbiol. 2006, 72, 4382–4387. [Google Scholar] [CrossRef] [PubMed]
- Portmann, C.; Blom, J.F.; Gademann, K.; Juttner, F. Aerucyclamides A and B : Isolation and Synthesis of Toxic Ribosomal Heterocyclic Peptides from the Cyanobacterium Microcystis aeruginosa PCC 7806. J. Nat. Prod. 2008, 71, 1193–1196. [Google Scholar] [CrossRef] [PubMed]
- Portmann, C.; Blom, J.F.; Kaiser, M.; Brun, R.; Ju, F.; Gademann, K. Isolation of Aerucyclamides C and D and Structure Revision of Microcyclamide 7806A : Heterocyclic Ribosomal Peptides from Microcystis aeruginosa PCC 7806 and Their Antiparasite Evaluation. J. Nat. Prod. 2008, 71, 1891–1896. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Ishida, K.; Quillardet, P.; Bouchier, C.; Hertweck, C.; Marsac, N.T.D.; Dittmann, E. Microcyclamide Biosynthesis in Two Strains of Microcystis aeruginosa : From Structure to Genes and Vice Versa. Appl. Environ. Microbiol. 2008, 74, 1791–1797. [Google Scholar] [CrossRef] [PubMed]
- Milne, B.F.; Long, P.F.; Starcevic, A.; Hranueli, D.; Jaspars, M. Spontaneity in the patellamide biosynthetic pathway. Org. Biomol. Chem. 2006, 4, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Pierce, E.; McIntosh, J.; Schmidt, E.W.; Nair, S.K. Structures of Cyanobactin Maturation Enzymes Define a Family of Transamidating Proteases. Chem. Biol. 2012, 19, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- Donia, M.S.; Hathaway, B.J.; Sudek, S.; Haygood, M.G.; Rosovitz, M.J.; Ravel, J.; Schmidt, E.W. Natural combinatorial peptide libraries in cyanobacterial symbionts of marine ascidians. Nat. Chem. Biol. 2006, 2, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Houssen, W.E.; Wright, S.H.; Kalverda, A.P.; Thompson, G.S.; Kelly, S.M.; Jaspars, M. Solution Structure of the Leader Sequence of the Patellamide Precursor Peptide, PatE1-34. ChemBioChem 2010, 11, 1867–1873. [Google Scholar] [CrossRef] [PubMed]
- Sardar, D.; Pierce, E.; McIntosh, J.A.; Schmidt, E.W. Recognition Sequences and Substrate Evolution in Cyanobactin Biosynthesis. ACS Synth. Biol. 2015, 4, 167–176. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Schmidt, E.W. Marine Molecular Machines : Heterocyclization in Cyanobactin Biosynthesis. ChemBioChem 2010, 11, 1413–1421. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Donia, M.S.; Schmidt, E.W. Insights into heterocyclization from two highly similar enzymes. J. Am. Chem. Soc. 2010, 132, 4089–4091. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Donia, M.S.; Nair, S.K.; Schmidt, E.W. Enzymatic Basis of Ribosomal Peptide Prenylation in Cyanobacteria. J. Am. Chem. Soc. 2011, 133, 13698–13705. [Google Scholar] [CrossRef] [PubMed]
- Koehnke, J.; Bent, A.; Houssen, W.E.; Zollman, D.; Morawitz, F. The mechanism of patellamide macrocyclization revealed by the characterization of the PatG macrocyclase domain. Nat. Struct. Mol. Biol. 2012, 19, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Cotter, P.D.; Ross, R.P.; Hill, C. Bacteriocins—A viable alternative to antibiotics. Nat. Rev. Microbiol. 2013, 11, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Koehnke, J.; Bent, A.F.; Houssen, W.E.; Mann, G.; Jaspars, M.; Naismith, J.H. The structural biology of patellamide biosynthesis. Curr. Opin. Struct. Biol. 2014, 29, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Koehnke, J.; Bent, A.F.; Zollman, D.; Smith, K.; Houssen, W.E.; Zhu, X.; Mann, G.; Lebl, T.; Scharff, R.; Shirran, S.; et al. The Cyanobactin Heterocyclase Enzyme : A Processive Adenylase That Operates with a Defined Order of Reaction. Angew. Chem. Int. Ed. 2013, 52, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; McIntosh, J.; Hathaway, B.J.; Schmidt, E.W. Using marine natural products to discover a protease that catalyses peptide macrocyclization of diverse substrates. J. Am. Chem. Soc. 2009, 131, 2122–2124. [Google Scholar] [CrossRef] [PubMed]
- Majmudar, J.; Gibbs, R. Pericyclic prenylation: Peptide modification through a Claisen rearrangement. ChemBioChem 2011, 122723–122726. [Google Scholar] [CrossRef] [PubMed]
- Bent, A.F.; Koehnke, J.; Smith, M.C.M.; Jaspars, M.; Naismith, J.H. Structure of PatF from Prochloron didemni. Acta Crystallogr. 2013, 69, 618–623. [Google Scholar] [CrossRef]
- Melby, J.; Li, X.; Mitchell, D. Orchestration of enzymatic processing by thiazole/oxazole-modified microcin dehydrogenases. Biochemistry (Moscow) 2014, 53, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Houssen, W.E.; Bent, A.F.; MCewan, A.R.; Pieiller, N.; Tabudravu, J.; Koehnke, J.; Mann, G.; Adaba, R.I.; Thomas, L.; Hawas, U.W.; et al. An Efficient Method for the In Vitro Production of Azol(in)e-Based Cyclic Peptides. Angew. Chem. Int. Ed. 2014, 53, 14171–14174. [Google Scholar] [CrossRef] [PubMed]
- Mann, G.; Koehnke, J.; Bent, A.F.; Graham, R.; Houssen, W.; Jaspars, M.; Schwarz-Linek, U.; Naismith, J.H. The structure of the cyanobactin domain of unknown function from PatG in the patellamide gene cluster. Acta Crystallogr. 2014, F70, 1597–1603. [Google Scholar] [CrossRef] [PubMed]
- Rinehart, K.L.; Kishore, V.; Bible, K.C.; Sakai, P.; Sullins, D.W.; Li, K.M. Didemnins and Tunichlorin: Novel Natural Products from the Marine Tunicate Trididemnum solidum. J. Nat. Prod. 1988, 51, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, Y.; Endo, M.; Nakagawa, H.; Nakanishi, T.; Mizukawa, J. A new cyclic peptide, ascidiacyclamide, isolated from ascidian. J. Chem. Soc. Chem. Commun. 1983, 323. [Google Scholar] [CrossRef]
- Hamada, Y.; Shibata, M.; Shioiri, T. New methods and reagents in organic synthesis. 58. : A synthesis of patellamide a, a cytotoxic cyclic peptide from a tunicate. Revision of its proposed structure. Tetrahedron Lett. 1985, 26, 5155–5158. [Google Scholar] [CrossRef]
- Ishida, T.; Inoue, M.; Hamada, Y.; Kato, S.; Shioiri, T. X-Ray crystal structure of ascidiacyclamide, a cytotoxic cyclic peptide from ascidian. J. Chem. Soc. Chem. Commun. 1987, 370–371. [Google Scholar] [CrossRef]
- Degnan, B.M.; Hawkins, C.J.; Lavin, M.F.; Mccaffrey, E.J.; Parry, D.L.; Brenk, A.L.V.D.; Watterst, D.J. New Cyclic Peptides with Cytotoxic Activity from the Ascidian Lissoclinum patella. J. Med. Chem. 1989, 32, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, F.J.; Ksebati, M.B.; Chang, J.S.; Wang, J.L.; Hossain, B.M.; van de Helm, D. Cyclic Peptides from the Ascidian Lissoclinum patella: Conformational Analysis of patellamide D by X-ray Analysis and Molecular, modeling. J. Org. Chem. 1989, 54, 3463–3472. [Google Scholar] [CrossRef]
- Biskupiak, J.E.; Ireland, C.M. Absolute configuration of thiazole amino acids in peptides. J. Org. Chem. 1983, 48, 2302–2304. [Google Scholar] [CrossRef]
- Williams, D.E.; Moore, R.E. The Structure of Ulithiacyclamide B. Antitumor Evaluation of Cyclic Peptides and Macrolides from Lissoclinum Patella. J. Nat. Prod. 1989, 52, 732–739. [Google Scholar] [CrossRef] [PubMed]
- McDonald, L.A.; Ireland, C. Patellamide E: A new cyclic peptide from the ascidian Lissoclinum patella. J. Nat. Prod. 1992, 55, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.A.; Gustafson, K.R.; Cardellina, J.H.; Boyd, M.R. Patellamide F, a new cytotoxic cyclic peptide from the colonial ascidian Lissoclinum patella. J. Nat. Prod. 1995, 58, 594–597. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Do, T.; Schmitz, F.J.; Andrusevich, V.; Engel, M.H. New Cyclic Peptides from the Ascidian Lissoclinum patella. J. Nat. Prod. 1998, 61, 1547–1551. [Google Scholar] [CrossRef] [PubMed]
- Wipf, P.; Uto, Y. Total Synthesis and Revision of Stereochemistry of the Marine Metabolite Trunkamide A. J. Org. Chem. 2000, 65, 1037–1049. [Google Scholar] [CrossRef] [PubMed]
- Caba, J.M.; Rodriguez, I.M.; Manzanares, I.; Giralt, E.; Albericio, F. Solid-Phase Total Synthesis of Trunkamide A1. J. Org. Chem. 2001, 66, 7568–7574. [Google Scholar] [CrossRef] [PubMed]
- McKeever, B.; Pattenden, G. Total synthesis of the prenylated cyclopeptide trunkamide A, a cytotoxic metabolite from Lissoclinum sp. Tetrahedron Lett. 2001, 42, 2573–2577. [Google Scholar] [CrossRef]
- Salvatella, X.; Caba, J.M.; Albericio, F.; Giralt, E. Solution Structure of the Antitumor Candidate Trunkamide A by 2D NMR and Restrained Simulated Annealing Methods. J. Org. Chem. 2003, 68, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Shioiri, T.; Hamada, Y.; Kato, S.; Shibata, M.; Kondo, Y. Cytotoxic activity of cyclic peptides of marine origin and their derivates: Importance of oxazoline functions. Biochem. Pharmacol. 1987, 3, 4181–4185. [Google Scholar] [CrossRef]
- Kohda, K.; Ohta, Y.; Yokoyama, Y.; Kawazoe, Y.; Kato, T.; Suzumura, Y.; Hamada, Y.; Shioiri, T. Mechanistic aspects of the cytocidal action of ulithiacyclamide on mouse leukemia L1210 cells in vitro. Biochem. Pharmacol. 1990, 38, 4497–4500. [Google Scholar]
- Kohda, K.; Ohta, Y.; Kawazoe, Y.; Kato, T.; Suzumura, Y.; Hamada, Y.; Shioiri, T. Ulicyclamide is cytotoxic against L1210 cells in vitro and inhibits both DNA and RNA syntheses. Biochem. Pharmacol. 1989, 38, 4500–4502. [Google Scholar] [PubMed]
- Houssen, W.E.; Jaspars, M. Azole-Based Cyclic Peptides from the Sea Squirt Lissoclinum Patella : Old Scaffolds, New Avenues. ChemBioChem 2010, 11, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Bowden, B.F.; Gravalos, D.G. Cyclic Hepta-Peptide Derivate from Colonial Ascidians Lissoclinum sp. U.S. Patent 6,025,466, 15 February 2000. [Google Scholar]
- Zafrir-ilan, E.; Carmeli, S. Two new microcyclamides from a water bloom of the cyanobacterium. Tetrahedron Lett. 2010, 51, 6602–6604. [Google Scholar] [CrossRef]
- Raveh, A.; Carmeli, S. Aeruginazole A, a novel thiazole-containing cyclopeptide from the cyanobacterium Microcystis sp. Org. Lett. 2010, 12, 3536–3539. [Google Scholar] [CrossRef] [PubMed]
- Adiv, S.; Pierce, E.; McIntosh, J.; Schmidt, E.W.; Nair, S.K. New aeruginazoles. A group of thiazole containing cyclic peptides from Microcystis aeruginosa. Chem. Biol. 2012, 19, 1411–1422. [Google Scholar] [CrossRef]
- Ishida, K.; Matsuda, H.; Murakami, M.; Yamaguchi, K. Kawaguchipeptin B, an antibacterial cyclic undecapeptide from the cyanobacterium Microcystis aeruginosa. J. Nat. Prod. 1997, 60, 724–726. [Google Scholar] [CrossRef] [PubMed]
- Todorova, A.K.; Juttner, F.; Linden, A.; Pluss, T.; von Philipsborn, W. Nostocyclamide: A new macrocyclic, thiazole-containing allelochemical from Nostoc sp. 31. J. Org. Chem. 1995, 60, 7891–7895. [Google Scholar] [CrossRef]
- Juttner, F.; Todorova, A.K.; Walch, N.; von Philipsborn, W. Nostocyclamide M: A cyanobacterial cyclic peptide with allelopathic activity from Nostoc 31. Phytochemistry 2001, 57, 613–619. [Google Scholar] [CrossRef]
- Bertram, A.; Pattenden, G. Marine metabolites: Metal binding and metal complexes of azole-based cyclic peptides of marine origin. Nat. Prod. Rep. 2007, 1, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Comba, P. Strains and stresses in coordination compounds. Coord. Chem. Rev. 1999, 182, 343–371. [Google Scholar] [CrossRef]
- Comba, P.; Dovalil, N.; Gahan, L.R.; Hanson, G.R.; Westphal, M. Cyclic peptide marine metabolites and CuII. R. Soc. Chem. 2014, 43, 1935–1956. [Google Scholar] [CrossRef] [PubMed]
- Comba, P.; Dovalil, N.; Hanson, G.R.; Linti, G. Synthesis and Cu II Coordination Chemistry of a Patellamide Derivative : Consequences of the Change from the Natural Thiazole/Oxazoline to the Artificial Imidazole Heterocycles. Inorg. Chem. 2011, 50, 5165–5174. [Google Scholar] [CrossRef] [PubMed]
- Van den Brenk, A.L.; Byriel, K.A.; Fairlie, D.P.; Gahan, L.R.; Hanson, G.R.; Hawkins, C.J.; Jones, A.; Kennard, H.L.; Moubaraki, B.; Murray, K.S. Crystal Structure, Electrospray Ionization Mass Spectrometry, Electron Paramagnetic Resonance, and Magnetic Susceptibility of Cu2(ascidH2)(1,2-u-CO3)(H2O)2).2H2O, the Bis(copper(II)) Complex of Ascidiacyclamide (ascidH4), a Cyclic Peptide Isolated from the Ascidian lissoclinum patella. Inorg. Chem. 1994, 33, 3549–3557. [Google Scholar] [CrossRef]
- Van den Brenk, A.L.; Fairlie, D.P.; Hanson, G.R.; Gahan, L.R.; Hawkins, C.J.; Jones, A. Binding of copper(II) to the Cyclic Octapeptide Patellamide D. Inorg. Chem. 1994, 33, 2280–2289. [Google Scholar] [CrossRef]
- Freeman, D.J.; Pattenden, G.; Drake, A.F.; Siligardi, G. Marine metabolites and metal ion chelation. Circular dichroism studies of metal binding to Lissoclinum cyclopeptides. J. Chem. Soc. Perkin Trans. 1998, 2, 129–135. [Google Scholar] [CrossRef]
- Morris, L.A.; Jaspars, M.; Kettenes-van den Bosh, J.J.; Versuis, K.; Heck, A.J.R.; Kelly, S.M.; Prince, N.C. Metal Binding of Lissoclinum patella metabolites. Part 1: Patellamides A, C and ulithiacyclamide A. Tetrahedron 2001, 57, 3185–3197. [Google Scholar] [CrossRef]
- Morris, L.A.; Milne, B.F.; Jaspars, M.; Kettenes-van den Bosh, J.J.; Versuis, K.; Heck, A.J.R.; Kelly, S.M.; Prince, N.C. Metal Binding of Lissoclinum patella metabolites. Part 2: Lissoclinamides 9 and 10. Tetrahedron 2001, 57, 3199–3207. [Google Scholar] [CrossRef]
- Comba, P.; Gahan, L.R.; Hanson, G.R.; Maeder, M.; Westphal, M. Carbonic anhydrase activity of dinuclear CuII complexes with patellamide model ligands. R. Soc. Chem. 2014, 43, 3144–3152. [Google Scholar]
- Wipf, P. Synthetic Studies of Biologically Active Marine Cyclopeptides. Chem. Rev. 1995, 95, 2115–2134. [Google Scholar] [CrossRef]
- Driggers, E.M.; Hale, J.; Lee, J.; Terrent, N.K. The exploration of macrocycles for drug discovery—An underexploited structural class. Nat. Rev. Drug Discov. 2008, 7, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Tianero, M.D.B.; Donia, M.S.; Young, T.S.; Schultz, P.G.; Schmidt, E.W. Ribosomal Route to Small-Molecule Diversity. J. Am. Chem. Soc. 2012, 134, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Ruffner, D.E.; Schmidt, E.W.; Heemstra, J.R. Assessing the combinatorial potential of the RiPP cyanobactin tru pathway. ACS Synth. Biol. 2015, 4, 482–492. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, J.A.; Robertson, C.R.; Agarwal, V.; Nair, S.K.; Bulaj, G.W.; Schmidt, E.W. Circular Logic: Nonribosomal Peptide-like Macrocyclization with a Ribosomal Peptide Catalyst. J. Am. Chem. Soc. 2010, 132, 15499–15501. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Ito, Y.; Kato, Y.; Tsunoda, S.; Suga, H. One-Pot Synthesis of Azoline-Containing Peptides in a Cell-free Translation System Integrated with a Posttranslational Cyclodehydratase. Chem. Biol. 2014, 21, 766–774. [Google Scholar] [CrossRef] [PubMed]
- Koehnke, J.; Mann, G.; Bent, A.F.; Ludewig, H.; Shirran, S.; Botting, C.; Lebl, T.; Houssen, W.E.; Jaspars, M.; Naismith, H. Structural analysis of leader peptide binding enables leader-free cyanobactin process. Nat. Chem. Biol. 2015, 11, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Sardar, D.; Lin, Z.; Schmidt, E.W. Modularity of RiPP Enzymes Enables Designed Synthesis of Decorated Peptides. Chem. Biol. 2015, 22, 907–916. [Google Scholar] [CrossRef] [PubMed][Green Version]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).