
Leveraging Hot Spots of TEAD–Coregulator Interactions in the Design of Direct Small Molecule Protein-Protein Interaction Disruptors Targeting Hippo Pathway Signaling



Abstract
1. Introduction
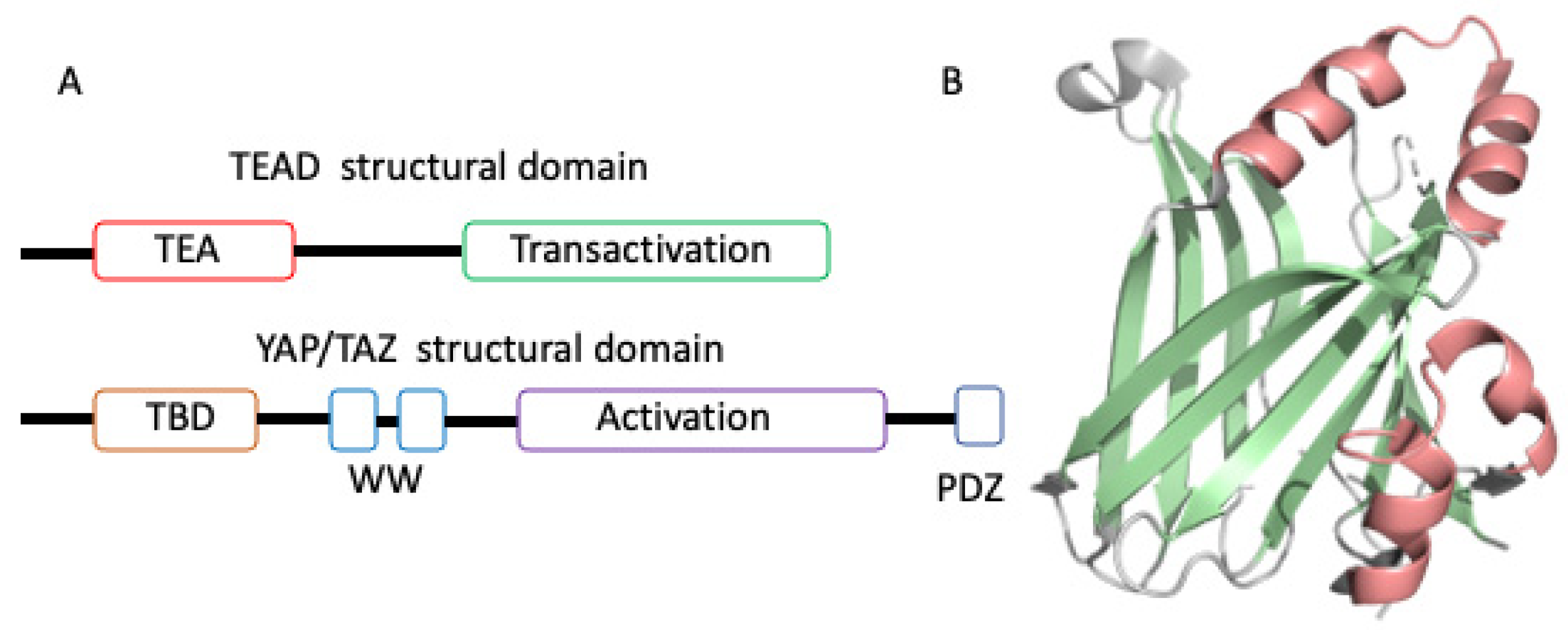
2. Overview of TEAD Transactivation Domain Structures
3. Strategies Employed in Targeting TEAD
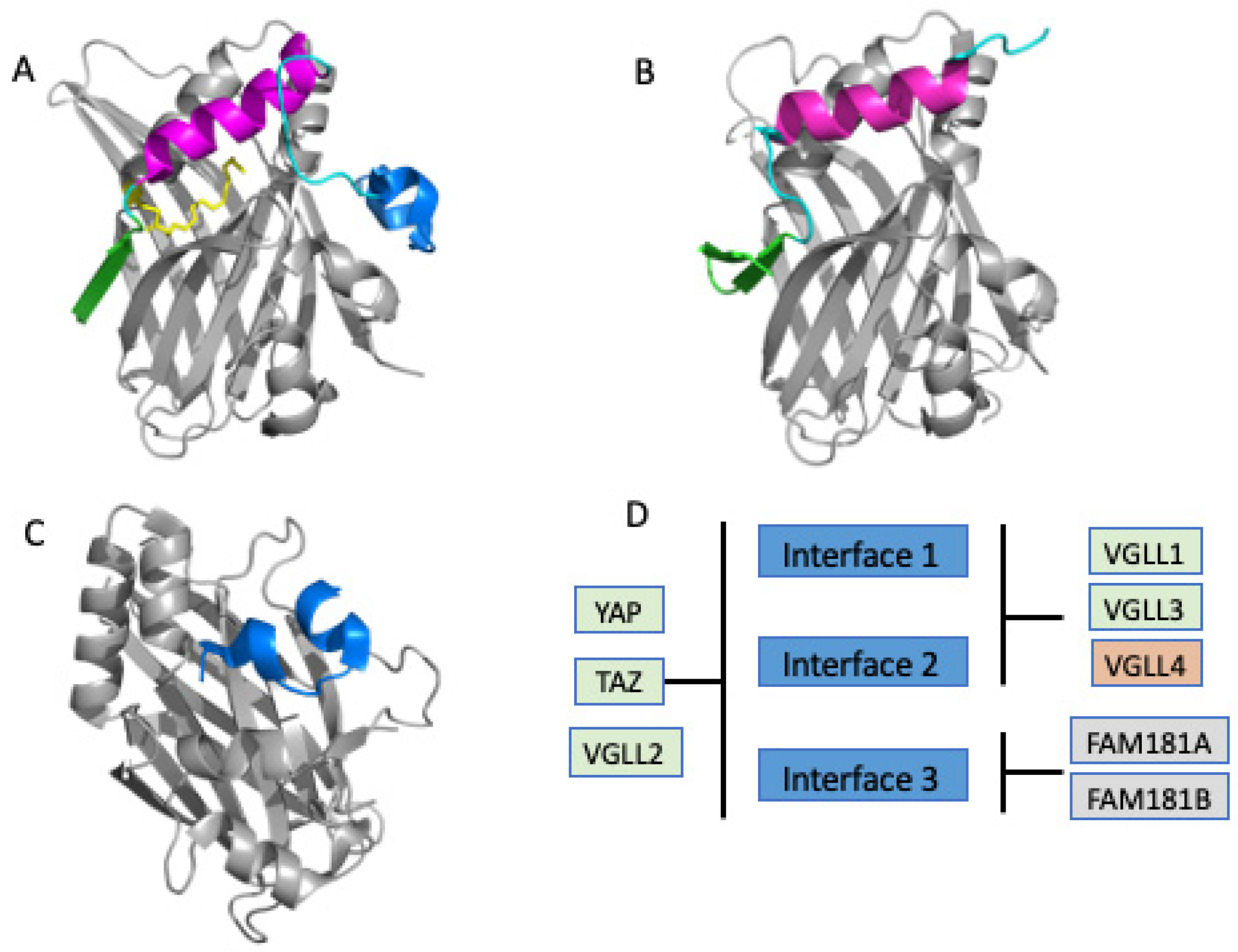
3.1. Direct PPIDs Need to Target either Interface 2 or Interface 3 but Not Both
3.2. Peptide-Based Small Molecule Inhibitors
3.3. Direct Non-Peptide Small Molecule Inhibitors Targeting Interface 2 or 3
4. Interface Hot-Spots That Can Be Tapped for Drug Discovery

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Structure | Screening Method | Validation Method | Binding | Reference |
|---|---|---|---|---|---|
| Fragment 1 |  | Thermal shift assay | Crystal structure | >300 μM; ITCInterface 2 | [72] |
| BY03 |  | Molecular docking | 9.4 μM; SPR Interface 2 | [68] | |
| Fragment compound 1 |  | Molecular docking | Crystal structure | 180 μM; NMR Interface 2 and 3 | [62] |
| Trisubstituted pyrazoles |  | Pharmacophore hopping | Crystal structure | Interface 2 | [67] |
| CPD 3.1 |  | Molecular docking | 12 μM, ITC Interface 3 | [69] | |
| Novartis Compound 4 |  | Molecular docking | Crystal structure | 0.15 μM, TR-FRET Interface 3 | [62] |
| Inventiva Compound 4 |  | Fragment screening and HTS | Interface 3 | [66] |
| Protein | Mutation | Location | Effect | Method | Phenotype | Ref. | |
|---|---|---|---|---|---|---|---|
| hYAP | Leu65Ala | Interface 2 | P1 | Destabilizes YAP-TEAD complex (∆∆G 2.24 kcal/mol) | Surface Plasmon Resonance | [70] | |
| hYAP | Leu68Ala | Interface 2 | P2 | Destabilizes YAP-TEAD complex (∆∆G 1.92 kcal/mol) | Surface Plasmon Resonance | [70] | |
| hYAP | Phe69Ala | Interface 2 | P3 | Destabilizes YAP-TEAD complex (∆∆G 3.48 kcal/mol) | Surface Plasmon Resonance | [70] | |
| mVgll1 | Met40Val | Interface 2 | other | TEAD4 interaction was reduced by 9-fold | TR-FRET | [47] | |
| mVgll1 | His68Leu | Interface 2 | P2 | Dramatic reduction in its ability to interact with TEAD | TR-FRET | [52] | |
| mVgll1 | Ala72Val | Interface 2 | P4 | TEAD interaction was reduced by 16-fold | TR-FRET | [52] | |
| mVgll1 | Arg71Ala | Interface 2 | P2 | Dramatic reduction in its ability to interact with TEAD | TR-FRET | [52] | |
| mVgll4 | Leu242A | Interface 2 | P4 | Dramatic reduction in its ability to interact with TEAD | Surface Plasmon Resonance | [65] | |
| hTEAD4 | Phe337Ala | Interface 2 | P2 | >90% reduction in the YAP-TEAD interaction; YAP interaction was reduced by 11-fold | Co-IP; Surface Plasmon Resonance | Decreased number of YAP/TEAD-driven soft agar colonies | [42,70] |
| hTEAD4 | Ser336Ala | Interface 2 | P2 | Vgll1 interaction was reduced by more than 20-fold | TR-FRET | [47] | |
| hTEAD4 | Lys376Ala | Interface 2 | P3 | YAP interaction was reduced by 11-fold | Surface Plasmon Resonance | [70] | |
| hTEAD4 | Leu380Ala | Interface 2 | P3, P4 | YAP interaction was reduced by 9-fold | Surface Plasmon Resonance | [70] | |
| hTEAD4 | Val389Ala | Interface 2 | P2, P3 | YAP interaction was reduced by 17-fold | Surface Plasmon Resonance | [70] | |
| hTEAD2 | Glu267Arg | Interface 3 | >90% reduction in the YAP-TEAD interaction | GST-pull down | [40] | ||
| hTEAD4 | Glu263Ala | Interface 3 | Destabilizes YAP-TEAD complex (∆∆G 1.19 kcal/mol) | Surface Plasmon Resonance | [73] | ||
| hTEAD2 | Ile274Ala | Interface 3 | >90% reduction in the YAP-TEAD interaction | GST-pull down | [40] | ||
| hTEAD1 hTEAD2 hTEAD4 | Tyr421His Tyr442His Tyr429His | Destabilizes YAP/TAZ-TEAD complex (∆∆G > 2.9 kcal/mol) | Surface Plasmon Resonance | [74] | |||
| mTEAD1 | Tyr410His | Interface 3 | >90% reduction in the YAP-TEAD interaction | GST-pull down | Loss of transcriptional activity | [75] | |
| hTEAD4 | Tyr429Ala | Co-IP | Decreased number of YAP/TEAD-driven soft agar colonies | [42] | |||
| hTEAD2 | Lys277Glu | Interface 3 | >90% reduction in the YAP-TEAD interaction | GST-pull down | [40] | ||
| hTEAD2 | Trp303Ala | Interface 3 | >90% reduction in the YAP-TEAD interaction | GST-pull down | Decreased number of YAP/TEAD-driven soft agar colonies | [42] | |
| hTEAD4 | Trp299Ala | Co-IP | |||||
| hTEAD2 | Leu444Ala | Interface 3 | >90% reduction in the YAP-TEAD interaction | GST-pull down | [40] | ||
| hTEAD4 | Lys297Ala | Interface 3 | >90% reduction in the YAP-TEAD interaction | Co-IP | Decreased number of YAP/TEAD-driven soft agar colonies | [42] | |
| hYAP | Met86Ala | Interface 3 | TEAD4 interaction was reduced by 87-fold | TR-FRET | [70] | ||
| hYAP | Arg89Ala | Interface 3 | Destabilizes YAP-TEAD complex (∆∆G 4.34 kcal/mol) | Surface Plasmon Resonance | [70] | ||
| hYAP | Phe95Ala | Interface 3 | Destabilizes YAP-TEAD complex (∆∆G 4.31 kcal/mol) | Surface Plasmon Resonance | [70] | ||
| hYAP | Leu91Ala | Interface 3 | Destabilizes YAP-TEAD complex (∆∆G 4.4 kcal/mol) | Surface Plasmon Resonance | [70] | ||
| hTAZ | Trp43Ala | Interface 3 | TEAD4 interaction was reduced by 457-fold | TR-FRET | [53] | ||
| hTAZ | Lys46Ala | Interface 3 | TEAD4 interaction was reduced by 29-fold | TR-FRET | [53] |
5. Conclusions and Future Considerations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ma, S.; Meng, Z.; Chen, R.; Guan, K.-L. The Hippo Pathway: Biology and Pathophysiology. Annu. Rev. Biochem. 2019, 88, 577–604. [Google Scholar] [CrossRef] [PubMed]
- Piccolo, S.; Dupont, S.; Cordenonsi, M. The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 2014, 94, 1287–1312. [Google Scholar] [CrossRef]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef]
- Hilman, D.; Gat, U. The evolutionary history of YAP and the hippo/YAP pathway. Mol. Biol. Evol. 2011, 28, 2403–2417. [Google Scholar] [CrossRef]
- Hao, Y.; Chun, A.; Cheung, K.; Rashidi, B.; Yang, X. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J. Biol. Chem. 2008, 283, 5496–5509. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.; Guan, K.-L. The YAP and TAZ transcription co-activators: Key downstream effectors of the mammalian Hippo pathway. Semin. Cell Dev. Biol. 2012, 23, 785–793. [Google Scholar] [CrossRef]
- Liu, A.M.; Wong, K.-F.; Jiang, X.; Qiao, Y.; Luk, J.M. Regulators of mammalian Hippo pathway in cancer. Biochim. Biophys. Acta Rev. Cancer 2012, 1826, 357–364. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Guan, K.-L. Mechanisms of Hippo pathway regulation. Genes Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Lei, Q.; Guan, K.-L. The Hippo-YAP pathway in organ size control and tumorigenesis: An updated version. Genes Dev. 2010, 24, 862–874. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Xu, Q.; Wang, S.; Zhang, W.; Liu, M.; Liang, S.; Zhu, H.; Xu, N. Nuclear accumulation of Yes-Associated Protein (YAP) maintains the survival of doxorubicin-induced senescent cells by promoting survivin expression. Cancer Lett. 2016, 375, 84–91. [Google Scholar] [CrossRef]
- Zhao, B.; Ye, X.; Yu, J.; Li, L.; Li, W.; Li, S.; Yu, J.; Lin, J.D.; Wang, C.-Y.; Chinnaiyan, A.M.; et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008, 22, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Liu, Y.; Gao, H.; Meng, F.; Yang, S.; Lou, G. Clinical significance of yes-associated protein overexpression in cervical carcinoma: The differential effects based on histotypes. Int. J. Gynecol. Cancer 2013, 23, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Tanas, M.R.; Ma, S.; Jadaan, F.O.; Ng, C.K.Y.; Weigelt, B.; Reis-Filho, J.S.; Rubin, B.P. Mechanism of action of a WWTR1(TAZ)-CAMTA1 fusion oncoprotein. Oncogene 2015, 35, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Striedinger, K.; VandenBerg, S.R.; Baia, G.S.; McDermott, M.W.; Gutmann, D.H.; Lal, A. The Neurofibromatosis 2 Tumor Suppressor Gene Product, Merlin, Regulates Human Meningioma Cell Growth by Signaling through YAP. Neoplasia 2008, 10, 1204–1212. [Google Scholar] [CrossRef] [PubMed]
- Kurppa, K.J.; Liu, Y.; To, C.; Zhang, T.; Fan, M.; Vajdi, A.; Knelson, E.H.; Xie, Y.; Lim, K.; Cejas, P.; et al. Treatment-Induced Tumor Dormancy through YAP-Mediated Transcriptional Reprogramming of the Apoptotic Pathway. Cancer Cell 2020, 37, 104–122.e12. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Chang, M.T.; Vissers, J.H.; Dey, A.; Harvey, K.F. The Hippo Pathway as a Driver of Select Human Cancers. Trends Cancer 2020, 6, 781–796. [Google Scholar] [CrossRef]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e10. [Google Scholar] [CrossRef]
- Drexler, R.; Fahy, R.; Küchler, M.; Wagner, K.C.; Reese, T.; Ehmke, M.; Feyerabend, B.; Kleine, M.; Oldhafer, K.J. Association of subcellular localization of TEAD transcription factors with outcome and progression in pancreatic ductal adenocarcinoma. Pancreatology 2021, 21, 170–179. [Google Scholar] [CrossRef]
- Tang, J.-Y.; Yu, C.-Y.; Bao, Y.-J.; Chen, L.; Chen, J.; Yang, S.-L.; Chen, H.-Y.; Hong, J.; Fang, J.-Y. TEAD4 promotes colorectal tumorigenesis via transcriptionally targeting YAP1. Cell Cycle 2018, 17, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, C.D.; Yi, C. YAP/TAZ Signaling and Resistance to Cancer Therapy. Trends Cancer 2019, 5, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-H.; Mullett, S.J.; Stewart, A.F.R. Vgl-4, a novel member of the vestigial-like family of transcription cofactors, regulates alpha1-adrenergic activation of gene expression in cardiac myocytes. J. Biol. Chem. 2004, 279, 30800–30806. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Tajonar, A.; Maehr, R.; Hu, G.; Sneddon, J.B.; Rivera-Feliciano, J.; Cohen, D.E.; Elledge, S.J.; Melton, D.A. Brief Report: VGLL4 Is a Novel Regulator of Survival in Human Embryonic Stem Cells. Stem Cells 2013, 31, 2833–2841. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, H.; Withers, H.G.; Yang, N.; Denson, K.E.; Mussell, A.L.; Truskinovsky, A.; Fan, Q.; Gelman, I.H.; Frangou, C.; et al. VGLL4 Selectively Represses YAP-Dependent Gene Induction and Tumorigenic Phenotypes in Breast Cancer. Sci. Rep. 2017, 7, 6190. [Google Scholar] [CrossRef]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef]
- Holden, J.; Cunningham, C. Targeting the Hippo Pathway and Cancer through the TEAD Family of Transcription Factors. Cancers 2018, 10, 81. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Rubin, B.P. Protein-Protein Interaction Disruptors of the YAP/TAZ-TEAD Transcriptional Complex. Molecules 2020, 25, 6001. [Google Scholar] [CrossRef]
- Barry, E.R.; Simov, V.; Valtingojer, I.; Venier, O. Recent Therapeutic Approaches to Modulate the Hippo Pathway in Oncology and Regenerative Medicine. Cells 2021, 10, 2715. [Google Scholar] [CrossRef]
- Liberelle, M.; Toulotte, F.; Renault, N.; Gelin, M.; Allemand, F.; Melnyk, P.; Guichou, J.-F.; Cotelle, P. Toward the Design of Ligands Selective for the C-Terminal Domain of TEADs. J. Med. Chem. 2022, 65, 5926–5940. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Kumar, R.; Rubin, B.P.; Hong, W. Therapeutic targeting of TEAD transcription factors in cancer. Trends Biochem. Sci. 2023. [Google Scholar] [CrossRef] [PubMed]
- Pobbati, A.V.; Hong, W. A combat with the YAP/TAZ-TEAD oncoproteins for cancer therapy. Theranostics 2020, 10, 3622–3635. [Google Scholar] [CrossRef]
- Lou, J.; Lu, Y.; Cheng, J.; Zhou, F.; Yan, Z.; Zhang, D.; Meng, X.; Zhao, Y. A chemical perspective on the modulation of TEAD transcriptional activities: Recent progress, challenges, and opportunities. Eur. J. Med. Chem. 2022, 243, 114684. [Google Scholar] [CrossRef]
- Xie, J.; Wang, Y.; Ai, D.; Yao, L.; Jiang, H. The role of the Hippo pathway in heart disease. FEBS J. 2021, 289, 5819–5833. [Google Scholar] [CrossRef] [PubMed]
- Dey, A.; Varelas, X.; Guan, K.-L. Targeting the Hippo pathway in cancer, fibrosis, wound healing and regenerative medicine. Nat. Rev. Drug Discov. 2020, 19, 480–494. [Google Scholar] [CrossRef]
- Sahu, M.R.; Mondal, A.C. The emerging role of Hippo signaling in neurodegeneration. J. Neurosci. Res. 2020, 98, 796–814. [Google Scholar] [CrossRef]
- Jin, J.; Zhao, X.; Fu, H.; Gao, Y. The Effects of YAP and Its Related Mechanisms in Central Nervous System Diseases. Front. Neurosci. 2020, 14, 595. [Google Scholar] [CrossRef]
- Moya, I.M.; Halder, G. Hippo–YAP/TAZ signalling in organ regeneration and regenerative medicine. Nat. Rev. Mol. Cell Biol. 2019, 20, 211–226. [Google Scholar] [CrossRef]
- Tian, W.; Yu, J.; Tomchick, D.R.; Pan, D.; Luo, X. Structural and functional analysis of the YAP-binding domain of human TEAD2. Proc. Natl. Acad. Sci. USA 2010, 107, 7293–7298. [Google Scholar] [CrossRef]
- Feichtinger, M.; Sara, T.; Platzer, G.; Mateos, B.; Bokhovchuk, F.; Chene, P.; Konrat, R. (1)H, (13)C, (15)N resonance assignment of human YAP 50-171 fragment. Biomol. NMR Assign. 2018, 12, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chan, S.W.; Zhang, X.; Walsh, M.; Lim, C.J.; Hong, W.; Song, H. Structural basis of YAP recognition by TEAD4 in the Hippo pathway. Genes Dev. 2010, 24, 290–300. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, B.; Wang, P.; Chen, F.; Dong, Z.; Yang, H.; Guan, K.-L.; Xu, Y. Structural insights into the YAP and TEAD complex. Genes Dev. 2010, 24, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Bokhovchuk, F.; Mesrouze, Y.; Delaunay, C.; Martin, T.; Villard, F.; Meyerhofer, M.; Fontana, P.; Zimmermann, C.; Erdmann, D.; Furet, P.; et al. Identification of FAM181A and FAM181B as new interactors with the TEAD transcription factors. Protein Sci. 2020, 29, 509–520. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Chan, S.W.; Lee, I.; Song, H.; Hong, W. Structural and Functional Similarity between the Vgll1-TEAD and the YAP-TEAD Complexes. Structure 2012, 20, 1135–1140. [Google Scholar] [CrossRef]
- Chan, P.; Han, X.; Zheng, B.; DeRan, M.; Yu, J.; Jarugumilli, G.K.; Deng, H.; Pan, D.; Luo, X.; Wu, X. Autopalmitoylation of TEAD proteins regulates transcriptional output of the Hippo pathway. Nat. Chem. Biol. 2016, 12, 282–289. [Google Scholar] [CrossRef]
- Mesrouze, Y.; Hau, J.C.; Erdmann, D.; Zimmermann, C.; Fontana, P.; Schmelzle, T.; Chène, P. The Surprising Features of the TEAD4-Vgll1 Protein-Protein Interaction. Chembiochem 2014, 15, 537–542. [Google Scholar] [CrossRef]
- Mesrouze, Y.; Aguilar, G.; Bokhovchuk, F.; Martin, T.; Delaunay, C.; Villard, F.; Meyerhofer, M.; Zimmermann, C.; Fontana, P.; Wille, R.; et al. A new perspective on the interaction between the Vg/VGLL1-3 proteins and the TEAD transcription factors. Sci. Rep. 2020, 10, 17442. [Google Scholar] [CrossRef]
- Bokhovchuk, F.; Mesrouze, Y.; Meyerhofer, M.; Fontana, P.; Zimmermann, C.; Viliard, F.; Erdmann, D.; Kallen, J.; Scheufler, C.; Velez-Vega, C.; et al. N-terminal β-strand in YAP is critical for stronger binding to scalloped relative to TEAD transcription factor. Protein Sci. 2023, 32, 1. [Google Scholar] [CrossRef]
- Simon, E.; Faucheux, C.; Zider, A.; Thézé, N.; Thiébaud, P. From vestigial to vestigial-like: The Drosophila gene that has taken wing. Dev. Genes. Evol. 2016, 226, 297–315. [Google Scholar] [CrossRef]
- Pobbati, A.V.; Han, X.; Hung, A.W.; Weiguang, S.; Huda, N.; Chen, G.-Y.; Kang, C.; Chia, C.S.B.; Luo, X.; Hong, W.; et al. Targeting the Central Pocket in Human Transcription Factor TEAD as a Potential Cancer Therapeutic Strategy. Structure 2015, 23, 2076–2086. [Google Scholar] [CrossRef] [PubMed]
- Mesrouze, Y.; Erdmann, D.; Fontana, P.; Meyerhofer, M.; Zimmermann, C.; Schmelzle, T.; Chène, P. Different Recognition of TEAD Transcription Factor by the Conserved B-strand:loop:a-helix Motif of the TEAD Binding Site of YAP and VGLL1. Chemistryselect 2016, 1, 2993–2997. [Google Scholar] [CrossRef]
- Hau, J.C.; Erdmann, D.; Mesrouze, Y.; Furet, P.; Fontana, P.; Zimmermann, C.; Schmelzle, T.; Hofmann, F.; Chene, P. The TEAD4-YAP/TAZ protein-protein interaction: Expected similarities and unexpected differences. Chembiochem 2013, 14, 1218–1225. [Google Scholar] [CrossRef]
- Noland, C.L.; Gierke, S.; Schnier, P.D.; Murray, J.; Sandoval, W.N.; Sagolla, M.; Dey, A.; Hannoush, R.N.; Fairbrother, W.J.; Cunningham, C.N. Palmitoylation of TEAD Transcription Factors Is Required for Their Stability and Function in Hippo Pathway Signaling. Structure 2016, 24, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Holden, J.K.; Crawford, J.J.; Noland, C.L.; Schmidt, S.; Zbieg, J.R.; Lacap, J.A.; Zang, R.; Miller, G.M.; Zhang, Y.; Beroza, P.; et al. Small Molecule Dysregulation of TEAD Lipidation Induces a Dominant-Negative Inhibition of Hippo Pathway Signaling. Cell Rep. 2020, 31, 107809. [Google Scholar] [CrossRef]
- Lu, W.; Wang, J.; Li, Y.; Tao, H.; Xiong, H.; Lian, F.; Gao, J.; Ma, H.; Lu, T.; Zhang, D.; et al. Discovery and biological evaluation of vinylsulfonamide derivatives as highly potent, covalent TEAD autopalmitoylation inhibitors. Eur. J. Med. Chem. 2019, 184, 111767. [Google Scholar] [CrossRef]
- Gibault, F.; Sturbaut, M.; Coevoet, M.; Pugniere, M.; Burtscher, A.; Allemand, F.; Melnyk, P.; Hong, W.; Rubin, B.P.; Pobbati, A.V.; et al. Design, Synthesis and Evaluation of a Series of 1,5-Diaryl-1,2,3-triazole-4-carbohydrazones as Inhibitors of the YAP-TAZ/TEAD Complex. ChemMedChem 2021, 16, 2823–2844. [Google Scholar] [CrossRef]
- Feichtinger, M.; Beier, A.; Migotti, M.; Schmid, M.; Bokhovchuk, F.; Chène, P.; Konrat, R. Long-range structural preformation in yes-associated protein precedes encounter complex formation with TEAD. iScience 2022, 25, 104099. [Google Scholar] [CrossRef]
- Olejniczak, E.T.; Hajduk, P.J.; Marcotte, P.A.; Nettesheim, D.G.; Meadows, R.P.; Edalji, R.; Holzman, T.F.; Fesik, S.W. Stromelysin Inhibitors Designed from Weakly Bound Fragments: Effects of Linking and Cooperativity. J. Am. Chem. Soc. 1997, 119, 5828–5832. [Google Scholar] [CrossRef]
- Scott, D.E.; Coyne, A.G.; Hudson, S.A.; Abell, C. Fragment-Based Approaches in Drug Discovery and Chemical Biology. Biochemistry 2012, 51, 4990–5003. [Google Scholar] [CrossRef]
- Zhang, Z.; Lin, Z.; Zhou, Z.; Shen, H.C.; Yan, S.F.; Mayweg, A.V.; Xu, Z.; Qin, N.; Wong, J.C.; Zhang, Z.; et al. Structure-Based Design and Synthesis of Potent Cyclic Peptides Inhibiting the YAP-TEAD Protein-Protein Interaction. ACS Med. Chem. Lett. 2014, 5, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Bordas, V.; Le Douget, M.; Salem, B.; Mesrouze, Y.; Imbach-Weese, P.; Sellner, H.; Voegtle, M.; Soldermann, N.; Chapeau, E.; et al. The First Class of Small Molecules Potently Disrupting the YAP-TEAD Interaction by Direct Competition. ChemMedChem 2022, 17, e202200303. [Google Scholar] [CrossRef]
- Crook, Z.R.; Sevilla, G.P.; Della Friend, D.; Brusniak, M.-Y.; Bandaranayake, A.D.; Clarke, M.; Gewe, M.; Mhyre, A.J.; Baker, D.; Strong, R.K.; et al. Mammalian display screening of diverse cystine-dense peptides for difficult to drug targets. Nat. Commun. 2017, 8, 2244. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Salem, B.; Mesrouze, Y.; Schmelzle, T.; Lewis, I.; Kallen, J.; Chène, P. Structure-based design of potent linear peptide inhibitors of the YAP-TEAD protein-protein interaction derived from the YAP omega-loop sequence. Bioorganic Med. Chem. Lett. 2019, 29, 2316–2319. [Google Scholar] [CrossRef]
- Adihou, H.; Gopalakrishnan, R.; Förster, T.; Guéret, S.M.; Gasper, R.; Geschwindner, S.; García, C.C.; Karatas, H.; Pobbati, A.V.; Vazquez-Chantada, M.; et al. A protein tertiary structure mimetic modulator of the Hippo signalling pathway. Nat. Commun. 2020, 11, 5425. [Google Scholar] [CrossRef]
- Crawford, J.J.; Bronner, S.M.; Zbieg, J.R. Hippo pathway inhibition by blocking the YAP/TAZ–TEAD interface: A patent review. Expert Opin. Ther. Patents 2018, 28, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Sturbaut, M.; Bailly, F.; Coevoet, M.; Sileo, P.; Pugniere, M.; Liberelle, M.; Magnez, R.; Thuru, X.; Chartier-Harlin, M.-C.; Melnyk, P.; et al. Discovery of a cryptic site at the interface 2 of TEAD—Towards a new family of YAP/TAZ-TEAD inhibitors. Eur. J. Med. Chem. 2021, 226, 113835. [Google Scholar] [CrossRef]
- Kim, J.; Lim, H.; Moon, S.; Cho, S.Y.; Kim, M.; Park, J.H.; Park, H.W.; No, K.T. Hot Spot Analysis of YAP-TEAD Protein-Protein Interaction Using the Fragment Molecular Orbital Method and Its Application for Inhibitor Discovery. Cancers 2021, 13, 4246. [Google Scholar] [CrossRef]
- Smith, S.A.; Sessions, R.B.; Shoemark, D.K.; Williams, C.; Ebrahimighaei, R.; McNeill, M.C.; Crump, M.P.; McKay, T.R.; Harris, G.; Newby, A.C.; et al. Antiproliferative and Antimigratory Effects of a Novel YAP–TEAD Interaction Inhibitor Identified Using in Silico Molecular Docking. J. Med. Chem. 2019, 62, 1291–1305. [Google Scholar] [CrossRef]
- Mesrouze, Y.; Bokhovchuk, F.; Meyerhofer, M.; Fontana, P.; Zimmermann, C.; Martin, T.; Delaunay, C.; Erdmann, D.; Schmelzle, T.; Chene, P. Dissection of the interaction between the intrinsically disordered YAP protein and the transcription factor TEAD. eLife 2017, 6, e25068. [Google Scholar] [CrossRef]
- Kaan, H.Y.K.; Chan, S.W.; Tan, S.K.J.; Guo, F.; Lim, C.J.; Hong, W.; Song, H. Crystal structure of TAZ-TEAD complex reveals a distinct interaction mode from that of YAP-TEAD complex. Sci. Rep. 2017, 7, 2035. [Google Scholar] [CrossRef]
- Kaan, H.Y.K.; Sim, A.Y.L.; Tan, S.K.J.; Verma, C.; Song, H. Targeting YAP/TAZ-TEAD protein-protein interactions using fragment-based and computational modeling approaches. PLoS ONE 2017, 12, e0178381. [Google Scholar] [CrossRef] [PubMed]
- Mesrouze, Y.; Bokhovchuk, F.; Izaac, A.; Meyerhofer, M.; Zimmermann, C.; Fontana, P.; Schmelzle, T.; Erdmann, D.; Furet, P.; Kallen, J.; et al. Adaptation of the bound intrinsically disordered protein YAP to mutations at the YAP:TEAD interface. Protein Sci. 2018, 27, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Bokhovchuk, F.; Mesrouze, Y.; Izaac, A.; Meyerhofer, M.; Zimmermann, C.; Fontana, P.; Schmelzle, T.; Erdmann, D.; Furet, P.; Kallen, J.; et al. Molecular and structural characterization of a TEAD mutation at the origin of Sveinsson’s chorioretinal atrophy. FASEB J. 2020, 34, 2381–2398. [Google Scholar] [CrossRef]
- Kitagawa, M. A Sveinsson’s chorioretinal atrophy-associated missense mutation in mouse Tead1 affects its interaction with the co-factors YAP and TAZ. Biochem. Biophys. Res. Commun. 2007, 361, 1022–1026. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, B.; Pobbati, A.V.; Rubin, B.P.; Stauffer, S. Leveraging Hot Spots of TEAD–Coregulator Interactions in the Design of Direct Small Molecule Protein-Protein Interaction Disruptors Targeting Hippo Pathway Signaling. Pharmaceuticals 2023, 16, 583. https://doi.org/10.3390/ph16040583
Zhao B, Pobbati AV, Rubin BP, Stauffer S. Leveraging Hot Spots of TEAD–Coregulator Interactions in the Design of Direct Small Molecule Protein-Protein Interaction Disruptors Targeting Hippo Pathway Signaling. Pharmaceuticals. 2023; 16(4):583. https://doi.org/10.3390/ph16040583
Chicago/Turabian StyleZhao, Bin, Ajaybabu V. Pobbati, Brian P. Rubin, and Shaun Stauffer. 2023. "Leveraging Hot Spots of TEAD–Coregulator Interactions in the Design of Direct Small Molecule Protein-Protein Interaction Disruptors Targeting Hippo Pathway Signaling" Pharmaceuticals 16, no. 4: 583. https://doi.org/10.3390/ph16040583
APA StyleZhao, B., Pobbati, A. V., Rubin, B. P., & Stauffer, S. (2023). Leveraging Hot Spots of TEAD–Coregulator Interactions in the Design of Direct Small Molecule Protein-Protein Interaction Disruptors Targeting Hippo Pathway Signaling. Pharmaceuticals, 16(4), 583. https://doi.org/10.3390/ph16040583