
RIG-I Deficiency Promotes Obesity-Induced Insulin Resistance

 , ,
, ,  ,
,  and
and
Abstract
:1. Introduction
2. Results
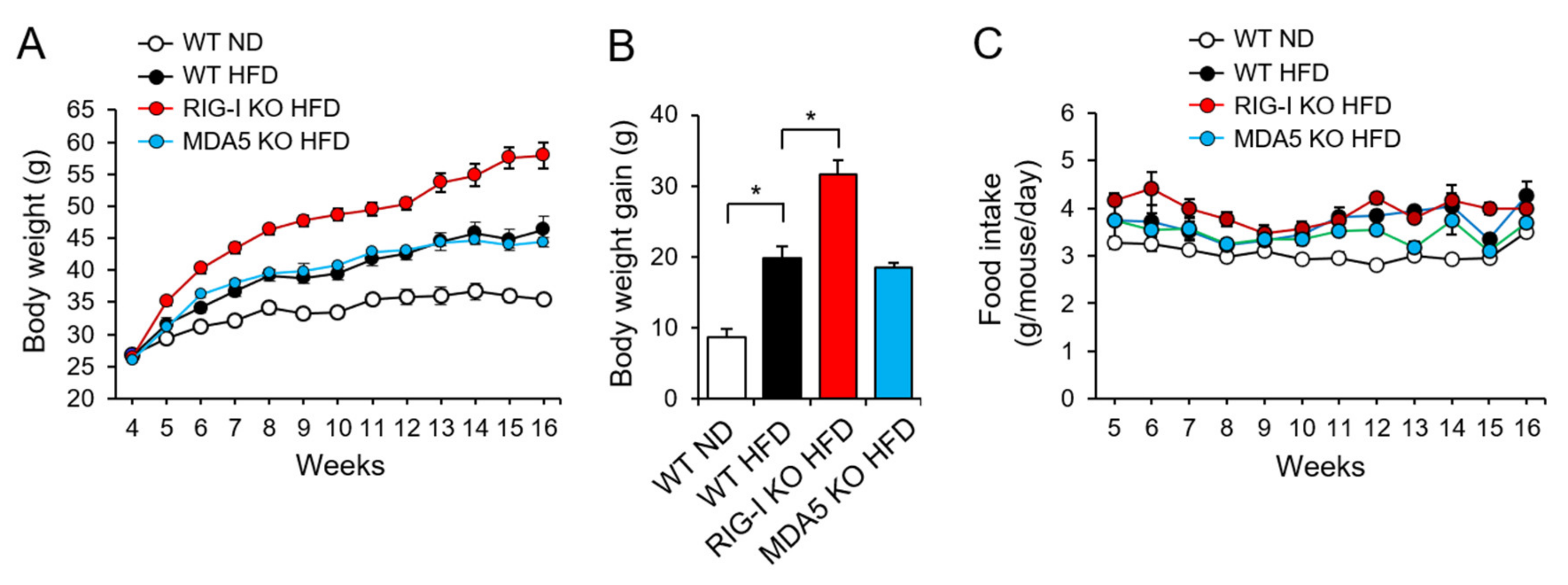
2.1. RIG-I Deficiency Enhances High-Fat Diet-Induced Obesity in Mice
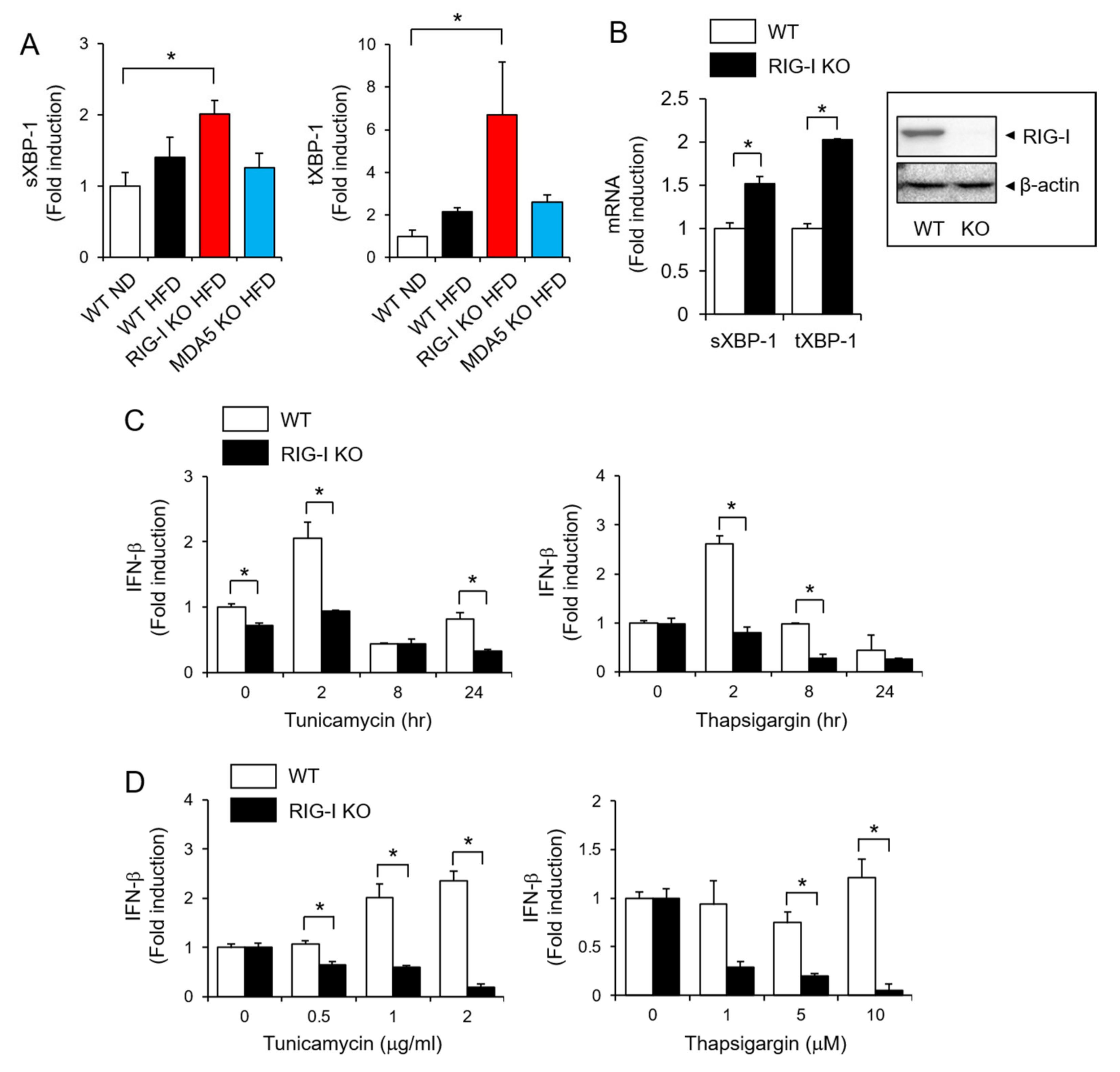
2.2. RIG-I Deficiency Aggravates High-Fat Diet-Induced Insulin Resistance
2.3. RIG-I Is Required for IFN-β Production in Response to ER Stress
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Cell Culture
4.3. Reagents
4.4. A High Fat Diet-Induced Obesity Mouse Model
4.5. Analysis of Whole-Body Composition
4.6. Histological Analysis
4.7. Intraperitoneal Glucose Tolerance Test (IPGTT)
4.8. Quantitative Real-Time Polymerase Chain Reaction (qPCR) Analysis
4.9. Immunoblotting Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gong, T.; Liu, L.; Jiang, W.; Zhou, R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 2020, 20, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Zhao, L.; Youn, H.S.; Weatherill, A.R.; Tapping, R.; Feng, L.; Lee, W.H.; Fitzgerald, K.A.; Hwang, D.H. Saturated fatty acid activates but polyunsaturated fatty acid inhibits Toll-like receptor 2 dimerized with Toll-like receptor 6 or 1. J. Biol. Chem. 2004, 279, 16971–16979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.K.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Therapeutic regulation of the NLRP3 inflammasome in chronic inflammatory diseases. Arch. Pharm. Res. 2021, 44, 16–35. [Google Scholar] [CrossRef]
- Yang, G.; Lee, H.E.; Moon, S.J.; Ko, K.M.; Koh, J.H.; Seok, J.K.; Min, J.K.; Heo, T.H.; Kang, H.C.; Cho, Y.Y.; et al. Direct Binding to NLRP3 Pyrin Domain as a Novel Strategy to Prevent NLRP3-Driven Inflammation and Gouty Arthritis. Arthritis Rheumatol. 2020, 72, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Choi, N.; Yang, G.; Jang, J.H.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Loganin Alleviates Gout Inflammation by Suppressing NLRP3 Inflammasome Activation and Mitochondrial Damage. Molecules 2021, 26, 1071. [Google Scholar] [CrossRef]
- Schlee, M. Master sensors of pathogenic RNA-RIG-I like receptors. Immunobiology 2013, 218, 1322–1335. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Saito, T.; Hirai, R.; Loo, Y.M.; Owen, D.; Johnson, C.L.; Sinha, S.C.; Akira, S.; Fujita, T.; Gale, M., Jr. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 2007, 104, 582–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Li, G.; Zhong, H.; Chen, M.; Chen, T.; Gao, L.; Wu, H.; Guo, J. RIG-I inhibits pancreatic beta cell proliferation through competitive binding of activated Src. Sci. Rep. 2016, 6, 28914. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, G.; Xu, Z.G.; Tu, H.; Hu, F.; Dai, J.; Chang, Y.; Chen, Y.; Lu, Y.; Zeng, H.; et al. Lactate Is a Natural Suppressor of RLR Signaling by Targeting MAVS. Cell 2019, 178, 176–189.e115. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, M.; Qadri, I.; Rahman, S.M.; Schroeder-Gloeckler, J.; Janssen, R.C.; Friedman, J.E. C/EBPbeta is AMP kinase sensitive and up-regulates PEPCK in response to ER stress in hepatoma cells. Mol. Cell. Endocrinol. 2011, 331, 102–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Liao, Y.; Zhou, L.; Peng, S.; Chen, H.; Yuan, Z. Amplified RLR signaling activation through an interferon-stimulated gene-endoplasmic reticulum stress-mitochondrial calcium uniporter protein loop. Sci. Rep. 2016, 6, 20158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Wieser, V.; Adolph, T.E.; Grander, C.; Grabherr, F.; Enrich, B.; Moser, P.; Moschen, A.R.; Kaser, S.; Tilg, H. Adipose type I interferon signalling protects against metabolic dysfunction. Gut 2018, 67, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Alsaggar, M.; Mills, M.; Liu, D. Interferon beta overexpression attenuates adipose tissue inflammation and high-fat diet-induced obesity and maintains glucose homeostasis. Gene Ther. 2017, 24, 60–66. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.C.; Damen, M.; Moreno-Fernandez, M.E.; Stankiewicz, T.E.; Cappelletti, M.; Alarcon, P.C.; Oates, J.R.; Doll, J.R.; Mukherjee, R.; Chen, X.; et al. Type I interferon sensing unlocks dormant adipocyte inflammatory potential. Nat. Commun. 2020, 11, 2745. [Google Scholar] [CrossRef] [PubMed]
- Ghazarian, M.; Revelo, X.S.; Nohr, M.K.; Luck, H.; Zeng, K.; Lei, H.; Tsai, S.; Schroer, S.A.; Park, Y.J.; Chng, M.H.Y.; et al. Type I Interferon Responses Drive Intrahepatic T cells to Promote Metabolic Syndrome. Sci. Immunol. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Salazar Hernandez, M.A.; Auen, T.; Mucka, P.; Lee, J.; Ozcan, U. PGC-1alpha functions as a co-suppressor of XBP1s to regulate glucose metabolism. Mol. Metab. 2018, 7, 119–131. [Google Scholar] [CrossRef]
- Zhou, Y.; Lee, J.; Reno, C.M.; Sun, C.; Park, S.W.; Chung, J.; Lee, J.; Fisher, S.J.; White, M.F.; Biddinger, S.B.; et al. Regulation of glucose homeostasis through a XBP-1-FoxO1 interaction. Nat. Med. 2011, 17, 356–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G., Jr.; Ozcan, U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef] [Green Version]
- Manuel, A.M.; Walla, M.D.; Faccenda, A.; Martin, S.L.; Tanis, R.M.; Piroli, G.G.; Adam, J.; Kantor, B.; Mutus, B.; Townsend, D.M.; et al. Succination of Protein Disulfide Isomerase Links Mitochondrial Stress and Endoplasmic Reticulum Stress in the Adipocyte During Diabetes. Antioxid. Redox Signal. 2017, 27, 1281–1296. [Google Scholar] [CrossRef] [PubMed]
- Balakumar, M.; Raji, L.; Prabhu, D.; Sathishkumar, C.; Prabu, P.; Mohan, V.; Balasubramanyam, M. High-fructose diet is as detrimental as high-fat diet in the induction of insulin resistance and diabetes mediated by hepatic/pancreatic endoplasmic reticulum (ER) stress. Mol. Cell. Biochem. 2016, 423, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, M.; Liew, C.W.; Lu, S.; Hu, J.; Martinez, R.; Hambro, B.; Kennedy, R.T.; Kulkarni, R.N. X-box binding protein 1 is essential for insulin regulation of pancreatic alpha-cell function. Diabetes 2013, 62, 2439–2449. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A.; Turner, M.J.; DeLay, M.L.; Klenk, E.I.; Sowders, D.P.; Colbert, R.A. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur. J. Immunol. 2008, 38, 1194–1203. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Liu, Y.P.; Sha, H.; Chen, H.; Qi, L.; Smith, J.A. XBP-1 couples endoplasmic reticulum stress to augmented IFN-beta induction via a cis-acting enhancer in macrophages. J. Immunol. 2010, 185, 2324–2330. [Google Scholar] [CrossRef] [Green Version]
- Van Eyck, L.; De Somer, L.; Pombal, D.; Bornschein, S.; Frans, G.; Humblet-Baron, S.; Moens, L.; de Zegher, F.; Bossuyt, X.; Wouters, C.; et al. Brief Report: IFIH1 Mutation Causes Systemic Lupus Erythematosus With Selective IgA Deficiency. Arthritis Rheumatol. 2015, 67, 1592–1597. [Google Scholar] [CrossRef] [Green Version]
- Rutsch, F.; MacDougall, M.; Lu, C.; Buers, I.; Mamaeva, O.; Nitschke, Y.; Rice, G.I.; Erlandsen, H.; Kehl, H.G.; Thiele, H.; et al. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am. J. Hum. Genet. 2015, 96, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Jang, M.A.; Kim, E.K.; Now, H.; Nguyen, N.T.; Kim, W.J.; Yoo, J.Y.; Lee, J.; Jeong, Y.M.; Kim, C.H.; Kim, O.H.; et al. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am. J. Hum. Genet. 2015, 96, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frietze, K.K.; Brown, A.M.; Das, D.; Franks, R.G.; Cunningham, J.L.; Hayward, M.; Nickels, J.T., Jr. Lipotoxicity reduces DDX58/Rig-1 expression and activity leading to impaired autophagy and cell death. Autophagy 2021, 1–19. [Google Scholar] [CrossRef]
- Charni-Natan, M.; Goldstein, I. Protocol for Primary Mouse Hepatocyte Isolation. STAR Protoc. 2020, 1, 100086. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, H.J.; Yang, J.W.; Park, J.H.; Choi, E.S.; Lim, C.S.; Lee, S.; Han, C.Y. Endoplasmic Reticulum Stress Increases DUSP5 Expression via PERK-CHOP Pathway, Leading to Hepatocyte Death. Int. J. Mol. Sci. 2019, 20, 4369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, T.L.; Chen, P.S.; Li, R.H.; Yuan, S.S.; Su, I.J.; Hung, J.H. Induction of apurinic endonuclease 1 overexpression by endoplasmic reticulum stress in hepatoma cells. Int. J. Mol. Sci. 2014, 15, 12442–12457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offor, U.; Naidu, E.C.S.; Ogedengbe, O.O.; Aniekan, P.I.; Azu, O.O. Momordica charantia mitigates hepatic injury following adjuvant treatment with antiretroviral drugs in diabetic animal models. Toxicol. Res. 2020, 36, 37–44. [Google Scholar] [CrossRef]
- Lee, J.K.; Lee, H.E.; Yang, G.; Kim, K.B.; Kwack, S.J.; Lee, J.Y. Para-phenylenediamine, an oxidative hair dye ingredient, increases thymic stromal lymphopoietin and proinflammatory cytokines causing acute dermatitis. Toxicol. Res. 2020, 36, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Seok, J.K.; Hong, E.H.; Yang, G.; Lee, H.E.; Kim, S.E.; Liu, K.H.; Kang, H.C.; Cho, Y.Y.; Lee, H.S.; Lee, J.Y. Oxidized Phospholipids in Tumor Microenvironment Stimulate Tumor Metastasis via Regulation of Autophagy. Cells 2021, 10, 558. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 4-Week | 8-Week | 12-Week | |||||||
|---|---|---|---|---|---|---|---|---|---|
| WT ND | 646.5 | ± | 42.79 | 838.3 | ± | 46.96 | 1018 | ± | 60.07 |
| WT HFD | 868.7 | ± | 54.32 * | 1227 | ± | 48.95 * | 1282 | ± | 71.26 * |
| RIG KO HFD | 856.4 | ± | 80.41 * | 1406 | ± | 86.43 *,† | 1483 | ± | 101.4 *,† |
| MDA KO HFD | 802.7 | ± | 73.32 * | 1188 | ± | 33.24 * | 1175 | ± | 59.71 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, G.; Lee, H.E.; Seok, J.K.; Kang, H.C.; Cho, Y.-Y.; Lee, H.S.; Lee, J.Y. RIG-I Deficiency Promotes Obesity-Induced Insulin Resistance. Pharmaceuticals 2021, 14, 1178. https://doi.org/10.3390/ph14111178
Yang G, Lee HE, Seok JK, Kang HC, Cho Y-Y, Lee HS, Lee JY. RIG-I Deficiency Promotes Obesity-Induced Insulin Resistance. Pharmaceuticals. 2021; 14(11):1178. https://doi.org/10.3390/ph14111178
Chicago/Turabian StyleYang, Gabsik, Hye Eun Lee, Jin Kyung Seok, Han Chang Kang, Yong-Yeon Cho, Hye Suk Lee, and Joo Young Lee. 2021. "RIG-I Deficiency Promotes Obesity-Induced Insulin Resistance" Pharmaceuticals 14, no. 11: 1178. https://doi.org/10.3390/ph14111178
APA StyleYang, G., Lee, H. E., Seok, J. K., Kang, H. C., Cho, Y.-Y., Lee, H. S., & Lee, J. Y. (2021). RIG-I Deficiency Promotes Obesity-Induced Insulin Resistance. Pharmaceuticals, 14(11), 1178. https://doi.org/10.3390/ph14111178