
Atrial Fibrillation in Diabetes: Pathogenesis and Targeted Rhythm Control Strategies

 , , , , , , and
, , , , , , and 
Abstract
1. Introduction
2. Methods
3. Pathophysiologic Nexus Between Diabetes and Atrial Fibrillation
3.1. Metabolic Remodeling
3.2. Inflammatory Signaling and Oxidative Stress
3.3. Fibrosis and Extracellular Matrix Remodeling
3.4. Electrophysiological Remodeling and Ion Channel Dysfunction
3.5. Mitochondrial Dysfunction and Energetic Impairment
3.6. Autonomic Dysfunction
4. Emerging Molecular Targets for Rhythm Control in Diabetes-Associated AF
4.1. Anti-Inflammatory Pathways
4.2. Anti-Fibrotic Signaling
4.3. Ion Channel and Calcium Handling Modulators
4.4. Mitochondria-Targeted Therapeutics
5. Modulation of AF Substrate by SGLT2 Inhibitors and GLP-1 Receptor Agonists
5.1. SGLT2 Inhibitors
5.2. GLP-1 Receptor Agonists
6. Multi-Omics Approaches to Unravel Molecular Networks in Diabetes-Associated AF
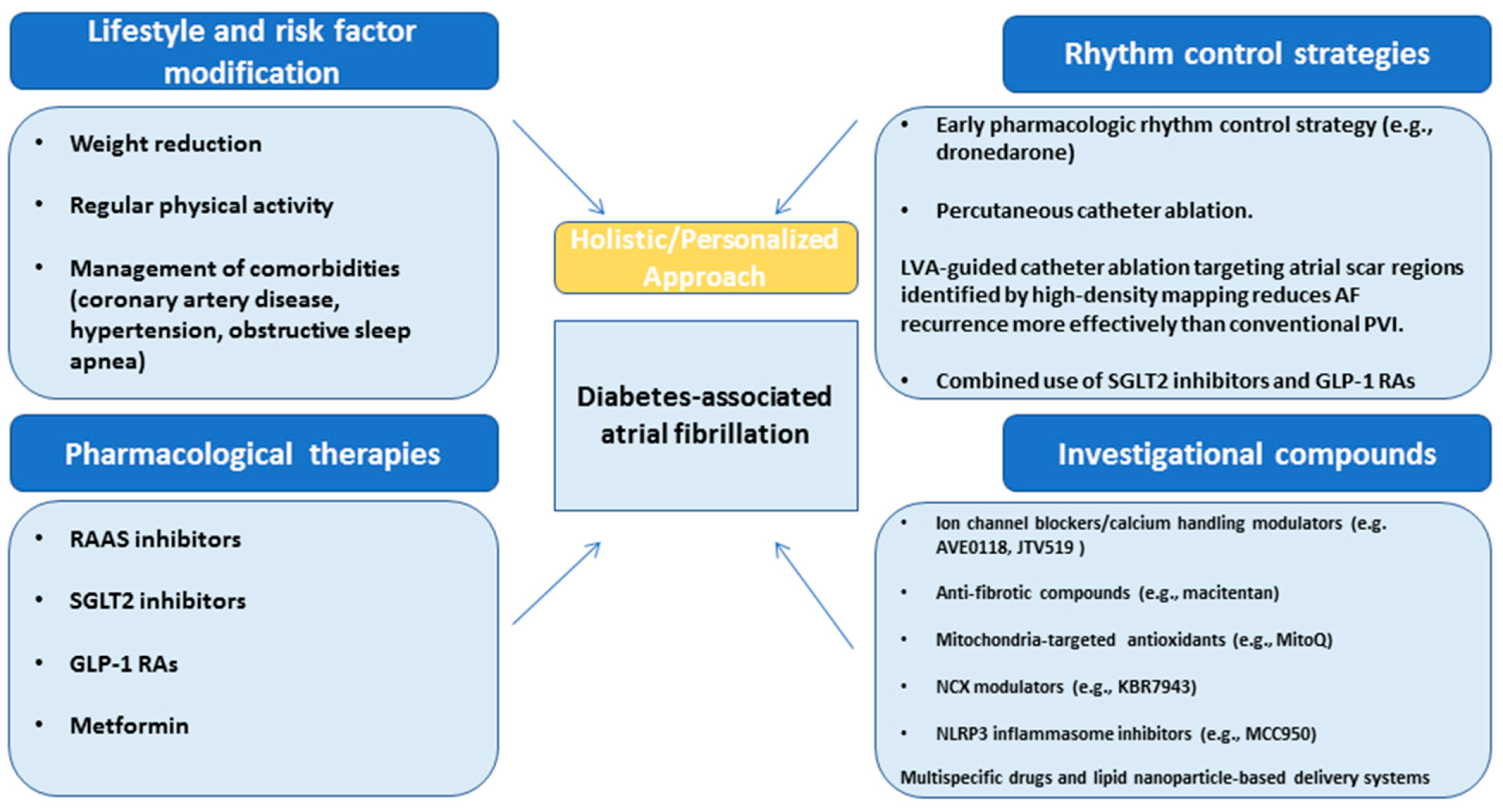
7. Translational Perspective: Precision Rhythm Control in Individuals with Diabetes
8. Future Directions and Knowledge Gaps
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- GBD 2021 Diabetes Collaborators. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the Global Burden of Disease Study 2021. Lancet 2023, 402, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Levy, D.; Vaziri, S.M.; D’Agostino, R.B.; Belanger, A.J.; Wolf, P.A. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham Study. JAMA 1994, 271, 840–844. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.; Zhou, J.; Veang, T.; Lin, Q.; Liu, Q. Global, regional, and national burden of atrial fibrillation and atrial flutter from 1990 to 2021: Sex differences and global burden projections to 2046—A systematic analysis of the Global Burden of Disease Study 2021. Europace 2025, 27, euaf027. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.Y.; Kotalczyk, A.; Boriani, G.; Marin, F.; Blomström-Lundqvist, C.; Potpara, T.S.; Fauchier, L.; Lip, G.Y.H.; ESC-EHRA EORP-AF Long-Term General Registry Investigators. Impact of diabetes on the management and outcomes in atrial fibrillation: An analysis from the ESC-EHRA EORP-AF Long-Term General Registry. Eur. J. Intern. Med. 2022, 103, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Ugowe, F.E.; Jackson, L.R.; Thomas, K.L. Atrial fibrillation and diabetes mellitus. Circ. Arrhythmia Electrophysiol. 2019, 12, e007351. [Google Scholar] [CrossRef] [PubMed]
- Dublin, S.; Glazer, N.L.; Smith, N.L.; Psaty, B.M.; Lumley, T.; Wiggins, K.L.; Page, R.L.; Heckbert, S.R. Diabetes mellitus, glycemic control, and risk of atrial fibrillation. J. Gen. Intern. Med. 2010, 25, 853–858. [Google Scholar] [CrossRef] [PubMed]
- Dahlqvist, S.; Rosengren, A.; Gudbjörnsdottir, S.; Pivodic, A.; Wedel, H.; Kosiborod, M.; Svensson, A.M.; Lind, M. Risk of atrial fibrillation in people with type 1 diabetes compared with matched controls from the general population: A prospective case-control study. Lancet Diabetes Endocrinol. 2017, 5, 799–807. [Google Scholar] [CrossRef] [PubMed]
- Proietti, M.; Romiti, G.F.; Basili, S. The case of diabetes mellitus and atrial fibrillation: Underlining the importance of non-cardiovascular comorbidities. Eur. J. Intern. Med. 2022, 103, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Echouffo-Tcheugui, J.B.; Shrader, P.; Thomas, L.; Gersh, B.J.; Kowey, P.R.; Mahaffey, K.W.; Singer, D.E.; Hylek, E.M.; Go, A.S.; Peterson, E.D.; et al. Care Patterns and Outcomes in Atrial Fibrillation Patients With and Without Diabetes: ORBIT-AF Registry. J. Am. Coll. Cardiol. 2017, 70, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Hsu, J.C.; Yang, Y.Y.; Chuang, S.L.; Yu, C.C.; Lin, L.Y. Higher long-term visit-to-visit glycemic variability predicts new-onset atrial fibrillation in patients with diabetes mellitus. Cardiovasc. Diabetol. 2021, 20, 148. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Zhang, N.; Korantzopoulos, P.; Letsas, K.P.; Cheng, M.; Di, F.; Tse, G.; Liu, T.; Li, G. Serum Glycated Hemoglobin Level as a Predictor of Atrial Fibrillation: A Systematic Review with Meta-analysis and Meta-regression. PLoS ONE 2017, 12, e0170955. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Pamporis, K.; Siontis, K.C.; Theofilis, P.; Samaras, A.; Patoulias, D.; Stachteas, P.; Karagiannidis, E.; Stavropoulos, G.; Tzikas, A.; et al. Major clinical outcomes in symptomatic vs. asymptomatic atrial fibrillation: A meta-analysis. Eur. Heart J. 2025, 46, 1189–1202. [Google Scholar] [CrossRef] [PubMed]
- Pamporis, K.; Karakasis, P.; Sagris, M.; Theofilis, P.; Milaras, N.; Pantelidaki, A.; Mourouzis, I.; Fragakis, N.; Vlachos, K.; Kordalis, A.; et al. Prevalence of asymptomatic atrial fibrillation and risk factors associated with asymptomatic status: A systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2025, zwaf138. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Theofilis, P.; Sagris, M.; Pamporis, K.; Stachteas, P.; Sidiropoulos, G.; Vlachakis, P.K.; Patoulias, D.; Antoniadis, A.P.; Fragakis, N. Artificial Intelligence in Atrial Fibrillation: From Early Detection to Precision Therapy. J. Clin. Med. 2025, 14, 2627. [Google Scholar] [CrossRef] [PubMed]
- Creta, A.; Providência, R.; Adragão, P.; de Asmundis, C.; Chun, J.; Chierchia, G.; Defaye, P.; Schmidt, B.; Anselme, F.; Finlay, M.; et al. Impact of type-2 diabetes mellitus on the outcomes of catheter ablation of atrial fibrillation (European observational multicentre study). Am. J. Cardiol. 2020, 125, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Truong, T.; Black-Maier, E.; Green, C.; Campbell, K.B.; Barnett, A.S.; Febre, J.; Loring, Z.; Al-Khatib, S.M.; Atwater, B.D.; et al. Catheter ablation of atrial fibrillation in patients with diabetes mellitus. Heart Rhythm O2 2020, 1, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Luo, D.; Yang, G.; Huang, W.; Tang, Y.; Xu, B.; He, G.; Yang, Y.; He, J.; Sun, H.; et al. The Effect of Non-insulin-based Insulin Resistance Indices on the Prediction of Recurrence in Patients with Atrial Fibrillation Undergoing Radiofrequency Catheter Ablation. Cardiovasc. Diabetol. 2024, 23, 291. [Google Scholar] [CrossRef] [PubMed]
- Manolis, A.A.; Manolis, T.A.; Melita, H.; Manolis, A.S. Sodium-glucose cotransporter type 2 inhibitors and cardiac arrhythmias. Trends Cardiovasc. Med. 2023, 33, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Stoll, L.; Lo, J.C. GLP-1 Receptor Agonists, the Holy Grail Preventing Atrial Fibrillation in Patients With T2D? JACC Basic Transl. Sci. 2023, 8, 937–938. [Google Scholar] [CrossRef] [PubMed]
- Stachteas, P.; Nasoufidou, A.; Karagiannidis, E.; Patoulias, D.; Karakasis, P.; Alexiou, S.; Samaras, A.; Zormpas, G.; Stavropoulos, G.; Tsalikakis, D.; et al. The Role of Sodium Glucose Co-Transporter 2 Inhibitors in Atrial Fibrillation: A Comprehensive Review. J. Clin. Med. 2024, 13, 5408. [Google Scholar] [CrossRef] [PubMed]
- Gumprecht, J.; Lip, G.Y.H.; Sokal, A.; Średniawa, B.; Mitręga, K.; Stokwiszewski, J.; Wierucki, Ł.; Rajca, A.; Rutkowski, M.; Zdrojewski, T.; et al. Relationship between diabetes mellitus and atrial fibrillation prevalence in the Polish population: A report from the Non-invasive Monitoring for Early Detection of Atrial Fibrillation (NOMED-AF) prospective cross-sectional observational study. Cardiovasc. Diabetol. 2021, 20, 128. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Choi, J.; Choi, E.K.; Lee, H.; Han, M.; Ahn, H.J.; Kwon, S.; Lee, S.W.; Han, K.D.; Oh, S.; et al. Early rhythm control on diabetes-related complications and mortality in patients with type 2 diabetes mellitus and atrial fibrillation. Diabetes Res. Clin. Pract. 2023, 206, 111020. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Tzeis, S.; Pamporis, K.; Schuermans, A.; Theofilis, P.; Milaras, N.; Tsiachris, D.; Efremidis, M.; Antoniadis, A.P.; Fragakis, N. Impact of Catheter Ablation Timing According to Duration of Atrial Fibrillation History on Arrhythmia Recurrences and Clinical Outcomes: A Meta-Analysis. EP Europace 2025, 27, euaf110. [Google Scholar] [CrossRef] [PubMed]
- Bode, D.; Pronto, J.R.D.; Schiattarella, G.G.; Voigt, N. Metabolic remodelling in atrial fibrillation: Manifestations, mechanisms and clinical implications. Nat. Rev. Cardiol. 2024, 21, 682–700. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Nattel, S.; Nanthakumar, K. Linking cellular energy state to atrial fibrillation pathogenesis: Potential role of adenosine monophosphate–activated protein kinase. Heart Rhythm 2020, 17, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.; Jin, L.; Gong, H.; Zheng, Q. Electro-metabolic coupling in atrial fibrillation: A deeper understanding of the metabolic driver. Biomed. Pharmacother. 2024, 180, 117536. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bai, F.; Liu, N.; Ouyang, F.; Liu, Q. The Warburg effect: A new insight into atrial fibrillation. Clin. Chim. Acta 2019, 499, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Ning, Z.H.; Xie, Z.; Ou, Y.; Yang, J.Y.; Liu, Y.X.; Huang, H.; Tang, H.F.; Jiang, Z.S.; Hu, H.J. SIRT3/AMPK Signaling Pathway Regulates Lipid Metabolism and Improves Vulnerability to Atrial Fibrillation in Dahl Salt-sensitive Rats. Am. J. Hypertens. 2024, 37, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Gao, F.; Gong, H.; Fu, Y.; Liu, B.; Qin, X.; Zheng, Q. Intermittent fasting attenuates obesity-related atrial fibrillation via SIRT3-mediated insulin resistance mitigation. Biochim. Biophys. Acta Mol. Basis Dis. 2023, 1869, 166638. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, K.; Nzirorera, C.; Kienesberger, P.C. Lipid metabolism and signaling in cardiac lipotoxicity. Biochim. Biophys. Acta 2016, 1861, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Trappe, K.; Thomas, D.; Bikou, O.; Kelemen, K.; Lugenbiel, P.; Voss, F.; Becker, R.; Katus, H.A.; Bauer, A. Suppression of persistent atrial fibrillation by genetic knockdown of caspase 3: A pre-clinical pilot study. Eur. Heart J. 2013, 34, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.; Li, N.; Dobrev, D. Role of inflammatory signaling in atrial fibrillation. Int. J. Cardiol. 2019, 287, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Van Wagoner, D.R.; Nattel, S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ. J. 2015, 79, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Yamashita, T.; Sekiguchi, A.; Tsuneda, T.; Sagara, K.; Takamura, M.; Kaneko, S.; Aizawa, T.; Fu, L.T. AGEs-RAGE system mediates atrial structural remodeling in the diabetic rat. J. Cardiovasc. Electrophysiol. 2008, 19, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Goldin, A.; Beckman, J.A.; Schmidt, A.M.; Creager, M.A. Advanced glycation end products. Circulation 2006, 114, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Cha, S.J.; Park, J.H.; Shin, J.H.; Lim, Y.H.; Park, H.C.; Shin, J.; Kim, C.K.; Park, J.K. Association between Insulin Resistance and Risk of Atrial Fibrillation in Non-diabetics. Eur. J. Prev. Cardiol. 2020, 27, 1934–1941. [Google Scholar] [CrossRef] [PubMed]
- Fontes, J.D.; Lyass, A.; Massaro, J.M.; Rienstra, M.; Dallmeier, D.; Schnabel, R.B.; Wang, T.J.; Vasan, R.S.; Lubitz, S.A.; Magnani, J.W.; et al. Insulin resistance and atrial fibrillation (from the Framingham Heart Study). Am. J. Cardiol. 2012, 109, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Garg, P.K.; Biggs, M.L.; Kaplan, R.; Kizer, J.R.; Heckbert, S.R.; Mukamal, K.J. Fasting and Post-glucose Load Measures of Insulin Resistance and Risk of Incident Atrial Fibrillation: The Cardiovascular Health Study. Nutr. Metab. Cardiovasc. 2018, 28, 716–721. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.H.; Chang, G.J.; Lai, Y.J.; Chen, W.J.; Chang, S.H.; Hung, L.M.; Kuo, C.T.; Yeh, Y.H. Atrial fibrillation and its arrhythmogenesis associated with insulin resistance. Cardiovasc. Diabetol. 2019, 18, 125. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Lip, G.Y.H.; Apostolakis, S. Inflammation in atrial fibrillation. J. Am. Coll. Cardiol. 2012, 60, 2263–2270. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Mondragón, R.; Galindo, C.A.; Avila, G. Role of TGF-beta on cardiac structural and electrical remodeling. Vasc. Health Risk Manag. 2008, 4, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Ziolo, M.T.; Mohler, P.J. Defining the role of oxidative stress in atrial fibrillation and diabetes. J. Cardiovasc. Electrophysiol. 2015, 26, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Veleva, T.; Scott, L., Jr.; Cao, S.; Li, L.; Chen, G.; Jeyabal, P.; Pan, X.; Alsina, K.M.; Abu-Taha, I.; et al. Enhanced Cardiomyocyte NLRP3 Inflammasome Signaling Promotes Atrial Fibrillation. Circulation 2018, 138, 2227–2242. [Google Scholar] [CrossRef] [PubMed]
- Hajer, D.R.; van Haeften, T.W.; Visseren, F.L.J. Adipose tissue dysfunction in obesity, diabetes, and vascular diseases. Eur. Heart J. 2008, 29, 2959–2971. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Zhao, W.; Thomson, J.K.; Gao, X.; DeMarco, D.M.; Carrillo, E.; Chen, B.; Wu, X.; Ginsburg, K.S.; Bakhos, M.; et al. Stress Signaling JNK2 Crosstalk With CaMKII Underlies Enhanced Atrial Arrhythmogenesis. Circ. Res. 2018, 122, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Bare, D.J.; DeSantiago, J.; Zhao, W.; Mei, Y.; Chen, Z.; Ginsburg, K.; Solaro, R.J.; Wolska, B.M.; Bers, D.M.; et al. JNK2, a Newly-Identified SERCA2 Enhancer, Augments an Arrhythmic [Ca2+] SR Leak-Load Relationship. Circ. Res. 2021, 128, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Biernacka, A.; Cavalera, M.; Wang, J.; Russo, I.; Shinde, A.; Kong, P.; Gonzalez-Quesada, C.; Rai, V.; Dobaczewski, M.; Lee, D.W.; et al. Smad3 Signaling Promotes Fibrosis While Preserving Cardiac and Aortic Geometry in Obese Diabetic Mice. Circ. Heart Fail. 2015, 8, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Makary, S.; Voigt, N.; Maguy, A.; Wakili, R.; Nishida, K.; Harada, M.; Dobrev, D.; Nattel, S. Differential protein kinase C isoform regulation and increased constitutive activity of acetylcholine-regulated potassium channels in atrial remodeling. Circ. Res. 2011, 109, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Theofilis, P.; Vlachakis, P.K.; Korantzopoulos, P.; Patoulias, D.; Antoniadis, A.P.; Fragakis, N. Atrial fibrosis in Atrial Fibrillation: Mechanistic Insights, Diagnostic Challenges, and Emerging Therapeutic Targets. Int. J. Mol. Sci. 2024, 26, 209. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Vlachakis, P.K.; Theofilis, P.; Ktenopoulos, N.; Patoulias, D.; Fyntanidou, B.; Antoniadis, A.P.; Fragakis, N. Atrial Cardiomyopathy in Atrial Fibrillation: A Multimodal Diagnostic Framework. Diagnostics 2025, 15, 1207. [Google Scholar] [CrossRef] [PubMed]
- Polyakova, V.; Miyagawa, S.; Szalay, Z.; Risteli, J.; Kostin, S. Atrial extracellular matrix remodelling in patients with atrial fibrillation. J. Cell Mol. Med. 2008, 12, 189–208. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Frangogiannis, N.G. Diabetes-associated cardiac fibrosis: Cellular effectors, molecular mechanisms and therapeutic opportunities. J. Mol. Cell Cardiol. 2016, 90, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Abe, I.; Teshima, Y.; Kondo, H.; Kaku, H.; Kira, S.; Ikebe, Y.; Saito, S.; Fukui, A.; Shinohara, T.; Yufu, K.; et al. Association of Fibrotic Remodeling and Cytokines/chemokines Content in Epicardial Adipose Tissue with Atrial Myocardial Fibrosis in Patients with Atrial Fibrillation. Heart Rhythm 2018, 15, 1717–1727. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Fu, H.; Li, J.; Yang, W.; Cheng, L.; Liu, T. Hyperglycemia aggravates atrial interstitial fibrosis, ionic remodeling and vulnerability to atrial fibrillation in diabetic rabbits. Anadolu Kardiyol Derg 2012, 12, 543. Available online: https://link.gale.com/apps/doc/A352493933/AONE?u=anon~2d8b3dec&sid=googleScholar&xid=f5ae61e2 (accessed on 1 July 2025). [CrossRef] [PubMed]
- Watanabe, M.; Yokoshiki, H.; Mitsuyama, H.; Mizukami, K.; Ono, T.; Tsutsui, H. Conduction and refractory disorders in the diabetic atrium. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H86. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Liu, X.; Li, X.; Yang, F.; Wang, R. Effects of Electrical Remodeling on Atrial Fibrillation in Diabetes Mellitus. Rev. Cardiovasc. Med. 2023, 24, 3. [Google Scholar] [CrossRef] [PubMed]
- Polina, I.; Jansen, H.J.; Li, T.; Moghtadaei, M.; Bohne, L.J.; Liu, Y.; Krishnaswamy, P.; Egom, E.E.; Belke, D.D.; Rafferty, S.A.; et al. Loss of insulin signaling may contribute to atrial fibrillation and atrial electrical remodeling in type 1 diabetes. Proc. Natl. Acad. Sci. USA 2020, 117, 7990–8000. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Jiang, Y.; Xue, G.; Yuan, Y.; Zhu, H.; Zhan, L.; Zhuang, Y.; Huang, Q.; Shi, L.; Zhao, Y.; et al. Increase of late sodium current contributes to enhanced susceptibility to atrial fibrillation in diabetic mice. Eur. J. Pharmacol. 2019, 857, 172444. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Belardinelli, L. Basal late sodium current is a significant contributor to the duration of action potential of guinea pig ventricular myocytes. Physiol. Rep. 2017, 5, e13295. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Liu, H.H.; Zhang, L.; Zhang, X.L.; Zhang, J.; Li, F.; Zhao, N.; Zhang, Z.Y.; Kong, Q.; Liu, X.Y.; et al. Advanced glycation end products downregulate connexin 43 and connexin 40 in diabetic atrial myocytes via the AMPK pathway. Diabetes Metab. Syndr. Obes. 2023, 16, 3045–3056. [Google Scholar] [CrossRef] [PubMed]
- De Vos, C.B.; Weijs, B.; Crijns, H.J.; Cheriex, E.C.; Palmans, A.; Habets, J.; Prins, M.H.; Pisters, R.; Nieuwlaat, R.; Tieleman, R.G. Atrial tissue Doppler imaging for prediction of new-onset atrial fibrillation. Heart 2009, 95, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Demir, K.; Avci, A.; Kaya, Z.; Marakoglu, K.; Ceylan, E.; Yilmaz, A.; Ersecgin, A.; Armutlukuyu, M.; Altunkeser, B.B. Assessment of atrial electromechanical delay and P-wave dispersion in patients with type 2 diabetes mellitus. J. Cardiol. 2016, 67, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Rudokas, M.W.; McKay, M.; Toksoy, Z.; Eisen, J.N.; Bögner, M.; Young, L.H.; Akar, F.G. Mitochondrial network remodeling of the diabetic heart: Implications to ischemia related cardiac dysfunction. Cardiovasc. Diabetol. 2024, 23, 261. [Google Scholar] [CrossRef] [PubMed]
- Fossier, L.; Panel, M.; Butruille, L.; Colombani, S.; Azria, L.; Woitrain, E.; Decoin, R.; Torrente, A.G.; Thireau, J.; Lacampagne, A.; et al. Enhanced Mitochondrial Calcium Uptake Suppresses Atrial Fibrillation Associated With Metabolic Syndrome. J. Am. Coll. Cardiol. 2022, 80, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Xie, W.; Betzenhauser, M.; Reiken, S.; Chen, B.X.; Wronska, A.; Marks, A.R. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2012, 111, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Jeganathan, J.; Saraf, R.; Mahmood, F.; Pal, A.; Bhasin, M.K.; Huang, T. Mitochondrial Dysfunction in Atrial Tissue of Patients Developing Postoperative Atrial Fibrillation. Ann. Thorac. Surg. 2017, 104, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Abel, E.D. Mitochondria in the diabetic heart. Cardiovasc. Res. 2010, 88, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Tadic, M.; Cuspidi, C. Type 2 diabetes mellitus and atrial fibrillation: From mechanisms to clinical practice. Arch. Cardiovasc. Dis. 2015, 108, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Solhjoo, S.; Millare, B.; Plank, G.; Abraham, M.R.; Cortassa, S.; Trayanova, N.; O’Rourke, B. Effects of regional mitochondrial depolarization on electrical propagation: Implications for arrhythmogenesis. Circ. Arrhythm. Electrophysiol. 2014, 7, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Mason, F.E.; Pronto, J.R.D.; Alhussini, K.; Alhussini, K.; Maack, C.; Voigt, N. Cellular and mitochondrial mechanisms of atrial fibrillation. Basic. Res. Cardiol. 2020, 115, 72. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, M.; Stevens, M.J. Cardiovascular autonomic neuropathies as complications of diabetes mellitus. Nat. Rev. Endocrinol. 2012, 8, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.W.; Lee, T.I.; Lin, Y.K.; Chen, Y.C.; Kao, Y.H.; Chen, Y.J. Effect of antidiabetic drugs on the risk of atrial fibrillation: Mechanistic insights from clinical evidence and translational studies. Cell Mol. Life Sci. 2021, 78, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Almorós, A.; Casado Cerrada, J.; Álvarez-Sala Walther, L.-A.; Méndez Bailón, M.; Lorenzo González, Ó. Atrial Fibrillation and Diabetes Mellitus: Dangerous Liaisons or Innocent Bystanders? J. Clin. Med. 2023, 12, 2868. [Google Scholar] [CrossRef] [PubMed]
- Karam, B.S.; Chavez-Moreno, A.; Koh, W.; Akar, J.G.; Akar, F.G. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc. Diabetol. 2017, 16, 120. [Google Scholar] [CrossRef] [PubMed]
- Ajoolabady, A.; Nattel, S.; Lip, G.Y.H.; Ren, J. Inflammasome Signaling in Atrial Fibrillation: JACC state-of-the-art review. J. Am. Coll. Cardiol. 2022, 79, 2349–2366. [Google Scholar] [CrossRef] [PubMed]
- Niskala, A.; Heijman, J.; Dobrev, D.; Jespersen, T.; Saljic, A. Targeting the NLRP3 inflammasome signaling for the management of atrial fibrillation. Br. J. Pharmacol. 2024, 181, 4939–4957. [Google Scholar] [CrossRef] [PubMed]
- Deftereos, S.G.; Beerkens, F.J.; Shah, B.; Giannopoulos, G.; Vrachatis, D.A.; Giotaki, S.G.; Siasos, G.; Nicolas, J.; Arnott, C.; Patel, S.; et al. Colchicine in Cardiovascular Disease: In-Depth Review. Circulation 2022, 145, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Saljic, A.; Heijman, J.; Dobrev, D. Emerging Antiarrhythmic Drugs for Atrial Fibrillation. Int. J. Mol. Sci. 2022, 23, 4096. [Google Scholar] [CrossRef] [PubMed]
- Conen, D.; Ke Wang, M.; Popova, E.; Chan, M.T.V.; Landoni, G.; Cata, J.P.; Reimer, C.; McLean, S.R.; Srinathan, S.K.; Reyes, J.C.T.; et al. Effect of Colchicine on Perioperative Atrial Fibrillation and Myocardial Injury after Non-Cardiac Surgery in Patients Undergoing Major Thoracic Surgery (COP-AF): An International Randomised Trial. Lancet 2023, 402, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Krisai, P.; Blum, S.; Schnabel, R.B.; Sticherling, C.; Kühne, M.; von Felten, S.; Ammann, P.; Pruvot, E.; Albert, C.M.; Conen, D.; et al. Canakinumab After Electrical Cardioversion in Patients With Persistent Atrial Fibrillation: A Pilot Randomized Trial. Circ. Arrhythm. Electrophysiol. 2020, 13, e008197. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.; Zhang, C.; Wu, G.; Wan, W.; Liang, J.; Liu, X.; Liu, D.; Yang, B. Pinocembrin attenuates autonomic dysfunction and atrial fibrillation susceptibility via inhibition of the NF-κB/TNF-α pathway in a rat model of myocardial infarction. Int. Immunopharmacol. 2019, 77, 105926. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.; Xie, Q.; Hu, T.; Yao, Q.; Zhao, J.; Wu, Q.; Tang, Q. S100A8/A9 in Myocardial Infarction: A Promising Biomarker and Therapeutic Target. Front. Cell Dev. Biol. 2020, 8, 603902. [Google Scholar] [CrossRef] [PubMed]
- Jakobsson, G.; Papareddy, P.; Andersson, H.; Mulholland, M.; Bhongir, R.; Ljungcrantz, I.; Engelbertsen, D.; Björkbacka, H.; Nilsson, J.; Manea, A.; et al. Therapeutic S100A8/A9 Blockade Inhibits Myocardial and Systemic Inflammation and Mitigates Sepsis-Induced Myocardial Dysfunction. Crit. Care 2023, 27, 374. [Google Scholar] [CrossRef] [PubMed]
- Sweat, M.E.; Pu, W.T. Genetic and Molecular Underpinnings of Atrial Fibrillation. npj Cardiovasc. Health 2024, 1, 35. [Google Scholar] [CrossRef] [PubMed]
- Liaw, L.; Birk, D.E.; Ballas, C.B.; Whitsitt, J.S.; Davidson, J.M.; Hogan, B.L. Altered Wound Healing in Mice Lacking a Functional Osteopontin Gene (Spp1). J. Clin. Investig. 1998, 101, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Momin, N.; Pabel, S.; Rudra, A.; Kumowski, N.; Lee, I.H.; Mentkowski, K.; Yamazoe, M.; Stengel, L.; Muse, C.G.; Seung, H.; et al. Therapeutic Spp1 Silencing in TREM2+ Cardiac Macrophages Suppresses Atrial Fibrillation. bioRxiv 2024. [Google Scholar] [CrossRef]
- Du, L.; Qin, M.; Yi, Y.; Chen, X.; Jiang, W.; Zhou, L.; Zhang, D.; Xu, K.; Yang, Y.; Li, C.; et al. Eplerenone Prevents Atrial Fibrosis via the TGF-β Signaling Pathway. Cardiology 2017, 138, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Takemoto, Y.; Ramirez, R.J.; Kaur, K.; Salvador-Montañés, O.; Ponce-Balbuena, D.; Ramos-Mondragón, R.; Ennis, S.R.; Guerrero-Serna, G.; Berenfeld, O.; Jalife, J. Eplerenone Reduces Atrial Fibrillation Burden Without Preventing Atrial Electrical Remodeling. J. Am. Coll. Cardiol. 2017, 70, 2893–2905. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, W.; Yang, B.; Han, W.; Dong, D.; Xue, J.; Li, B.; Yang, S.; Sheng, L. Effects of Cilazapril on Atrial Electrical, Structural and Functional Remodeling in Atrial Fibrillation Dogs. J. Electrocardiol. 2007, 40, 100.e1–100.e6. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, K.; Nakashima, H.; Urata, H.; Gondo, N.; Arakawa, K.; Saku, K. Effects of Angiotensin II Type 1 Receptor Antagonist on Electrical and Structural Remodeling in Atrial Fibrillation. J. Am. Coll. Cardiol. 2003, 41, 2197–2204. [Google Scholar] [CrossRef] [PubMed]
- Kapelios, C.J.; Murrow, J.R.; Nührenberg, T.G.; Montoro Lopez, M.N. Effect of Mineralocorticoid Receptor Antagonists on Cardiac Function in Patients with Heart Failure and Preserved Ejection Fraction: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Heart Fail. Rev. 2019, 24, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Patoulias, D.; Popovic, D.S.; Pamporis, K.; Theofilis, P.; Nasoufidou, A.; Stachteas, P.; Samaras, A.; Tzikas, A.; Giannakoulas, G.; et al. Effects of Mineralocorticoid Receptor Antagonists on New-Onset or Recurrent Atrial Fibrillation: A Bayesian and Frequentist Network Meta-Analysis of Randomized Trials. Curr. Probl. Cardiol. 2024, 49, 102742. [Google Scholar] [CrossRef] [PubMed]
- Pabon, M.A.; Filippatos, G.; Claggett, B.L.; Miao, M.Z.; Desai, A.S.; Jhund, P.S.; Henderson, A.; Brinker, M.; Schloemer, P.; Hofmeister, L.; et al. Finerenone Reduces New-Onset Atrial Fibrillation Across the Spectrum of Cardio-Kidney-Metabolic Syndrome: The FINE-HEART Pooled Analysis. J. Am. Coll. Cardiol. 2025, 85, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Wolke, C.; Bukowska, A.; Goette, A.; Lendeckel, U. Redox Control of Cardiac Remodeling in Atrial Fibrillation. Biochim. Biophys. Acta 2015, 1850, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Goette, A.; Bukowska, A.; Dobrev, D.; Pfeiffenberger, J.; Morawietz, H.; Strugala, D.; Wiswedel, I.; Röhl, F.W.; Wolke, C.; Bergmann, S.; et al. Acute Atrial Tachyarrhythmia Induces Angiotensin II type 1 Receptor-mediated Oxidative Stress and Microvascular Flow Abnormalities in the Ventricles. Eur. Heart J. 2009, 30, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Paw, M.; Kusiak, A.A.; Nit, K.; Litewka, J.J.; Piejko, M.; Wnuk, D.; Sarna, M.; Fic, K.; Stopa, K.B.; Hammad, R.; et al. Hypoxia Enhances Anti-fibrotic Properties of Extracellular Vesicles Derived from HiPSCs via the MiR302b-3p/TGFβ/SMAD2 Axis. BMC Med. 2023, 21, 412. [Google Scholar] [CrossRef] [PubMed]
- Hai, Z.; Wu, Y.; Ning, Z. Salidroside Attenuates Atrial Fibrosis and Atrial Fibrillation Vulnerability Induced by Angiotensin-II through Inhibition of LOXL2-TGF-B1-Smad2/3 Pathway. Heliyon 2023, 9, e21220. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, A.; Nikonova, Y.; Wolke, C.; Lendeckel, U.; Kockskämper, J.; Goette, A. Anti-Inflammatory Effects of Endothelin Receptor Blockade in Left Atrial Tissue of Spontaneously Hypertensive Rats. Int. J. Cardiol. Heart Vasc. 2022, 42, 101088. [Google Scholar] [CrossRef] [PubMed]
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 2010, 121, 2407–2418. [Google Scholar] [CrossRef] [PubMed]
- Ruf, L.; Bukowska, A.; Gardemann, A.; Goette, A. Coagulation Factor Xa Has No Effects on the Expression of PAR1, PAR2, and PAR4 and No Proinflammatory Effects on HL-1 Cells. Cells 2023, 12, 2849. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, A.; Zacharias, I.; Weinert, S.; Skopp, K.; Hartmann, C.; Huth, C.; Goette, A. Coagulation factor Xa Induces an Inflammatory Signalling by Activation of Protease-activated Receptors in Human Atrial Tissue. Eur. J. Pharmacol. 2013, 718, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, A.; Schild, L.; Bornfleth, P.; Peter, D.; Wiese-Rischke, C.; Gardemann, A.; Isermann, B.; Walles, T.; Goette, A. Activated Clotting Factor X Mediates Mitochondrial Alterations and Inflammatory Responses via Protease-activated Receptor Signaling in Alveolar Epithelial Cells. Eur. J. Pharmacol. 2020, 869, 172875. [Google Scholar] [CrossRef] [PubMed]
- Spronk, H.M.; De Jong, A.M.; Verheule, S.; De Boer, H.C.; Maass, A.H.; Lau, D.H.; Rienstra, M.; van Hunnik, A.; Kuiper, M.; Lumeij, S.; et al. Hypercoagulability Causes Atrial Fibrosis and Promotes Atrial Fibrillation. Eur. Heart J. 2017, 38, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Theofilis, P.; Milaras, N.; Vlachakis, P.K.; Patoulias, D.; Karamitsos, T.; Antoniadis, A.P.; Fragakis, N. Epigenetic Drivers of Atrial Fibrillation: Mechanisms, Biomarkers, and Therapeutic Targets. Int. J. Mol. Sci. 2025, 26, 5253. [Google Scholar] [CrossRef] [PubMed]
- Shan, H.; Zhang, Y.; Lu, Y.; Zhang, Y.; Pan, Z.; Cai, B.; Wang, N.; Li, X.; Feng, T.; Hong, Y.; et al. Downregulation of miR-133 and miR-590 Contributes to Nicotine-induced Atrial Remodelling in Canines. Cardiovasc. Res. 2009, 83, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yuan, B.; Xu, Y.; Zhou, N.; Zhang, R.; Lu, L.; Feng, Z. MiR-208b/miR-21 Promotes the Progression of Cardiac Fibrosis Through the Activation of the TGF-β1/Smad-3 Signaling Pathway: An in vitro and in vivo Study. Front. Cardiovasc. Med. 2022, 9, 924629. [Google Scholar] [CrossRef] [PubMed]
- Rubiś, P.; Totoń-Żurańska, J.; Kołton-Wróż, M.; Wołkow, P.; Pitera, E.; Wiśniowska-Śmiałek, S.; Dziewięcka, E.; Garlitski, A.; Podolec, P. Twelve-month kinetics of circulating fibrosis-linked microRNAs (miR-21, miR-29, miR-30, and miR-133a) and the relationship with extracellular matrix fibrosis in dilated cardiomyopathy. Arch. Med. Sci. 2019, 18, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Afzal, J.; Vakrou, S.; Greenland, G.V.; Talbot, C.C., Jr.; Hebl, V.B.; Guan, Y.; Karmali, R.; Tardiff, J.C.; Leinwand, L.A.; et al. Differences in microRNA-29 and Pro-fibrotic Gene Expression in Mouse and Human Hypertrophic Cardiomyopathy. Front. Cardiovasc. Med. 2019, 6, 170. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, N.W.E.; Kawasaki, M.; Nariswari, F.A.; Fabrizi, B.; Neefs, J.; van der Made, I.; Wesselink, R.; van Boven, W.J.P.; Driessen, A.H.G.; Jongejan, A.; et al. MicroRNAs in atrial fibrillation target genes in structural remodelling. Cell Tissue Res. 2023, 394, 497–514. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Y.; Han, J.; Li, Y.; Xie, C.; Xie, L.; Shi, J.; Zhang, J.; Yang, B.; Chen, D.; et al. Integrated analysis of microRNA and mRNA expression profiles in the left atrium of patients with nonvalvular paroxysmal atrial fibrillation: Role of miR-146b-5p in atrial fibrosis. Heart Rhythm 2015, 12, 1018–1026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Hu, X.; Henning, R.H.; Brundel, B.J. Keeping up the balance: Role of HDACs in cardiac proteostasis and therapeutic implications for atrial fibrillation. Cardiovasc. Res. 2016, 109, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Zhang, R.; Mo, B.; Chen, L.; Liu, L.; Yu, Y.; Cao, W.; Fang, G.; Wan, Y.; Gu, Y.; et al. EZH2 as a novel therapeutic target for atrial fibrosis and atrial fibrillation. J. Mol. Cell Cardiol. 2019, 135, 119–133. [Google Scholar] [CrossRef] [PubMed]
- van Ouwerkerk, A.F.; Hall, A.W.; Kadow, Z.A.; Lazarevic, S.; Reyat, J.S.; Tucker, N.R.; Nadadur, R.D.; Bosada, F.M.; Bianchi, V.; Ellinor, P.T.; et al. Epigenetic and Transcriptional Networks Underlying Atrial Fibrillation. Circ. Res. 2020, 127, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Chen, L.; Li, F.; Cao, Y.; Li, D.; Xiong, Q.; Ling, Z. Biomimetic nanoparticles loaded lutein functionalized by macrophage membrane for targeted amelioration pressure overload-induced cardiac fibrosis. Biomed. Pharmacother. 2023, 167, 115579. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.N.; Lee, S.Y.; Lee, S.; Youn, H.; Im, H.J. Lipid nanoparticles for delivery of RNA therapeutics: Current status and the role of in vivo imaging. Theranostics 2022, 12, 7509–7531. [Google Scholar] [CrossRef] [PubMed]
- Jardin, B.; Epstein, J.A. Emerging mRNA therapies for cardiac fibrosis. Am. J. Physiol. Cell Physiol. 2024, 326, C107–C111. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wakili, R.; Xiao, J.; Wu, C.T.; Luo, X.; Clauss, S.; Dawson, K.; Qi, X.; Naud, P.; Shi, Y.F.; et al. Detailed Characterization of MicroRNA Changes in a Canine Heart Failure Model: Relationship to Arrhythmogenic Structural Remodeling. J. Mol. Cell. Cardiol. 2014, 77, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, Y.; Tu, D.; Liu, X.; Niu, S.; Suo, Y.; Liu, T.; Li, G.; Liu, C. Role of NLRP3-inflammasome/caspase-1/galectin-3 Pathway on Atrial Remodeling in Diabetic Rabbits. J. Cardiovasc. Transl. Res. 2020, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Rao, F.; Lian, F.; Yang, H.; Kuang, S.; Wu, S.; Deng, C.; Xue, Y. Mechanism of electrical remodeling of atrial myocytes and its influence on susceptibility to atrial fibrillation in diabetic rats. Life Sci. 2019, 239, 116903. [Google Scholar] [CrossRef] [PubMed]
- Allessie, M.; Ausma, J.; Schotten, U. Electrical, contractile and structural remodeling during atrial fibrillation. Cardiovasc. Res. 2002, 54, 230–246. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Burstein, B.; Dobrev, D. Atrial remodeling and atrial fibrillation: Mechanisms and implications. Circ. Arrhythm. Electrophysiol. 2008, 1, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Jost, N.; Kohajda, Z.; Kristóf, A.; Kovács, P.P.; Husti, Z.; Juhász, V.; Kiss, L.; Varró, A.; Virág, L.; Baczkó, I. Atrial remodeling and novel pharmacological strategies for antiarrhythmic therapy in atrial fibrillation. Curr. Med. Chem. 2011, 18, 3675–3694. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Dobrev, D. The multidimensional role of calcium in atrial fibrillation pathophysiology: Mechanistic insights and therapeutic opportunities. Eur. Heart J. 2012, 33, 1870–1877. [Google Scholar] [CrossRef] [PubMed]
- Wirth, K.J.; Paehler, T.; Rosenstein, B.; Knobloch, K.; Maier, T.; Frenzel, J.; Brendel, J.; Busch, A.E.; Bleich, M. Atrial effects of the novel K(+)-channel-blocker AVE0118 in anesthetized pigs. Cardiovasc. Res. 2003, 60, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Christ, T.; Wettwer, E.; Voigt, N.; Hála, O.; Radicke, S.; Matschke, K.; Várro, A.; Dobrev, D.; Ravens, U. Pathology-specific effects of the IKur/Ito/IK,ACh blocker AVE0118 on ion channels in human chronic atrial fibrillation. Br. J. Pharmacol. 2008, 154, 1619–1630. [Google Scholar] [CrossRef] [PubMed]
- Oros, A.; Volders, P.G.; Beekman, J.D.; van der Nagel, T.; Vos, M.A. Atrial-specific drug AVE0118 is free of torsades de pointes in anesthetized dogs with chronic complete atrioventricular block. Heart Rhythm 2006, 3, 1339–1345. [Google Scholar] [CrossRef] [PubMed]
- de Haan, S.; Greiser, M.; Harks, E.; Blaauw, Y.; van Hunnik, A.; Verheule, S.; Allessie, M.; Schotten, U. AVE0118, blocker of the transient outward current (I(to)) and ultrarapid delayed rectifier current (I(Kur)), fully restores atrial contractility after cardioversion of atrial fibrillation in the goat. Circulation 2006, 114, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Blaauw, Y.; Schotten, U.; van Hunnik, A.; Neuberger, H.R.; Allessie, M.A. Cardioversion of persistent atrial fibrillation by a combination of atrial specific and non-specific class III drugs in the goat. Cardiovasc. Res. 2007, 75, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Almquist, J.; Wallman, M.; Jacobson, I.; Jirstrand, M. Modeling the effect of Kv1.5 block on the canine action potential. Biophys. J. 2010, 99, 2726–2736. [Google Scholar] [CrossRef] [PubMed]
- Shiroshita-Takeshita, A.; Ford, J.; Madge, D.; Pinnock, R.; Nattel, S. Electrophysiological and atrial antiarrhythmic effects of a novel IKur/Kv1.5 blocker in dogs with atrial tachycardia remodeling. Heart Rhythm 2006, 3, S183. [Google Scholar] [CrossRef]
- Ford, J.; Milnes, J.; Wettwer, E.; Christ, T.; Rogers, M.; Sutton, K.; Madge, D.; Virag, L.; Jost, N.; Horvath, Z.; et al. Human electrophysiological and pharmacological properties of XEN-D0101: A novel atrial-selective Kv1.5/IKur inhibitor. J. Cardiovasc. Pharmacol. 2013, 61, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.; Milnes, J.; El Haou, S.; Wettwer, E.; Loose, S.; Matschke, K.; Tyl, B.; Round, P.; Ravens, U. The positive frequency-dependent electrophysiological effects of the IKur inhibitor XEN-D0103 are desirable for the treatment of atrial fibrillation. Heart Rhythm 2016, 13, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Flenner, F.; Friedrich, F.W.; Ungeheuer, N.; Christ, T.; Geertz, B.; Reischmann, S.; Wagner, S.; Stathopoulou, K.; Söhren, K.D.; Weinberger, F.; et al. Ranolazine antagonizes catecholamine-induced dysfunction in isolated cardiomyocytes, but lacks long-term therapeutic effects in vivo in a mouse model of hypertrophic cardiomyopathy. Cardiovasc. Res. 2016, 109, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Sag, C.M.; Mallwitz, A.; Wagner, S.; Hartmann, N.; Schotola, H.; Fischer, T.H.; Ungeheuer, N.; Herting, J.; Shah, A.M.; Maier, L.S.; et al. Enhanced late INa induces proarrhythmogenic SR Ca leak in a CaMKII-dependent manner. J. Mol. Cell. Cardiol. 2014, 76, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Poulet, C.; Wettwer, E.; Grunnet, M.; Jespersen, T.; Fabritz, L.; Matschke, K.; Knaut, M.; Ravens, U. Late Sodium Current in Human Atrial Cardiomyocytes from Patients in Sinus Rhythm and Atrial Fibrillation. PLoS ONE 2015, 10, e0131432. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Ma, J.; Li, H.; Wang, C.; Grandi, E.; Zhang, P.; Luo, A.; Bers, D.M.; Shryock, J.C.; Belardinelli, L. Late sodium current contributes to the reverse rate-dependent effect of IKr inhibition on ventricular repolarization. Circulation 2011, 123, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Ravens, U. Antiarrhythmic therapy in atrial fibrillation. Pharmacol. Ther. 2010, 128, 129–145. [Google Scholar] [CrossRef] [PubMed]
- De Ferrari, G.M.; Maier, L.S.; Mont, L.; Schwartz, P.J.; Simonis, G.; Leschke, M.; Gronda, E.; Boriani, G.; Darius, H.; Guillamón Torán, L.; et al. Ranolazine in the treatment of atrial fibrillation: Results of the dose-ranging RAFFAELLO (Ranolazine in Atrial Fibrillation Following An ELectricaL CardiOversion) study. Heart Rhythm 2015, 12, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Reiffel, J.A.; Camm, A.J.; Belardinelli, L.; Zeng, D.; Karwatowska-Prokopczuk, E.; Olmsted, A.; Zareba, W.; Rosero, S.; Kowey, P.; the HARMONY Investigators. The HARMONY Trial: Combined Ranolazine and Dronedarone in the Management of Paroxysmal Atrial Fibrillation: Mechanistic and Therapeutic Synergism. Circ. Arrhythm. Electrophysiol. 2015, 8, 1048–1056. [Google Scholar] [CrossRef] [PubMed]
- Kanter, H.L.; Saffitz, J.E.; Beyer, E.C. Cardiac myocytes express multiple gap junction proteins. Circ. Res. 1992, 70, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.M.; Rodefeld, M.E.; Green, K.; Beyer, E.C.; Saffitz, J.E. Gap junction protein phenotypes of the human heart and conduction system. J. Cardiovasc. Electrophysiol. 1995, 6, 813–822. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, E. Cardiac ionic currents and acute ischemia: From channels to arrhythmias. Physiol. Rev. 1999, 79, 917–1017. [Google Scholar] [CrossRef] [PubMed]
- Spach, M.S.; Heidlage, J.F.; Barr, R.C.; Dolber, P.C. Cell size and communication: Role in structural and electrical development and remodeling of the heart. Heart Rhythm 2004, 1, 500–515. [Google Scholar] [CrossRef] [PubMed]
- Jost, N.; Christ, T.; Magyar, J. New Strategies for the Treatment of Atrial Fibrillation. Pharmaceuticals 2021, 14, 926. [Google Scholar] [CrossRef] [PubMed]
- Pogwizd, S.M. Clinical potential of sodium-calcium exchanger inhibitors as antiarrhythmic agents. Drugs 2003, 63, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Tóth, A.; Kiss, L.; Varró, A.; Nánási, P.P. Potential therapeutic effects of Na+/Ca2+ exchanger inhibition in cardiac diseases. Curr. Med. Chem. 2009, 16, 3294–3321. [Google Scholar] [CrossRef] [PubMed]
- Kovács, P.P.; Simon, J.; Christ, T.; Wettwer, E.; Varró, A.; Ravens, U. NCX current is increased in human chronic atrial fibrillation: A possible explanation for contractile dysfunction? Cardiovasc. Res. 2010, 87, S50. [Google Scholar]
- Elias, C.L.; Lukas, A.; Shurraw, S.; Scott, J.; Omelchenko, A.; Gross, G.J.; Hnatowich, M.; Hryshko, L.V. Inhibition of Na+/Ca2+ exchange by KB-R7943: Transport mode selectivity and antiarrhythmic consequences. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H1334–H1345. [Google Scholar] [CrossRef] [PubMed]
- Birinyi, P.; Tóth, A.; Jóna, I.; Acsai, K.; Almássy, J.; Nagy, N.; Prorok, J.; Gherasim, I.; Papp, Z.; Hertelendi, Z.; et al. The Na+/Ca2+ exchange blocker SEA0400 fails to enhance cytosolic Ca2+ transient and contractility in canine ventricular cardiomyocytes. Cardiovasc. Res. 2008, 78, 476–484. [Google Scholar] [CrossRef] [PubMed]
- Lebek, S.; Pichler, K.; Reuthner, K.; Trum, M.; Tafelmeier, M.; Mustroph, J.; Camboni, D.; Rupprecht, L.; Schmid, C.; Maier, L.S.; et al. Enhanced CaMKII-Dependent Late INa Induces Atrial Proarrhythmic Activity in Patients with Sleep-disordered Breathing. Circ. Res. 2020, 126, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Nattel, S.; Heijman, J.; Zhou, L.; Dobrev, D. Molecular basis of atrial fibrillation pathophysiology and therapy: A translational perspective. Circ. Res. 2020, 127, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Chelu, M.G.; Sarma, S.; Sood, S.; Wang, S.; van Oort, R.J.; Skapura, D.G.; Li, N.; Santonastasi, M.; Müller, F.U.; Schmitz, W.; et al. Calmodulin kinase II-mediated sarcoplasmic reticulum Ca2+ leak promotes atrial fibrillation in mice. J. Clin. Investig. 2009, 119, 1940–1951. [Google Scholar] [CrossRef] [PubMed]
- Vest, J.A.; Wehrens, X.H.; Reiken, S.R.; Lehnart, S.E.; Dobrev, D.; Chandra, P.; Danilo, P.; Ravens, U.; Rosen, M.R.; Marks, A.R. Defective cardiac ryanodine receptor regulation during atrial fibrillation. Circulation 2005, 111, 2025–2032. [Google Scholar] [CrossRef] [PubMed]
- Dincer, U.D. Cardiac ryanodine receptor in metabolic syndrome: Is JTV519 (K201) future therapy? Diabetes Metab. Syndr. Obes. 2012, 5, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Nassif, M.E.; Windsor, S.L.; Tang, F.; Khariton, Y.; Husain, M.; Inzucchi, S.E.; McGuire, D.K.; Pitt, B.; Scirica, B.M.; Austin, B.; et al. Dapagliflozin effects on biomarkers, symptoms, and functional status in patients with heart failure with reduced ejection fraction: The DEFINE-HF trial. Circulation 2019, 140, 1463–1476. [Google Scholar] [CrossRef] [PubMed]
- Mauriello, A.; Correra, A.; Molinari, R.; Del Vecchio, G.E.; Tessitore, V.; D’Andrea, A.; Russo, V. Mitochondrial Dysfunction in Atrial Fibrillation: The Need for a Strong Pharmacological Approach. Biomedicines 2024, 12, 2720. [Google Scholar] [CrossRef] [PubMed]
- Chatfield, K.C.; Sparagna, G.C.; Chau, S.; Phillips, E.K.; Ambardekar, A.V.; Aftab, M.; Mitchell, M.B.; Sucharov, C.C.; Miyamoto, S.D.; Stauffer, B.L. Elamipretide Improves Mitochondrial Function in the Failing Human Heart. JACC Basic Transl. Sci. 2019, 4, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Sulaimon, L.A.; Afolabi, L.O.; Adisa, R.A.; Ayankojo, A.G.; Afolabi, M.O.; Adewolu, A.M.; Wan, X. Pharmacological significance of MitoQ in ameliorating mitochondria-related diseases. Adv. Redox Res. 2022, 5, 100037. [Google Scholar] [CrossRef]
- Muszyński, P.; Bonda, T.A. Mitochondrial Dysfunction in Atrial Fibrillation—Mechanisms and Pharmacological Interventions. J. Clin. Med. 2021, 10, 2385. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.S.; Kim, J.H.; Min, K.N.; Moon, J.A.; Roh, T.C.; Lee, M.J.; Lee, K.W.; Min, J.E.; Lee, Y.M. KL1333, a novel NAD+ modulator, improves energy metabolism and mitochondrial dysfunction in MELAS fibroblasts. Front. Neurol. 2018, 9, 552. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Patoulias, D.; Kassimis, G.; Koufakis, T.; Klisic, A.; Doumas, M.; Fragakis, N.; Rizzo, M. Therapeutic Potential of Sodium-glucose Co-transporter-2 Inhibitors and Glucagon-like peptide-1 Receptor Agonists for Patients with Acute Coronary Syndrome: A Review of Clinical Evidence. Curr. Pharm. Des. 2024, 30, 2109–2119. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Pamporis, K.; Stachteas, P.; Patoulias, D.; Bougioukas, K.I.; Fragakis, N. Efficacy and safety of sodium-glucose cotransporter-2 inhibitors in heart failure with mildly reduced or preserved ejection fraction: An overview of 36 systematic reviews. Heart Fail. Rev. 2023, 28, 1033–1051. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Popovic, D.S.; Patoulias, D.; Koufakis, T.; Papanas, N.; Fragakis, N.; Rizzo, M. The Effect of Sodium-glucose Cotransporter Inhibitors on Renal Function as Adjunctive to Insulin in Adults with Type 1 diabetes: An Updated Multilevel Meta-analysis of Randomized Controlled Trials. Diabetes Ther. 2024, 15, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, N.; Nikolaou, P.E.; Karakasis, P.; Stachteas, P.; Fragakis, N.; Andreadou, I. Endothelial protection by sodium-glucose cotransporter 2 inhibitors: A literature review of in vitro and in vivo studies. Int. J. Mol. Sci. 2024, 25, 7274. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, Y.; Kokubo, H.; Bumdelger, B.; Yoshizumi, M.; Yamamotoya, T.; Matsunaga, Y.; Ueda, K.; Inoue, Y.; Inoue, M.K.; Fujishiro, M.; et al. The SGLT2 inhibitor Luseogliflozin Rapidly Normalizes Aortic mRNA Levels of Inflammation-related but not Lipid-Metabolism-Related Genes and Suppresses Atherosclerosis in Diabetic ApoE KO mice. Int. J. Mol. Sci. 2017, 18, 1704. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Zhan, F.; He, M.; Liu, Z.; Song, X. Anti-inflammatory effects of sodium-glucose co-transporter 2 inhibitors on atherosclerosis. Vascul. Pharmacol. 2020, 133–134, 106779. [Google Scholar] [CrossRef] [PubMed]
- Garvey, W.T.; Van Gaal, L.; Leiter, L.A.; Vijapurkar, U.; List, J.; Cuddihy, R.; Ren, J.; Davies, M.J. Effects of canagliflozin versus glimepiride on adipokines and inflammatory biomarkers in type 2 diabetes. Metabolism 2018, 85, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Homayr, A.; Juni, R.P.; Spin, E.L.; Kerindongo, R.; Boomsma, M.; Hollmann, M.W.; Preckel, B.; Koolwijk, P.; van Hinsbergh, V.W.M.; et al. Empagliflozin and Dapagliflozin Reduce ROS Generation and Restore NO Bioavailability in Tumor Necrosis Factor α-stimulated Human Coronary Arterial Endothelial Cells. Cell. Physiol. Biochem. 2019, 53, 865–886. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Lee, S.G.; Kim, S.H.; Kim, J.H.; Choi, E.; Cho, W.; Rim, J.H.; Hwang, I.; Lee, C.J.; Lee, M.; et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat. Commun. 2020, 11, 2127. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Aizawa, Y.; Yuasa, S.; Kishi, S.; Fuse, K.; Fujita, S.; Ikeda, Y.; Kitazawa, H.; Takahashi, M.; Sato, M.; et al. The effect of dapagliflozin treatment on epicardial adipose tissue volume. Cardiovasc. Diabetol. 2018, 17, 6. [Google Scholar] [CrossRef] [PubMed]
- Stachteas, P.; Karakasis, P.; Patoulias, D.; Clemenza, F.; Fragakis, N.; Rizzo, M. The effect of sodium-glucose co-transporter-2 inhibitors on markers of subclinical atherosclerosis. Ann. Med. 2023, 55, 2304667. [Google Scholar] [CrossRef] [PubMed]
- Olgar, Y.; Turan, B. A sodium-glucose cotransporter 2 (SGLT2) inhibitor dapagliflozin comparison with insulin shows important effects on Zn2+-transporters in cardiomyocytes from insulin-resistant metabolic syndrome rats through inhibition of oxidative stress. Can. J. Physiol. Pharmacol. 2019, 97, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Schauer, A.; Adams, V.; Kämmerer, S.; Langner, E.; Augstein, A.; Barthel, P.; Männel, A.; Fabig, G.; Alves, P.K.N.; Günscht, M.; et al. Empagliflozin improves diastolic function in HFpEF by restabilizing the mitochondrial respiratory chain. Circ. Heart Fail. 2024, 17, e011107. [Google Scholar] [CrossRef] [PubMed]
- Packer, M. Critical Reanalysis of the Mechanisms Underlying the Cardiorenal Benefits of SGLT2 Inhibitors and Reaffirmation of the Nutrient Deprivation Signaling/Autophagy Hypothesis. Circulation 2022, 146, 1383–1405. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Zhang, M.; Suo, M.; Liu, D.; Wang, X.; Liu, M.; Pan, J.; Jin, T.; An, F. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF-β/Smad signalling in type 2 diabetic rats. J. Cell. Mol. Med. 2021, 25, 7642–7659. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Shiou, Y.L.; Jhuo, S.J.; Chang, C.Y.; Liu, P.L.; Jhuang, W.J.; Dai, Z.K.; Chen, W.Y.; Chen, Y.F.; Lee, A.S. The sodium-glucose co-transporter 2 inhibitor empagliflozin attenuates cardiac fibrosis and improves ventricular hemodynamics in hypertensive heart failure rats. Cardiovasc. Diabetol. 2019, 18, 45. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, X.; Wang, Q. Effects and mechanisms of SGLT2 inhibitors on the NLRP3 inflammasome, with a focus on atherosclerosis. Front. Endocrinol. 2022, 13, 992937. [Google Scholar] [CrossRef] [PubMed]
- Scisciola, L.; Cataldo, V.; Taktaz, F.; Fontanella, R.A.; Pesapane, A.; Ghosh, P.; Franzese, M.; Puocci, A.; De Angelis, A.; Sportiello, L.; et al. Anti-inflammatory role of SGLT2 inhibitors as part of their anti-atherosclerotic activity: Data from basic science and clinical trials. Front. Cardiovasc. Med. 2022, 9, 1008922. [Google Scholar] [CrossRef] [PubMed]
- Dyck, J.R.B.; Sossalla, S.; Hamdani, N.; Coronel, R.; Weber, N.C.; Light, P.E.; Zuurbier, C.J. Cardiac mechanisms of the beneficial effects of SGLT2 inhibitors in heart failure: Evidence for potential off-target effects. J. Mol. Cell. Cardiol. 2022, 167, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Andersen, A.; Bagger, J.I.; Sørensen, S.K.; Baldassarre, M.P.A.; Pedersen-Bjergaard, U.; Forman, J.L.; Gislason, G.; Lindhardt, T.B.; Knop, F.K.; Vilsbøll, T. Associations of hypoglycemia, glycemic variability and risk of cardiac arrhythmias in insulin-treated patients with type 2 diabetes: A prospective, observational study. Cardiovasc. Diabetol. 2021, 20, 241. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Teshima, Y.; Fukui, A.; Kondo, H.; Nishio, S.; Nakagawa, M.; Saikawa, T.; Takahashi, N. Glucose fluctuations increase the incidence of atrial fibrillation in diabetic rats. Cardiovasc. Res. 2014, 104, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Paasche, A.; Wiedmann, F.; Kraft, M.; Seibertz, F.; Herlt, V.; Blochberger, P.L.; Jávorszky, N.; Beck, M.; Weirauch, L.; Seeger, T.; et al. Acute antiarrhythmic effects of SGLT2 inhibitors—Dapagliflozin lowers the excitability of atrial cardiomyocytes. Basic Res. Cardiol. 2024, 119, 93–112. [Google Scholar] [CrossRef] [PubMed]
- Philippaert, K.; Kalyaanamoorthy, S.; Fatehi, M.; Long, W.; Soni, S.; Byrne, N.J.; Barr, A.; Singh, J.; Wong, J.; Palechuk, T.; et al. Cardiac late sodium channel current is a molecular target for the sodium/glucose cotransporter 2 inhibitor empagliflozin. Circulation 2021, 143, 2188–2204. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.S.; Singh, T.; Newby, D.E.; Singh, J. Sodium-glucose co-transporter 2 inhibitor therapy: Mechanisms of action in heart failure. Heart 2021, 107, 1032–1038. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.E.; Petersenn, F.; Hackbarth, J.; Pfeiffer, J.; Gampp, H.; Frey, N.; Lugenbiel, P.; Thomas, D.; Rahm, A.K. Electrophysiological Effects of the Sodium-glucose Co-transporter-2 (SGLT2) Inhibitor Dapagliflozin on Human Cardiac Potassium Channels. Int. J. Mol. Sci. 2024, 25, 5701. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.I.; Chen, Y.C.; Lin, Y.K.; Chung, C.C.; Lu, Y.Y.; Kao, Y.H.; Chen, Y.J. Empagliflozin Attenuates Myocardial Sodium and Calcium Dysregulation and Reverses Cardiac Remodeling in Streptozotocin-induced Diabetic Rats. Int. J. Mol. Sci. 2019, 20, 1680. [Google Scholar] [CrossRef] [PubMed]
- Mustroph, J.; Wagemann, O.; Lücht, C.M.; Trum, M.; Hammer, K.P.; Sag, C.M.; Lebek, S.; Tarnowski, D.; Reinders, J.; Perbellini, F.; et al. Empagliflozin reduces Ca/calmodulin-dependent kinase II activity in isolated ventricular cardiomyocytes. ESC Heart Fail. 2018, 5, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Zhan, G.; Wang, X.; Wang, X.; Li, J.; Tang, Y.; Bi, H.; Yang, X.; Xia, Y. Dapagliflozin: A sodium-glucose cotransporter 2 inhibitor, attenuates angiotensin II-induced atrial fibrillation by regulating atrial electrical and structural remodeling. Eur. J. Pharmacol. 2024, 978, 176712. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Lip, G.Y.H.; Feng, Q.; Fei, Y.; Tse, Y.K.; Wu, M.Z.; Ren, Q.W.; Tse, H.F.; Cheung, B.Y.; Yiu, K.H. Sodium-Glucose Cotransporter 2 Inhibitors (SGLT2i) and Cardiac Arrhythmias: A Systematic Review and Meta-analysis. Cardiovasc. Diabetol. 2021, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, Y.; Wang, Z.; Liu, D.; Mao, S.; Liang, B. The effectiveness of SGLT2 inhibitor in the incidence of atrial fibrillation/atrial flutter in patients with type 2 diabetes mellitus/heart failure: A systematic review and meta-analysis. J. Thorac. Dis. 2022, 14, 1620–1637. [Google Scholar] [CrossRef] [PubMed]
- Zelniker, T.A.; Bonaca, M.P.; Furtado, R.H.M.; Mosenzon, O.; Kuder, J.F.; Murphy, S.A.; Bhatt, D.L.; Leiter, L.A.; McGuire, D.K.; Wilding, J.P.H.; et al. Effect of Dapagliflozin on Atrial Fibrillation in Patients with Type 2 Diabetes Mellitus: Insights From the DECLARE-TIMI 58 trial. Circulation 2020, 141, 1227–1234. [Google Scholar] [CrossRef] [PubMed]
- Younis, A.; Arous, T.; Klempfner, R.; Kharsa, A.; McNitt, S.; Schleede, S.; Polonski, B.; Abdallah, Z.; Buttar, R.; Bodurian, C.; et al. Effect of sodium glucose cotransporter 2 inhibitors on atrial tachy-arrhythmia burden in patients with cardiac implantable electronic devices. J. Cardiovasc. Electrophysiol. 2023, 34, 1595–1604. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and Cardiovascular Outcomes in Patients with Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [PubMed]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Fragakis, N.; Patoulias, D.; Theofilis, P.; Sagris, M.; Koufakis, T.; Vlachakis, P.K.; Rangraze, I.R.; El Tanani, M.; Tsioufis, K.; et al. The Emerging Role of Glucagon-like Peptide-1 Receptor Agonists in the Management of Obesity-Related Heart Failure with Preserved Ejection Fraction: Benefits beyond What Scales Can Measure? Biomedicines 2024, 12, 2112. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Zhang, W.; Zhang, D.; Ren, G.; Wang, P.; Gao, L.; Chen, H.; Ding, C. Comparison of the effect of glucose-lowering agents on the risk of atrial fibrillation: A network meta-analysis. Heart Rhythm 2021, 18, 1090–1096. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Niwano, S.; Niwano, H.; Fukaya, H.; Murakami, M.; Kishihara, J.; Satoh, A.; Yoshizawa, T.; Ishizue, N.; Igarashi, T.; et al. Liraglutide suppresses atrial electrophysiological changes. Heart Vessel. 2019, 34, 1389–1393. [Google Scholar] [CrossRef] [PubMed]
- Bohne, L.J.; Jansen, H.J.; Dorey, T.W.; Daniel, I.M.; Jamieson, K.L.; Belke, D.D.; McRae, M.D.; Rose, R.A. Glucagon-Like Peptide-1 Protects Against Atrial Fibrillation and Atrial Remodeling in Type 2 Diabetic Mice. JACC Basic Transl. Sci. 2023, 8, 922–936. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, G.Y.; Maeng, H.J.; Kim, H.; Bae, J.H.; Kim, K.M.; Lim, S. Effects of glucagon-like peptide-1 analogue and fibroblast growth factor 21 combination on the atherosclerosis-related process in a type 2 diabetes mouse model. Endocrinol. Metab. 2021, 36, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.; Khurana, A.; Bhatti, J.S.; Weiskirchen, R.; Navik, U. Glucagon-like peptide 1 and fibroblast growth factor-21 in non-alcoholic steatohepatitis: An experimental to clinical perspective. Pharmacol. Res. 2022, 184, 106426. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, S.H. Anti-inflammatory role of glucagon-like peptide 1 receptor agonists and its clinical implications. Ther. Adv. Endocrinol. Metab. 2024, 15, 20420188231222367. [Google Scholar] [CrossRef] [PubMed]
- Mehdi, S.F.; Pusapati, S.; Anwar, M.S.; Lohana, D.; Kumar, P.; Nandula, S.A.; Nawaz, F.K.; Tracey, K.; Yang, H.; LeRoith, D.; et al. Glucagon-like peptide-1: A multi-faceted anti-inflammatory agent. Front. Immunol. 2023, 14, 1148209. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lin, Y.; Wang, S.; Zhang, L.; Guo, L. GLP-1 Inhibits High-Glucose-Induced Oxidative Injury of Vascular Endothelial Cells. Sci. Rep. 2017, 7, 8008. [Google Scholar] [CrossRef] [PubMed]
- Mangmool, S.; Hemplueksa, P.; Parichatikanond, W.; Chattipakorn, N. Epac is required for GLP-1R-mediated inhibition of oxidative stress and apoptosis in cardiomyocytes. Mol. Endocrinol. 2015, 29, 583–596. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Jun, H.S. Effects of Glucagon-Like Peptide-1 on Oxidative Stress and Nrf2 Signaling. Int. J. Mol. Sci. 2017, 19, 26. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Mangmool, S.; Parichatikanond, W. Multifaceted Roles of GLP-1 and Its Analogs: A Review on Molecular Mechanisms with a Cardiotherapeutic Perspective. Pharmaceuticals 2023, 16, 836. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fan, S.; Xiong, Q.; Niu, Y.; Zhang, X.; Qin, J.; Shi, Y.; Zhang, L. Glucagon-like peptide-1 attenuates cardiac hypertrophy via the AngII/AT1R/ACE2 and AMPK/mTOR/p70S6K pathways. Acta Biochim. Biophys. Sin. 2021, 53, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Nreu, B.; Dicembrini, I.; Tinti, F.; Sesti, G.; Mannucci, E.; Monami, M. Major cardiovascular events, heart failure, and atrial fibrillation in patients treated with glucagon-like peptide-1 receptor agonists: An updated meta-analysis of randomized controlled trials. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, F.C.; Yen, K.C.; Chao, T.F.; Chen, S.W.; Chan, Y.H.; Chu, P.H. New-Onset Atrial Fibrillation in Patients With Type 2 Diabetes Treated With Novel Glucose-Lowering Therapies. J. Clin. Endocrinol. Metab. 2022, 107, 2493–2499. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Fragakis, N.; Patoulias, D.; Theofilis, P.; Kassimis, G.; Karamitsos, T.; El-Tanani, M.; Rizzo, M. Effects of Glucagon-Like Peptide 1 Receptor Agonists on Atrial Fibrillation Recurrence After Catheter Ablation: A Systematic Review and Meta-Analysis. Adv. Ther. 2024, 41, 3749–3756. [Google Scholar] [CrossRef] [PubMed]
- Christophersen, I.E.; Rienstra, M.; Roselli, C.; Yin, X.; Geelhoed, B.; Barnard, J.; Lin, H.; Arking, D.E.; Smith, A.V.; Albert, C.M.; et al. Large-Scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat. Genet. 2017, 49, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Ito, K.; Ito, M.; Zou, Z.; Kubota, M.; Nomura, S.; Matsunaga, H.; Koyama, S.; Ieki, H.; Akiyama, M.; et al. Cross-ancestry genome-wide analysis of atrial fibrillation unveils disease biology and enables cardioembolic risk prediction. Nat. Genet. 2023, 55, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.W.; Chen, J.J.; Wang, C.H.; Chang, S.N.; Chiu, F.C.; Huang, P.S.; Chua, S.K.; Chuang, E.Y.; Tsai, C.T. Identification of a new genetic locus associated with atrial fibrillation in the Taiwanese population by genome-wide and transcriptome-wide association studies. EP Eur. 2025, 27, euaf042. [Google Scholar] [CrossRef] [PubMed]
- Harati, H.; Zanetti, D.; Rao, A.; Gustafsson, S.; Perez, M.; Ingelsson, E.; Knowles, J.W. No evidence of a causal association of type 2 diabetes and glucose metabolism with Atrial Fibrillation. Diabetologia 2019, 62, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Steenman, M. Insight into Atrial Fibrillation through Analysis of the Coding Transcriptome in Humans. Biophys. Rev. 2020, 12, 817–826. [Google Scholar] [CrossRef] [PubMed]
- Li, T.L.; Zhu, N.N.; Yin, Z.; Sun, J.; Guo, J.P.; Yuan, H.T.; Shi, X.M.; Guo, H.Y.; Li, S.X.; Shan, Z.L. Transcriptomic analysis of epicardial adipose tissue reveals the potential crosstalk genes and immune relationship between type 2 diabetes mellitus and atrial fibrillation. Gene 2024, 920, 148528. [Google Scholar] [CrossRef] [PubMed]
- Menezes Junior, A.D.S.; Ferreira, L.C.; Barbosa, L.J.V.; Silva, D.M.E.; Saddi, V.A.; Silva, A.M.T.C. Circulating MicroRNAs as Specific Biomarkers in Atrial Fibrillation: A Meta-Analysis. Noncoding RNA 2023, 9, 13. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Chen, H.; Ge, D.; Xu, Y.; Xu, H.; Yang, Y.; Gu, M.; Zhou, Y.; Zhu, J.; Ge, T.; et al. Mir-21 Promotes Cardiac Fibrosis After Myocardial Infarction Via Targeting Smad7. Cell Physiol. Biochem. 2017, 42, 2207–2219. [Google Scholar] [CrossRef] [PubMed]
- Duisters, R.F.; Tijsen, A.J.; Schroen, B.; Leenders, J.J.; Lentink, V.; van der Made, I.; Herias, V.; van Leeuwen, R.E.; Schellings, M.W.; Barenbrug, P.; et al. miR-133 and miR-30 regulate connective tissue growth factor: Implications for a role of microRNAs in myocardial matrix remodeling. Circ. Res. 2009, 104, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Nie, J.; Han, Y.; Ni, L. Epigenetic Mechanism and Therapeutic Implications of Atrial Fibrillation. Front. Cardiovasc. Med. 2022, 8, 763824. [Google Scholar] [CrossRef] [PubMed]
- Vigorelli, V.; Resta, J.; Bianchessi, V.; Lauri, A.; Bassetti, B.; Agrifoglio, M.; Pesce, M.; Polvani, G.; Bonalumi, G.; Cavallotti, L.; et al. Abnormal DNA Methylation Induced by Hyperglycemia Reduces CXCR4 Gene Expression in CD34+ Stem Cells. J. Am. Heart Assoc. 2019, 8, e010012. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.E.; El-Osta, A. Epigenetics: Mechanisms and implications for diabetic complications. Circ. Res. 2010, 107, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Nural-Guvener, H.F.; Zakharova, L.; Nimlos, J.; Popovic, S.; Mastroeni, D.; Gaballa, M.A. HDAC class I inhibitor, Mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation. Fibrogenesis Tissue Repair 2014, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Atzemian, N.; Androutsopoulou, V.; Portokallidou, K.; Leonidis, G.; Pallikarou, M.; Ragia, G.; Karangelis, D.; Mikroulis, D.; Manolopoulos, V. Insights into Type 2 Diabetes Mellitus in Atrial Fibrillation Patients: A Proteomics Approach. Eur. Heart J. 2024, 45 (Suppl. S1), ehae666.446. [Google Scholar] [CrossRef]
- Mayr, M.; Yusuf, S.; Weir, G.; Chung, Y.L.; Mayr, U.; Yin, X.; Ladroue, C.; Madhu, B.; Roberts, N.; De Souza, A.; et al. Combined Metabolomic and Proteomic Analysis of Human Atrial Fibrillation. J. Am. Coll. Cardiol. 2008, 51, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Rohun, J.; Dudzik, D.; Raczak-Gutknecht, J.; Wabich, E.; Młodziński, K.; Markuszewski, M.J.; Daniłowicz-Szymanowicz, L. Metabolomics in Atrial Fibrillation: Unlocking Novel Biomarkers and Pathways for Diagnosis, Prognosis, and Personalized Treatment. J. Clin. Med. 2024, 14, 34. [Google Scholar] [CrossRef] [PubMed]
- Van Gelder, I.C.; Rienstra, M.; Bunting, K.V.; Casado-Arroyo, R.; Caso, V.; Crijns, H.J.G.M.; De Potter, T.J.R.; Dwight, J.; Guasti, L.; Hanke, T.; et al. 2024 ESC Guidelines for the Management of Atrial Fibrillation Developed in Collaboration with the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2024, 45, 3314–3414. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Wu, L.S.; Chiou, M.J.; Liu, J.R.; Yu, K.H.; Kuo, C.F.; Wen, M.S.; Chen, W.J.; Yeh, Y.H.; See, L.C. Association of Metformin with Lower Atrial Fibrillation Risk among Patients with Type 2 Diabetes Mellitus: A Population-Based Dynamic Cohort and In Vitro Studies. Cardiovasc. Diabetol. 2014, 13, 123. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Viollet, B. Metformin: Update on Mechanisms of Action and Repurposing Potential. Nat. Rev. Endocrinol. 2023, 19, 460–476. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Liu, Y.; Tu, T.; Li, B.; Xiao, Y.; Ma, Y.; Qin, F.; Xie, J.; Zhou, S.; Liu, Q. Metformin Regulates Lipid Metabolism in a Canine Model of Atrial Fibrillation through AMPK/PPAR-α/VLCAD Pathway. Lipids Health Dis. 2019, 18, 109. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Po, S.S.; Zhang, B.; Bai, F.; Li, J.; Qin, F.; Liu, N.; Sun, C.; Xiao, Y.; Tu, T.; et al. Metformin Regulates Adiponectin Signalling in Epicardial Adipose Tissue and Reduces Atrial Fibrillation Vulnerability. J. Cell. Mol. Med. 2020, 24, 7751–7766. [Google Scholar] [CrossRef] [PubMed]
- Nantsupawat, T.; Wongcharoen, W.; Chattipakorn, S.C.; Chattipakorn, N. Effects of Metformin on Atrial and Ventricular Arrhythmias: Evidence from Cell to Patient. Cardiovasc. Diabetol. 2020, 19, 198. [Google Scholar] [CrossRef] [PubMed]
- Shimano, M.; Tsuji, Y.; Inden, Y.; Kitamura, K.; Uchikawa, T.; Harata, S.; Nattel, S.; Murohara, T. Pioglitazone, a peroxisome proliferator-activated receptor-gamma activator, attenuates atrial fibrosis and atrial fibrillation promotion in rabbits with congestive heart failure. Heart Rhythm 2008, 5, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Chao, T.F.; Leu, H.B.; Huang, C.C.; Chen, J.W.; Chan, W.L.; Lin, S.J.; Chen, S.A. Thiazolidinediones can prevent new onset atrial fibrillation in patients with non-insulin dependent diabetes. Int. J. Cardiol. 2012, 156, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Park, S.Y.; Kim, B.; Min, C.; Cho, W.; Yon, D.K.; Kim, J.Y.; Han, K.D.; Rhee, E.J.; Lee, W.Y.; et al. Association between antidiabetic drugs and the incidence of atrial fibrillation in patients with type 2 diabetes: A nationwide cohort study in South Korea. Diabetes Res. Clin. Pract. 2023, 198, 110626. [Google Scholar] [CrossRef] [PubMed]
- Pallisgaard, J.L.; Brooks, M.M.; Chaitman, B.R.; Boothroyd, D.B.; Perez, M.; Hlatky, M.A.; Bypass Angioplasty Revascularization Investigation 2 Diabetes Study Group. Thiazolidinediones and Risk of Atrial Fibrillation Among Patients with Diabetes and Coronary Disease. Am. J. Med. 2018, 131, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.S.; Yang, F.Y.; Chen, H.Y.; Jong, G.P. Antihyperglycemic drugs use and new-onset atrial fibrillation: A population-based nested case control study. PLoS ONE 2018, 13, e0197245. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, G.; Chang, C.; Chou, O.H.I.; Lee, S.; Leung, K.S.K.; Wong, W.T.; Liu, T.; Wai, A.K.C.; Cheng, S.H.; et al. Metformin versus sulphonylureas for new onset atrial fibrillation and stroke in type 2 diabetes mellitus: A population-based study. Acta Diabetol. 2022, 59, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Yang, F.Y.; Jong, G.P.; Liou, Y.S. Antihyperglycemic drugs use and new-onset atrial fibrillation in elderly patients. Eur. J. Clin. Investig. 2017, 47, 388–393. [Google Scholar] [CrossRef] [PubMed]
- Patoulias, D.I.; Boulmpou, A.; Teperikidis, E.; Katsimardou, A.; Siskos, F.; Doumas, M.; Papadopoulos, C.E.; Vassilikos, V. Cardiovascular efficacy and safety of dipeptidyl peptidase-4 inhibitors: A meta-analysis of cardiovascular outcome trials. World J. Cardiol. 2021, 13, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Sagris, M.; Patoulias, D.; Koufakis, T.; Theofilis, P.; Klisic, A.; Fragakis, N.; El Tanani, M.; Rizzo, M. Mitigating Increased Cardiovascular Risk in Patients with Obstructive Sleep Apnea Using GLP-1 Receptor Agonists and SGLT2 Inhibitors: Hype or Hope? Biomedicines 2024, 12, 2503. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Patoulias, D.; Fragakis, N.; Bernal-López, M.R.; Gómez-Huelgas, R. Glucagon-like peptide-1 receptor agonists and sodium-glucose cotransporter-2 inhibitors combination therapy versus monotherapy and major adverse cardiovascular events: Do the benefits add up? Eur. J. Intern. Med. 2024, 130, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Karakasis, P.; Theofilis, P.; Patoulias, D.; Vlachakis, P.K.; Pamporis, K.; Sagris, M.; Ktenopoulos, N.; Kassimis, G.; Antoniadis, A.P.; Fragakis, N. Sodium-Glucose Cotransporter 2 Inhibitors in Aortic Stenosis: Toward a Comprehensive Cardiometabolic Approach. Int. J. Mol. Sci. 2025, 26, 4494. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Kang, B.; Zhou, J. Sodium glucose cotransporter 2 inhibitors with cardiac arrhythmias in patients with type 2 diabetes mellitus: A systematic review and meta-analysis of randomized placebo-controlled trials. Clin. Res. Cardiol. 2024, 113, 910–923. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.; Stegmann, C.; Kosiuk, J.; Dinov, B.; Richter, S.; Arya, A.; Müssigbrodt, A.; Sommer, P.; Hindricks, G.; Bollmann, A. Predictors, management, and outcome of cardioversion failure early after atrial fibrillation ablation. Europace 2018, 20, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Grönberg, T.; Hartikainen, J.E.; Nuotio, I.; Biancari, F.; Vasankari, T.; Nikkinen, M.; Ylitalo, A.; Airaksinen, K.E. Can we predict the failure of electrical cardioversion of acute atrial fibrillation? The FinCV study. Pacing Clin. Electrophysiol. 2015, 38, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Soran, H.; Banerjee, M.; Mohamad, J.B.; Adam, S.; Ho, J.H.; Ismaeel, S.M.; Dhage, S.; Syed, A.A.; Abdulla, I.M.A.; Younis, N.; et al. Risk Factors for Failure of Direct Current Cardioversion in Patients with Type 2 Diabetes Mellitus and Atrial Fibrillation. Biomed Res. Int. 2018, 2018, 5936180. [Google Scholar] [CrossRef] [PubMed]
- Kriz, R.; Freynhofer, M.K.; Weiss, T.W.; Egger, F.; Gruber, S.C.; Eisenburger, P.; Wojta, J.; Huber, K.; Koch, J. Safety and efficacy of pharmacological cardioversion of recent-onset atrial fibrillation: A single-center experience. Am. J. Emerg. Med. 2016, 34, 1486–1490. [Google Scholar] [CrossRef] [PubMed]
- Ergene, U.; Ergene, O.; Cete, Y.; Fowler, J.; Nazli, C.; Oktay, C. Predictors of success in the conversion of new-onset atrial fibrillation using oral propafenone. Eur. J. Emerg. Med. 1998, 5, 425–428. [Google Scholar] [CrossRef]
- Handelsman, Y.; Bunch, T.J.; Rodbard, H.W.; Steinberg, B.A.; Thind, M.; Bigot, G.; Konigsberg, L.; Wieloch, M.; Kowey, P.R. Impact of dronedarone on patients with atrial fibrillation and diabetes: A sub-analysis of the ATHENA and EURIDIS/ADONIS studies. J. Diabetes Complicat. 2022, 36, 108227. [Google Scholar] [CrossRef] [PubMed]
- Walters, T.E.; Nisbet, A.; Morris, G.M.; Tan, G.; Mearns, M.; Teo, E.; Lewis, N.; Ng, A.; Gould, P.; Lee, G.; et al. Progression of atrial remodeling in patients with high-burden atrial fibrillation: Implications for early ablative intervention. Heart Rhythm 2016, 13, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.H.; Yu, H.T.; Park, Y.J.; Kim, T.H.; Joung, B.; Lee, M.H.; Pak, H.N. Diabetes Mellitus Is an Independent Risk Factor for a Stiff Left Atrial Physiology After Catheter Ablation for Atrial Fibrillation. Front. Cardiovasc. Med. 2022, 9, 828478. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Gaspar, T.; Schönbauer, R.; Wójcik, M.; Fiedler, L.; Roithinger, F.X.; Martinek, M.; Pürerfellner, H.; Kirstein, B.; Richter, U.; et al. Low-Voltage Myocardium-Guided Ablation Trial of Persistent Atrial Fibrillation. NEJM Evid. 2022, 1, EVIDoa2200141. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A.; Testani, J.M. SGLT2 inhibitors and diuretics in heart failure: Clicking reset on the renal volume setpoint? Eur. Heart J. 2023, 44, 2944–2946. [Google Scholar] [CrossRef] [PubMed]
- Solomon, S.D.; Ostrominski, J.W.; Wang, X.; Shah, S.J.; Borlaug, B.A.; Butler, J.; Davies, M.J.; Kitzman, D.W.; Verma, S.; Abildstrøm, S.Z.; et al. STEP-HFpEF Trial Committees and Investigators. Effect of Semaglutide on Cardiac Structure and Function in Patients With Obesity-Related Heart Failure. J. Am. Coll. Cardiol. 2024, 84, 1587–1602. [Google Scholar] [CrossRef] [PubMed]
- Kantharia, B.K.; Lip, G.Y.H.; Martin, D.T. Alterations in atrial electrogram amplitude as steady sinus rhythm transitions to paroxysmal atrial fibrillation during continuous monitoring in patients with implantable cardiac devices: Insights from the IMPACT study. J. Cardiovasc. Electrophysiol. 2021, 32, 1357–1363. [Google Scholar] [CrossRef] [PubMed]
- Scirica, B.M.; Bhatt, D.L.; Braunwald, E.; Steg, P.G.; Davidson, J.; Hirshberg, B.; Ohman, P.; Frederich, R.; Wiviott, S.D.; Hoffman, E.B.; et al. SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N. Engl. J. Med. 2013, 369, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Deshaies, R.J. Multispecific drugs herald a new era of biopharmaceutical innovation. Nature 2020, 580, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Molla, G.; Bitew, M. Revolutionizing Personalized Medicine: Synergy with Multi-Omics Data Generation, Main Hurdles, and Future Perspectives. Biomedicines 2024, 12, 2750. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Compound | Target | Proposed Mechanism of Action | Major Effects |
|---|---|---|---|
| Anti-inflammatory compounds | |||
| MCC950 | NLRP3 inflammasome | Selective inhibitor of NLRP3 inflammasome by blocking ASC oligomerization | Alleviates fibrosis and AF in animal models [77]. |
| Colchicine | NLRP3 inflammasome/ Chemotaxis | Inhibits β-tubulin polymerization into microtubules | Mixed evidence; COP-AF trial showed no significant AF reduction in patients who underwent major thoracic surgery [77,80]. |
| CY-09 | NLRP3 inflammasome | Inhibits NLRP3 oligomerization by binding to ATPase domain | Under investigation [77]. |
| RRx-001 | NLRP3 inflammasome | Inhibitor of NEK7 binding to LRR of the NLRP3 inflammasome | Under investigation [77]. |
| JC-121, JC-124 | NLRP3 inflammasome | Inhibitors of NLRP3 inflammasome activation and ASC aggregation | Under investigation [77]. |
| Canakinumab | Pro-inflammatory cytokines | Monoclonal IL-1β neutralizing antibody | CONVERT-AF demonstrated a trend towards reduced AF-occurrence after cardioversion [82]. |
| Pinocembrin | NF-κB | Abundant flavonoid suppresses inflammatory and fibrotic responses | Reduces atrial fibrosis, autonomic and electrical remodeling, and inflammation in MI animal model [83]. |
| Tasquinimod | NF-κB | Inhibitor of the S100A8/A9 interaction and TLR4 signaling. | Enhances LV systolic function and suppresses pro-inflammatory cytokine expression in mice model [85]. |
| Anti-fibrotic compounds | |||
| Finerenone | TGF-β, Galectin-3, CTGF | Inhibit profibrotic signaling and atrial remodeling | It reduces risk of new-onset AF/AFL across the cardio–kidney–metabolic spectrum [95]. |
| Macitentan | Endothelin-1 | Endothelin-1 receptor antagonist | Decreased atrial endothelin 1 levels and prevented pacing-induced increases in pro-endothelin 1 mRNA [100]. |
| Ion channel blockers/calcium handling modulators | |||
| AVE0118 | IKur current | IKur, Ito, and IK,ACh blocker | Prolonged atrial refractory period without ventricular effects in experimental models [126,127,128,129]. |
| XEN-D0101 | IKur current | IKur blocker | Prolonged atrial refractory period without ventricular effects in experimental models [133,134]. |
| KN-93 analogs | CaMKII, L-type calcium channels | Inhibits CaMKII and affects CaV1.3 and CaV1.2 calcium channels | It prevents pacing-induced AF in both in vitro and in vivo mouse models [154] |
| JTV519 | RyR2 stabilizer | Inhibits IK,Ach and IKur in atrial muscle cells | Under investigation [156]. |
| Ranolazine | INa,L current | Selective inhibitor of INa,L | Shown to reduce AF risk in clinical studies [135,136,137,138,139,140,141]. |
| NCX modulators | |||
| KBR7943 | NCX | A selective NCX blocker; also inhibits Ito, IK1, INa, and ICa,L. | Under investigation [150]. |
| SEA-0400 | NCX | A selective NCX blocker; additionally blocks ICa,L | Under investigation [151]. |
| Mitochondria-targeted antioxidants | |||
| MitoQ | Mitochondria | Mitochondria-targeted antioxidant | Decreases aortic stiffness and improves endothelial function; mitigates atrial remodeling by neutralizing ROS [160]. |
| Trimetazidine | Mitochondria | Metabolic modulator; activates mitochondrial complex I, promotes biogenesis, and reduces oxidative stress. | Prevents atrial structural remodeling [161]. |
| KL1333 | Mitochondria | NAD+ modulator, enhances the activity of AMPK/SIRT1/PGC-1a axis | Under investigation [162]. |
| Glucose-lowering drugs | |||
| SGLT2 inhibitors | SGLT2 protein | Exert antiarrhythmic effects via anti-inflammatory, antifibrotic, antioxidant, and electrophysiological modulation; enhance mitochondrial function, Ca2+ handling, and metabolic efficiency. | Significantly reduce AF risk and embolic stroke, independent of glycemic status. Reduction in atrial tachyarrhythmias in patients with cardiac implantable electronic devices [191,192,193,194]. |
| GLP-1 RAs | Glucagon-like peptide-1 receptor | Exert antifibrotic, anti-inflammatory, antioxidant, and metabolic modulation; improve atrial electrophysiology and mitochondrial function. | Reduce major cardiovascular events and neutral AF risk overall, but may lower AF recurrence post-ablation; less effective than SGLT2is in AF prevention [195,196,210,211,212] |
| Metformin | Gluconeogenesis | Pleiotropic effects via antioxidant, anti-inflammatory, and AMPK-driven metabolic and electrophysiological modulation | Reduces AF burden [231,232,233,234]. |
| TZDs | PPAR-γ receptors | Enhances insulin sensitivity via activation of PPAR-γ | Attenuates atrial fibrosis and shortens interatrial activation time, potentially decreasing AF susceptibility [235] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigoriou, K.; Karakasis, P.; Pamporis, K.; Theofilis, P.; Patoulias, D.; Karagiannidis, E.; Fyntanidou, B.; Antoniadis, A.P.; Fragakis, N. Atrial Fibrillation in Diabetes: Pathogenesis and Targeted Rhythm Control Strategies. Curr. Issues Mol. Biol. 2025, 47, 559. https://doi.org/10.3390/cimb47070559
Grigoriou K, Karakasis P, Pamporis K, Theofilis P, Patoulias D, Karagiannidis E, Fyntanidou B, Antoniadis AP, Fragakis N. Atrial Fibrillation in Diabetes: Pathogenesis and Targeted Rhythm Control Strategies. Current Issues in Molecular Biology. 2025; 47(7):559. https://doi.org/10.3390/cimb47070559
Chicago/Turabian StyleGrigoriou, Konstantinos, Paschalis Karakasis, Konstantinos Pamporis, Panagiotis Theofilis, Dimitrios Patoulias, Efstratios Karagiannidis, Barbara Fyntanidou, Antonios P. Antoniadis, and Nikolaos Fragakis. 2025. "Atrial Fibrillation in Diabetes: Pathogenesis and Targeted Rhythm Control Strategies" Current Issues in Molecular Biology 47, no. 7: 559. https://doi.org/10.3390/cimb47070559
APA StyleGrigoriou, K., Karakasis, P., Pamporis, K., Theofilis, P., Patoulias, D., Karagiannidis, E., Fyntanidou, B., Antoniadis, A. P., & Fragakis, N. (2025). Atrial Fibrillation in Diabetes: Pathogenesis and Targeted Rhythm Control Strategies. Current Issues in Molecular Biology, 47(7), 559. https://doi.org/10.3390/cimb47070559