1. Introduction
Propyl gallate (
n-propyl gallate,
n-propyl 3,4,5-trihydroxybenzoate, PG, compound
III, CAS Registry number: 121-79-9) is an important synthetic substance widely used in cosmetics, foods, pharmaceuticals, and some other fields [
1,
2,
3]. It is used as an effective antioxidant in cosmetics to stabilize vitamins, essential oils, perfumes, as well as fats and oils [
1]. In foods, PG has been employed as an additive (E310) since 1948 to protect fats, oils, and fat-containing food from peroxide-induced rancidity [
1]. At a concentration of up to 0.1%, PG is used as a strong preservative and stabilizer in various medicinal preparations approved by FDA [
4,
5]. In addition to its antioxidant activity, PG exhibits anti-inflammatory, anti-angiogenic, and anti-tumor properties [
2,
3,
4,
5,
6].
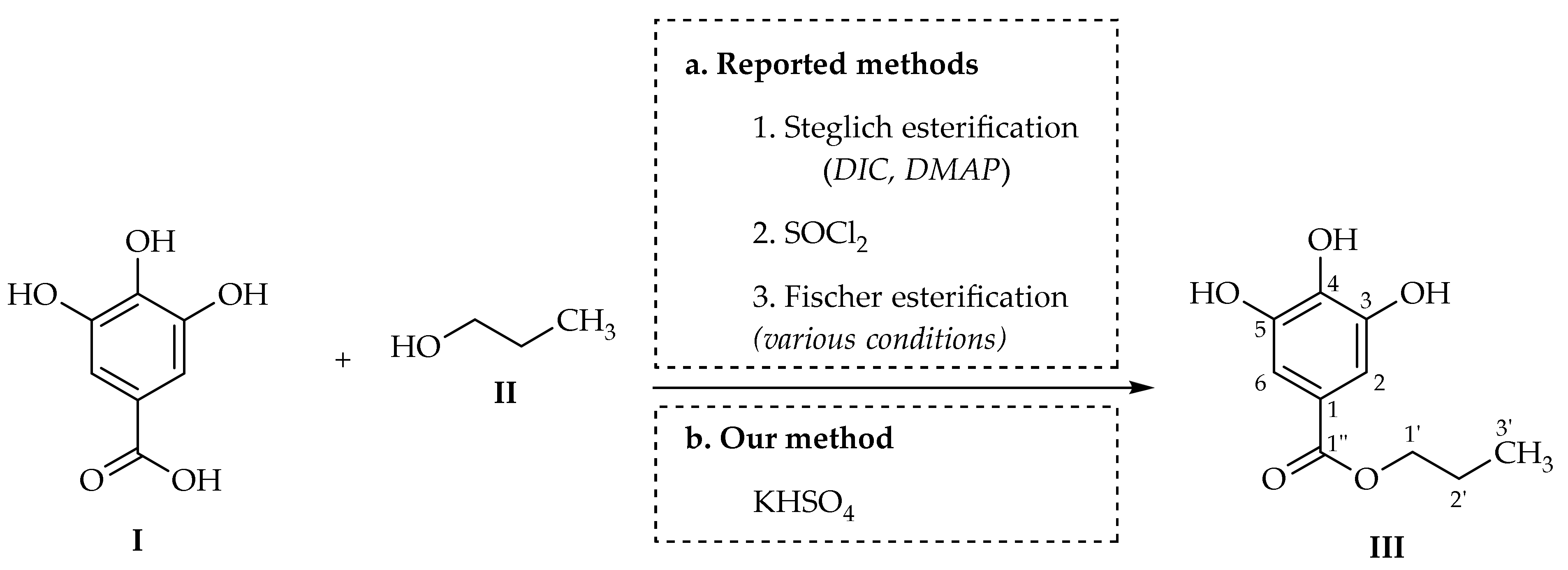
PG is not a natural compound and can only be obtained via chemical synthesis. In practice, PG is prepared by either biological (enzymatic) or chemical methods [
2,
7,
8,
9,
10,
11,
12,
13]. The latter have been dominant, focusing on the reaction between gallic acid (
I) and
n-propanol (propan-1-ol,
II) in different conditions as illustrated in
Scheme 1 [
1,
2,
3,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23,
24,
25,
26,
27].
Experimentally, there are three main approaches to form PG from gallic acid and
n-propanol. The first approach is via a typical Steglich esterification with
N,
N′-di
isopropylcarbodiimide (DIC) as the coupling reagent and 4-dimethylaminopyridine (DMAP) as the catalyst [
14]. However, urea generated by DIC as a by-product can sometimes be difficult to remove, especially when scaling up. In the second approach, thionyl chloride is used as an additive to convert gallic acid to galloyl chloride which in turn readily reacts in situ with
n-propanol to form PG [
15,
16,
17]. This approach requires rigorously anhydrous conditions, i.e., dried gallic acid, freshly distilled thionyl chloride and total exclusion of water or the use of anhydrous solvents. The third approach utilizes direct Fischer esterification in the presence of various homogenous and heterogenous catalysts, including concentrated sulfuric acid (H
2SO
4),
p-toluenesulfonic acid (
p-TsOH),
p-toluenesulfonic acid and sulfamic acid (
p-TsOH + H
2NSO
3H), perchloric acid (HClO
4), perchloric acid and sulfamic acid (HClO
4 + H
2NSO
3H), ionic liquid
N-methyl-2-pyrrolidonium hydrogensulfate ([Hnmp]HSO
4), brominated sulfonic acid resin, mordenite (a zeolite mineral with orthorhombic structure containing calcium, sodium, potassium, aluminum, and silicate), and tetramethyl cucurbit[6]uril-phosphomolybdic acid (TMeQ [6]-PMA) (
Table 1) [
18,
19,
20,
21,
22,
23,
24,
25,
26,
27].
In recent years, the study and development of heterogenous catalysts has received significant interest in different areas of organic transformations including the synthesis of PG. Heterogenous catalysts obtain many noticeable advantages, including their reusability, higher reaction rate and selectivity, easy product/catalyst separation, and affordability [
28,
29,
30,
31]. Among them, potassium hydrogen sulfate (potassium bisulfate, KHSO
4) has emerged as an inexpensive, green, non-toxic, and easy to handle catalyst displaying high level of efficiency and reusability in many organic preparations [
32]. However, the synthesis of PG using KHSO
4 as a catalyst has not been yet reported. In relation to the spectroscopic characterization of PG, the current data are not complete and restricted to MS, IR,
1H, and
13C NMR data [
33,
34,
35], while data on the pharmacological studies and physicochemical properties have been constantly updated [
34,
35,
36].
Herein, we report an efficient, convenient, and economical synthetic procedure to obtain PG by Fischer esterification using KHSO4 as the sole heterogenous catalyst. At the same time, we provide a more complete spectral data set updating missing data on DEPT and 2D NMR (HSQC and HMBC) spectra for this compound.
2. Results and Discussion
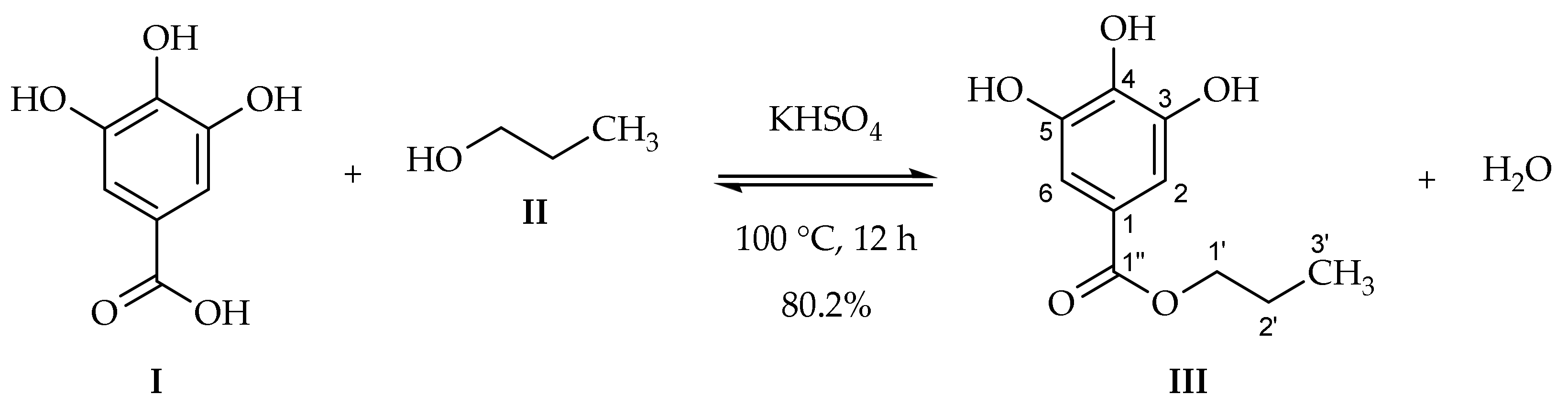
The target compound
III was synthesized in 80.2% of yield from gallic acid monohydrate (
I) and
n-propanol (
II) using potassium hydrogen sulfate as the heterogenous acidic catalyst (
Scheme 2).
A thorough search in Reaxys and SciFinder databases has returned a range of different methods for the preparation of PG. These methods are classified according to the usage of chemical catalysts or additives and summarized in
Table 1. Our synthetic procedure was based on [
2,
25,
26] in entries 3, 9, and 10 of
Table 1. These methods were straightforward and offered a green pathway to obtain the final products without a using additional organic solvents and column chromatography.
Using traditional catalysts such as concentrated sulfuric acid (H
2SO
4),
p-toluenesulfonic acid (
p-TsOH), and perchloric acid (HClO
4), its mixture with sulfamic acid (H
2NSO
3H) afforded PG in moderate to excellent (56.5–94.0%, entries 3–7,
Table 1) [
18,
19,
20,
21,
22,
23,
24]. However, these reactive catalysts can cause equipment corrosion problems, especially on a large scale. In addition, their use was usually accompanied by toxic and expensive solvents such as dichloromethane and benzene, as well as column chromatography to purify final products [
19,
20,
21,
24]. In [
2], ionic liquid
N-methyl-2-pyrrolidonium hydrogensulfate ([Hnmp]HSO
4) was used as an efficient catalyst to afford PG in 89.8% of yield (entry 8,
Table 1). However, the preparation of this ionic liquid is quite complicated, and its regeneration requires high energy to evaporate water from the filtrate. Recently, three [
25,
26,
27] have demonstrated the synthesis of PG with excellent yield (up to 98.0%) and purity (up to 99.95%) using heterogenous catalysts, i.e. modified sulfonic acid resin, mordenite, and tetramethyl cucurbit[6]uril-phosphomolybdic acid (entries 9–11,
Table 1). However, these catalysts needed to be activated before any reaction using hard conditions such as chlorination, bromination [
25], calcination at 400–500 °C under inert atmosphere [
26], or fabricated into nanocubes [
27].
Although KHSO
4 has been widely used in various organic transformations [
32,
37,
38], there are few publications on its application for Fischer esterification. One of the most related example was mentioned in [
39], in which it was used as an efficient catalyst for the preparation of butyl paraben in 92.9% yield from
p-hydroxybenzoic acid and
n-butyl alcohol.
Our experiments have showed that KHSO
4 can be used as an inexpensive, eco-friendly, and easy to handle catalyst for synthesizing PG under mild conditions. The optimized reaction conditions were investigated and presented in
Table 2,
Table 3,
Table 4,
Table 5 and
Table 6.
We first screened the influence of temperature on PG yield (
Table 2). Increasing temperature from 70 to 100 °C resulted in an increase of yield from 51.2 to 68.1%. However, temperatures above 100 °C seemed to have a detrimental effect on the yield (62.9%, entry 4,
Table 2). We thus maintained the temperature at 100 °C for further reaction optimization. Second, in relation to the reaction time, we found that there was a slight change in product yield when running the reaction for 10 to 12 h (64.0–67.9%. entries 1–3,
Table 3). Longer reaction time of up to 13 and 14 h returned 68.1% of product (entries 4 and 5,
Table 3). This indicated that the reaction has reached a plateau and longer reaction time does not improve the yield. As there was only a slight change in product yield between 12 and 13 h reacting time (67.9% vs. 68.1%, entries 3 and 4,
Table 3), and no starting material was detected by TLC after 12 h, we decided that 12 h was the standard reacting time for our protocol. We then investigated the suitable mole ratio between
n-propanol and gallic acid, and found that the ratio (12.0:1, entry 4,
Table 4) offered the best product yield of 72.2%. Finally, we screened for the suitable equivalent of KHSO
4 to be used in this reaction, and identified the mole ratio of 0.4:1 (KHSO
4: gallic acid, entry 4,
Table 5) to be the one affording the best product yield of 80.2%.
Notably, the recovery of the KHSO
4 was straightforward by filtration or decantation following by drying at 100 °C for 3 h. Up to 85.0% of dried KHSO
4 was recovered after the first use and it was further reused for three consecutive times with only slight variation in the yields of the final product (data not shown). The reaction work-up was performed as indicated in [
2,
18,
25,
26], including distillation (under or without vacuum condition), crystallization/precipitation in water and subsequent filtration to remove water-soluble impurities (starting gallic acid and a small amount of the catalyst dissolved in the filtrate). Using this protocol at a larger scale of 26.58 mmol gallic acid returned the product with similar yield (80.2%). This yield is comparable to those reported in the literature using traditional catalysts [
2,
18,
19,
21,
22,
23,
24]. The purity of the product was confirmed by differential scanning calorimetry (DSC) as one of the most widely used thermal analysis technique in the chemical and pharmaceutical industries. The purity was 99.60% without an additional refining, which both met the requirements of US Pharmacopeia (98.0–102.0%) [
40] and the European Pharmacopoeia (97.0–103.0%) [
41].
MS, FT-IR,
1H- And
13C-NMR data for compound
III were in entire accordance with those reported in the literature [
2,
14,
15,
21,
23,
27,
33]. HR-MS data showed a molecular ion peak at
m/
z 211.0622 ([M-H]
−), which indicated molecular formula of PG C
10H
12O
5. The absorption band on the infrared spectrum, 1687 cm
−1, demonstrated the presence of ester C=O group in compound
III. A characteristic triplet (H-3’, δ 0.95 ppm), a multiplet (H-2’, δ 1.67 ppm) and a triplet (H-1’, δ 4.11 ppm) splitting pattern and integration indicated that propyl esterification had successfully taken place. The signals of three hydroxyl protons were overlapped as a broad and unsymmetrical singlet at downfield 9.19 ppm. One aromatic singlet at 6.96 ppm with the integral of 2 demonstrated two characteristic symmetrical aromatic protons (H-2 and H-6). The results were also supported by the
13C signals at 165.9 (C-1’), 65.4 (C-1’), 21.7 (C-2’), and 10.4 ppm (C-3’), which verified the success of the esterification.
All new obtained data by DEPT and 2D NMR experiments are in entire agreement with the structure of the title compound
III (
Table 7). In addition, powder X-ray diffraction (PXRD) data were also collected as an effort to provide a comprehensive spectral data set for a polymorphic form of the title compound
III obtained by our method (
Table 8).
This method of obtaining PG can be useful for extensive research on the application of potassium hydrogen sulfate as a catalyst in Fischer esterification of a wide range of carboxylic acids, especially at industrial scale, due to its feasibility, environment friendliness, economy and affordability. In addition, the updated 2D-NMR and PXRD data can support studies on PG metabolites in various medical and pharmaceutical studies of this compound.


 ,
, 




{kind=link}
{kind=link}