
JNK Inhibition Overcomes Resistance of Metastatic Tetraploid Cancer Cells to Irradiation-Induced Apoptosis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
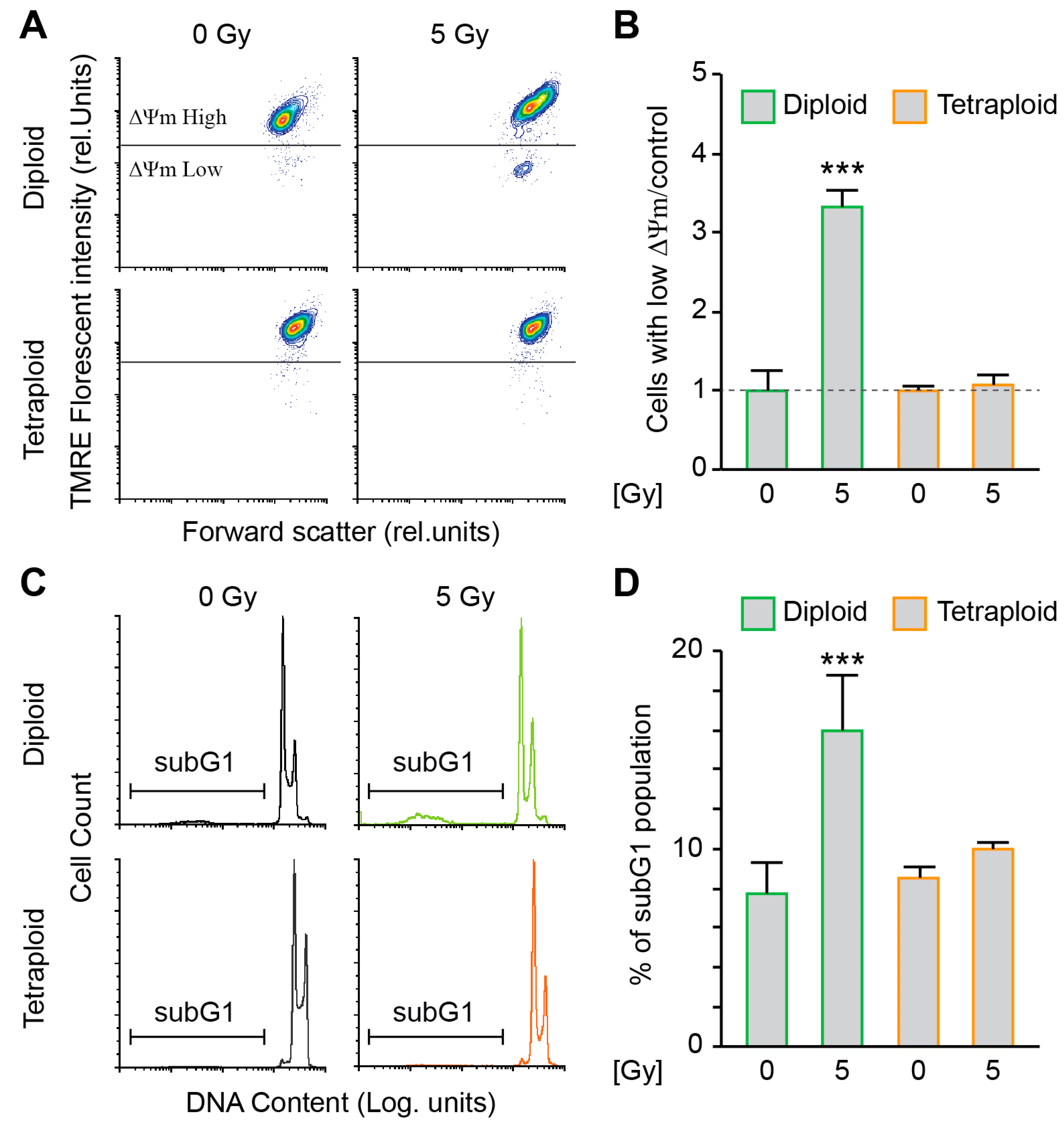
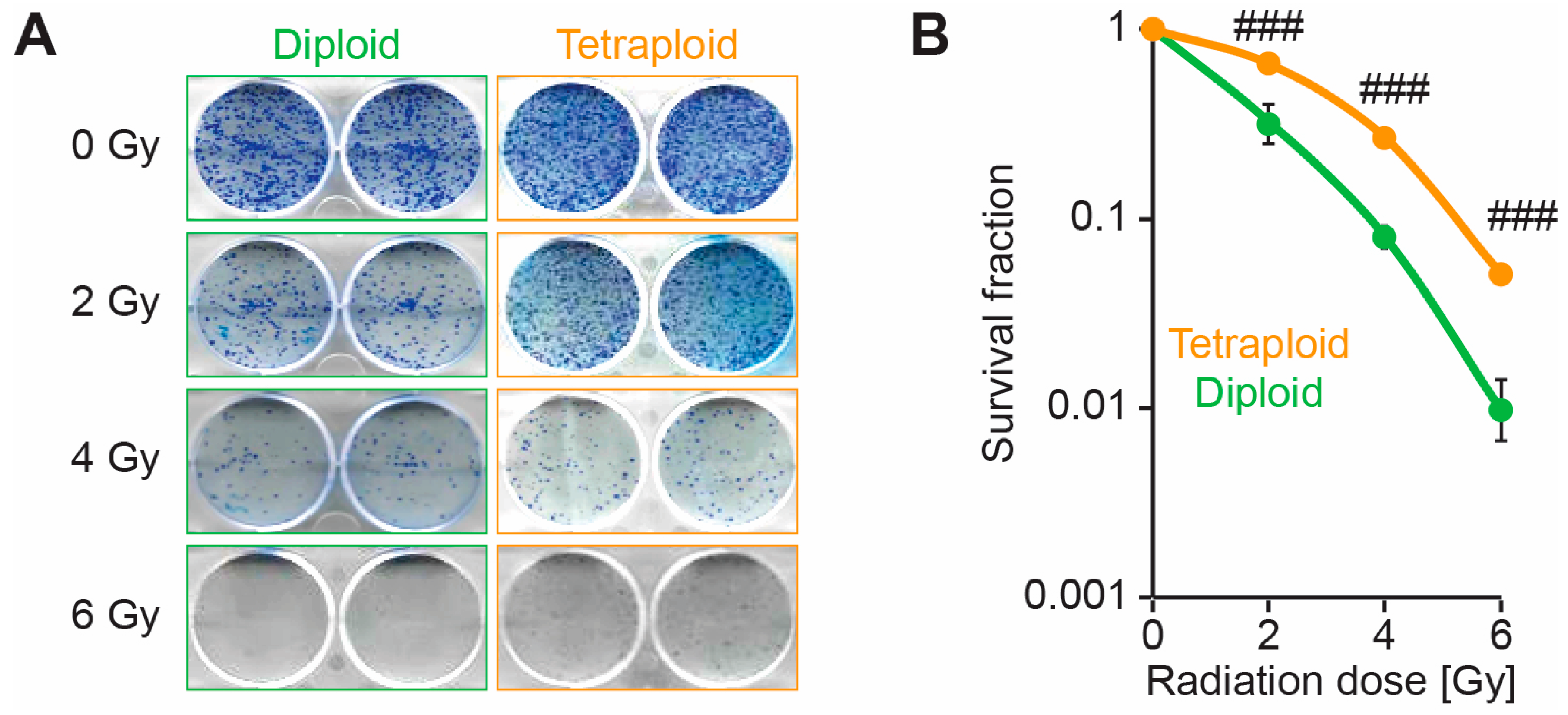
2.1. Metastatic Tetraploid Cells Are More Resistant to DNA Damage Induced by Radiation
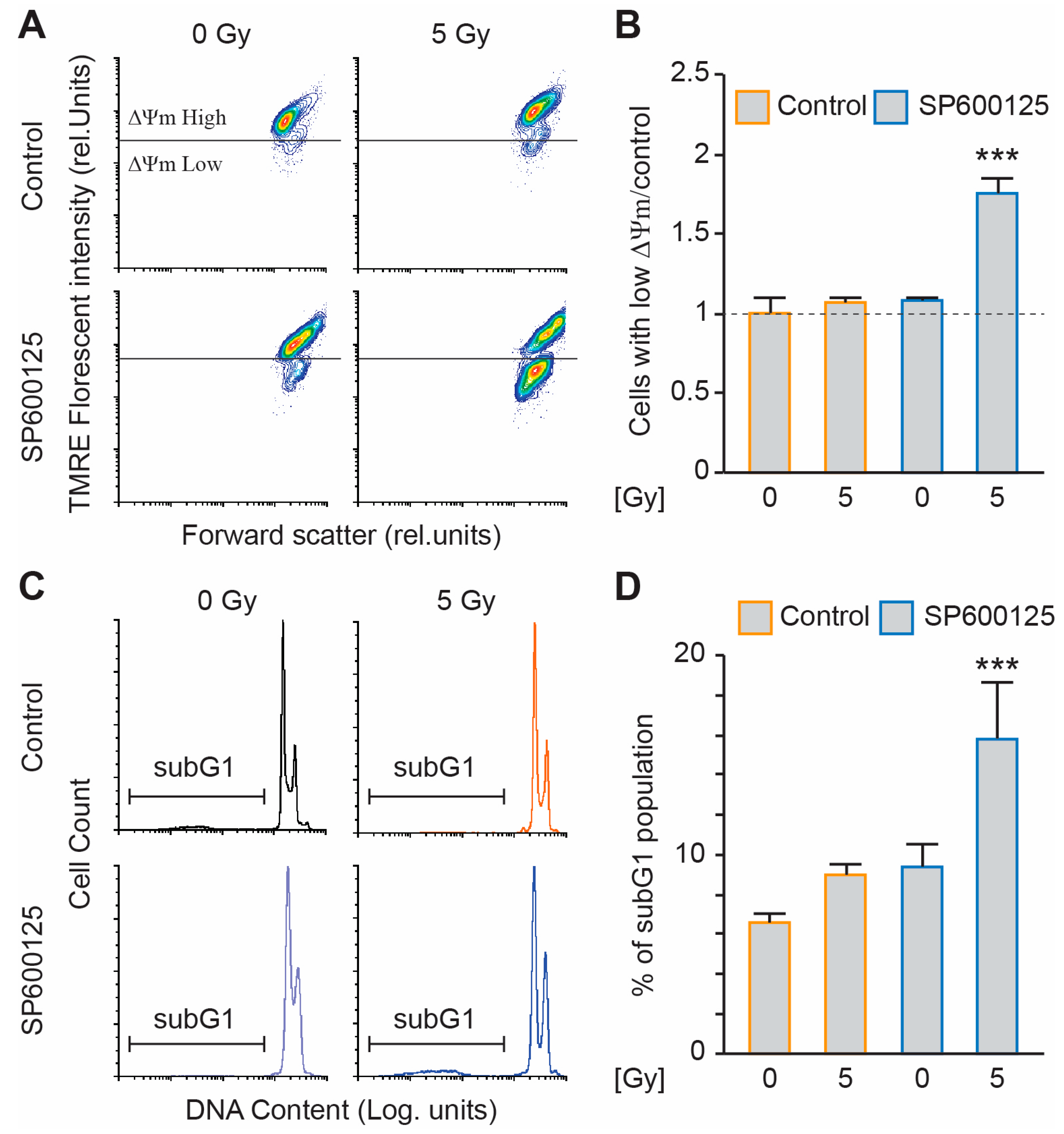
2.2. SP600125 Administration Sensitises Metastatic Tetraploid Cells to Radiation
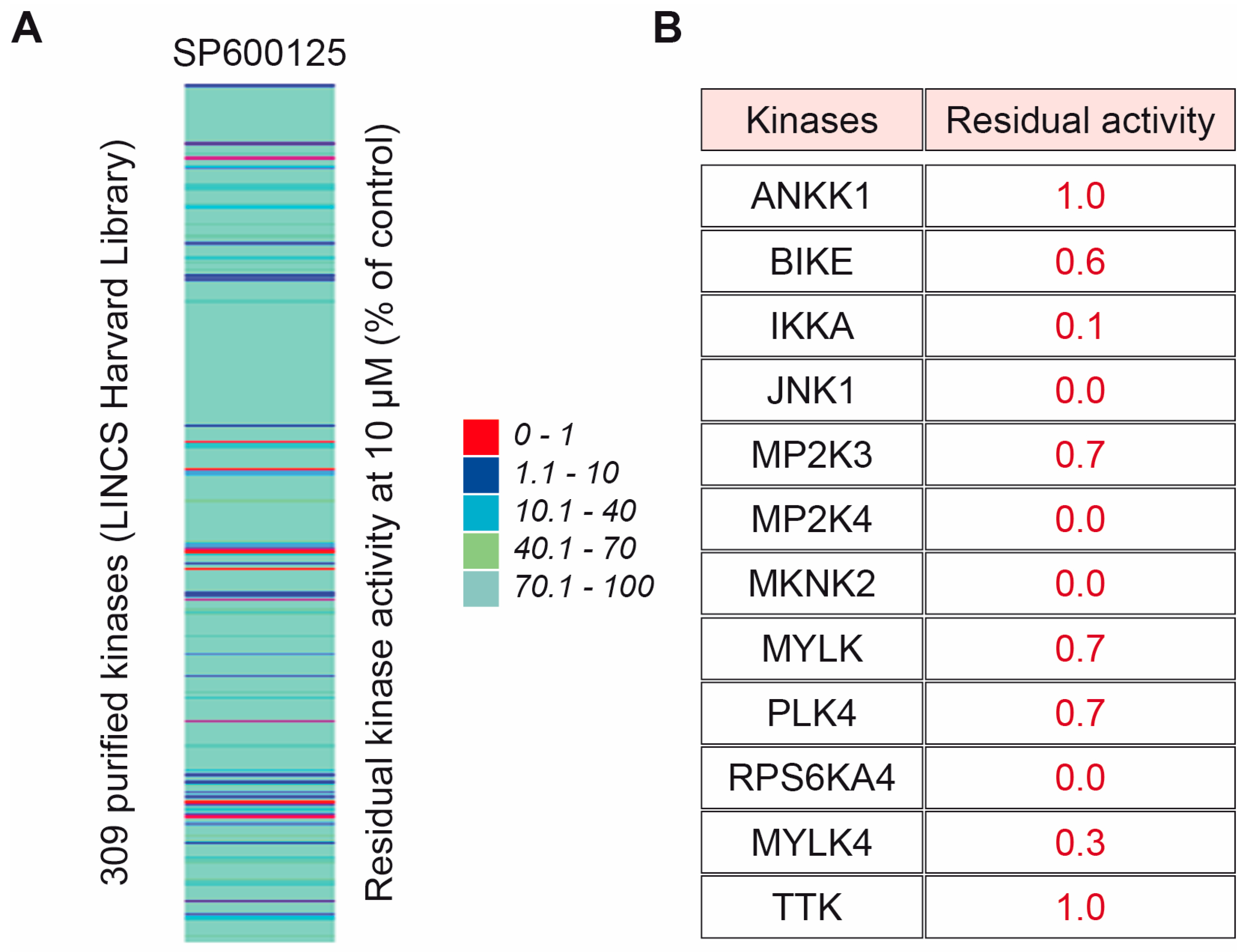
2.3. SP600125 Target the JNK Pathway to Sensitise Metastatic Tetraploid Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Culture Conditions and Reagents
4.2. Ionising Radiation (IR)
4.3. Clonogenic Survival Assay
4.4. Cytofluorometric Studies
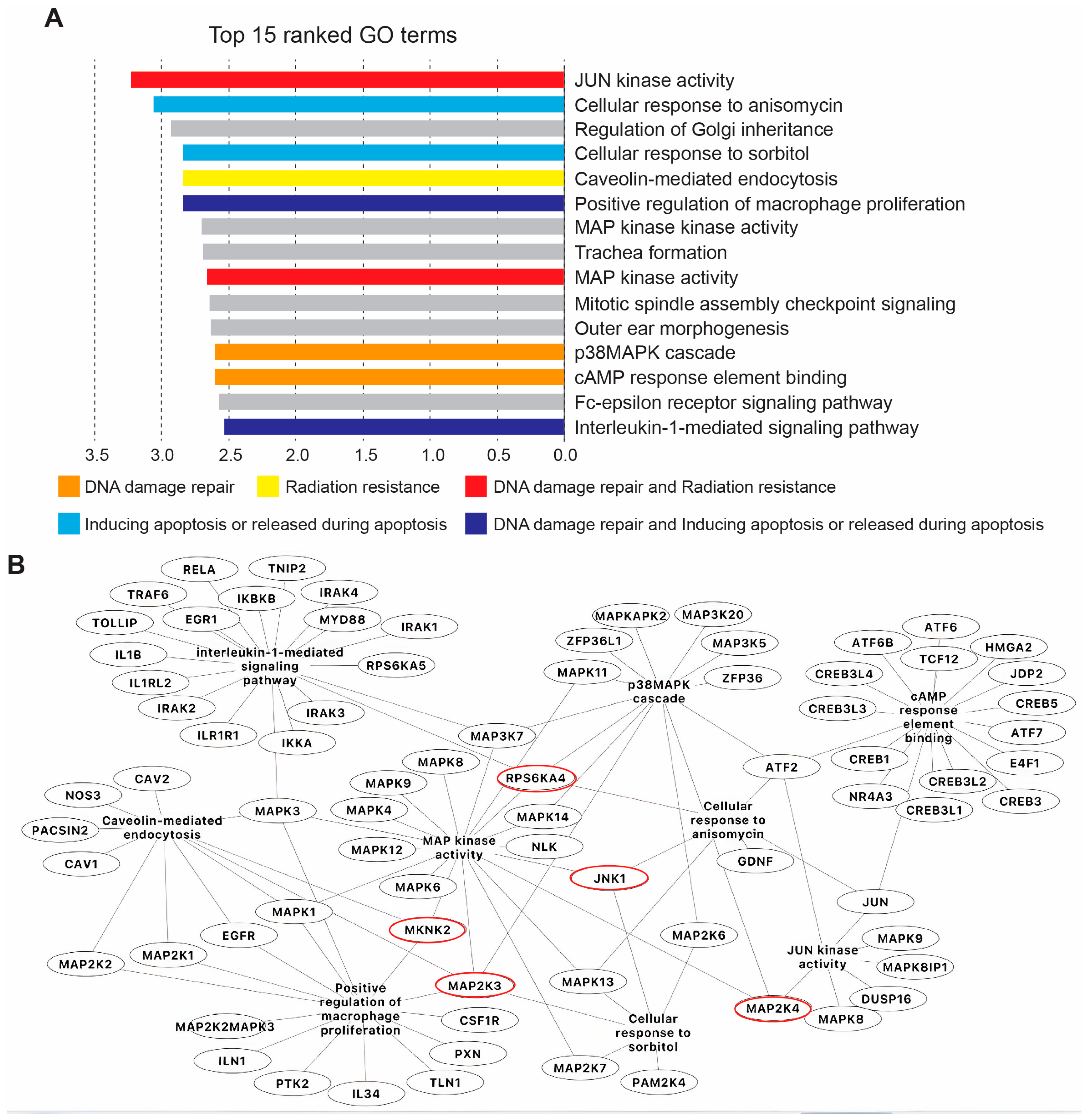
4.5. Pathway Enrichment Analysis and Network Construction
4.6. Statistical Procedures
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Storchova, Z.; Pellman, D. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 2004, 5, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Manic, G.; Senovilla, L.; Kroemer, G.; Galluzzi, L. Karyotypic Aberrations in Oncogenesis and Cancer Therapy. Trends Cancer 2015, 1, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Galluzzi, L.; Senovilla, L.; Criollo, A.; Jemaa, M.; Castedo, M.; Kroemer, G. Illicit survival of cancer cells during polyploidization and depolyploidization. Cell Death Differ. 2011, 18, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Storchova, Z.; Kuffer, C. The consequences of tetraploidy and aneuploidy. J. Cell Sci. 2008, 121, 3859–3866. [Google Scholar] [CrossRef]
- Ganem, N.J.; Storchova, Z.; Pellman, D. Tetraploidy, aneuploidy and cancer. Curr. Opin. Genet. Dev. 2007, 17, 157–162. [Google Scholar] [CrossRef]
- Duelli, D.; Lazebnik, Y. Cell fusion: A hidden enemy? Cancer Cell 2003, 3, 445–448. [Google Scholar] [CrossRef]
- Pawelek, J.M. Tumour-cell fusion as a source of myeloid traits in cancer. Lancet Oncol. 2005, 6, 988–993. [Google Scholar] [CrossRef]
- Senovilla, L.; Vitale, I.; Galluzzi, L.; Vivet, S.; Joza, N.; Younes, A.B.; Rello-Varona, S.; Castedo, M.; Kroemer, G. p53 represses the polyploidization of primary mammary epithelial cells by activating apoptosis. Cell Cycle 2009, 8, 1380–1385. [Google Scholar] [CrossRef]
- Vitale, I.; Jemaa, M.; Senovilla, L.; Galluzzi, L.; Rello-Varona, S.; Metivier, D.; Ripoche, H.; Lazar, V.; Dessen, P.; Castedo, M.; et al. Involvement of p38alpha in the mitotic progression of p53(-/-) tetraploid cells. Cell Cycle 2010, 9, 2823–2829. [Google Scholar] [CrossRef]
- Vitale, I.; Senovilla, L.; Jemaa, M.; Michaud, M.; Galluzzi, L.; Kepp, O.; Nanty, L.; Criollo, A.; Rello-Varona, S.; Manic, G.; et al. Multipolar mitosis of tetraploid cells: Inhibition by p53 and dependency on Mos. EMBO J. 2010, 29, 1272–1284. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Jemaa, M.; Daams, R.; Charfi, S.; Mertens, F.; Huber, S.M.; Massoumi, R. Tetraploidization Increases the Motility and Invasiveness of Cancer Cells. Int. J. Mol. Sci. 2023, 24, 13926. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Coquelle, A.; Vitale, I.; Vivet, S.; Mouhamad, S.; Viaud, S.; Zitvogel, L.; Kroemer, G. Selective resistance of tetraploid cancer cells against DNA damage-induced apoptosis. Ann. N. Y. Acad. Sci. 2006, 1090, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Dai, H.; Zhou, M.; Li, X.; Liu, C.; Guo, Z.; Wu, X.; Wu, J.; Wang, C.; Zhong, J.; et al. Polyploid cells rewire DNA damage response networks to overcome replication stress-induced barriers for tumour progression. Nat. Commun. 2012, 3, 815. [Google Scholar] [CrossRef]
- Galofre, C.; Gonul Geyik, O.; Asensio, E.; Wangsa, D.; Hirsch, D.; Parra, C.; Saez, J.; Molla, M.; Yuce, Z.; Castells, A.; et al. Tetraploidy-Associated Genetic Heterogeneity Confers Chemo-Radiotherapy Resistance to Colorectal Cancer Cells. Cancers 2020, 12, 1118. [Google Scholar] [CrossRef]
- Zhou, T.; Zhang, L.Y.; He, J.Z.; Miao, Z.M.; Li, Y.Y.; Zhang, Y.M.; Liu, Z.W.; Zhang, S.Z.; Chen, Y.; Zhou, G.C.; et al. Review: Mechanisms and perspective treatment of radioresistance in non-small cell lung cancer. Front. Immunol. 2023, 14, 1133899. [Google Scholar] [CrossRef]
- Jemaa, m. Mitotic Spindle as Therapeutic Target for Tetraploid Cancer Cells. Eurasian J. Med. Oncol. 2021, 5, 205–208. [Google Scholar] [CrossRef]
- Schmidt, M.; Budirahardja, Y.; Klompmaker, R.; Medema, R.H. Ablation of the spindle assembly checkpoint by a compound targeting Mps1. EMBO Rep. 2005, 6, 866–872. [Google Scholar] [CrossRef]
- Chu, M.L.; Chavas, L.M.; Douglas, K.T.; Eyers, P.A.; Tabernero, L. Crystal structure of the catalytic domain of the mitotic checkpoint kinase Mps1 in complex with SP600125. J. Biol. Chem. 2008, 283, 21495–21500. [Google Scholar] [CrossRef]
- Jemaa, M.; Vitale, I.; Kepp, O.; Berardinelli, F.; Galluzzi, L.; Senovilla, L.; Marino, G.; Malik, S.A.; Rello-Varona, S.; Lissa, D.; et al. Selective killing of p53-deficient cancer cells by SP600125. EMBO Mol. Med. 2012, 4, 500–514. [Google Scholar] [CrossRef]
- Kusakabe, K.; Ide, N.; Daigo, Y.; Tachibana, Y.; Itoh, T.; Yamamoto, T.; Hashizume, H.; Hato, Y.; Higashino, K.; Okano, Y.; et al. Indazole-based potent and cell-active Mps1 kinase inhibitors: Rational design from pan-kinase inhibitor anthrapyrazolone (SP600125). J. Med. Chem. 2013, 56, 4343–4356. [Google Scholar] [CrossRef] [PubMed]
- Jemaa, M.; Galluzzi, L.; Kepp, O.; Castedo, M.; Rello-Varona, S.; Vitale, I.; Kroemer, G. Transgenerational cell fate profiling: A method for the graphical presentation of complex cell cycle alterations. Cell Cycle 2013, 12, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Parashar, B.; Chen, W.C.; Herman, J.M.; Potters, L. Disease Site-Specific Guidelines for Curative Radiation Treatment During ‘Limited Surgery’ and ‘Hospital Avoidance’: A Radiation Oncology Perspective From the Epicenter of COVID-19 Pandemic. Cureus 2020, 12, e8190. [Google Scholar] [CrossRef] [PubMed]
- Picco, V.; Pages, G. Linking JNK Activity to the DNA Damage Response. Genes. Cancer 2013, 4, 360–368. [Google Scholar] [CrossRef]
- Yacoub, A.; Park, J.S.; Qiao, L.; Dent, P.; Hagan, M.P. MAPK dependence of DNA damage repair: Ionizing radiation and the induction of expression of the DNA repair genes XRCC1 and ERCC1 in DU145 human prostate carcinoma cells in a MEK1/2 dependent fashion. Int. J. Radiat. Biol. 2001, 77, 1067–1078. [Google Scholar] [CrossRef]
- Phong, M.S.; Van Horn, R.D.; Li, S.; Tucker-Kellogg, G.; Surana, U.; Ye, X.S. p38 mitogen-activated protein kinase promotes cell survival in response to DNA damage but is not required for the G(2) DNA damage checkpoint in human cancer cells. Mol. Cell Biol. 2010, 30, 3816–3826. [Google Scholar] [CrossRef]
- Cohen, I.; Rider, P.; Vornov, E.; Tomas, M.; Tudor, C.; Wegner, M.; Brondani, L.; Freudenberg, M.; Mittler, G.; Ferrando-May, E.; et al. IL-1alpha is a DNA damage sensor linking genotoxic stress signaling to sterile inflammation and innate immunity. Sci. Rep. 2015, 5, 14756. [Google Scholar] [CrossRef]
- de Sousa, M.M.L.; Bjoras, K.O.; Hanssen-Bauer, A.; Solvang-Garten, K.; Otterlei, M. p38 MAPK signaling and phosphorylations in the BRCT1 domain regulate XRCC1 recruitment to sites of DNA damage. Sci. Rep. 2017, 7, 6322. [Google Scholar] [CrossRef]
- Lee, A.J.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Birkbak, N.J.; Futreal, P.A.; Downward, J.; Szallasi, Z.; Tomlinson, I.P.; Howell, M.; et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011, 71, 1858–1870. [Google Scholar] [CrossRef]
- Kuznetsova, A.Y.; Seget, K.; Moeller, G.K.; de Pagter, M.S.; de Roos, J.A.; Durrbaum, M.; Kuffer, C.; Muller, S.; Zaman, G.J.; Kloosterman, W.P.; et al. Chromosomal instability, tolerance of mitotic errors and multidrug resistance are promoted by tetraploidization in human cells. Cell Cycle 2015, 14, 2810–2820. [Google Scholar] [CrossRef]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef] [PubMed]
- Jemaa, M.; Abassi, Y.; Kifagi, C.; Fezai, M.; Daams, R.; Lang, F.; Massoumi, R. Reversine inhibits Colon Carcinoma Cell Migration by Targeting JNK1. Sci. Rep. 2018, 8, 11821. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Moon, D.O.; Kim, M.O.; Kang, C.H.; Lee, J.D.; Choi, Y.H.; Kim, G.Y. JNK inhibitor SP600125 promotes the formation of polymerized tubulin, leading to G2/M phase arrest, endoreduplication, and delayed apoptosis. Exp. Mol. Med. 2009, 41, 665–677. [Google Scholar] [CrossRef]
- Mili, D.; Abid, K.; Rjiba, I.; Kenani, A. Effect of SP600125 on the mitotic spindle in HeLa Cells, leading to mitotic arrest, endoreduplication and apoptosis. Mol. Cytogenet. 2016, 9, 86. [Google Scholar] [CrossRef]
- Grassi, E.S.; Vezzoli, V.; Negri, I.; Labadi, A.; Fugazzola, L.; Vitale, G.; Persani, L. SP600125 has a remarkable anticancer potential against undifferentiated thyroid cancer through selective action on ROCK and p53 pathways. Oncotarget 2015, 6, 36383–36399. [Google Scholar] [CrossRef]
- Nickoloff, J.A.; Boss, M.K.; Allen, C.P.; LaRue, S.M. Translational research in radiation-induced DNA damage signaling and repair. Transl. Cancer Res. 2017, 6, S875–S891. [Google Scholar] [CrossRef]
- Vaishnav, D.; Jambal, P.; Reusch, J.E.; Pugazhenthi, S. SP600125, an inhibitor of c-jun N-terminal kinase, activates CREB by a p38 MAPK-mediated pathway. Biochem. Biophys. Res. Commun. 2003, 307, 855–860. [Google Scholar] [CrossRef]
- Yan, H.; He, L.; Lv, D.; Yang, J.; Yuan, Z. The Role of the Dysregulated JNK Signaling Pathway in the Pathogenesis of Human Diseases and Its Potential Therapeutic Strategies: A Comprehensive Review. Biomolecules 2024, 14, 243. [Google Scholar] [CrossRef]
- Lee, C.H.; Sidik, K.; Chin, K.V. Role of cAMP-dependent protein kinase in the regulation of DNA repair. Cancer Lett. 2001, 169, 51–58. [Google Scholar] [CrossRef]
- Li, C.H.; Lim, S.H.; Ryu, H.H.; Moon, K.S.; Jung, T.Y.; Jung, S. Enhancement of radiosensitivity by inhibition of c-Jun N-terminal kinase activity in a Lewis lung carcinoma-bearing subcutaneous tumor mouse model. Oncol. Rep. 2016, 36, 3397–3404. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Lim, S.H.; Jeong, Y.I.; Ryu, H.H.; Jung, S. Synergistic Effects of Radiotherapy With JNK Inhibitor-Incorporated Nanoparticle in an Intracranial Lewis Lung Carcinoma Mouse Models. IEEE Trans. Nanobiosci. 2023, 22, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.Y.; Clark, J.J.; Telisak, M.; Hansen, M.R. Inhibition of c-Jun N-terminal kinase activity enhances vestibular schwannoma cell sensitivity to gamma irradiation. Neurosurgery 2013, 73, 506–516. [Google Scholar] [CrossRef]
- Werwein, E.; Dzuganova, M.; Usadel, C.; Klempnauer, K.H. B-Myb switches from Cyclin/Cdk-dependent to Jnk- and p38 kinase-dependent phosphorylation and associates with SC35 bodies after UV stress. Cell Death Dis. 2013, 4, e511. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jemaà, M.; Setti Boubaker, N.; Kerkeni, N.; M. Huber, S. JNK Inhibition Overcomes Resistance of Metastatic Tetraploid Cancer Cells to Irradiation-Induced Apoptosis. Int. J. Mol. Sci. 2025, 26, 1209. https://doi.org/10.3390/ijms26031209
Jemaà M, Setti Boubaker N, Kerkeni N, M. Huber S. JNK Inhibition Overcomes Resistance of Metastatic Tetraploid Cancer Cells to Irradiation-Induced Apoptosis. International Journal of Molecular Sciences. 2025; 26(3):1209. https://doi.org/10.3390/ijms26031209
Chicago/Turabian StyleJemaà, Mohamed, Nouha Setti Boubaker, Nesrine Kerkeni, and Stephan M. Huber. 2025. "JNK Inhibition Overcomes Resistance of Metastatic Tetraploid Cancer Cells to Irradiation-Induced Apoptosis" International Journal of Molecular Sciences 26, no. 3: 1209. https://doi.org/10.3390/ijms26031209
APA StyleJemaà, M., Setti Boubaker, N., Kerkeni, N., & M. Huber, S. (2025). JNK Inhibition Overcomes Resistance of Metastatic Tetraploid Cancer Cells to Irradiation-Induced Apoptosis. International Journal of Molecular Sciences, 26(3), 1209. https://doi.org/10.3390/ijms26031209