
Novel Thiourea and Oxime Ether Isosteviol-Based Anticoagulants: MD Simulation and ADMET Prediction

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Molecular Dynamic (MD) Simulation and Analysis
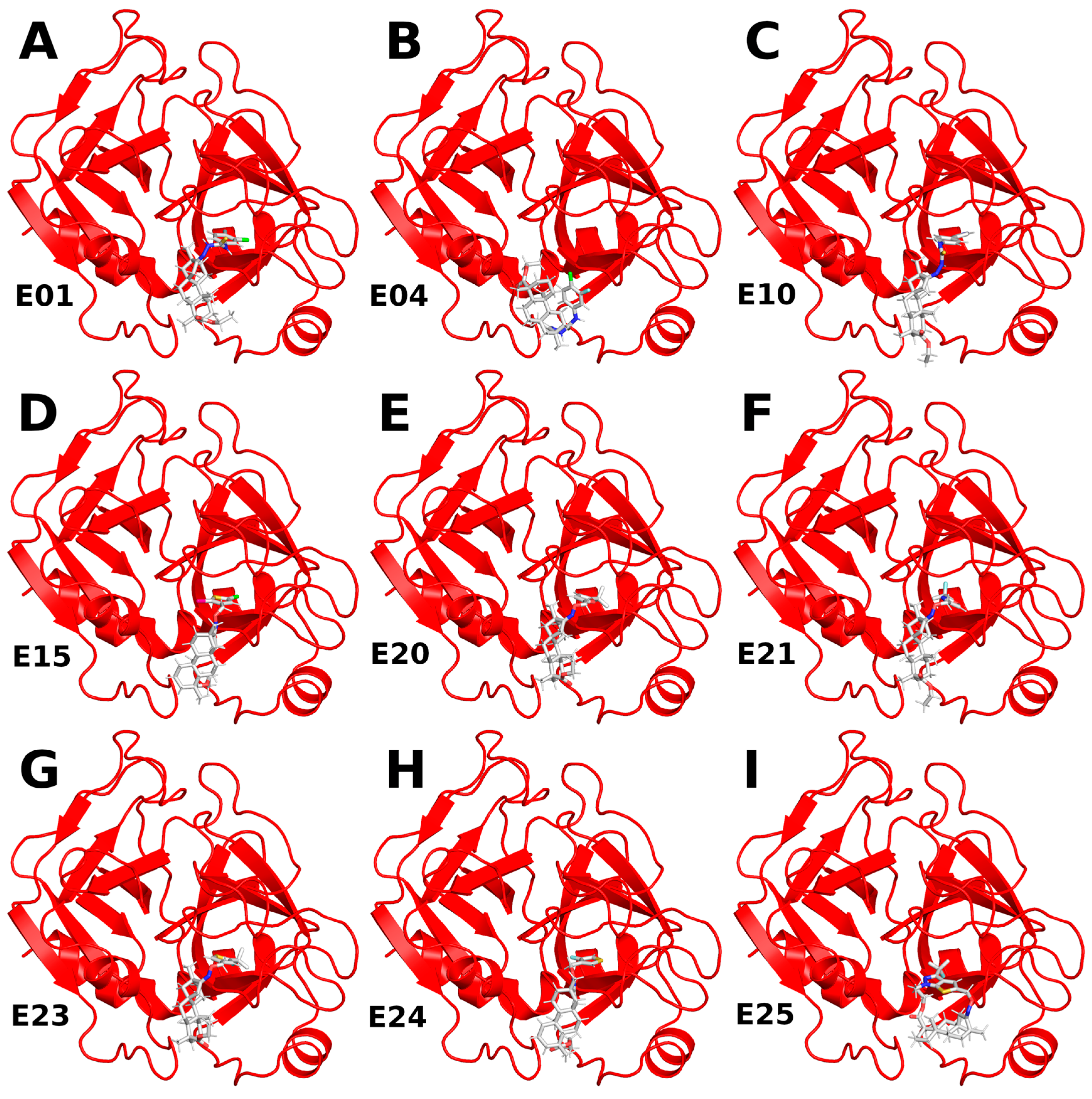
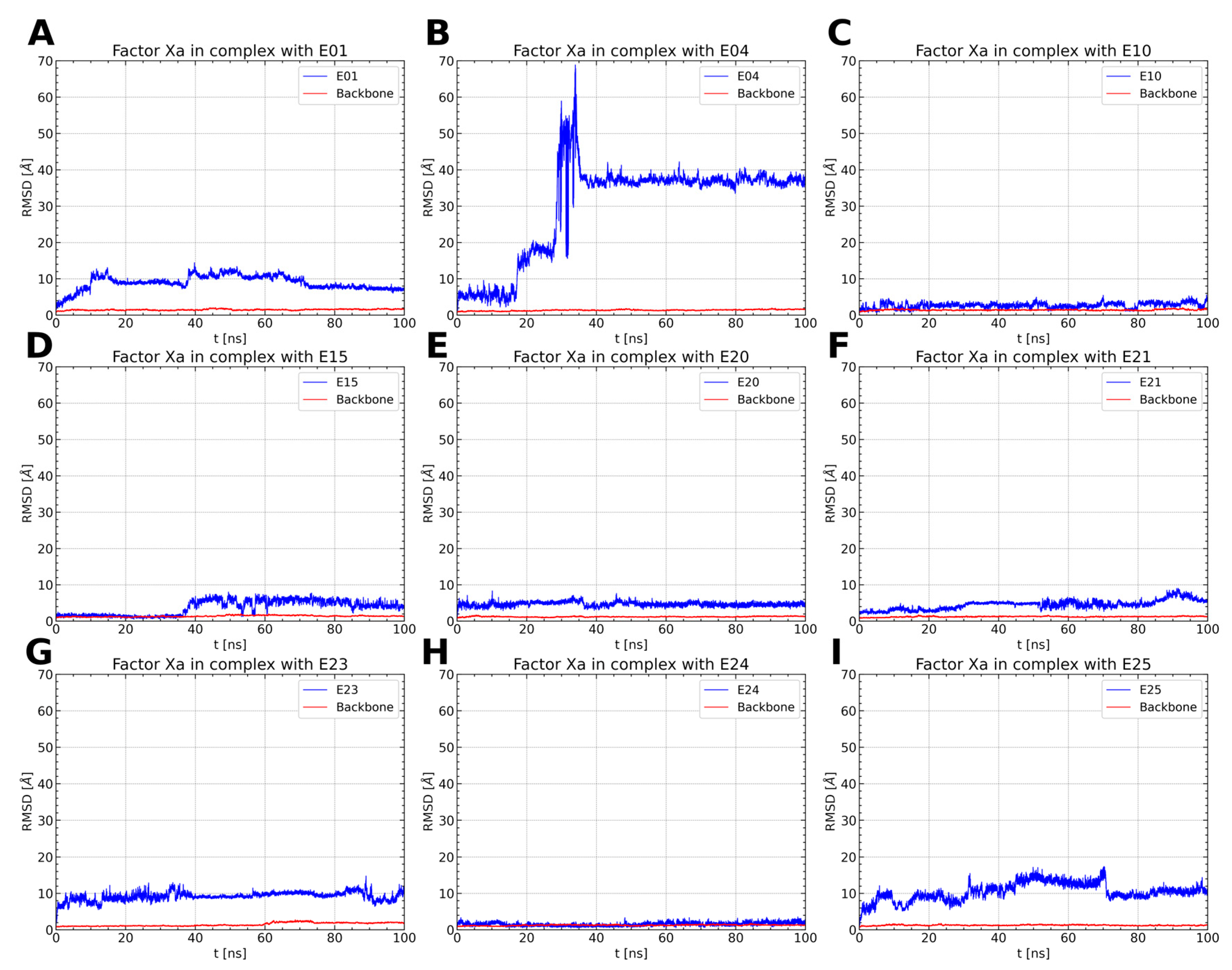
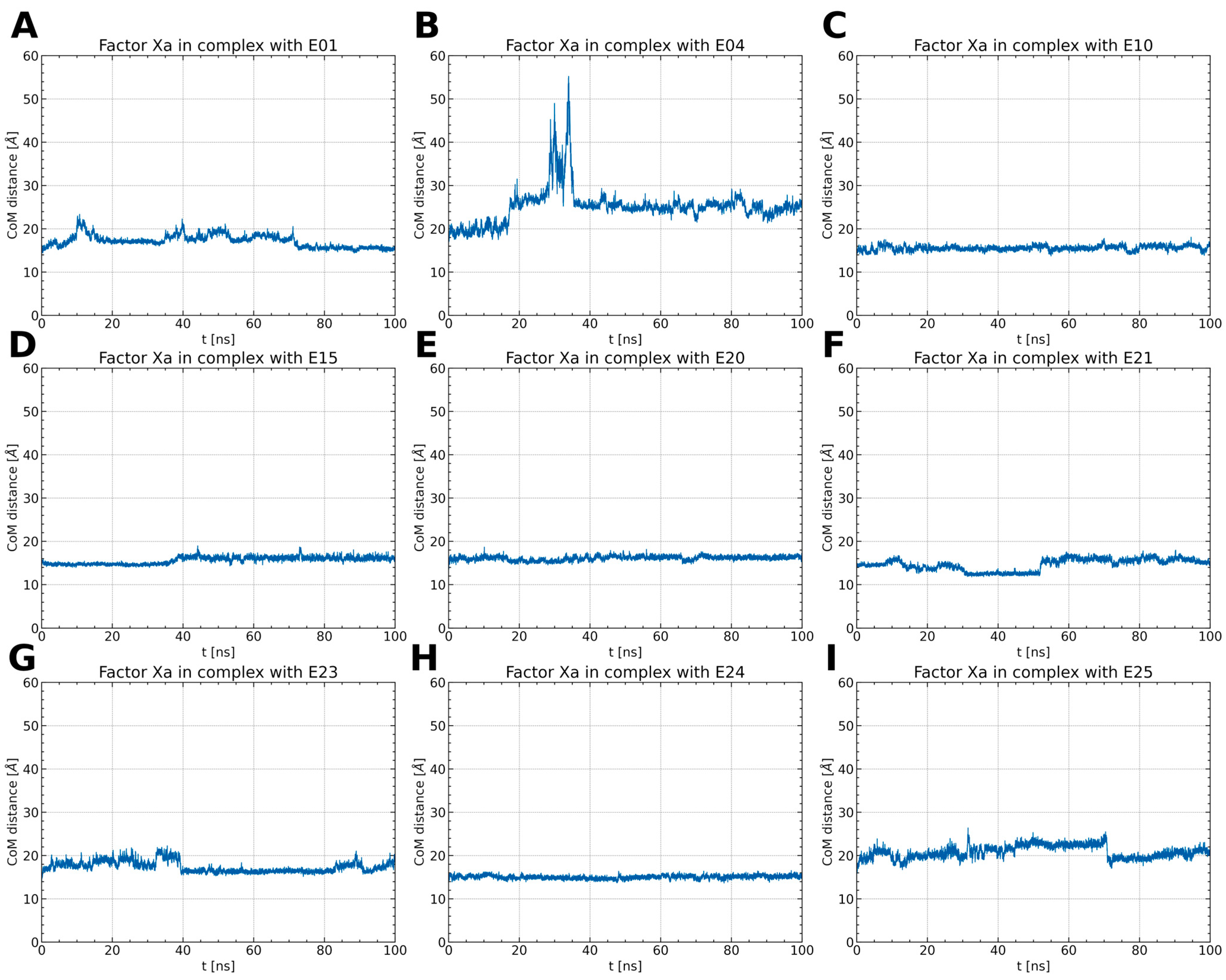
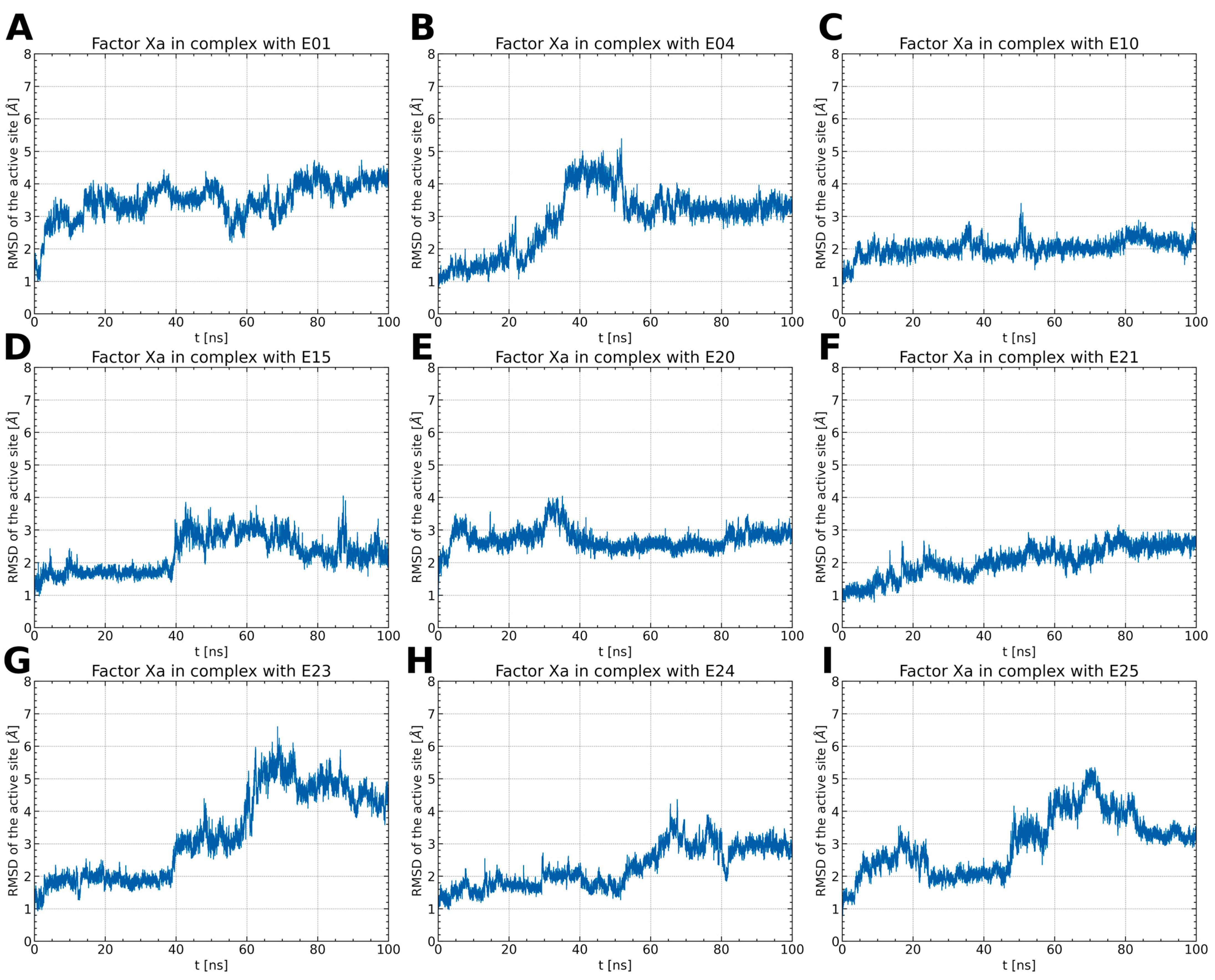
2.1.1. ISV Derivative Complexes with FXa Exhibit Variable Stability
2.1.2. ISV Derivatives Exhibit Different Conformational Dynamics
2.1.3. ISV Derivatives Exhibit a Similar Pattern of Interaction to FDA-Approved FXa Inhibitors
2.2. ADMET prediction
2.3. Limitations of the Present Study
3. Materials and Methods
3.1. MD Simulation
3.2. ADMET Properties Prediction
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mariño-Ocampo, N.; Rodríguez, D.F.; Guerra Díaz, D.; Zúñiga-Núñez, D.; Duarte, Y.; Fuentealba, D.; Zacconi, F.C. Direct Oral FXa Inhibitors Binding to Human Serum Albumin: Spectroscopic, Calorimetric, and Computational Studies. Int. J. Mol. Sci. 2023, 24, 4900. [Google Scholar] [CrossRef]
- Saviano, A.; Brigida, M.; Petruzziello, C.; Candelli, M.; Gabrielli, M.; Ojetti, V. Gastrointestinal Bleeding Due to NOACs Use: Exploring the Molecular Mechanisms. Int. J. Mol. Sci. 2022, 23, 13955. [Google Scholar] [CrossRef]
- Zheng, W.; Dai, X.; Xu, B.; Tian, W.; Shi, J. Discovery and Development of Factor Xa Inhibitors (2015–2022). Front. Pharmacol. 2023, 14, 5880. [Google Scholar] [CrossRef] [PubMed]
- Steffel, J.; Collins, R.; Antz, M.; Cornu, P.; Desteghe, L.; Haeusler, K.G.; Oldgren, J.; Reinecke, H.; Roldan-Schilling, V.; Rowell, N.; et al. 2021 European Heart Rhythm Association Practical Guide on the Use of Non-Vitamin K Antagonist Oral Anticoagulants in Patients with Atrial Fibrillation. Europace 2021, 23, 1612–1676. [Google Scholar] [CrossRef]
- Urquhart, L. Top Companies and Drugs by Sales in 2022. Nat. Rev. Drug Discov. 2023, 22, 260. [Google Scholar] [CrossRef] [PubMed]
- Schwarb, H.; Tsakiris, D.A. New Direct Oral Anticoagulants (DOAC) and Their Use Today. Dent. J. 2016, 4, 5. [Google Scholar] [CrossRef]
- Rodríguez, D.F.; Durán-Osorio, F.; Duarte, Y.; Olivares, P.; Moglie, Y.; Dua, K.; Zacconi, F.C. Green by Design: Convergent Synthesis, Computational Analyses, and Activity Evaluation of New FXa Inhibitors Bearing Peptide Triazole Linking Units. Pharmaceutics 2021, 14, 33. [Google Scholar] [CrossRef] [PubMed]
- Santana-Romo, F.; Lagos, C.F.; Duarte, Y.; Castillo, F.; Moglie, Y.; Maestro, M.A.; Charbe, N.; Zacconi, F.C. Innovative Three-Step Microwave-Promoted Synthesis of N-Propargyltetrahydroquinoline and 1,2,3-Triazole Derivatives as a Potential Factor Xa (FXA) Inhibitors: Drug Design, Synthesis, and Biological Evaluation. Molecules 2020, 25, 491. [Google Scholar] [CrossRef] [PubMed]
- Gackowski, M.; Madriwala, B.; Koba, M. In Silico Design, Docking Simulation, and ANN-QSAR Model for Predicting the Anticoagulant Activity of Thiourea Isosteviol Compounds as FXa Inhibitors. Chem. Pap. 2023, 77, 7027–7044. [Google Scholar] [CrossRef]
- Gackowski, M.; Madriwala, B.; Studzińska, R.; Koba, M. Novel Isosteviol-Based FXa Inhibitors: Molecular Modeling, In Silico Design and Docking Simulation. Molecules 2023, 28, 4977. [Google Scholar] [CrossRef]
- Becker, R.C. Factor Xa Inhibitors: Critical Considerations for Clinical Development and Testing. J. Thromb. Thrombolysis 2021, 52, 397–402. [Google Scholar] [CrossRef]
- Sakano, T.; Mahamood, M.I.; Yamashita, T.; Fujitani, H. Molecular Dynamics Analysis to Evaluate Docking Pose Prediction. Biophys. Physicobiol. 2016, 13, 181–194. [Google Scholar] [CrossRef]
- Du, Q.; Qian, Y.; Yao, X.; Xue, W. Elucidating the Tight-Binding Mechanism of Two Oral Anticoagulants to Factor Xa by Using Induced-Fit Docking and Molecular Dynamics Simulation. J. Biomol. Struct. Dyn. 2020, 38, 625–633. [Google Scholar] [CrossRef]
- Verhoef, D.; Visscher, K.M.; Vosmeer, C.R.; Cheung, K.L.; Reitsma, P.H.; Geerke, D.P.; Bos, M.H.A. Engineered Factor Xa Variants Retain Procoagulant Activity Independent of Direct Factor Xa Inhibitors. Nat. Commun. 2017, 8, 528. [Google Scholar] [CrossRef]
- Nagata, T.; Yoshino, T.; Haginoya, N.; Yoshikawa, K.; Nagamochi, M.; Kobayashi, S.; Komoriya, S.; Yokomizo, A.; Muto, R.; Yamaguchi, M.; et al. Discovery of N-[(1R,2S,5S)-2-{[(5-Chloroindol-2-Yl)Carbonyl]Amino}-5-(Dimethylcarbamoyl)Cyclohexyl]-5-Methyl-4,5,6,7-Tetrahydrothiazolo [5,4-c]Pyridine-2-Carboxamide Hydrochloride: A Novel, Potent and Orally Active Direct Inhibitor of Factor Xa. Bioorg. Med. Chem. 2009, 17, 1193–1206. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- SwissADME. Available online: http://www.swissadme.ch/index.php (accessed on 15 December 2023).
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.E.A.; Al-hussain, S.A.; Masand, V.H.; Sabnani, M.K.; Samad, A. Mechanistic and Predictive Qsar Analysis of Diverse Molecules to Capture Salient and Hidden Pharmacophores for Anti-thrombotic Activity. Int. J. Mol. Sci. 2021, 22, 8352. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Colombo, E.; Tenconi, M.; Baldessin, L.; Corsini, A. Drug-Drug Interactions of Direct Oral Anticoagulants (DOACs): From Pharmacological to Clinical Practice. Pharmaceutics 2022, 14, 1120. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Diabetes and Digestive and Kidney Diseases. LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2023. [Google Scholar]
- Gao, X. Areas of Application and Limits of Molecular Dynamics. Fluid Mech. Open Access 2021, 8, 2476. [Google Scholar]
- Durrant, J.D.; McCammon, J.A. Molecular Dynamics Simulations and Drug Discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef]
- Davis, A.M.; Riley, R.J. Predictive ADMET Studies, the Challenges and the Opportunities. Curr. Opin. Chem. Biol. 2004, 8, 378–386. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins Struct. Funct. Bioinform. 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Sousa Da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Structure | IUPAC Name | Binding Free Energy [kcal/mol] | Molar Mass [g/mol] |
|---|---|---|---|---|
| E01 |  | ethyl (4R,4aS,6aR,9S,11aR,11bS,E)-8-(2-((3-chloro-4-fluorophenyl)carbamothioyl)hydrazineylidene)-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate | −8.8 | 548.16 |
| E04 |  | ethyl (4R,4aS,6aR,9S,11aR,11bS,E)-8-(2-((4-chloro-3-fluorophenyl)carbamothioyl)hydrazineylidene)-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate | −8.5 | 548.16 |
| E10 |  | ethyl (4R,4aS,6aR,9S,11aR,11bS,E)-4,9,11b-trimethyl-8-(2-((4-(oxazol-5-yl)phenyl)carbamothioyl)hydrazineylidene)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate | −8.8 | 562.77 |
| E15 |  | ethyl (4R,4aS,6aR,9S,11aR,11bS,E)-8-(((2,5-dichlorothiophen-3-yl)methoxy)imino)-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate | −8.3 | 526.56 |
| E20 |  | ethyl (4R,4aS,6aR,9S,11aR,11bS,E)-4,9,11b-trimethyl-8-(((5-(trifluoromethyl)furan-2-yl)methoxy)imino)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate | −8.1 | 509.60 |
| E21 |  | ethyl (4R,4aS,6aR,9S,11aR,11bS,E)-4,9,11b-trimethyl-8-(((2-(trifluoromethyl)oxazol-4-yl)methoxy)imino)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate | −8.3 | 510.59 |
| E23 | 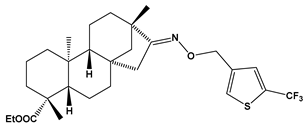 | ethyl (4R,4aS,6aR,9S,11aR,11bS,E)-4,9,11b-trimethyl-8-(((5-(trifluoromethyl)thiophen-3-yl)methoxy)imino)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate | −8.1 | 525.67 |
| E24 |  | ethyl (4R,4aS,6aR,9S,11aR,11bS,E)-8-(((4-fluorothiophen-3-yl)methoxy)imino)-4,9,11b-trimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate | −8.2 | 475.66 |
| E25 |  | ethyl (4R,4aS,6aR,9S,11aR,11bS,E)-4,9,11b-trimethyl-8-(((1-methyl-3-(trifluoromethyl)-1H-thieno[2,3-c]pyrazol-5-yl)methoxy)imino)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate | −8.2 | 579.72 |
| Docking Pose | Conformation from MD Simulation | ||
|---|---|---|---|
| First Cluster | Second Cluster | Third Cluster | |
| E10 | 1.21 Å | - | - |
| E15 | 1.63 Å | 0.44 Å | - |
| E20 | 1.06 Å | 1.84 Å | 1.67 Å |
| E24 | 0.45 Å | - | - |
| Complexes | Hydrogen Bonds | ||||
|---|---|---|---|---|---|
| Donor | Acceptor | Occupancy [%] | Distance ± SD [Å] | Angle ± SD [°] | |
| E10 | E10 (N3) | G216 (O) | 22.42 | 3.00 ± 0.32 | 28.04 ± 11.27 |
| E15 | G216 (N) | E15 (N) | 28.63 | 4.22 ± 0.85 | 20.37 ± 11.18 |
| Y99 (N) | E15 (O1) | 26.23 | 9.60 ± 5.23 | 40.26 ± 23.62 | |
| E20 | - | - | - | - | - |
| E24 | G216 (N) | E24 (N) | 40.10 | 3.56 ± 0.32 | 16.1 ± 7.93 |
| Ligand | Hydrophobic Interactions | ||
|---|---|---|---|
| Y99 | F174 | W215 | |
| E10 | 96.94% | 88.55% | 99.89% |
| E15 | 99.56% | 57.40% | 99.74% |
| E20 | 99.96% | 26.10% | 99.98% |
| E24 | 99.01% | 81.44% | 99.98% |
| Compound | Molecular Weight | Num. Rotatable Bonds | Num. H-Bond Acceptors | Num. H-Bond Donors | TPSA [Ų] 1 | Consensus Log Po/w | Lipinski | Bioavailability Score | Water Solubility [log mol/L] | Synthetic Accessibility | Intestinal Absorption [% Absorbed] | P-Glycoprotein Substrate | VDss 2 [log L/kg] | BBB Permeability 3 [log BB] | CYP2D6 Substrate | CYP2D6 Inhibitor | CYP3A4 Substrate | CYP3A4 Inhibitor | CYP1A2 Inhibitor | CYP2C19 Inhibitor | CYP2C9 Inhibitor | Total Clearance [log ml/min/kg] | AMES 4 Toxicity | Max. Tolerated Dose [log mg/kg/day] | Oral Rat Chronic Toxicity [0.644] | Hepatotoxicity |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E01 | 548.16 | 7 | 4 | 2 | 94.81 | 6.69 | No | 0.17 | −5.198 | 6.63 | 92.59 | Yes | −0.211 | −0.233 | No | No | Yes | Yes | No | No | No | −0.711 | No | −0.264 | 0.644 | No |
| E04 | 548.16 | 7 | 4 | 2 | 94.81 | 6.70 | No | 0.17 | −5.322 | 6.67 | 92.56 | Yes | −0.202 | −0.233 | No | No | Yes | Yes | No | No | No | −0.712 | No | −0.243 | 0.627 | No |
| E10 | 562.77 | 8 | 5 | 2 | 120.84 | 6.01 | No | 0.17 | −5.463 | 6.96 | 94.29 | Yes | 0.377 | −0.086 | No | No | Yes | No | No | No | No | −0.682 | No | −0.02 | 0.356 | No |
| E15 | 526.56 | 6 | 4 | 0 | 76.13 | 7.38 | No | 0.17 | −5.792 | 6.78 | 94.58 | Yes | −0.081 | −0.13 | No | No | Yes | No | No | No | No | −0.247 | No | 0.746 | 0.481 | No |
| E20 | 509.60 | 7 | 8 | 0 | 61.03 | 6.52 | No | 0.17 | −5.917 | 6.90 | 96.36 | Yes | 0.035 | −0.195 | No | No | Yes | No | No | No | No | −0.107 | No | 0.257 | 0.711 | No |
| E21 | 510.59 | 7 | 9 | 0 | 73.92 | 5.96 | No | 0.17 | −5.623 | 6.69 | 96.81 | Yes | −0.19 | −0.949 | No | No | Yes | Yes | No | No | No | −0.093 | No | 0.129 | 0.806 | No |
| E23 | 525.67 | 7 | 7 | 0 | 76.13 | 7.24 | No | 0.17 | −5.673 | 6.91 | 94.42 | Yes | −0.177 | −0.12 | No | No | Yes | No | No | No | No | −0.363 | No | 0.523 | 0.588 | No |
| E24 | 475.66 | 6 | 5 | 0 | 76.13 | 6.39 | Yes | 0.55 | −5.248 | 6.71 | 96.60 | No | −0.232 | −0.196 | No | No | Yes | No | No | No | No | −0.249 | No | 0.643 | 0.972 | No |
| E25 | 579.72 | 7 | 8 | 0 | 93.95 | 7.11 | No | 0.17 | −5.509 | 7.14 | 94.72 | Yes | −0.185 | −0.281 | No | No | Yes | Yes | No | No | No | −0.331 | No | 0.379 | 0.54 | No |
| R 5 | 435.88 | 6 | 5 | 1 | 116.42 | 2.29 | Yes | 0.55 | −4.382 | 3.63 | 92.80 | Yes | −0.687 | −1.022 | No | No | Yes | Yes | No | Yes | Yes | 0.296 | Yes | −0.232 | 1.125 | Yes |
| A 6 | 459.50 | 5 | 5 | 1 | 110.76 | 2.30 | Yes | 0.55 | −4.181 | 3.48 | 88.96 | Yes | −0.14 | −0.985 | No | No | Yes | Yes | No | Yes | Yes | 0.247 | No | −0.119 | 1.276 | Yes |
| E 7 | 548.06 | 10 | 7 | 3 | 164.87 | 1.35 | No | 0.17 | −3.377 | 5.04 | 72.09 | Yes | −0.243 | −1.082 | No | No | No | Yes | No | No | No | 0.474 | No | 0.109 | 2.471 | Yes |
| B 8 | 451.91 | 9 | 5 | 3 | 107.41 | 3.22 | Yes | 0.55 | −4.313 | 3.05 | 76.59 | Yes | −0.08 | −1.276 | No | No | Yes | Yes | No | Yes | Yes | 0.257 | No | 0.685 | 1.181 | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gackowski, M.; Jędrzejewski, M.; Medicharla, S.S.; Kondabala, R.; Madriwala, B.; Mądra-Gackowska, K.; Studzińska, R. Novel Thiourea and Oxime Ether Isosteviol-Based Anticoagulants: MD Simulation and ADMET Prediction. Pharmaceuticals 2024, 17, 163. https://doi.org/10.3390/ph17020163
Gackowski M, Jędrzejewski M, Medicharla SS, Kondabala R, Madriwala B, Mądra-Gackowska K, Studzińska R. Novel Thiourea and Oxime Ether Isosteviol-Based Anticoagulants: MD Simulation and ADMET Prediction. Pharmaceuticals. 2024; 17(2):163. https://doi.org/10.3390/ph17020163
Chicago/Turabian StyleGackowski, Marcin, Mateusz Jędrzejewski, Sri Satya Medicharla, Rajesh Kondabala, Burhanuddin Madriwala, Katarzyna Mądra-Gackowska, and Renata Studzińska. 2024. "Novel Thiourea and Oxime Ether Isosteviol-Based Anticoagulants: MD Simulation and ADMET Prediction" Pharmaceuticals 17, no. 2: 163. https://doi.org/10.3390/ph17020163
APA StyleGackowski, M., Jędrzejewski, M., Medicharla, S. S., Kondabala, R., Madriwala, B., Mądra-Gackowska, K., & Studzińska, R. (2024). Novel Thiourea and Oxime Ether Isosteviol-Based Anticoagulants: MD Simulation and ADMET Prediction. Pharmaceuticals, 17(2), 163. https://doi.org/10.3390/ph17020163