Abstract
Cryptic species are morphologically indistinguishable but possess genetically distinct taxa. Alternative splicing (AS) regulates physiological processes, thereby facilitating ecological adaptation and evolution. To explore the sex-specific differences in transcriptional regulation among cryptic species, we profiled both AS and gene expression in two cryptic species of Wiebesia pumilae (WPDZ19 and WPHS), which differ in Wolbachia infection status. The results showed that 101 and 71 differentially alternatively spliced genes (DASs) were identified in female and male groups, respectively. Functional enrichment revealed that female DASs were significantly enriched in mitotic cell cycle process, cytoskeleton organization, cellular component organization, and DNA damage. On the other hand, male DASs were predominantly related to actin, cytoskeleton, and muscle development. Gene set enrichment analysis of DASs also revealed that the regulation of mitotic nuclear division and meiotic nuclear division were enriched in female and male groups, respectively. We identified 4509 DEGs in females and 3645 in males, with minimal overlap between DASs and DEGs. Moreover, RT-PCR has been used to validate the key genes. Our results revealed sexually divergent transcriptional regulation patterns between WPDZ19 and WPHS, suggesting a potential association with Wolbachia persistence. Our findings provide new insights into the study of adaptive evolution of cryptic species.
1. Introduction
Cryptic species represent biologically distinct taxa that exhibit minimal or no morphological differentiation despite significant genetic divergence [1]. Cryptic species have been identified in diverse groups, including mammals [2], fish [3], birds [4], amphibians [5], and insects [6,7]. Cryptic parasites and nematode species have also been identified [8,9,10]. Insects are a large group of animals, accounting for approximately half of all living species on the planet and are important for estimating global biodiversity [11]. However, the diversity of insect species is likely underestimated due to the existence of cryptic species. The emergence of insect cryptic species might be related to intrinsic characteristics and external environmental conditions, such as pheromone recognition, mating time, preference, endosymbiosis, and environmental adaptation [12,13,14,15]. The study of cryptic species is of great significance for ecological adaptation, species evolution, and biodiversity.
The adaptive ability of organisms to environmental challenges fundamentally relies on molecular plasticity, particularly through mechanisms that expand proteomic diversity [16]. Among these, transcriptional regulation is one of the most common forms of molecular regulation in organisms and is closely related to differences among species and adaptability. Alternative splicing (AS) represents the predominant transcriptional regulatory mechanism in eukaryotes, dramatically amplifying genomic coding capacity and functional complexity [17,18]. Five principal AS types exist: skipped exons (SEs), exon mutual exclusions (MXEs), optional 5′ splice sites (A5SSs), optional 3′ splice sites (A3SSs), and retained introns (RIs) [19]. Notably, RI provides a rapid evolutionary pathway for coding variation, offering particular advantages for ecological adaptation over short evolutionary timescales [20], while novel exon incorporation often incurs deleterious frameshifts under purifying selection [21]. AS promotes ecological adaptation and is an important foundation for adaptive evolution and speciation, especially on a short time scale [21,22]. It has been demonstrated that AS plays crucial adaptive roles across diverse organisms. In fishes, differential splicing patterns contribute to the ecological divergence between freshwater and marine species, facilitating their habitat-specific adaptations [23,24]. Body lice exhibit distinctive AS events in genes associated with nervous system, salivary gland, trachea, ovarian follicle cells, and transcriptional regulation, driving functional divergence from head lice [25]. Environmental plasticity is similarly mediated by alternative splicing, as evidenced in Aedes aegypti mosquitoes where temperature-responsive splicing variants of digestive, iron storage, redox regulation, and immune-related genes shape physiological responses to environmental variation [26].
Emerging evidence highlights the functional significance of sex-specific splicing across diverse taxa. In the egg parasitoid Trichogramma dendrolimi Matsumura, the identification of sex-determining genes and their sexually specific splicing variants provides novel molecular markers for sex identification of immature T. dendrolimi and biological pest control [27]. In birds, the AS of sex differences leads to phenotypic differences, and when the changes in gene expression levels are functionally limited, sex-specific splicing serves as an alternative mechanism for sexual adaptation [28]. Remarkably, differential AS and gene expression show little overlap in some physiological regulation, which have been well characterized in Drosophila [29], aphids [30], and salmonid fish [31]. The AS of Down syndrome cell adhesion molecule (Dscam) in Laodelphax striatellus affects the infection amount of Wolbachia in the host [32]. In Ostrinia scapulalis, Wolbachia manipulates the host’s sex by interfering with the sex-specific AS process or sex-determination mechanism of the host gene Osdsx [33,34]. Although Illumina RNA-seq can provide some AS information, its short-read lengths pose challenges for accurate isoform identification and complex splicing event detection. In contrast, isoform-sequencing (Iso-Seq) enables full-length transcript capture, offering more reliable technical support for precise AS analysis.
Fig wasps (Chalcidoidea, Hymenoptera) are distinguished by their unique development and life activities, which are closely related to the fig syconia of fig trees (Ficus, Moraceae). Pollinating fig wasps pollinate fig trees. These pollinators and fig trees have undergone approximately 75 million years of co-evolution, and they are considered one of the classical models of mutualism and provide important materials for co-evolutionary studies [35]. The pollinating fig wasp Wiebesia pumilae of Ficus pumila comprises three cryptic species (Wiebesia sp. 1, 2, and 3) [36]. Notably, Wiebesia sp. 1 exhibits a 100% infection rate with Wolbachia, while Wiebesia sp. 2 and 3 are uninfected [37]. Cryptic fig wasp species with natural differences in Wolbachia infection and limited genetic variation due to recent divergence provide excellent models for studying cryptic species differentiation and speciation. Previous research has revealed sex-specific effects of Wolbachia on gene expression in Ceratosolen solmsi, the pollinating fig wasp of Ficus hispida. In females, differentially expressed genes were primarily enriched in biological processes such as cellular metabolism, meiosis, and signal transduction. In males, the affected processes were more related to immune stress, cytoskeleton organization, and oxidation reduction [38]. Furthermore, Wolbachia infection has significantly reduced the mitochondrial genetic diversity of the wasps through selective sweeps [39]. Although previous studies have revealed overall sex-specific differences in host gene expression, the underlying regulatory mechanisms, particularly at the post-transcriptional and transcriptional levels, remain largely unexplored.
In this study, we focused on two cryptic species of the fig wasp Wiebesia pumilae that are morphologically indistinguishable yet genetically distinct, with their most striking difference being their infection status with the endosymbiont Wolbachia. We aimed to analyze the sex-specific regulation patterns in the transcriptomes of cryptic fig wasp species by investigating both alternative splicing and gene expression, thereby providing molecular evidence for the study of cryptic species differentiation.
2. Materials and Methods
2.1. Sample Preparation and Identification
The morphospecies Wiebesia pumilae is known to include three cryptic species (Wiebesia sp. 1, 2, and 3) [36]. We collected W. pumilae fig wasp samples from Huangshan City, Anhui province, China (designated as WPHS), and Danzhou City, Hainan province, China (designated as WPDZ19). The fig syconia were picked from the field and placed in a climate incubator at a temperature of 27 °C and a relative humidity of 50% to cultivate and collect fig wasps.
The whole genomic DNA of fig wasp samples was extracted in a sterilized environment for Wolbachia infection status and cryptic species identification. The infection status of Wolbachia was determined by PCR amplification of three molecular markers: (1) the surface protein gene (wsp) with the primers wsp81F (5′-TGGTCCAATAAGTGATGAAGAAAC-3′) and wsp691R (5′-AAAAATTAAACGCTACTCCA-3′) [40]; (2) the 16S rRNA gene with the primers 16SwolF (5′-TTGTAGCCTGCTATGGTATAACT-3′) and 16SwolR (5′-GAATAGGTATGATTTTCATGT-3′) [41]; (3) the ftsZ gene with the primers ftsZF (5′-TACTGACTGTTGGAGTTGTAACTAACGCGT-3′) and ftsZR (5′-TGCCAGTTGCAAGAACAGAAACTCTAACTC-3′) [42]. The mitochondrial gene cytochrome oxidase I (COI) was amplified using primers LCO1490 (5′-GGTCAACAAATCATAAAGATATTGG-3′) and HCO2198 (5′-TAAACTTCAGGGTGACCAAAAAATCA-3′) to examine the effectiveness of genomic extraction. PCR amplification was conducted in a 25 μL reaction mixture containing 12.5 μL of 2× Super Mix (TransGen, Beijing, China), 1 μL of template DNA, 0.5 μL each of forward and reverse primers, and 10.5 μL of sterile nuclease-free water. The thermal cycling protocol consisted of an initial denaturation at 94 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °C for 30 s, with a final extension at 72 °C for 10 min. For each PCR run, a no-template control was included to monitor contamination, alongside positive control from a confirmed positive sample. The PCR products were detected on a 1% agarose gel and visualized using GelStain (TransGen, Beijing, China). The amplification products were sequenced by Sangon Biotech (Shanghai, China) Company using the same primers as those for PCR amplification.
For cryptic species identification, we used the COI gene as a DNA barcode. We combined our own samples with sequences from Chen et al. [36] and conducted a phylogenetic analysis using PhyloSuite software (version 1.2.3) [43]. Specifically, multiple sequence alignment was performed with MAFFT (version 7.037), followed by sequence processing with Gblocks (version 0.91b), and finally, a MrBayes phylogenetic tree was reconstructed.
2.2. RNA Extraction
The preparation of each RNA sample involved approximately 30 fig wasps. For Iso-seq, total RNA was isolated using the standard TRIzol protocol per the manufacturer’s instructions. The integrity of the RNA was determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). Total RNA samples with a RIN value ≥ 8 were used for cDNA library construction, fulfilling the input material requirements for subsequent PacBio SMRT sequencing. For Illumina RNA-seq, four groups of samples were set up (WPDZ19_F, WPDZ19_M, WPHS_F, and WPHS_M), with three biological replicates in each group. Total RNA was isolated using the EasyPure RNA kit (TransGen, Beijing, China) according to the manufacturer’s instructions. The NanoDrop ND-1000 Spectrophotometer (Nano-Drop Technologies, Wilmington, DE, USA) was used to confirm adequate RNA concentration and A260/A280 ratio.
2.3. Library Construction and Sequencing
For full-length transcriptome sequencing, cDNA synthesis was performed using the Clontech SMARTer PCR cDNA Synthesis Kit (Takara Biotechnology, Dalian, China) starting with 4 μg of high-quality total RNA per sample. Following the manufacturer’s protocol, the SMARTer technology was employed to synthesize first-strand cDNA, which was subsequently amplified to generate double-stranded cDNA. A SMRTbell library was then constructed from 1 μg of the purified cDNA using the Pacific Biosciences SMRTbell Template Prep Kit (Pacific Biosciences of California, Menlo Park, CA, USA), which involved steps of blunt ligation and adapter ligation. The prepared SMRTbell templates were annealed with sequencing primers and bound to polymerases using the Sequel II Binding Kit (Pacific Biosciences of California, USA). Finally, the library was sequenced on the Pacific Bioscience Sequel II platform to generate continuous long reads (CLR).
For high-throughput Illumina RNA-seq, sequencing libraries were constructed using the NEBNext® Ultra™ RNA Library Prep Kit for Illumina® (NEB, Ipswich, MA, USA). Briefly, 3 μg of total RNA per sample was used as input. The poly(A) mRNA was purified and fragmented, followed by first-strand and second-strand cDNA synthesis. The cDNA fragments were then end-repaired, adenylated at the 3′ ends, and ligated with NEBNext adapters. The products were purified and enriched by PCR amplification to finalize the library construction. The quality and size distribution of the resulting libraries were assessed using the Agilent Bioanalyzer 2100 system (Agilent Technologies, USA). Libraries meeting the quality thresholds were subjected to sequencing on the Illumina Hiseq 2500 platform (Illumina of San Diego, San Diego, CA, USA), ultimately generating approximately 125 bp paired-end reads.
2.4. Data Processing and Analysis
For the data of Iso-seq, the raw data may contain connector sequences, sequences with lengths that deviate from those in the constructed database, sequencing errors, and low-quality bases. SMRTlink (version 9.0.0.92188) was used for quality control of the raw data. Sequences of low quality and short reads were removed; they were then classified, clustered, and corrected to obtain high-quality full-length sequences. After quality control of the sequencing data, the results were statistically analyzed, and the data volume, N50, average GC content, and length distribution were calculated. The complete insertion sequence was a full-length transcript sequence that included a 5′ primer, 3′ primer, and poly(A) sequence before the 3′ primer. Isoseq3 was used to remove poly(A) and chimeric sequences from full-length sequences to obtain full-length consistent non-chimeric transcript sequences. Clustering and error correction were performed to obtain high-quality transcripts with an accuracy greater than 0.99. GMAP software (gmap–2021–12–17) [44] was used to align high-quality transcript sequences with the reference genome. The distribution of full-length transcripts in the reference genome was calculated using a bin size of 10 kb, and the number of reads in each bin was calculated.
For the Illumina RNA-seq data analysis, raw sequencing reads were first processed to ensure data quality. Specifically, adaptor sequences and low-quality reads were filtered out using Trimmomatic (version 0.36) [45]. All subsequent analyses were performed using these high-quality cleaned reads. The cleaned reads were then aligned to the reference genome using Hisat2 (version 2–2.1.0) [46] with default parameters. Based on the alignment results, the read counts mapped to each gene were accurately quantified using Htseq (version 2.0.1). Finally, the read counts were normalized to Fragments Per Kilobase of transcript per Million mapped fragments (FPKM) values to obtain and compare the gene expression levels across samples.
2.5. Identification of Alternative Splicing Events and Differentially Expressed Genes
AS events were classified into five different types: skipped exons (SEs), mutually exclusive exons (MXEs), alternative 5′ splice sites (A5SSs), alternative 3′ splice sites (A3SSs), and retained introns (RIs). AStalavista (version 4.0.1) [47] was used to analyze AS events. This method effectively distinguished exon–intron structures, and AS types at the transcriptome level were statistically analyzed. FLAIR [48] was used to screen differential alternative splicing genes (DASs) (p-value < 0.05).
Gene expression level among the various samples were analyzed based on the short-read datasets generated by the Illumina RNA-seq. The expression level of each gene was quantified and normalized using the FPKM method. DESeq2 (version 1.20.0) was used to perform differential expression analysis between comparison groups, utilizing a negative binomial model with triplicate biological replicates. Genes with an absolute value of log2 fold change (log2FC) greater than or equal to 1 and an adjusted q-value of less than 0.05, as determined by the Benjamini–Hochberg procedure for multiple testing correction, were statistically defined as differentially expressed genes (DEGs).
Gene function was annotated using multiple databases, including the NCBI non-redundant protein sequences (Nr), protein family (Pfam), Clusters of Orthologous Groups of proteins (KOG/COG), Swiss-Prot (a manually annotated and reviewed protein sequence database), Gene Ontology (GO), and KEGG Ortholog (KO). For functional enrichment analysis, GO and KEGG databases were performed. The R software (version 4.4.2) was used to perform gene set enrichment analysis (GSEA), which evaluates DAS sets by calculating their enrichment score (ES) within a ranked gene list and assesses statistical significance through permutation testing, thereby identifying specific biological processes [49]. We quantified the expression levels of DASs using Illumina RNA-seq data and examined their potential biological functions through GSEA. The R software was used to present the results.
2.6. Integrative Analysis of Alternative Splicing and Gene Expression
To further investigate the relationship between alternative splicing and gene expression, we constructed a scatter plot combining DASs with DEGs. Each DAS was assigned a p-value to indicate the statistical significance of alternative splicing differences, while each DEG was associated with a log2FC value to quantify the magnitude of gene expression change. The distribution of DASs and DEGs was visualized using R software, with −log2(p-value) and −log2FC as the two-dimensional axes. Among the DASs and DEGs, overlapping genes were extracted. Functional enrichment of overlapping genes was performed through GO database.
2.7. RT-PCR Validation
Semi-quantitative RT-PCR was performed to validate the DASs and DEGs. Total RNA was extracted according to the protocols of TransZol Up Plus RNA Kit (TransGen, Beijing, China). cDNA was prepared using EasyScript® First-Strand cDNA Synthesis SuperMix Kit (TransGen, Beijing, China) with random primers. The first-strand cDNA was synthesized by incubation at 25 °C for 10 min, followed by 42 °C for 20 min, with a final heating step at 85 °C for 5 s. The cDNA products were used as DNA templates for amplifying the target genes. The target gene was amplified with specific primers (Table S1). The PCR was performed under the following conditions: initial denaturation at 94 °C for 5 min, followed by 35 cycles of 94 °C for 30 s, 51~55 °C for 30 s, and 72 °C for 30~60 s (depending on target genes), and a final extension at 72 °C for 10 min. The PCR products were analyzed by 1% agarose gel electrophoresis, with target bands identified under UV transillumination. Each verification experiment includes two independent biological replicates.
3. Results
3.1. Distribution of Cryptic Species and Transcriptome Sequencing Data Statistics
After examining the amplification of wsp, 16S rRNA, and ftsZ genes for fig wasp, we detected WPHS was Wolbachia-infected (with wsp, 16S rRNA, and ftsZ genes), and WPDZ19 was Wolbachia-uninfected (without wsp, 16S rRNA, and ftsZ genes) (Figure S1), which were consistent with the previous research results [36]. The samples WPHS were identified as Wiebesia sp. 1, and WPDZ19 were identified as Wiebesia sp. 2, according to the MrBayes phylogenetic tree of the COI gene (Figure S2). The Wolbachia infection is fixed in WPHS. The collected fig wasps included both female and male individuals. Our samples included Wolbachia-uninfected female (WPDZ19_F), Wolbachia-uninfected male (WPDZ19_M), Wolbachia-infected female (WPHS_F), and Wolbachia-infected male (WPHS_M). Separate comparison groups were set for each sex, including female group (WPDZ19_F vs. WPHS_F) and male group (WPDZ19_M vs. WPHS_M).
The RNA of all samples was mixed for full-length transcriptome sequencing using the PacBio Sequel platform. SMRT sequencing yielded 26,946,060, 24,424,934, 27,240,059, and 14,595,712 subreads from WPDZ19_F, WPDZ19_M, WPHS_F, and WPHS_M, respectively. The mean length of subreads of the four samples ranged from 1708.59 to 2603.96 bp; the N50 of subreads ranged from 1981 to 2784 bp; and the average GC content of subreads ranged from 40% to 41% (Figure S3A–D; Table S2). After self-correction and merging, 397,115, 362,774, 687,479, and 234,158 circular consensus sequences (CCSs) with a mean length of 2088.83 bp, 2072.59 bp, 2619.69 bp, and 1855.15 bp were formed from WPDZ19_F, WPDZ19_M, WPHS_F, and WPHS_M, respectively (Table S2).
Using GMAP, high-quality full-length transcripts were mapped to the reference genome with >99% alignment rate across all samples (Table S3). Unmatched sequences accounted for <1%, while multi-mapped reads represented 0.21–1.09% (Figure S3E; Table S3). Strand-specific alignment showed balanced distribution between forward (49.03–55.06%) and reverse chains (43.17–50.53%). The distribution of reads in the reference genome was calculated using a bin size of 10 kb, and showed predominant distribution on chromosomes 1–6, correlating with chromosome length (Figure S3F).
The Illumina RNA-seq data analysis included four experimental groups, WPDZ19_F, WPDZ19_M, WPHS_F, and WPHS_M, with three biological replicates per group. The raw reads number ranged from 42.74 to 61.65 million per sample, and the clean reads number ranged from 42.35 to 60.74 million per sample, showing minimal loss during data filtering (Table S4). All samples exhibited high sequencing quality with Q30 scores exceeding 90.18%. The mapping rate of all samples to the reference genome exceeded 92.46%. Notably, the WPHS groups demonstrated superior alignment efficiency with mapping rates ranging from 95.34 to 96.05%. These results indicate the sequencing data possessed sufficient depth and quality for reliable downstream transcriptomic analysis.
3.2. Identification of AS Events
AS generates mRNA isoform diversity in eukaryotes through five primary event types: SEs, A5SSs, A3SSs, MXEs, and RIs. In WPDZ19_F, 2099 AS events were identified, predominantly containing 34.35% SEs, 26.39% A3SSs, 19.44% A5SSs, 15.82% RIs, and 4.00% MXEs (Figure 1A). In WPDZ19_M, 1827 AS events were identified, including 35.52% SEs, 18.77% A5SSs, 21.46% A3SSs, 5.15% MXEs, and 19.10% RIs (Figure 1B). WPHS_F displayed 4468 AS events, primarily comprising 31.42% SEs, 26.34% A3SSs, 22.25% A5SSs, 16.65% RIs, and 3.33% MXEs (Figure 1C). WPHS_M showed 879 AS events, including 33.68% SEs, 19.00% A5SSs, 20.59% A3SSs, 3.41% MXEs, and 23.32% RIs (Figure 1D). Comparative analysis showed SEs were the most abundant AS type, whereas MXEs were the least type. Notably, sex-specific AS patterns were observed, with WPHS_F exhibiting significantly more AS events than WPDZ19_F, while WPHS_M had fewer than WPDZ19_M, suggesting sex-biased splicing regulation.
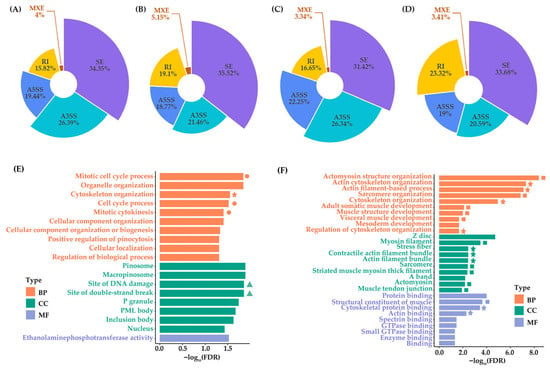
Figure 1.
Statistics of AS events and GO functional enrichment of DASs. AS events of genes from WPDZ19_F (A), WPDZ19_M (B), WPHS_F (C), and WPHS_M (D) were calculated. SE represents skipped exons; A5SS represents alternative 5′ splice sites; A3SS represents alternative 3′ splice sites; MXE represents mutually exclusive exons; and RI represents retained introns. (E) GO enrichment analysis of DASs from the female group. (F) GO enrichment analysis of DASs from the male group. The top 10 most significant terms are presented. The stars in the figure indicate actin, cytoskeleton-related terms, the squares represent muscle-related terms, the circles indicate terms related to the cell cycle, and the triangles indicate terms related to DNA damage. BP represents biological process, CC represents cell component, and MF represents molecular function.
3.3. Functional Enrichment Analysis of DASs
In total, 101 and 71 DASs were identified in female and male group, respectively. GO functional enrichment of DASs reveals that the female DASs are enriched in biological processes mainly related to mitotic cell cycle-related terms (including mitotic cell cycle process, cell cycle process, and mitotic cytokinesis), organelle organization, cytoskeleton organization, cellular component organization, cellular component organization or biogenesis, regulation of biological process, cellular localization, and positive regulation of pinocytosis (FDR < 0.05) (Figure 1E, Table S5). Meanwhile, the male DASs are enriched in biological processes mainly related to actin, cytoskeleton, and muscle development-related terms (including actomyosin structure organization, actin cytoskeleton organization, actin filament-based process, sarcomere organization, cytoskeleton organization, regulation of cytoskeleton organization, adult somatic muscle development, muscle structure development, and visceral muscle development), mesoderm development (FDR < 0.05) (Figure 1F, Table S6). It is worth noting that in cellular component, terms related to DNA damage are enriched in female group, such as site of DNA damage and site of double-strand break (Figure 1E). Meanwhile, terms related to filament, sarcomere, actomyosin, and muscle are enriched in male group, including myosin filament, stress fiber, contractile actin filament bundle, actin filament bundle, sarcomere, striated muscle myosin thick filament, actomyosin, and muscle tendon junction (Figure 1F). In molecular function, structural constituent of muscle, cytoskeletal protein binding, actin binding are enriched in male group (Figure 1F).
GSEA of female DAS events coupled with gene expression data revealed the top 20 significant enrichment in nucleus and RNA-related biological functions (Table S7). The nucleus-related biological terms included nuclear speck, regulation of mitotic nuclear division, nuclear body, nuclear lumen, nucleoplasm, regulation of chromatin organization, and nuclear chromosome segregation, which were consistent with the mitotic cell cycle process enriched in the functional enrichment of the female DASs (Figure 1E and Figure 2A). The RNA-related biological terms included mRNA metabolic process, mRNA processing, RNA processing, RNA binding, RNA splicing via transesterification reactions, RNA splicing via transesterification reactions with bulged adenosine as nucleophile, mRNA splicing via spliceosome, and mRNA binding (Figure 2B). In addition, muscle system process, cellular component assembly involved in morphogenesis, exocytosis, Z disc, I band, and establishment or maintenance of cell polarity were also enriched. Male DAS events coupled with gene expression data revealed the top 20 significant enrichment in nucleus, growth, development, and reproduction-related biological functions (Table S7). The nucleus-related biological terms included nucleus, meiotic nuclear division, nuclear membrane, nuclear body, and nucleoplasm (Figure 2C). The growth, development, and reproduction-related biological terms included in utero-embryonic development, regulation of embryonic development, head development, positive regulation of growth, chordate embryonic development, regulation of reproductive process, oocyte differentiation, and oocyte development (Figure 2D). In addition, membrane-bounded organelle, intracellular protein transport, small molecule biosynthetic process, intracellular membrane-bounded organelle, membrane-enclosed lumen, organelle lumen, and intracellular organelle lumen were also enriched.
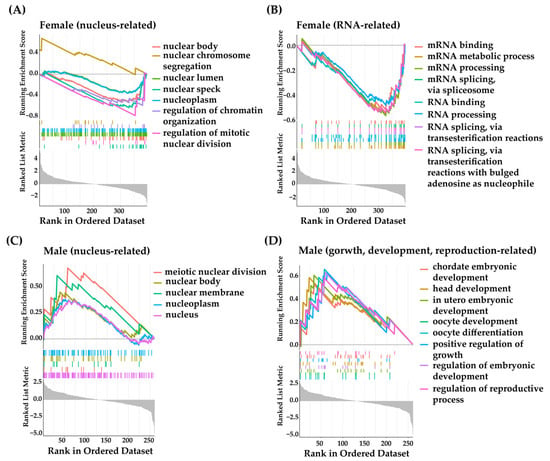
Figure 2.
GSEA of DASs. (A) Nucleus-related GO terms enriched in female group. (B) RNA-related GO terms enriched in female group. (C) Nucleus-related GO terms enriched in male group. (D) Gorwth, development, reproduction-related GO terms enriched in male group.
3.4. Identification and Functional Analysis of DEGs
Supported by Illumina RNA-seq data, genes exhibiting |log2FC| ≥ 1 and q-value < 0.05 were identified as significant DEGs. In the female group, 4509 genes showed significantly differential expression, including 2281 up-regulated and 2228 down-regulated (Figure 3A,B). On the other hand, the comparison of male group revealed 3645 DEGs, including 1862 up-regulated and 1783 down-regulated (Figure 3A,C).
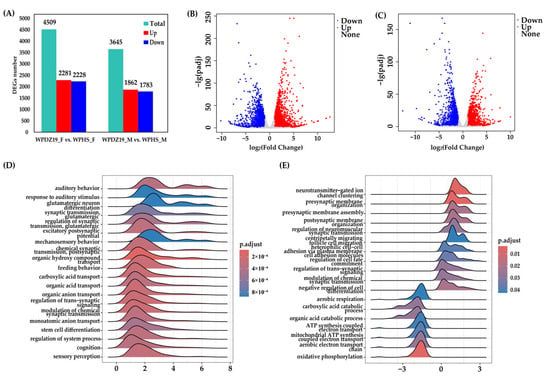
Figure 3.
Quantity statistics and functional enrichment of DEGs. (A) The quantity statistics of DEGs. (B) The volcanic plot of DEGs from the female group. (C) The volcanic plot of DEGs from the male group. (D) The top 20 most significant terms of GO enrichment analysis of DEGs by GSEA from the female group. (E) The top 20 most significant terms of GO enrichment analysis of DEGs by GSEA from the male group.
GSEA was performed to identify gene sets with subtle but biologically significant expression differences in female and male groups, revealing coordinated changes across gene sets. The top 20 gene sets were presented. The female group were primarily enriched in synaptic transmission, transport, sensory perception, behavior, regulation of system process, cognition, and stem cell differentiation—all of which were upregulated in Wolbachia-infected females (Figure 3D, Table S8). The male group were mainly distributed in synaptic transmission, energy supply, metabolism, negative regulation of cell differentiation, regulation of cell fate commitment, heterophilic cell–cell adhesion via plasma membrane cell adhesion molecules, and centripetally migrating follicle cell migration (Figure 3E, Table S8). Among these, synaptic transmission (7 terms), negative regulation of cell differentiation, regulation of cell fate commitment, heterophilic cell–cell adhesion via plasma membrane cell adhesion molecules, and centripetal migration of follicular cells were upregulated in Wolbachia-infected males, whereas energy supply (5 terms) and metabolism (2 terms) were downregulated.
3.5. Integrative Analysis of AS and Gene Expression
To further determine the relationship between alternative splicing and gene expression, we selected the overlapping genes from both DASs and DEGs. A total of 30 overlapping genes were obtained from the female group (29.70% of DASs; Figure 4A) and 21 from the male group (29.58% of DASs; Figure 4B). The set of DASs largely does not overlap with the set of DEGs. Furthermore, the female overlapping genes were annotated in mRNA binding, and the male overlapping genes were related to mesoderm development, visceral muscle development, skeletal muscle tissue development, sister chromatid cohesion, actomyosin structure organization, mitotic chromosome condensation, muscle attachment, etc., which are consistent with the functional results of DASs (Table S9, Figure 1F).
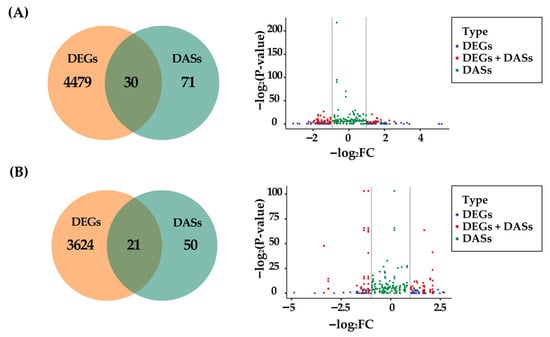
Figure 4.
Overlapping analysis of DASs and DEGs. (A) Venn diagram and scatter plot of DASs and DEGs in the female group. (B) Venn diagram and scatter plot of DASs and DEGs in the male group. In the scatter plot, the horizontal axes represent the value of −log2FC, and the vertical axes represent the value of −log2(p-value).
3.6. Verification of DASs and DEGs
Among the overlapping genes (genes that are both differentially alternatively spliced and differentially expressed) in the female group, we validated a mRNA binding-related gene, Wpum_In_07455, which plays a crucial role in synaptic transmission. By aligning full-length transcripts to the reference genome (Figure 5A), we detected two exon-skipping events in Wpum_In_07455, including exons of 96 bp and 72 bp in length. Semi-quantitative RT-PCR experiments further validated these differential alternative splicing events with two independent biological replicates. For the verification experiment of the 96 bp exon, in WPDZ19_F, the band of the transcript with exon skipping (488 bp) was brighter than that of the transcript with exon retention (584 bp), while in WPHS_F, only the band of the transcript with exon skipping (488 bp) was detected (Figure 5B). For the 72 bp exon, both WPDZ19_F and WPHS_F showed bands of transcripts with exon skipping (510 bp) and exon retention (582 bp) (Figure 5C). However, in WPDZ19_F, the band of the exon-skipped transcript was brighter, while in WPHS_F, the bands of both transcripts were comparable. Furthermore, motif prediction revealed that the 96 bp exon-skipped transcript lacked a conserved motif site, whereas no significant functional motifs were predicted in the 72 bp exon (Figure 5D).
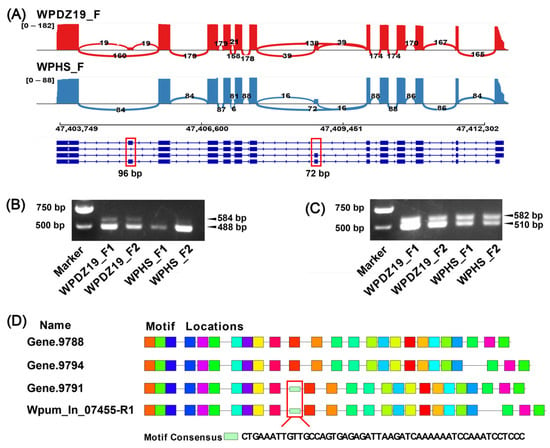
Figure 5.
Alternative splicing events analysis and RT-PCR gene verification. (A) The different alternative splicing types of Wpum_In_07455 gene in female group. Blocks represent exons and lines represent introns. The thickness of the curves in red and blue crossing two splicing junction sites represents the coverage degree of PacBio full-length sequencing reads, and the number of junction reads is marked in the middle of the splicing site. The RNA coverage is marked on the left by the number of reads. (B,C) Two exon-skipping events with 96 bp and 72 bp, respectively, in Wpum_In_07455 were verified by RT-PCR. (D) Motif prediction among the four transcripts of the Wpum_In_07455 gene. Different colored boxes represent different motifs.
Among the overlapping genes in the male group, we validated a key gene, Wpum_In_16800. Transcript structure analysis revealed a 105 bp exon skipping event in this gene. Notably, the exon-skipped transcript showed higher abundance in WPDZ19_M compared to the exon-retained isoform, while no exon-retained transcripts were predicted in WPHS_M (Figure S4A). Further analysis demonstrated that this 105 bp-exon contains a conserved motif (Figure S4A). Semi-quantitative RT-PCR results confirmed this differential alternative splicing pattern: both bands of the transcripts with exon skipping (580 bp) and exon retention (685 bp) were detected in WPDZ19_M and WPHS_M, with a higher brightness of the bands of exon-skipped transcript (580 bp) (Figure S4B). Notably, the exon-retained transcript (685 bp) showed greater abundance in WPHS_M compared to WPDZ19_M (Figure S4B).
4. Discussion
In the process of biological adaptation, AS and gene expression regulation are of great importance [17,50,51]. AS is an important post-transcriptional regulatory mechanism, and the number and types of AS events determine the complexity and diversity of gene expression products [52]. The AS of genes in the butterfly Bicyclus anynana mediates its seasonal plasticity and environmental adaptation [53]. AS of a carboxyl/choline esterase gene (TcCCE23) enhances the fenpropathrin tolerance of Tetranychus cinnabarinus [54]. Our study was the first to integrate the third-generation single-molecule real-time full-length transcriptome sequencing and Illumina high-throughput short-read transcriptome sequencing to elucidate sexual divergence of gene transcription and expression between the two cryptic fig wasp species of W. pumilae.
4.1. Functional Divergence of Sex-Specific DASs
Sex-specific DASs were detected, and there were more DASs in female group (101) than those in male group (71). The functional profile of DASs in females is consistent with the possibility of an association with Wolbachia persistence (such as mitotic cell cycle process and cytoskeleton organization). This endosymbiont has acquired the ability to precisely regulate key phases of the host’s cell cycle [55]. Cell cycle arrest diminished Wolbachia level, providing evidence that Wolbachia persistence requires active cell cycle regulation [56]. The significant enrichment of mitotic cell cycle regulatory genes in female group may suggest Wolbachia’s intervention in host cell division [57,58]. It has been reported that Wolbachia manipulated host reproductive cell mitosis to enhance fecundity and change the number of apoptotic nurse cells in a caspase-dependent manner to provide nutrition for oogenesis [59,60]. GSEA of female DASs revealed significant enrichment of regulation of mitotic nuclear division, chromatin organization, and nuclear chromosome segregation, all of which are closely associated with cell cycle regulation [61]. These results are consistent with the GO functional enrichment analysis, suggesting a potential link between Wolbachia infection and alterations in the mitotic cell cycle process of female hosts [56,57,58].
The DASs from male group mainly enriched in biological processes of actin, cytoskeleton, actomyosin, sarcomere, and muscle-related terms. Actin and cytoskeleton-related genes serve as crucial mediators in interaction between Wolbachia and hosts [62,63]. Wolbachia has been shown to utilize the host actin cytoskeleton to mediate its persistence and transmission [64,65]. Sarcomere is the basic unit of skeletal and cardiac striated muscles [66]. In Drosophila, Wolbachia infection upregulates genes related to muscle contraction, including sarcomere organization, actin filament organization, and myofibril assembly, which may directly affect the muscle function of the host, involving motor ability and reproductive behavior [67]. Wolbachia is known to infect muscles in host, and to increase locomotion in mosquitoes [68,69,70]. The male fig wasps are known to use abdominal inflexion during mating. Therefore, changes in muscle function could potentially reshape movement patterns relevant to mating and reproduction of males. GSEA of males DASs revealed that the most significant enrichment are biological functions related to the nucleus, growth, development, and reproduction (Figure 2C,D). Notably, the enriched terms meiotic nuclear division and regulation of reproductive process are associated with reproductive biology, suggesting a potential functional link with Wolbachia infection [71].
4.2. Functional Divergence of Sex-Specific DEGs
The female group showed more DEGs (4509) than the male group (3645) (Figure 3A). GSEA of DEGs revealed distinct sex-specific gene expression patterns. Previous studies have reported that Wolbachia infection affects host functions such as development [72], synaptic activity [73], and behavior [74]. For example, in Wolbachia-infected Aedes aegypti mosquito cells, the expression of synaptic vesicle membrane proteins was significantly upregulated [73]. In WPHS_F, the upregulation of gene sets associated with synaptic transmission, transport, sensory perception, behavior, regulation of system process, cognition, and stem cell differentiation (Figure 3D), suggests that WPHS_F may broadly enhance neuronal function and developmental processes.
In contrast, the regulatory patterns in male DEGs were more complex. Compared with WPDZ19_M, WPHS_M upregulated genes related to synaptic transmission, negative regulation of cell differentiation, regulation of cell fate commitment, heterophilic cell–cell adhesion via plasma membrane cell adhesion molecules, and centripetal migration of follicular cells, while downregulated those involved in energy supply and metabolism (Figure 3E). This divergence implies a potential trade-off in WPHS_M, where the organism may prioritize neuronal function and cellular remodeling at the expense of metabolic activity [73,75]. Notably, the shared upregulation of synaptic transmission in both sexes may suggest a conserved role of neuronal modulation [73]. These sex-specific expression changes are consistent with a model of physiological fine-tuning that benefits Wolbachia persistence. It is important to note, however, that this association does not establish a direct causal relationship, as host genetic factors may also contribute to the observed patterns.
4.3. Minimal Overlap Reveals Complementary Regulation Between AS and Gene Expression
To elucidate the coordinated regulatory relationship between alternative splicing and gene expression, we performed an intersection analysis of DASs and DEGs to identify core functional pathways under dual regulation [29,31,50]. Our findings revealed that DASs and DEGs rarely overlap, accounting for less than 30%, suggesting that these two regulatory mechanisms act on different cellular traits to complementarily alter organismal phenotype [76]. Our results are consistent with the findings in Drosophila and aphids, indicating that AS may have complementary regulatory effects on gene expression [29,30]. The overlapping genes of female group were only annotated in mRNA binding, while the male overlapping genes were related to mesoderm development, muscle development, and mitotic chromosome condensation, which are consistent with the functional enrichment analysis of DASs.
Our study compared the differences in alternative splicing and gene expression between two cryptic fig wasp species with distinct Wolbachia infection statuses, revealing sex-specific transcriptional regulation. Although we proposed that these differences might be potentially associated with Wolbachia infection, it remains necessary to consider the possibility that these differences could also be influenced by genetic background or stochastic variation. Importantly, our observed regulatory patterns are consistent with those in other host-Wolbachia systems. For instance, Wolbachia-mediated modulation of metabolism, oxidation reduction, immunity, and development has been reported in Drosophila [77], whitefly Bemisia tabaci [78], and spider Hylyphantes graminicola [79]. Similarly, we found DEGs involved in transportation, metabolism, and energy supply, and DASs enriched in cell cycle, mitosis, meiosis, actin, and cytoskeleton. This concordance across diverse systems enhances the credibility that our findings are linked to the endosymbiont infection. Nonetheless, the potential influence of host genetic background cannot be fully discounted. Future studies should employ designs that effectively control for this variable. For instance, antibiotic treatment to eliminate Wolbachia or microinjection to create novel infected lines would allow for a direct comparison under identical genetic backgrounds.
5. Conclusions
In this study, using two cryptic species of fig wasp W. pumilae as models, we systematically analyzed the differential patterns of AS and gene expression. Our analysis identified sexually divergent regulatory patterns, with 101 and 71 DASs in females and males, respectively. Functional enrichment revealed female-biased enrichment in mitotic cell cycle regulation, which is further confirmed by the results of gene set enrichment analysis. In contrast, male-biased DASs were related to actin, cytoskeleton, muscle development, and reproductive functions, consisting with the results of gene set enrichment analysis. Transcriptomic profiling further identified 4509 female- and 3645 male-biased DEGs, exhibiting minimal overlap with DASs. Our results revealed sexual divergence in alternative splicing and gene expression among cryptic species with different Wolbachia infection statuses, indicating the presence of sex-specific transcriptional regulation.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/d17100722/s1, Figure S1: Detection of Wolbachia infection status. The first lane in the electrophoretogram represents the D2000 Marker, followed by six biological replicates numbered 1–6. + indicates positive and negative control. The marker genes COI, wsp, 16S rRNA, and ftsZ are indicated on the right side of the figure; Figure S2: The MrBayes phylogenetic tree of COI gene. Figure S3: Length distribution of subreads of the full-length transcriptome sequencing data. The horizontal axes represent the length of subreads, and the vertical axes represent the number of subreads. (A) WPDZ19_F. (B) WPDZ19_M. (C) WPHS_F. (D) WPHS_M. (E) Genome mapping analysis and classification of filtered reads. The horizontal axis represents the percentage of reads, and the vertical axis represents the samples. (F) Distribution of reads in the genome. The outer circle indicates the chromosomes, and the next four circles from outside to inside correspond to WPDZ19_F, WPDZ19_M, WPHS_F, and WPHS_M. The statistics of reads were calculated using a bin size of 10 kb, and the read coverage of each bin was calculated. The log2 of this value was used to generate the plot. There was a median line in the center of each circle; above the median line indicates the alignment to the forward chain, and below the median indicates the alignment to the reverse chain. Figure S4: RT-PCR verification of Wpum_In_16800 gene. (A) The different alternative splicing types of Wpum_In_16800 gene in male group. Blocks represent exons and lines represent introns. The thickness of the curves in red and blue crossing two splicing junction sites represents the coverage degree of PacBio full-length sequencing reads, and the number of junction reads is marked in the middle of the splicing site. The RNA coverage is marked on the left by the number of reads. On the Wpum_In_16800 gene, the predicted motif on the 105 bp exon is shown. (B) The exon-skipping events with 105 bp in Wpum_In_16800 was verified by RT-PCR. Table S1: The specific primers used in RT-PCR validation; Table S2: Statistical results of Iso-seq data; Table S3: Statistics of the genome mapping results and classification of filtered reads; Table S4: Statistical results of Illumina RNA-seq data; Table S5: GO enrichment of DASs from the female group; Table S6: GO enrichment of DASs from the male group; Table S7: GO enrichment of DASs by GSEA; Table S8: The GSEA of DEGs; Table S9: The results of GO enrichment of overlapping genes between DASs and DEGs.
Author Contributions
Conceptualization, H.H. and J.L.; methodology, H.H., S.L. and B.G.; software, L.L. and Y.S.; validation, Y.S.; formal analysis, L.L.; investigation, H.H.; data curation, Y.S.; writing—original draft preparation, H.H. and S.L.; writing—review and editing, H.H., B.G. and J.L.; visualization, L.L. and B.G.; project administration, H.H. and J.L.; funding acquisition, H.H. and J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Natural Science Foundation of China (grant number 32200375 and 32301412), the Natural Science Foundation of Hebei Province, China (grant number C2024108006), and the Innovation Capacity Enhancement Plan Project of Xingtai City (2023ZZ084).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data will be made available on request. The raw RNA-seq data (Accession no. PRJCA039425) were uploaded to China National Center for Bioinformation (CNCB) (https://ngdc.cncb.ac.cn/; accessed on 30 April 2025).
Conflicts of Interest
Author Yalei Su was employed by the company Annoroad Gene Technology (Beiing) Company Limited by Shares, Beijing 100176, China. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Bickford, D.; Lohman, D.J.; Sodhi, N.S.; Ng, P.K.; Meier, R.; Winker, K.; Ingram, K.K.; Das, I. Cryptic species as a window on diversity and conservation. Trends Ecol. Evol. 2007, 22, 148–155. [Google Scholar] [CrossRef]
- Fennessy, J.; Bidon, T.; Reuss, F.; Kumar, V.; Elkan, P.; Nilsson, M.A.; Vamberger, M.; Fritz, U.; Janke, A. Multi-locus Analyses Reveal Four Giraffe Species Instead of One. Curr. Biol. 2016, 26, 2543–2549. [Google Scholar] [CrossRef]
- Li, C.; Jiang, S.; Schneider, K.; Jin, J.; Lin, H.; Wang, J.; Elmer, K.R.; Zhao, J. Cryptic species in White Cloud Mountain minnow, Tanichthys albonubes: Taxonomic and conservation implications. Mol. Phylogenet Evol. 2020, 153, 106950. [Google Scholar] [CrossRef]
- Saitoh, T.; Sugita, N.; Someya, S.; Iwami, Y.; Kobayashi, S.; Kamigaichi, H.; Higuchi, A.; Asai, S.; Yamamoto, Y.; Nishiumi, I. DNA barcoding reveals 24 distinct lineages as cryptic bird species candidates in and around the Japanese Archipelago. Mol. Ecol. Resour. 2015, 15, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Stuart, B.L.; Inger, R.F.; Voris, H.K. High level of cryptic species diversity revealed by sympatric lineages of Southeast Asian forest frogs. Biol. Lett. 2006, 2, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Naumenko, A.N.; Karagodin, D.A.; Yurchenko, A.A.; Moskaev, A.V.; Martin, O.I.; Baricheva, E.M.; Sharakhov, I.V.; Gordeev, M.I.; Sharakhova, M.V. Chromosome and Genome Divergence between the Cryptic Eurasian Malaria Vector-Species Anopheles messeae and Anopheles daciae. Genes 2020, 11, 165. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, M.; Carew, M.E.; Hoffmann, A.A. Molecular, morphological and behavioural data reveal the presence of a cryptic species in the widely studied Drosophila serrata species complex. J. Evol. Biol. 2004, 17, 430–442. [Google Scholar] [CrossRef] [PubMed]
- de León, G.P.; Nadler, S.A. What we don’t recognize can hurt us: A plea for awareness about cryptic species. J. Parasitol. 2010, 96, 453–464. [Google Scholar] [CrossRef]
- Derycke, S.; Remerie, T.; Backeljau, T.; Vierstraete, A.; Vanfleteren, J.; Vincx, M.; Moens, T. Phylogeography of the Rhabditis (Pellioditis) marina species complex: Evidence for long-distance dispersal, and for range expansions and restricted gene flow in the northeast Atlantic. Mol. Ecol. 2008, 17, 3306–3322. [Google Scholar] [CrossRef]
- Lee, M.R.; Canales-Aguirre, C.B.; Nuñez, D.; Pérez, K.; Hernández, C.E.; Brante, A. The identification of sympatric cryptic free-living nematode species in the Antarctic intertidal. PLoS ONE 2017, 12, e0186140. [Google Scholar] [CrossRef]
- Li, X.; Wiens, J.J. Estimating Global Biodiversity: The Role of Cryptic Insect Species. Syst. Biol. 2023, 72, 391–403. [Google Scholar] [CrossRef]
- Andrianto, E.; Kasai, A. Wolbachia in Black Spiny Whiteflies and Their New Parasitoid Wasp in Japan: Evidence of the Distinct Infection Status on Aleurocanthus camelliae Cryptic Species Complex. Insects 2022, 13, 788. [Google Scholar] [CrossRef]
- Bordenstein, S.R.; O’Hara, F.P.; Werren, J.H. Wolbachia-induced incompatibility precedes other hybrid incompatibilities in Nasonia. Nature 2001, 409, 707–710. [Google Scholar] [CrossRef] [PubMed]
- Hänniger, S.; Dumas, P.; Schöfl, G.; Gebauer-Jung, S.; Vogel, H.; Unbehend, M.; Heckel, D.G.; Groot, A.T. Genetic basis of allochronic differentiation in the fall armyworm. BMC Evol. Biol. 2017, 17, 68. [Google Scholar] [CrossRef] [PubMed]
- Hartke, J.; Sprenger, P.P.; Sahm, J.; Winterberg, H.; Orivel, J.; Baur, H.; Beuerle, T.; Schmitt, T.; Feldmeyer, B.; Menzel, F. Cuticular hydrocarbons as potential mediators of cryptic species divergence in a mutualistic ant association. Ecol. Evol. 2019, 9, 9160–9176. [Google Scholar] [CrossRef] [PubMed]
- Bludau, I.; Aebersold, R. Proteomic and interactomic insights into the molecular basis of cell functional diversity. Nat. Rev. Mol. Cell Biol. 2020, 21, 327–340. [Google Scholar] [CrossRef]
- Verta, J.P.; Jacobs, A. The role of alternative splicing in adaptation and evolution. Trends Ecol. Evol. 2022, 37, 299–308. [Google Scholar] [CrossRef]
- Chen, L.; Bush, S.J.; Tovar-Corona, J.M.; Castillo-Morales, A.; Urrutia, A.O. Correcting for differential transcript coverage reveals a strong relationship between alternative splicing and organism complexity. Mol. Biol. Evol. 2014, 31, 1402–1413. [Google Scholar] [CrossRef]
- Graveley, B.R. Alternative splicing: Increasing diversity in the proteomic world. Trends Genet. TIG 2001, 17, 100–107. [Google Scholar] [CrossRef]
- Singh, P.; Börger, C.; More, H.; Sturmbauer, C. The Role of Alternative Splicing and Differential Gene Expression in Cichlid Adaptive Radiation. Genome Biol. Evol. 2017, 9, 2764–2781. [Google Scholar] [CrossRef]
- Singh, P.; Ahi, E.P. The importance of alternative splicing in adaptive evolution. Mol. Ecol. 2022, 31, 1928–1938. [Google Scholar] [CrossRef]
- Luo, M.; Hu, J. Alternative splicing in parallel evolution and the evolutionary potential in sticklebacks. J. Anim. Ecol. 2024, 93, 1392–1405. [Google Scholar] [CrossRef]
- Rodríguez-Ramírez, C.E.; Peichel, C.L. The Role of Alternative Splicing in Marine-Freshwater Divergence in Threespine Stickleback. Genome Biol. Evol. 2025, 17, evaf105. [Google Scholar] [CrossRef] [PubMed]
- Howes, T.R.; Summers, B.R.; Kingsley, D.M. Dorsal spine evolution in Threespine Sticklebacks via a splicing change in MSX2A. BMC Biol. 2017, 15, 115. [Google Scholar] [CrossRef] [PubMed]
- Tovar-Corona, J.M.; Castillo-Morales, A.; Chen, L.; Olds, B.P.; Clark, J.M.; Reynolds, S.E.; Pittendrigh, B.R.; Feil, E.J.; Urrutia, A.O. Alternative Splice in Alternative Lice. Mol. Biol. Evol. 2015, 32, 2749–2759. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.G.; Fernandes, M.; Profeta, C.A.; Barbosa, R.C.; Murdock, C.C.; Martins, G.F.; Mendes, T.O. Temperature-dependent alternative splicing affects gene expression in Aedes aegypti mosquitoes midgut. Insect Mol. Biol. 2025, 13002. [Google Scholar] [CrossRef]
- Ning, S.F.; Huo, L.X.; Lv, L.; Wang, Y.; Zhang, L.S.; Che, W.N.; Dong, H.; Zhou, J.C. The identification and expression pattern of the sex determination genes and their sex-specific variants in the egg parasitoid Trichogramma dendrolimi Matsumura (Hymenoptera: Trichogrammatidae). Front. Physiol. 2023, 14, 1243753. [Google Scholar] [CrossRef]
- Rogers, T.F.; Palmer, D.H.; Wright, A.E. Sex-Specific Selection Drives the Evolution of Alternative Splicing in Birds. Mol. Biol. Evol. 2021, 38, 519–530. [Google Scholar] [CrossRef]
- Jakšić, A.M.; Schlötterer, C. The Interplay of Temperature and Genotype on Patterns of Alternative Splicing in Drosophila melanogaster. Genetics 2016, 204, 315–325. [Google Scholar] [CrossRef]
- Grantham, M.E.; Brisson, J.A. Extensive Differential Splicing Underlies Phenotypically Plastic Aphid Morphs. Mol. Biol. Evol. 2018, 35, 1934–1946. [Google Scholar] [CrossRef]
- Jacobs, A.; Elmer, K.R. Alternative splicing and gene expression play contrasting roles in the parallel phenotypic evolution of a salmonid fish. Mol. Ecol. 2021, 30, 4955–4969. [Google Scholar] [CrossRef]
- Zhang, F.; Li, Q.; Chen, X.; Huo, Y.; Guo, H.; Song, Z.; Cui, F.; Zhang, L.; Fang, R. Roles of the Laodelphax striatellus Down syndrome cell adhesion molecule in Rice stripe virus infection of its insect vector. Insect Mol. Biol. 2016, 25, 413–421. [Google Scholar] [CrossRef]
- Sugimoto, T.N.; Fujii, T.; Kayukawa, T.; Sakamoto, H.; Ishikawa, Y. Expression of a doublesex homologue is altered in sexual mosaics of Ostrinia scapulalis moths infected with Wolbachia. Insect Biochem. Mol. Biol. 2010, 40, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Herran, B.; Sugimoto, T.N.; Watanabe, K.; Imanishi, S.; Tsuchida, T.; Matsuo, T.; Ishikawa, Y.; Kageyama, D. Cell-based analysis reveals that sex-determining gene signals in Ostrinia are pivotally changed by male-killing Wolbachia. PNAS Nexus 2023, 2, pgac293. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhang, X.; Herre, E.A.; McKey, D.; Machado, C.A.; Yu, W.B.; Cannon, C.H.; Arnold, M.L.; Pereira, R.A.S.; Ming, R.; et al. Genomic evidence of prevalent hybridization throughout the evolutionary history of the fig-wasp pollination mutualism. Nat. Commun. 2021, 12, 718. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Compton, S.G.; Liu, M.; Chen, X.Y. Fig trees at the northern limit of their range: The distributions of cryptic pollinators indicate multiple glacial refugia. Mol. Ecol. 2012, 21, 1687–1701. [Google Scholar] [CrossRef]
- Zhang, Q.; Tong, X.; Li, Y.Y.; Sun, Q.; Gao, Y.; Zhang, S.H.; Wang, R.; Chen, X.Y. Presence of cryptic species in host insects forms a hierarchical Wolbachia infection pattern. Entomol. Gen. 2022, 42, 571–578. [Google Scholar] [CrossRef]
- Hou, H.-X.; Zhao, D.; Xiao, J.-H.; Huang, D.-W. Transcriptomic Analysis Reveals the Sexually Divergent Host–Wolbachia Interaction Patterns in a Fig Wasp. Microorganisms 2021, 9, 288. [Google Scholar] [CrossRef]
- Xiao, J.H.; Wang, N.X.; Murphy, R.W.; Cook, J.; Jia, L.Y.; Huang, D.W. Wolbachia infection and dramatic intraspecific mitochondrial DNA divergence in a fig wasp. Evolution 2012, 66, 1907–1916. [Google Scholar] [CrossRef]
- Zhou, W.; Rousset, F.; O’Neill, S. Phylogeny and PCR-based classification of Wolbachia strains using wsp gene sequences. Proc. Biol. Sci. 1998, 265, 509–515. [Google Scholar] [CrossRef]
- O’Neill, S.L.; Giordano, R.; Colbert, A.M.; Karr, T.L.; Robertson, H.M. 16S rRNA phylogenetic analysis of the bacterial endosymbionts associated with cytoplasmic incompatibility in insects. Proc. Natl. Acad. Sci. USA 1992, 89, 2699–2702. [Google Scholar] [CrossRef]
- Jeyaprakash, A.; Hoy, M.A. Long PCR improves Wolbachia DNA amplification: Wsp sequences found in 76% of sixty-three arthropod species. Insect Mol. Biol. 2000, 9, 393–405. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Wu, T.D.; Watanabe, C.K. GMAP: A genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics 2005, 21, 1859–1875. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Foissac, S.; Sammeth, M. ASTALAVISTA: Dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007, 35, W297–W299. [Google Scholar] [CrossRef]
- Tang, A.D.; Soulette, C.M.; van Baren, M.J.; Hart, K.; Hrabeta-Robinson, E.; Wu, C.J.; Brooks, A.N. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat. Commun. 2020, 11, 1438. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Innes, P.A.; Goebl, A.M.; Smith, C.C.R.; Rosenberger, K.; Kane, N.C. Gene expression and alternative splicing contribute to adaptive divergence of ecotypes. Heredity 2024, 132, 120–132. [Google Scholar] [CrossRef]
- Huang, Y.; Lack, J.B.; Hoppel, G.T.; Pool, J.E. Parallel and population-specific gene regulatory evolution in cold-adapted fly populations. Genetics 2021, 218, iyab077. [Google Scholar] [CrossRef] [PubMed]
- Mohr, C.; Hartmann, B. Alternative splicing in Drosophila neuronal development. J. Neurogenet. 2014, 28, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Steward, R.A.; de Jong, M.A.; Oostra, V.; Wheat, C.W. Alternative splicing in seasonal plasticity and the potential for adaptation to environmental change. Nat. Commun. 2022, 13, 755. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.; Zeng, X.; Han, H.; Yang, Y.; Zhang, Y.; He, L. Alternative splicing of a carboxyl/choline esterase gene enhances the fenpropathrin tolerance of Tetranychus cinnabarinus. Insect Sci. 2023, 30, 1255–1266. [Google Scholar] [CrossRef]
- 55 Porter, J.; Sullivan, W. The cellular lives of Wolbachia. Nat. Rev. Microbiol. 2023, 21, 750–766. [Google Scholar] [CrossRef]
- Fallon, A.M. Effects of mimosine on Wolbachia in mosquito cells: Cell cycle suppression reduces bacterial abundance. In Vitro Cell. Dev. Biol. Anim. 2015, 51, 958–963. [Google Scholar] [CrossRef]
- Fallon, A.M. Mitotically inactivated mosquito cells support robust Wolbachia infection and replication. In Vitro Cell. Dev. Biol. Anim. 2022, 58, 780–787. [Google Scholar] [CrossRef]
- Fallon, A.M. From Mosquito Ovaries to Ecdysone; from Ecdysone to Wolbachia: One Woman’s Career in Insect Biology. Insects 2022, 13, 756. [Google Scholar] [CrossRef]
- Guo, Y.; Khan, J.; Zheng, X.Y.; Wu, Y. Wolbachia increase germ cell mitosis to enhance the fecundity of Laodelphax striatellus. Insect Biochem. Mol. Biol. 2020, 127, 103471. [Google Scholar] [CrossRef]
- Fast, E.M.; Toomey, M.E.; Panaram, K.; Desjardins, D.; Kolaczyk, E.D.; Frydman, H.M. Wolbachia enhance Drosophila stem cell proliferation and target the germline stem cell niche. Science 2011, 334, 990–992. [Google Scholar] [CrossRef]
- Imoto, Y.; Yoshida, Y.; Yagisawa, F.; Kuroiwa, H.; Kuroiwa, T. The cell cycle, including the mitotic cycle and organelle division cycles, as revealed by cytological observations. J. Electron Microsc. 2011, 60 (Suppl. S1), S117–S136. [Google Scholar] [CrossRef] [PubMed]
- Mills, M.K.; McCabe, L.G.; Rodrigue, E.M.; Lechtreck, K.F.; Starai, V.J. Wbm0076, a candidate effector protein of the Wolbachia endosymbiont of Brugia malayi, disrupts eukaryotic actin dynamics. PLoS Pathog. 2023, 19, e1010777. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, K.B.; Martin, M.; Lesser, C.F.; Isberg, R.R.; Newton, I.L. Identification and Characterization of a Candidate Wolbachia pipientis Type IV Effector That Interacts with the Actin Cytoskeleton. mBio 2016, 7, e00622-16. [Google Scholar] [CrossRef] [PubMed]
- Nevalainen, L.B.; Layton, E.M.; Newton, I.L.G. Wolbachia Promotes Its Own Uptake by Host Cells. Infect. Immun. 2023, 91, e0055722. [Google Scholar] [CrossRef]
- Newton, I.L.; Savytskyy, O.; Sheehan, K.B. Wolbachia utilize host actin for efficient maternal transmission in Drosophila melanogaster. PLoS Pathog. 2015, 11, e1004798. [Google Scholar] [CrossRef]
- Hinks, A.; Hawke, T.J.; Franchi, M.V.; Power, G.A. The importance of serial sarcomere addition for muscle function and the impact of aging. J. Appl. Physiol. 2023, 135, 375–393. [Google Scholar] [CrossRef]
- Baião, G.C.; Schneider, D.I.; Miller, W.J.; Klasson, L. The effect of Wolbachia on gene expression in Drosophila paulistorum and its implications for symbiont-induced host speciation. BMC Genom. 2019, 20, 465. [Google Scholar] [CrossRef]
- Evans, O.; Caragata, E.P.; McMeniman, C.J.; Woolfit, M.; Green, D.C.; Williams, C.R.; Franklin, C.E.; O’Neill, S.L.; McGraw, E.A. Increased locomotor activity and metabolism of Aedes aegypti infected with a life-shortening strain of Wolbachia pipientis. J. Exp. Biol. 2009, 212, 1436–1441. [Google Scholar] [CrossRef]
- Pietri, J.E.; DeBruhl, H.; Sullivan, W. The rich somatic life of Wolbachia. MicrobiologyOpen 2016, 5, 923–936. [Google Scholar] [CrossRef]
- Rohkin Shalom, S.; Weiss, B.; Lalzar, M.; Kaltenpoth, M.; Chiel, E. Abundance and Localization of Symbiotic Bacterial Communities in the Fly Parasitoid Spalangia cameroni. Appl. Environ. Microbiol. 2022, 88, e0254921. [Google Scholar] [CrossRef]
- Miyata, M.; Konagaya, T.; Yukuhiro, K.; Nomura, M.; Kageyama, D. Wolbachia-induced meiotic drive and feminization is associated with an independent occurrence of selective mitochondrial sweep in a butterfly. Biol. Lett. 2017, 13, 20170153. [Google Scholar] [CrossRef]
- Duron, O.; Weill, M. Wolbachia infection influences the development of Culex pipiens embryo in incompatible crosses. Heredity 2006, 96, 493–500. [Google Scholar] [CrossRef]
- Hussain, M.; Qi, Z.; Asgari, S. Interaction of the Wolbachia surface protein with a novel pro-viral protein from Aedes aegypti. mBio 2025, 16, e0148624. [Google Scholar] [CrossRef]
- Bi, J.; Wang, Y.F. The effect of the endosymbiont Wolbachia on the behavior of insect hosts. Insect Sci. 2020, 27, 846–858. [Google Scholar] [CrossRef]
- Russell, S.L.; Castillo, J.R.; Sullivan, W.T. Wolbachia endosymbionts manipulate the self-renewal and differentiation of germline stem cells to reinforce fertility of their fruit fly host. PLoS Biol. 2023, 21, e3002335. [Google Scholar] [CrossRef]
- Salisbury, S.J.; Delgado, M.L.; Dalziel, A.C. Alternative splicing: An overlooked mechanism contributing to local adaptation? Mol. Ecol. 2021, 30, 4951–4954. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Zheng, Y.; Yu, W.J.; Fang, Y.; Mao, B.; Wang, Y.F. How do Wolbachia modify the Drosophila ovary? New evidences support the “titration-restitution” model for the mechanisms of Wolbachia-induced CI. BMC Genom. 2019, 20, 608. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Lu, Z.; Ma, Y.; Song, X.; Wang, D.; Wu, C.; Ma, X.; Shan, Y.; Ren, X.; Ma, Y. Impact of transinfection of Wolbachia from the planthopper Laodelphax striatellus on reproductive fitness and transcriptome of the whitefly Bemisia tabaci. J. Invertebr. Pathol. 2024, 207, 108230. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.C.; Wang, X.; Deng, C.; Yun, Y.L.; Zhao, Y.; Peng, Y. Transcriptome responses to elevated CO2 level and Wolbachia-infection stress in Hylyphantes graminicola (Araneae: Linyphiidae). Insect Sci. 2020, 27, 908–920. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).