
Design, Synthesis, In Vitro Anticancer Evaluation and Molecular Modelling Studies of 3,4,5-Trimethoxyphenyl-Based Derivatives as Dual EGFR/HDAC Hybrid Inhibitors


Abstract
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. In Vitro Anticancer Activity
Cell Viability Assay
Antiproliferative Activity
2.2.2. In Vitro Enzymatic Inhibitory Activity Assay
Epidermal Growth Factor Receptor Activity (EGFR-TK) Inhibition
In Vitro HDAC Inhibition Assay
2.2.3. Western Blot Assay
2.2.4. Apoptotic Markers Activation Assay
Caspase-3, Caspase-8, Bax and Bcl-2 Levels Assay
2.2.5. Flow Cytometric Cell Cycle Analysis
2.3. Docking Study
2.3.1. EGFR Docking Study
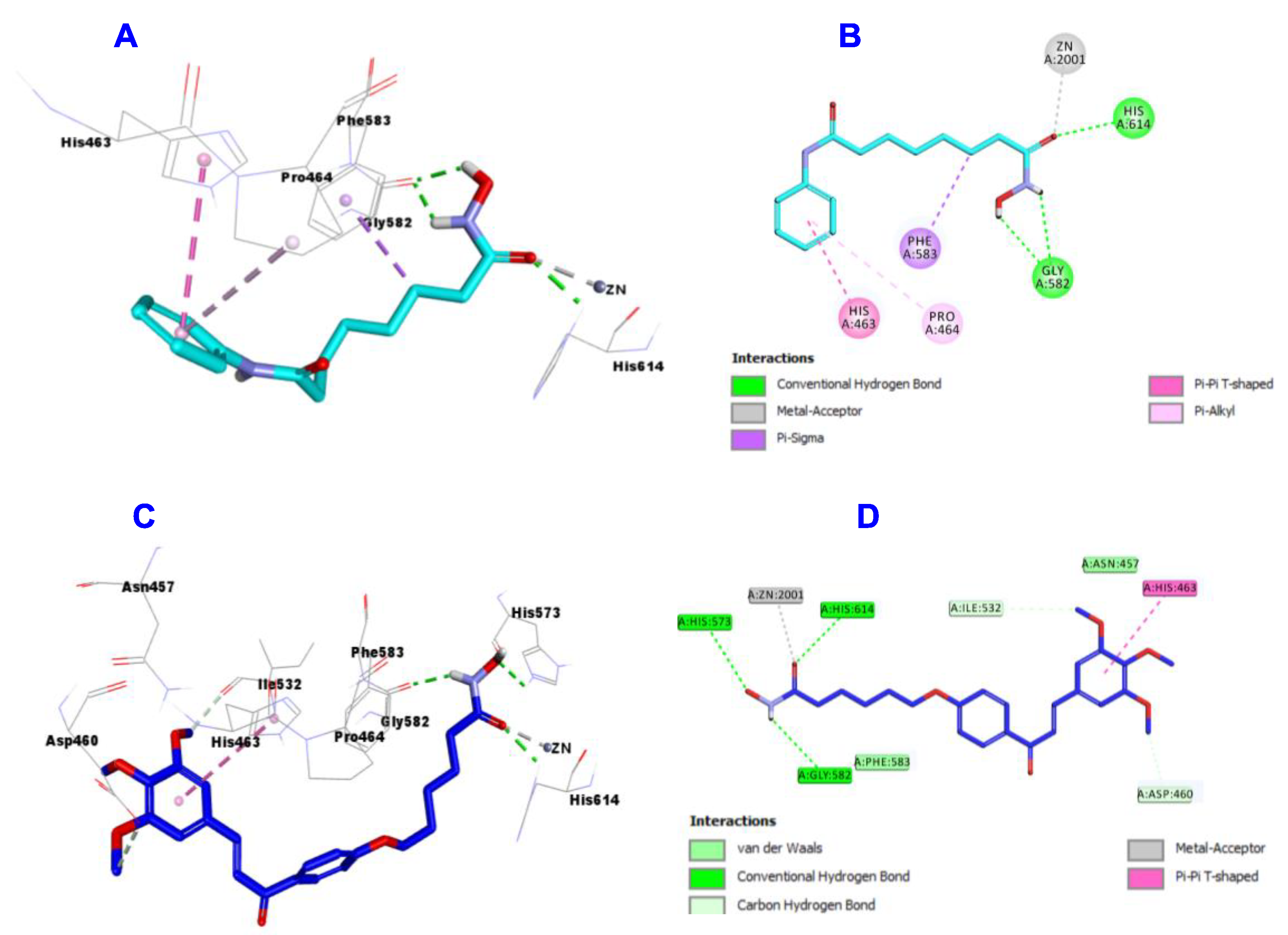
2.3.2. HDAC1 Docking Study
2.3.3. HDAC2 Docking Study
2.3.4. HDAC4 Docking Study
2.3.5. HDAC6 Docking Study
2.3.6. HDAC8 Docking Study
3. Experimental
3.1. Chemistry
3.1.1. General Procedure for Synthesis of Hybrids (2a–c)
(E)-5-(4-(3-(3,4,5-Trimethoxyphenyl)Acryloyl)Phenoxy)Pentanoic Acid (2a)
(E)-6-(4-(3-(3,4,5-Trimethoxyphenyl)Acryloyl)Phenoxy)Hexanoic Acid (2b)
(E)-7-(4-(3-(3,4,5-Trimethoxyphenyl)Acryloyl)Phenoxy)Heptanoic Acid (2c)
3.1.2. General Procedure for Synthesis of Hybrids (3a–c)
5-(4-(5-Cyano-6-Oxo-4-(3,4,5-Trimethoxyphenyl)-1,6-Dihydropyridin-2-yl)Phenoxy)Pentanoic Acid (3a)
6-(4-(5-Cyano-6-Oxo-4-(3,4,5-Trimethoxyphenyl)-1,6-Dihydropyridin-2-yl)Phenoxy) Hexanoic Acid (3b)
7-(4-(5-Cyano-6-Oxo-4-(3,4,5-Trimethoxyphenyl)-1,6-Dihydropyridin-2-yl)Phenoxy) Heptanoic Acid (3c)
3.1.3. General Procedure for Synthesis of Hybrids (4a–c)
(E)-N-Hydroxy-5-(4-(3-(3,4,5-Trimethoxyphenyl)Acryloyl)Phenoxy) Pentanamide (4a)
(E)-N-Hydroxy-6-(4-(3-(3,4,5-Trimethoxyphenyl)Acryloyl)Phenoxy) Hexanamide (4b)
(E)-N-Hydroxy-7-(4-(3-(3,4,5-Trimethoxyphenyl)Acryloyl)Phenoxy)Heptanamide (4c)
3.1.4. General Procedure for Synthesis of Hybrids (5a–c)
5-(4-(5-Cyano-6-Oxo-4-(3,4,5-Trimethoxyphenyl)-1,6-Dihydropyridin-2-yl)Phenoxy)-N-Hydroxypentanamide (5a)
6-(4-(5-Cyano-6-Oxo-4-(3,4,5-Trimethoxyphenyl)-1,6-Dihydropyridin-2-yl)Phenoxy)-N-Hydroxyhexanamide (5b)
7-(4-(5-Cyano-6-Oxo-4-(3,4,5-Trimethoxyphenyl)-1,6-dihydropyridin-2-yl)phenoxy)-N-Hydroxyheptanamide (5c)
3.2. Biological Evaluation
3.2.1. Cytotoxic Activity Using MTT Assay and Evaluation of IC50.
MTT Assay
Assay for Antiproliferative Effect
3.2.2. EGFR Inhibitory Assay
3.2.3. In Vitro HDAC Isoforms Inhibitory Activity
3.2.4. Western Blot Assay
3.2.5. Caspase-3 and 8 Activation Assay
3.2.6. Evaluation of Bax and Bcl-2 Expressions
3.2.7. Cell Apoptosis Assay
3.3. Docking Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mohamed, M.F.; Abuo-Rahma, G.E.-D.A. Molecular targets and anticancer activity of quinoline–chalcone hybrids: Literature review. RSC Adv. 2020, 10, 31139–31155. [Google Scholar] [CrossRef]
- Neganova, M.E.; Klochkov, S.G.; Aleksandrova, Y.R.; Aliev, G. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. Semin. Cancer Biol. 2020, 16, 1044–1057. [Google Scholar] [CrossRef]
- Stazi, G.; Fioravanti, R.; Mai, A.; Mattevi, A.; Valente, S. Histone deacetylases as an epigenetic pillar for the development of hybrid inhibitors in cancer. Curr. Opin. Chem. Biol. 2019, 50, 89–100. [Google Scholar] [CrossRef]
- Bass, A.K.A.; El-Zoghbi, M.S.; Nageeb, E.-S.M.; Mohamed, M.F.A.; Badr, M.; Abuo-Rahma, G.E.-D.A. Comprehensive review for anticancer hybridized multitargeting HDAC inhibitors. Eur. J. Med. Chem. 2021, 209, 112904. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Villanueva, J.; Matadamas-Martínez, F.; Yépez-Mulia, L.; Pérez-Koldenkova, V.; Leyte-Lugo, M.; Rodríguez-Villar, K.; Cortés-Benítez, F.; Macías-Jiménez, A.P.; González-Sánchez, I.; Romero-Velásquez, A.J.P. Synthesis and Cytotoxic Activity of Combretastatin A-4 and 2, 3-Diphenyl-2H-indazole Hybrids. Pharmaceuticals 2021, 14, 815. [Google Scholar] [CrossRef]
- Dobbelstein, M.; Moll, U. Targeting tumour-supportive cellular machineries in anticancer drug development. Nat. Rev. Drug Discov. 2014, 13, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Barone, I.; Giordano, C.; Bonofiglio, D.; Andò, S.; Catalano, S. Phosphodiesterase type 5 and cancers: Progress and challenges. Oncotarget 2017, 8, 99179–99202. [Google Scholar] [CrossRef]
- Werner, R.J.; Kelly, A.D.; Issa, J.J. Epigenetics and Precision Oncology. Cancer J. 2017, 23, 262–269. [Google Scholar] [CrossRef]
- Qin, J.; Wen, B.; Liang, Y.; Yu, W.; Li, H. Histone Modifications and their Role in Colorectal Cancer (Review). Pathol. Oncol. Res. 2020, 26, 2023–2033. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Han, Y.; Jiang, Q.; Wang, C.; Chen, X.; Li, X.; Xu, F.; Jiang, Y.; Wang, Q.; Xu, W. Trend of histone deacetylase inhibitors in cancer therapy: Isoform selectivity or multitargeted strategy. Med. Res. Rev. 2015, 35, 63–84. [Google Scholar] [CrossRef]
- Kelly, W.K.; O’Connor, O.A.; Krug, L.M.; Chiao, J.H.; Heaney, M.; Curley, T.; MacGregore-Cortelli, B.; Tong, W.; Secrist, J.P.; Schwartz, L.; et al. Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 3923–3931. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H.; Nakajima, H.; Hori, Y.; Fujita, T.; Nishimura, M.; Goto, T.; Okuhara, M. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968. I. Taxonomy, fermentation, isolation, physico-chemical and biological properties, and antitumor activity. J. Antibiot. 1994, 47, 301–310. [Google Scholar] [CrossRef]
- Coiffier, B.; Pro, B.; Prince, H.M.; Foss, F.; Sokol, L.; Greenwood, M.; Caballero, D.; Borchmann, P.; Morschhauser, F.; Wilhelm, M.; et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Novotny-Diermayr, V.; Hart, S.; Goh, K.C.; Cheong, A.; Ong, L.C.; Hentze, H.; Pasha, M.K.; Jayaraman, R.; Ethirajulu, K.; Wood, J.M. The oral HDAC inhibitor pracinostat (SB939) is efficacious and synergistic with the JAK2 inhibitor pacritinib (SB1518) in preclinical models of AML. Blood Cancer J. 2012, 2, e69. [Google Scholar] [CrossRef]
- Mottamal, M.; Zheng, S.; Huang, T.L.; Wang, G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules 2015, 20, 3898–3941. [Google Scholar] [CrossRef]
- Qiao, Z.; Ren, S.; Li, W.; Wang, X.; He, M.; Guo, Y.; Sun, L.; He, Y.; Ge, Y.; Yu, Q. Chidamide, a novel histone deacetylase inhibitor, synergistically enhances gemcitabine cytotoxicity in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2013, 434, 95–101. [Google Scholar] [CrossRef]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv. Cancer Res. 2018, 138, 183–211. [Google Scholar] [CrossRef]
- Abbass, S.A.; Hassan, H.A.; Mohamed, M.F.; Moustafa, G.A.; Abuo-Rahma, G.E.-D.A. Recent Prospectives of Anticancer Histone Deacetylase Inhibitors. J. Adv. Biomed. Pharm. Sci. 2019, 2, 135–151. [Google Scholar] [CrossRef]
- Guerra, F.S.; Rodrigues, D.A.; Fraga, C.A.M.; Fernandes, P.D. Novel Single Inhibitor of HDAC6/8 and Dual Inhibitor of PI3K/HDAC6 as Potential Alternative Treatments for Prostate Cancer. Pharmaceuticals 2021, 14, 387. [Google Scholar] [CrossRef] [PubMed]
- Luan, Y.; Li, J.; Bernatchez, J.A.; Li, R. Kinase and Histone Deacetylase Hybrid Inhibitors for Cancer Therapy. J. Med. Chem. 2019, 62, 3171–3183. [Google Scholar] [CrossRef]
- Fu, R.G.; Sun, Y.; Sheng, W.B.; Liao, D.F. Designing multi-targeted agents: An emerging anticancer drug discovery paradigm. Eur. J. Med. Chem. 2017, 136, 195–211. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Shibuya, M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 2013, 153, 13–19. [Google Scholar] [CrossRef]
- Teng, Y.; Lu, K.; Zhang, Q.; Zhao, L.; Huang, Y.; Ingarra, A.M.; Galons, H.; Li, T.; Cui, S.; Yu, P. Recent advances in the development of cyclin-dependent kinase 7 inhibitors. Eur. J. Med. Chem. 2019, 183, 111641. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed]
- Avizienyte, E.; Ward, R.A.; Garner, A.P. Comparison of the EGFR resistance mutation profiles generated by EGFR-targeted tyrosine kinase inhibitors and the impact of drug combinations. Biochem. J. 2008, 415, 197–206. [Google Scholar] [CrossRef]
- Zhou, N.; Xu, W.; Zhang, Y. Histone deacetylase inhibitors merged with protein tyrosine kinase inhibitors. Drug Discov. Ther. 2015, 9, 147–155. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, M.J.; Kim, D.E.; Jeong, I.G.; Choi, J.; Jang, S.; Lee, J.-H.; Ro, S.; Hwang, J.J.; Kim, C.-S. HDAC inhibitors synergize antiproliferative effect of sorafenib in renal cell carcinoma cells. Anticancer. Res. 2012, 32, 3161–3168. [Google Scholar]
- Nakagawa, T.; Takeuchi, S.; Yamada, T.; Ebi, H.; Sano, T.; Nanjo, S.; Ishikawa, D.; Sato, M.; Hasegawa, Y.; Sekido, Y. EGFR-TKI resistance due to BIM polymorphism can be circumvented in combination with HDAC inhibition. Cancer Res. 2013, 73, 2428–2434. [Google Scholar] [CrossRef]
- Chen, M.; Chen, C.; Wang, J.; Tsai, A.; Liou, J.; Pan, S.; Teng, C. The HDAC inhibitor, MPT0E028, enhances erlotinib-induced cell death in EGFR-TKI-resistant NSCLC cells. Cell Death Dis. 2013, 4, e810. [Google Scholar] [CrossRef]
- Chen, C.-H.; Chen, M.-C.; Wang, J.-C.; Tsai, A.-C.; Chen, C.-S.; Liou, J.-P.; Pan, S.-L.; Teng, C.-M. Synergistic interaction between the HDAC inhibitor, MPT0E028, and sorafenib in liver cancer cells in vitro and in vivo. Clin. Cancer Res. 2014, 20, 1274–1287. [Google Scholar] [CrossRef]
- Greve, G.; Schiffmann, I.; Pfeifer, D.; Pantic, M.; Schüler, J.; Lübbert, M. The pan-HDAC inhibitor panobinostat acts as a sensitizer for erlotinib activity in EGFR-mutated and-wildtype non-small cell lung cancer cells. BMC Cancer 2015, 15, 947. [Google Scholar] [CrossRef]
- Tanimoto, A.; Takeuchi, S.; Arai, S.; Fukuda, K.; Yamada, T.; Roca, X.; Ong, S.T.; Yano, S. Histone Deacetylase 3 Inhibition Overcomes BIM Deletion Polymorphism–Mediated Osimertinib Resistance in EGFR-Mutant Lung Cancer. Clin. Cancer Res. 2017, 23, 3139–3149. [Google Scholar] [CrossRef]
- Mahboobi, S.; Pilsl, B.; Sellmer, A. Generation and assessment of fusions between HDACi and TKi. In HDAC/HAT Function Assessment and Inhibitor Development; Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; Volume 1510, pp. 405–412. [Google Scholar] [CrossRef]
- Cai, X.; Zhai, H.X.; Wang, J.; Forrester, J.; Qu, H.; Yin, L.; Lai, C.J.; Bao, R.; Qian, C. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDc-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J. Med. Chem. 2010, 53, 2000–2009. [Google Scholar] [CrossRef]
- Press, M.F.; Lenz, H.-J. Egfr, her2 and vegf pathways. Drugs 2007, 67, 2045–2075. [Google Scholar] [CrossRef]
- Wang, J.; Pursell, N.W.; Samson, M.E.S.; Atoyan, R.; Ma, A.W.; Selmi, A.; Xu, W.; Cai, X.; Voi, M.; Savagner, P.; et al. Potential advantages of CUDC-101, a multitargeted HDAC, EGFR, and HER2 inhibitor, in treating drug resistance and preventing cancer cell migration and invasion. Mol. Cancer Ther. 2013, 12, 925–936. [Google Scholar] [CrossRef]
- Mohamed, M.F.A.; Shaykoon, M.S.A.; Abdelrahman, M.H.; Elsadek, B.E.M.; Aboraia, A.S.; Abuo-Rahma, G. Design, synthesis, docking studies and biological evaluation of novel chalcone derivatives as potential histone deacetylase inhibitors. Bioorg. Chem. 2017, 72, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, M.F.A.; Youssif, B.G.M.; Shaykoon, M.S.A.; Abdelrahman, M.H.; Elsadek, B.E.M.; Aboraia, A.S.; Abuo-Rahma, G.E.A. Utilization of tetrahydrobenzo [4,5] thieno [2,3-d] pyrimidinone as a cap moiety in design of novel histone deacetylase inhibitors. Bioorg. Chem. 2019, 91, 103127. [Google Scholar] [CrossRef]
- Al-Sanea, M.M.; Gotina, L.; Mohamed, M.F.; Parambi, D.G.T.; Gomaa, H.A.; Mathew, B.; Youssif, B.G.; Alharbi, K.S.; Elsayed, Z.M.; Abdelgawad, M.A. Design, Synthesis and Biological Evaluation of New HDAC1 and HDAC2 Inhibitors Endowed with Ligustrazine as a Novel Cap Moiety. Drug Des. Dev. Ther. 2020, 14, 497. [Google Scholar] [CrossRef]
- Ibrahim, T.S.; Sheha, T.A.; Abo-Dya, N.E.; AlAwadh, M.A.; Alhakamy, N.A.; Abdel-Samii, Z.K.; Panda, S.S.; Abuo-Rahma, G.E.-D.A.; Mohamed, M.F.A. Design, synthesis and anticancer activity of novel valproic acid conjugates with improved histone deacetylase (HDAC) inhibitory activity. Bioorg. Chem. 2020, 99, 103797. [Google Scholar] [CrossRef]
- Abou-Zied, H.A.; Youssif, B.G.M.; Mohamed, M.F.A.; Hayallah, A.M.; Abdel-Aziz, M. EGFR inhibitors and apoptotic inducers: Design, synthesis, anticancer activity and docking studies of novel xanthine derivatives carrying chalcone moiety as hybrid molecules. Bioorg. Chem. 2019, 89, 102997. [Google Scholar] [CrossRef]
- Abdelbaset, M.S.; Abdel-Aziz, M.; Ramadan, M.; Abdelrahman, M.H.; Abbas Bukhari, S.N.; Ali, T.F.S.; Abuo-Rahma, G.E.A. Discovery of novel thienoquinoline-2-carboxamide chalcone derivatives as antiproliferative EGFR tyrosine kinase inhibitors. Bioorg. Med. Chem. 2019, 27, 1076–1086. [Google Scholar] [CrossRef] [PubMed]
- Gayam, V.; Ravi, S. Cinnamoylated chloroquine analogues: A new structural class of antimalarial agents. Eur. J. Med. Chem. 2017, 135, 382–391. [Google Scholar] [CrossRef]
- Jasim, H.A.; Nahar, L.; Jasim, M.A.; Moore, S.A.; Ritchie, K.J.; Sarker, S.D. Chalcones: Synthetic Chemistry Follows Where Nature Leads. Biomolecules 2021, 11, 1203. [Google Scholar] [CrossRef]
- Sangwan, R.; Rajan, R.; Mandal, P.K. HDAC as onco target: Reviewing the synthetic approaches with SAR study of their inhibitors. Eur. J. Med. Chem. 2018, 158, 620–706. [Google Scholar] [CrossRef]
- Chen, C.; Hou, X.; Wang, G.; Pan, W.; Yang, X.; Zhang, Y.; Fang, H. Design, synthesis and biological evaluation of quinoline derivatives as HDAC class I inhibitors. Eur. J. Med. Chem. 2017, 133, 11–23. [Google Scholar] [CrossRef]
- Mahmood, T.; Yang, P.-C. Western blot: Technique, theory, and trouble shooting. N. Am. J. Med Sci. 2012, 4, 429–434. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis mediated by Fas and its related diseases. Nippon Ika Daigaku Zasshi 1997, 64, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Cory, S.; Adams, J.M. The Bcl2 family: Regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2002, 2, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Giaccia, A.J.; Kastan, M.B. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev. 1998, 12, 2973–2983. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Xu, G.; Yeung, S.-C.J. Cytochrome c Release Is Upstream to Activation of Caspase-9, Caspase-8, and Caspase-3 in the Enhanced Apoptosis of Anaplastic Thyroid Cancer Cells Induced by Manumycin and Paclitaxel. J. Clin. Endocrinol. Metab. 2001, 86, 4731–4740. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, S.N.A.; Jantan, I.; Unsal Tan, O.; Sher, M.; Naeem-Ul-Hassan, M.; Qin, H.-L. Biological activity and molecular docking studies of curcumin-related α, β-unsaturated carbonyl-based synthetic compounds as anticancer agents and mushroom tyrosinase inhibitors. J. Agric. Food Chem. 2014, 62, 5538–5547. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, R.B.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198. [Google Scholar] [CrossRef] [PubMed]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound № | Antiproliferative Activity IC50 ± SEM (μM) | |||
|---|---|---|---|---|
| MCF-7 | HepG2 | HCT116 | A549 | |
| 2a | 59.94 ± 3.23 | 88.41 ± 4.76 | 40.11 ± 2.16 | 44.37 ± 2.39 |
| 2b | 39.4 ± 2.12 | 30.35 ± 1.64 | 23.73 ± 1.28 | 33.74 ± 1.82 |
| 2c | 14.77 ± 0.8 | 17.12 ± 0.92 | 16.49 ± 0.89 | 22.67 ± 1.22 |
| 3a | 46.4 ± 2.54 | 65.82 ± 3.55 | 23.51 ± 1.27 | 31.03 ± 1.67 |
| 3b | 12.15 ± 0.65 | 16.23 ± 0.87 | 15.71 ± 0.85 | 15.29 ± 0.82 |
| 3c | 23.7 ± 1.28 | 21.09 ± 1.14 | 13.89 ± 0.75 | 21.31 ± 1.15 |
| 4a | 1.971 ± 0.11 | 3.619 ± 0.2 | 3.213 ± 0.17 | 2.067 ± 0.11 |
| 4b | 0.621 ± 0.03 | 0.536 ± 0.03 | 1.206 ± 0.07 | 0.797 ± 0.04 |
| 4c | 1.183 ± 0.06 | 2.536 ± 0.14 | 1.587 ± 0.09 | 1.934 ± 0.14 |
| 5a | 4.892 ± 0.26 | 3.456 ± 0.19 | 4.669 ± 0.25 | 2.297 ± 0.12 |
| 5b | 19.55 ± 1.05 | 28.34 ± 0.99 | 16.89 ± 3.71 | 18.78 ± 1.01 |
| 5c | 12.05 ± 0.65 | 27.64 ± 1.49 | 9.466 ± 0.51 | 8.577 ± 0.46 |
| SAHA | 2.43 ± 0.27 | 3.63 ± 0.24 | 2.53 ± 0.14 | 2.83 ± 0.13 |
| Gefitinib | 1.855 ± 0.13 | 2.848 ± 0.15 | 3.366 ± 0.18 | 1.439 ± 0.08 |
| Compd. № | EGFR | HDAC1 | HDAC2 | HDAC4 | HDAC6 | HDAC8 |
|---|---|---|---|---|---|---|
| 4a | 0.111 ± 0.002 | 0.121 | 0.119 | 6.685 | 0.086 | 6.354 |
| 4b | 0.063 ± 0.002 | 0.148 | 0.168 | 5.852 | 0.06 | 2.257 |
| 4c | 0.091 ± 0.001 | 0.07 | 0.277 | 8.716 | 0.113 | 5.015 |
| 5a | 0.214 ± 0.004 | 0.051 | 0.256 | 17.53 | 0.222 | 19.56 |
| Gefitinib | 0.044 ± 0.001 | nd | nd | nd | nd | nd |
| Staurosporine | 0.4 | nd | nd | nd | nd | nd |
| SAHA | nd | 0.037 | 0.112 | 4.062 | 0.019 | 1.133 |
| Compound № | Caspase-3 | Caspase-8 | Bax | Bcl-2 | ||||
|---|---|---|---|---|---|---|---|---|
| Conc (pg/mL) | Fold Change | Conc (ng/mL) | Fold Change | Conc (Pg/mL) | Fold Change | Conc (ng/mL) | Fold Change | |
| 4b | 483.2 ± 14.72 | 5.1 | 1.078 ± 0.046 | 3.15 | 398.9 ± 14.3 | 3.75 | 3.659 ± 0.09 | 0.42 |
| Staurosporine | 445.9 ± 15.39 | 4.71 | 1.343 ± 0.026 | 3.93 | 362.2 ± 9.61 | 3.4 | 3.146 ± 0.31 | 0.36 |
| Control | 94.61 ± 6.5 | 1 | 0.342 ± 0.038 | 1 | 106.5 ± 5.85 | 1 | 8.623 ± 0.19 | 1 |
| Compound | %G0–G1 | %S | %G2/M | %Pre-G1 | Comment |
|---|---|---|---|---|---|
| 4b/HepG2 | 53.04 | 39.11 | 7.85 | 47.21 | cell growth arrest at G1/S |
| cont. HepG2 | 42.97 | 36.58 | 20.45 | 2.16 |
| Compound | Apoptosis | Necrosis | ||
|---|---|---|---|---|
| Total | Early | Late | ||
| 4b/HepG2 | 47.21 | 9.33 | 24.67 | 13.21 |
| cont. HepG2 | 2.16 | 0.34 | 0.12 | 1.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibrahim, T.S.; Malebari, A.M.; Mohamed, M.F.A. Design, Synthesis, In Vitro Anticancer Evaluation and Molecular Modelling Studies of 3,4,5-Trimethoxyphenyl-Based Derivatives as Dual EGFR/HDAC Hybrid Inhibitors. Pharmaceuticals 2021, 14, 1177. https://doi.org/10.3390/ph14111177
Ibrahim TS, Malebari AM, Mohamed MFA. Design, Synthesis, In Vitro Anticancer Evaluation and Molecular Modelling Studies of 3,4,5-Trimethoxyphenyl-Based Derivatives as Dual EGFR/HDAC Hybrid Inhibitors. Pharmaceuticals. 2021; 14(11):1177. https://doi.org/10.3390/ph14111177
Chicago/Turabian StyleIbrahim, Tarek S., Azizah M. Malebari, and Mamdouh F. A. Mohamed. 2021. "Design, Synthesis, In Vitro Anticancer Evaluation and Molecular Modelling Studies of 3,4,5-Trimethoxyphenyl-Based Derivatives as Dual EGFR/HDAC Hybrid Inhibitors" Pharmaceuticals 14, no. 11: 1177. https://doi.org/10.3390/ph14111177
APA StyleIbrahim, T. S., Malebari, A. M., & Mohamed, M. F. A. (2021). Design, Synthesis, In Vitro Anticancer Evaluation and Molecular Modelling Studies of 3,4,5-Trimethoxyphenyl-Based Derivatives as Dual EGFR/HDAC Hybrid Inhibitors. Pharmaceuticals, 14(11), 1177. https://doi.org/10.3390/ph14111177