Abstract
Limb-Girdle Muscular Dystrophies (LGMDs) are genetically heterogeneous disorders primarily affecting proximal limb muscles. The most common form, LGMDR1, results from biallelic CAPN3 mutations encoding calpain-3, a muscle-specific protease. Recently, growing evidence implicates heterozygous CAPN3 variants in autosomal dominant disease (LGMDD4), with pathogenic mechanisms still incompletely understood. In a retrospective multicenter Italian study of patients harboring monoallelic CAPN3 variants (ClinicalTrials.gov NCT05956132), the p.Asp753Asn substitution was the most frequent change, detected in eight unrelated individuals. These patients, aged 6–80 years, exhibited a spectrum of presentations ranging from asymptomatic hyperCKemia and exertional myalgia to mild proximal weakness. Muscle biopsies showed mild, nonspecific myopathic changes, while calpain-3 expression was variably reduced. Structural modeling suggested that Asp753 may stabilize the Ca2+-bound conformation, with substitution potentially disrupting inter-domain interactions. Literature review identified 31 additional reports worldwide, confirming recurrence while highlighting marked phenotypic heterogeneity and limited clinical annotation. The aggregated evidence supports a pathogenic role for p.Asp753Asn, though the precise mechanism, potentially involving a dominant-negative effect, remains to be validated. These findings emphasize diagnostic challenges posed by single CAPN3 variants and underscore the need for integrated clinical, segregation, and functional studies to clarify pathogenic mechanisms, refine counseling, and guide patient-specific rehabilitation and therapeutic strategies.
1. Introduction
Limb-Girdle Muscular Dystrophies (LGMDs) comprise a genetically and clinically diverse group of disorders primarily affecting the muscles of the pelvic and shoulder girdles. The most prevalent form, LGMDR1 (formerly LGMD2A), arises from biallelic loss-of-function mutations in CAPN3 (MIM# 114240), encoding calpain-3, an intracellular cysteine protease primarily expressed in skeletal muscle [,,]. Calpain-3 is characterized by a modular structure with distinctive functional domains, which includes protease core domains PC1 and PC2, the calpain-type beta-sandwich (CBSW) domain and a C-terminal penta-EF-hand (PEF) domain which forms a stable functional dimer upon Ca2+ binding [,]. It also harbors sequences designated for autolytic and proteolytic activity and interaction with structural and regulatory muscle proteins, such as titin and filamin C [,]. Physiologically, calpain-3 plays a critical role in maintaining muscle homeostasis through sarcomere remodeling, proteostasis, and calcium signaling, processes essential for muscle repair and regeneration []. Current studies indicate that calpain-3 operates as an active homodimer, although it may also exist in different oligomeric states possibly modulated by cellular context or post-translational mechanisms, which in turn may affect its stability and function [,]. To date, over 500 mutations in CAPN3 have been identified, the majority of which are missense mutations [].
The pathogenesis of LGMDR1 involves a loss of calpain-3 expression and/or function. Importantly, loss-of-function mutations in CAPN3 do not always result in complete absence of calpain-3 protein, and expression levels do not consistently correlate with clinical severity, highlighting that pathogenicity is not solely dependent on protein abundance. Except in cases of complete absence or severely reduced calpain-3, Western blot (WB) is not a clear diagnostic indicator. In healthy muscle, a distinct pattern of bands is consistently observed by WB: the full-length protein at 94 kDa, a 30 kDa autolytic fragment, and ~60 kDa degradation products. In LGMDR1 patients, these banding patterns can vary significantly. Severely affected individuals often show loss of all bands, typically linked to null or truncating mutations [], while others may retain the 94 kDa band but lack the 30 and ~60 kDa fragments, indicating enzymatically inactive protein due to defective autolysis []. Therefore, the presence of calpain-3 protein does not necessarily indicate functional integrity, and accurate interpretation should integrate information from the analysis of all calpain-3 bands as a whole [].
While calpainopathies are classically inherited in an autosomal recessive manner, a small number of cases with autosomal dominant inheritance (LGMDD4) have also been reported and linked to specific CAPN3 mutations [,,,,,,,]. Clinically, LGMDD4 is often milder and later in onset than LGMDR1, with patients typically presenting in the third or fourth decade with limb-girdle weakness, often accompanied by myalgia and axial symptoms []. Notably, some of the mutations reported in compound heterozygous or homozygous form in patients with LGMDR1, have also been found in heterozygosity in individuals with dominantly inherited disease [,,]. In such cases, segregation analyses within affected families have demonstrated consistent transmission of muscle weakness with the heterozygous variant, confirming a dominant inheritance pattern. Research on the pathogenic mechanism of LGMDD4 has largely focused on loss-of-function mechanisms employing techniques such as co-transfection analyses or in silico simulations [,]. One proposed model for dominant-negative pathogenicity hypothesizes that mutant monomers combine with wild-type monomers, leading to faulty dimers [] which may be inactive or rapidly degraded, resulting in a level of wild-type calpain-3 insufficient to maintain muscle homeostasis. Interestingly, despite this hypothesis, protein expression in muscle biopsies from affected individuals often appears normal [,] or even lower than expected, assuming that wild-type and mutant monomers combine in a simple stoichiometric ratio [,]. Such mechanism remains speculative due to the current lack of reliable methods and biomarkers for confirming dominant pathogenicity in calpainopathies. The complexity of protein interactions, oligomerization states, and tissue-specific effects complicates the ability to assess the biological effect of single missense variants. Indeed, while nonsense, insertion/deletion, or splice-site mutations are likely to impair or eliminate calpain-3 function, the impact of missense or in-frame mutations on calpain-3 activity is not immediately predictable []. Many additional variants, beyond those described, may contribute individually to the development of LGMDD4. Indeed, a dominant form of calpainopathy may constitute a possible diagnosis in the relatively high proportion (~20%) of LGMD cases where only a single variant in CAPN3 is identified []. In sporadic cases, multiple reports of individuals with the same heterozygous CAPN3 variant and presenting with a compatible phenotype may provide circumstantial but compelling evidence for a dominant pathogenic effect. In this context, the missense variant c.2257G>A (p.Asp753Asn) emerged as a recurrent finding in our multicenter cohort of unrelated Italian patients with monoallelic changes in CAPN3. This variant has been extensively reported in both heterozygous and compound heterozygous states in literature [,,,,,,,,,,,,] and is currently classified as uncertain significance in the ClinVar database. In the present study, we aim to further investigate the potential role of the p.Asp753Asn substitution in the pathogenesis of LGMDD4 by consolidating existing literature and analyzing clinical and biochemical data from our cohort.
2. Results
2.1. Subjects
In a retrospective, observational, multicenter study (Ethics Committee Approval 2023.08; 6 June 2023, ClinicalTrials.gov NCT05956132), we gathered a cohort of 59 Italian patients heterozygous for CAPN3 variants. A comprehensive analysis of the entire dataset is beyond the scope of the present report and will be presented in a separate publication. Genetic analysis identified 39 distinct variants in total. The most frequent variant was c.2257G>A (p.Asp753Asn), located in exon 21, accounting for 13.5% of cases. This subset of patients represents the focus of the present study.
2.2. Clinical, Histopathological and Biochemical Features of p.Asp753Asn Patients
Demographic and clinical characteristics of the patients are summarized in Table 1.

Table 1.
Clinical and histopathological features of patients carrying the CAPN3 c.2257G>A (p.Asp753Asn) variant. Demographic, clinical, and pathological data are presented for eight individuals in our cohort. LL = lower limbs; UL = upper limbs; p = proximal; d = distal; R = right; L = left, and NP = not performed.
The study cohort consists of eight unrelated individuals, six males and two females, including one pediatric subject (6 years of age) and 7 adult cases with an age at last evaluation ranging from 52 to 80 years (mean age 63 ± 10 years). Age of onset in adult cases ranged between the fourth and the sixth decade of life (mean age 50.1 ± 7.1 years). All were found to be heterozygous for the p.Asp753Asn variant, with no additional variants identified in CAPN3. Although one patient (P8) carried a variant in another gene associated with muscular dystrophy (POMGNT2), it was considered unlikely to be causative. Family history was negative for all cases except P8, whose brother presented with pes cavus and hyperCKemia and whose daughter had hyperCKemia without clinical symptoms. This is suggestive of a possible dominant inheritance pattern, although segregation analysis could not be confirmed due to the absence of genetic testing in the affected relatives.
The clinical spectrum was very broad. Overall, five patients experienced muscular atrophy and weakness (P2, P3, P4, P7, P8), which was mainly distributed in proximal muscles of lower limbs determining progressive difficulty in walking and/or climbing stairs, frequent falls, or foot drop as initial symptoms. Interestingly two patients (P3 and P7) showed also a severe axial weakness with bent spine syndrome and one patient (P4) presented with a distal predominant involvement. One patient carried asymptomatic hyperCKemia at 56 years of age with normal neurological examination (P1). Three patients (P5, P6, P8) reported myalgia, exercise intolerance, or fatigue. Contractures and respiratory symptoms were present in one patient (P7) while cardiac involvement was not described in the entire cohort.
Creatine kinase (CK) levels ranged from normal to mild increased values (up to 5× normal, range normal-1100 U/L). Interestingly, no correlation was observed between muscle impairment and serum CK levels, as patients with muscular weakness could also present with normal CK values. Muscle MRI or CT scan was performed in 6/8 patients showing unremarkable pattern in 3 of them (P1, asymptomatic, and P4 and P5, showing weakness) and fibro-fatty replacement in 3 subjects (P3, P7, P8).
Muscle biopsy was performed in 6/8 patients and showed mild myopathic features in all of them with mild fiber size variability and few internal nuclei. Endomysial fibrosis was described in 3/6 samples (P1, P2, P3). Inflammatory infiltrates were absent. Degenerating muscle fibers, rimmed vacuoles, ring fibers, fibers with loss or reduction in oxidative enzyme activity and COX negative fibers were also observed occasionally (Figure 1A). Some nonspecific signs such as increase in PAS-positive material within the intermyofibrillar network, predominance of type I fibers, lobulated muscle fibers were described only in some samples but did not represent a key feature. WB analysis of calpain-3 performed in five specimens showed a normal expression of the protein in three patients (e.g., P2, Figure 1B) while in the other two calpain-3 expression was variably reduced (e.g., P3 and P4, Figure 1B).
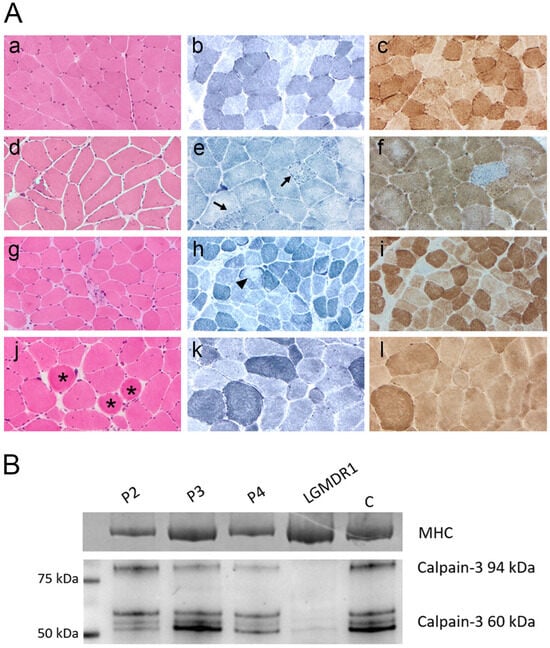
Figure 1.
(A) Muscle biopsy analysis. Representative morphological findings in patients P2, P3, and P4 compared with healthy control muscle (a–c). Panels (d–f), (g–i), and (j–l) correspond to patients P2, P3, and P4, respectively. H&E staining (d,g,j) showed marked fiber size variability, internal nuclei, rimmed vacuoles, endomysial fibrosis, and ring fibers (asterisks). NADH staining (e,h,k) revealed focal and irregular areas of reduced oxidative activity (arrows and arrowhead). COX staining identified occasional COX-negative fibers. Magnification: ×20. (B) Western blot analysis. Muscle biopsy samples from patients P2–P4 were analyzed by WB to assess calpain-3 expression. The blot shows the full-length 94 kDa calpain-3 protein and its autolytic/degradation fragments (~60 kDa) in comparison with a healthy control (C). Patient P2 showed calpain-3 expression within normal range, P3 exhibited a reduction in the 94 kDa band, and P4 displayed an overall reduction in all calpain-3 bands. A muscle biopsy from a patient diagnosed with LGMDR1 showing absence of the 94 kDa band and severe reduction in the autolytic/degradation fragments was included as a negative control. Total protein loading was confirmed by Coomassie blue staining of the myosin heavy chain (MHC) in the post-blotted gel.
2.3. Structural Modeling of the p.Asp753Asn Variant
In order to investigate the potential effect of the p.Asp753Asn mutation on calpain-3 structure and function, it is essential to rely on structural information about the protein. However, no full-length experimental structure of calpain-3 is currently available. Instead, structural data exist for the penta-EF-hand domain (PDB: 4OKH) and for several structures of the protease core domain (PDB: 6BJD, 6BDT, 6BKJ, 6BJP). Thus, for obtaining insights into the full-length protein in the Ca2+-bound and unbound states, homology modelling was performed. The templates chosen for both states were the M-calpain in Ca2+ bound and unbound states (PDB 3BOW and 1FKU), sharing a sequence identity of 52% with calpain 3. Then the mutation was mapped onto the modeled structures (Figure 2).
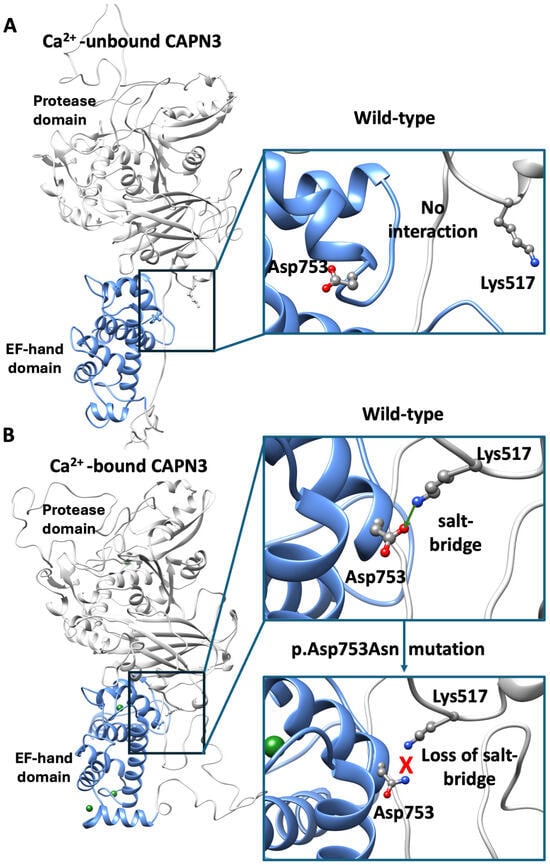
Figure 2.
Structural modeling of full-length calpain-3. (A) Homology model of full-length calpain-3 in the Ca2+-unbound state, Asp753 within the penta EF-domain (light blue) and Lys517 within the protease domain (gray) are highlighted. In the insight, the detailed location of both residues is shown. (B) Ca2+-bound model showing the formation of the Asp753–Lys517 salt bridge, consistent with Ca2+-induced conformational rearrangements. In the lower close-up view, the mutant model (p.Asp753Asn) illustrates the loss of the negative charge and the predicted breaking of the salt-bridge.
As a quality control step, our modeled structures were compared with the available per domain X-ray structures. For the Ca2+-bound state, the penta-EF-hand model showed an RMSD of 1 Å from 4OKH, while the protease core model displayed an RMSD of 0.7 Å from 6BGP, indicating high per-domain accuracy. The use of multiple templates allowed us to capture information about domain organization that is otherwise unavailable from isolated domain structures.
Mapping of Asp753 onto the model placed the residue within the PEF-hand domain, but outside of known Ca2+-binding sites, calmodulin-binding regions, or multimerization interfaces (Figure A1). Interestingly, in the Ca2+-bound model, Asp753 forms a salt bridge with Lys517 of the protease domain, a contact that is disrupted in the Ca2+-unbound state (Figure 2). This observation agrees with previous findings by Hanna et al. [], who reported that Ca2+ binding induces conformational rearrangements of PEF domains, driving broader domain reorganization (Supplementary Movie S1). This suggests that Asp753 may contribute to the stabilization of the Ca2+-bound conformation and its connection to the protease domain. Substitution of Asp753 with asparagine (p.Asp753Asn) likely removes a negative charge, potentially breaking the salt bridge with Lys517 and destabilizing inter-domain interactions.
2.4. The p.Asp753Asn Variant in CAPN3 Is Recurrent Across Unrelated Individuals Worldwide
To contextualize our findings and assess the broader significance of the p.Asp753Asn variant, we reviewed all available literature reports describing patients carrying this substitution. We identified 31 reports of the CAPN3 variant across 16 independent studies (Table A1). A substantial subset of these reports originated from large-scale sequencing initiatives, including targeted neuromuscular gene panels and whole-exome sequencing (WES) studies of undiagnosed disease cohorts from multiple countries [,,,]. Consequently, complete patient identifiers and detailed clinical annotations were often unavailable, making it difficult to distinguish between unique individuals, duplicate entries, or related family members. Demographic and phenotypic data were available for only 15 patients (~48%) with key information such as age at onset, family history, zygosity status, and detailed clinical features variably reported or absent. This paucity of clinical annotations significantly limits any robust genotype-phenotype correlation for the p.Asp753Asn substitution.
Genotyping data revealed that the p.Asp753Asn variant most frequently occurred in isolation, with no additional pathogenic CAPN3 mutations identified in the majority of cases (n = 14). However, compound heterozygous cases were also reported. For example, one individual was a compound heterozygous for p.Asp753Asn and the nonsense mutation c.967G>T (p.Glu323*) []. In two cases, two additional CAPN3 mutations were found in the same patient. Rubegni et al. reported a patient harboring c.1395-1397del (p.Leu465_Glu466del) and c.1453A>G (p.Met485Val) in cis, while p.Asp753Asn was present on the opposite allele []; in another report, c.1401_1403delGGA (p.Glu467del) was found on the same allele as p.Asp753Asn in a patient who carried c.967G>T (p.Glu323*) in trans []. Two individuals carried intronic variants or rare genomic variants whose clinical significance remains uncertain [,], and in two cases, causative variants were detected in other genes, suggesting an alternative molecular diagnosis [].
Where information was available, most individuals appeared to be of sporadic origin, with no reported family history of neuromuscular disease. Clinical presentation was heterogeneous: among the individuals with reported phenotypic features, varied widely from pediatric (1–3 years) to adult (21–55 years). Phenotypes ranged from asymptomatic hyperCKemia (n = 2) to classic LGMD with slowly progressive proximal muscle weakness (n = 8). Mild or nonspecific proximal weakness was reported in 12 cases. One case was explicitly described as not suggestive of LGMD [], and another with pseudometabolic features, myalgia and fatigue []. Notably, one individual presented with ataxia in the absence of classical muscular dystrophy features [].
Reported serum CK levels ranged from mildly elevated (~140 U/L) to over tenfold above the upper normal limit. Muscle imaging in one patient revealed selective involvement of posterior thigh and pelvic muscles []. WB data for calpain-3 were available for a limited subset of individuals (n = 8) and demonstrated variable findings. In one case, calpain-3 was absent [], while another showed marked reduction []. The majority of individuals, however, showed normal calpain-3 expression [,,]. One more detailed report described a reduction in the 94 kDa full-length band by 45.1%, with preservation of the 30 kDa autolytic fragment, a pattern indicative of retained proteolytic activity []. Consistent with this, functional assays of calpain-3 autolytic activity, performed in only two individuals, demonstrated preserved enzymatic function [].
3. Discussion
Interpreting missense variants remains one of the most complex challenges in medical genetics. These single amino acid substitutions can variably affect protein structure, stability, interaction dynamics, and enzymatic activity and are often subtle and context-dependent, eluding current in silico predictions. The p.Asp753Asn variant in CAPN3 is an illustrative example of this complexity. Although extensively reported in the literature data regarding its clinical, histopathological and biochemical features have been incomplete or inconsistently annotated, preventing a clear understanding of its pathogenic significance and mode of inheritance. The present study provides new and comprehensive insights into this variant through a multidisciplinary approach integrating clinical, histological, and structural analyses.
To address the complexity surrounding this variant, we compiled and integrated all available data on p.Asp753Asn carriers, combining findings from our cohort with reports from the literature. We observed that the frequent lack of detailed phenotypic annotation substantially hampers the establishment of a robust diagnostic framework for dominantly inherited calpainopathy. Moreover, the marked variability in both clinical manifestations and laboratory findings further underscores the challenges inherent in developing a reliable and standardized diagnostic algorithm for suspected LGMDD4. In our cohort, the clinical variability associated with this variant is evident: phenotypes ranged from asymptomatic hyperCKemia, to mild or moderate limb-girdle muscle weakness, to progressive weakness with contractures and respiratory involvement. Muscle biopsies showed a consistent but nonspecific myopathic pattern with low-grade structural alterations, while WB analyses demonstrated variable calpain-3 expression, normal in some, markedly reduced in others. Variability in calpain-3 protein levels may reflect not only the presence of the variant but also inter-individual differences and sample handling. Calpain-3 WB analyses have known limitations: for example, approximately 20% of individuals with pathogenic CAPN3 variants may have normal protein levels despite functional impairment []. Tissue handling and preservation can also affect results, as partial thawing or exposure to moisture may promote artifactual autolysis, reducing detectable full-length protein [,]. Mechanistically, calpain-3 exists in dynamic oligomeric states, including dimers and higher-order assemblies such as trimers via the PEF domain [,]. Mutant and wild-type proteins may interact within these complexes, potentially affecting stability, turnover, or detection on WB. Notably, intrafamilial variability in calpain-3 expression has also been seen in dominant families, highlighting that protein levels can differ even among genetically related individuals []. Our observations align with previous reports of LGMDD4 [,,] and reinforce the idea that diagnostic evaluation of suspected dominant calpainopathies should not rely solely on WB or CK levels. Individuals with single heterozygous CAPN3 mutations may benefit from improved diagnostic evaluation through functional and imaging approaches reported in the literature. It has been demonstrated that standardized muscle MRI of the pelvis and lower limbs improves diagnostic accuracy and is especially useful when biochemical results are unclear. It usually reveals selective involvement of paraspinal, gluteal, hamstring, and medial gastrocnemius muscles [,,]. Functional analyses such as those published by Vissing et al. [], in which co-expression and activity assays demonstrated loss of mutant calpain-3 function along with dominant-negative reduction of wild-type protein, offer a useful example of assays that could be integrated into diagnostic workflows. Incorporating these additional tests can greatly boost diagnostic confidence when WB and CK results are insufficiently informative. Ultimately, comprehensive clinical, biochemical, and genetic correlation is essential for accurate diagnosis, particularly when evaluating single heterozygous variants with uncertain significance.
The p.Asp753Asn variant results in the substitution of a charged polar amino acid with a non-charged polar residue at position 753, which lies within the EF-hand domain, crucial for calpain-3 homodimerization [,]. Although previous structural modeling did not predict any major steric hindrance or disruption of substrate binding, it was previously hypothesized that the change in charge could modify the bond with other calpain-3 monomers []. Here we show that, although residue Asp753 is indeed present in the dimerization domain, it is located far away from the dimerization interface (Figure A1), thus excluding a putatively direct effect of p.Asp753Asn mutant in the dimerization process. Instead, our structural modeling provides novel insights into the potential molecular mechanism underlying the pathogenicity of p.Asp753Asn. In the wild-type protein, Asp753 forms a stabilizing salt bridge with Lys517, which contributes to maintaining inter-domain stability during activation. Substitution with asparagine removes the negative charge, likely disrupting this electrostatic interaction and weakening the structural coupling between domains. This disruption may alter the dynamic equilibrium between inactive and active conformations of calpain-3, impairing autolytic processing or substrate recognition. Such an effect is consistent with a dominant-negative mechanism, whereby the mutant protein interferes with the activity or stability of the wild-type enzyme. While this hypothesis requires experimental validation, it aligns with the emerging concept that certain CAPN3 missense variants can exert dominant effects through altered dimerization or aberrant protease regulation. Although computational predictions and homology-based modeling cannot fully capture the complexity of calpain-3 dynamics, particularly in the absence of an experimentally resolved full-length structure, the consistency between our modeling data and the observed biochemical variability strengthens the plausibility of this structural interpretation. Future molecular dynamics simulations and in vitro functional assays will be necessary to confirm whether p.Asp753Asn perturbs calcium-dependent activation or dimer stability.
Recurrent detection of a given variant among unrelated affected individuals provides supportive evidence toward its pathogenic classification. Our literature review confirms that the p.Asp753Asn substitution is recurrently identified in unrelated individuals across diverse countries and diagnostic settings. Integration of our cohort data with published cases further demonstrates marked phenotypic variability, despite the frequent absence of detailed clinical information in many reports. A subset of patients presented atypical phenotypes, and at least two received an alternative diagnosis, underscoring how even mild or nonspecific manifestations, such as isolated CK elevation, may be overlooked when another disorder with more prominent clinical signs appears to better explain the presentation. Although LGMDD4 is typically a late-onset disease, a few pediatric-onset cases have been reported, including patients carrying c.643_663del21 or c.2440-1G>A variants and presenting with childhood-onset weakness and elevated CK []. This clinical overlap, combined with marked intrafamilial variability, suggests that genetic or environmental modifiers influence disease onset and progression and reinforces the notion of a spectrum of inheritance patterns including both dominant effects and contribution to recessive disease with a second pathogenic allele. Despite multiple independent reports, the precise molecular mechanism, particularly the hypothesized dominant-negative effect of p.Asp753Asn, remains unproven. Currently, no standardized or validated methods exist to directly demonstrate dominant-negative behavior in CAPN3-related disease. Functional studies capable of dissecting these effects at the cellular or molecular level are technically complex and rarely implemented in routine diagnostics. Consequently, many individuals carrying a single potentially pathogenic variant remain undiagnosed or misclassified.
The p.Asp753Asn variant exemplifies both the potential and the limitations of current genetic diagnostics. LGMDD4 represents a distinct and diagnostically challenging entity. Establishing pathogenicity requires integrated functional validation, family-based studies, and comprehensive phenotypic characterization. Future research should prioritize the development of standardized functional assays capable of detecting subtle alterations in calpain-3 activity and dimerization, alongside comprehensive phenotyping in larger, multicenter cohorts.
4. Materials and Methods
4.1. Subjects and Literature Review
The eight individuals carrying the c.2257G>A (p.Asp753Asn) variant included in the present study were drawn from a larger cohort of 59 patients with heterozygous CAPN3 variants referred to five Italian neuromuscular centers (ClinicalTrials.gov NCT05956132). Inclusion criteria comprised patients heterozygous for a single CAPN3 variant classified as pathogenic (class 5), likely pathogenic (class 4), or of uncertain significance (class 3), with no second CAPN3 variant, as confirmed by MLPA, mRNA analysis, targeted neuromuscular gene panels, or whole-exome sequencing (WES). Demographic and pseudonymized clinical data were collected from medical records and included functional muscle assessments, serum CK levels, WB analyses, muscle biopsy findings, and muscle MRI.
For the literature review, we analyzed all published reports of the CAPN3 c.2257G>A (p.Asp753Asn) variant, as documented ClinVar (accession no. VCV000281081.28). We identified 16 peer-reviewed publications originating from diverse geographic regions reporting a total of 31 cases carrying the p.Asp753Asn variant. These cases include both individuals with isolated heterozygosity and those with additional CAPN3 variants in either compound heterozygous or complex allelic configurations.
4.2. Sequencing Analysis
Whole genomic DNA was extracted from peripheral venous blood samples using a standard procedure. Patients in this study were analyzed with a dedicated NGS panel for muscle diseases. We used SureSelect technology (Agilent, Santa Clara, CA, USA) and SureDesign software v. 8.0 (https://earray.chem.agilent.com/suredesign/ (accessed on 20 January 2025)) to design a multi-exon amplicon panel containing a total of 298 genes known to be associated with limb-girdle muscular dystrophies (LGMD), rhabdomyolysis, and metabolic and distal myopathies; the panel offered gene coverage greater than 99%. To analyze the data obtained from our study, we used a routine bioinformatics pipeline, QIAGEN Clinical Insight (QCI) Interpret software v. 9.2.1.20231012. Interpretation of the variants identified by this analysis is based on current knowledge and the ACMG classification []. The identified variants were validated by standard PCR-based capillary Sanger sequencing. The presence of a second pathogenic variant was excluded by MLPA according to standard procedures or by analysis of mRNA extracted from muscle tissue as previously described [].
4.3. Calpain-3 Western Blot Analysis
Protein extracts were prepared from muscle biopsies of the patients as well as from healthy controls as described []. Briefly, comparable amounts of tissue from each sample were lysed in treatment buffer (0.125 mol/L Tris/HCl buffer, pH 6.4, 10% glycerol, 4% SDS, 4 mol/L urea), boiled for 2′ at 90 °C and centrifuged at 1300 rpm. An equal volume of 2× Laemmli Sample Buffer supplemented with 20% β-mercaptoethanol was added to each supernatant. Proteins were resolved on 4–15% TGX criterion midi gels in 1× TGS buffer for 2 h at 100 V. Dry transfer to a PDFV membrane was performed with the Trans-Blot Turbo Midi transfer pack for 20′ at 25 V. Gels were stained with SimplyBlue SafeStain. PVDF membranes were incubated with primary antibody (NCL-CALP-12A2, Leica Biosystems, Buccinasco, Italy) overnight at 4 °C. After incubation with HRP-conjugated anti-mouse secondary antibody (Life Technologies # 31450, Segrate, Italy) chemiluminescent detection was performed with Clarity™ Western ECL Substrate (BioRad # 1705060, Segrate, Italy) and acquisition was performed with the iBright Imaging System CL1000 (Thermo Fisher Scientific by Life Technologies, Segrate, Italy).
4.4. Structural Modeling
To be consistent with previous works [,] and to allow comparison, the calpain 3 structure was generated by using protein structure homology modelling using SWISSMODEL workspace [] and visualized by Chimera 1.19 software (UCSF) []. In order to assess the putative role of the Asp753Asn mutation, two models were generated: Ca2+ bound state and Ca2+ unbound state. The templates chosen for both cases were the M-calpain in Ca2+ bound and unbound states (PDB 3BOW and 1KFU, respectively), sharing a sequence identity of 52% with calpain 3. The movie was created by using the ‘Morph conformation’ program within the Chimera software []. To gain deeper insights into the multimer formation and putative effect of the mutation in the dimerization process, we have modelled the full-length dimer using Alphafold 2 (https://alphafoldserver.com/ (accessed on 25 February 2025)).
5. Conclusions
Dominant calpainopathies, particularly those linked to single missense variants such as p.Asp753Asn, remain a diagnostically challenging entity. Their pathogenicity cannot rely solely on variant type or zygosity but must be interpreted through functional studies, segregation analyses, and detailed phenotypic evaluation. Until integrated datasets become widely available, caution in the interpretation is warranted. Developing robust functional assays and standardized diagnostic algorithms will facilitate accurate genetic counseling and enable patient-specific rehabilitation strategies aimed at preserving muscle strength and function. Beyond diagnostics, these efforts will improve understanding of how CAPN3 variants perturb calpain-3 activity, stratify patients for future clinical trials, and ultimately guide the development of targeted therapies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms262311384/s1.
Author Contributions
Conceptualization, R.B.; methodology, R.B. and A.G.; validation, G.D. and D.C.; formal analysis, A.G.; investigation, G.D.; resources, F.M., D.R., A.R., D.L., A.M., L.M., G.V. and P.T.; data curation, D.C.; writing—original draft preparation, R.B. and G.D.; writing—review and editing, R.B., G.D., A.G., D.C., F.M. and G.V.; visualization, G.D. and A.G.; supervision, R.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Italian Ministry of Health (Ricerca Corrente).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of the Province of Venice and by IRCCS San Camillo (2023.08; 6 June 2023) and is registered on ClinicalTrials.gov (NCT05956132).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| LGMD | Limb-Girdle muscular dystrophy |
| WB | Western blot |
| WES | Whole exome sequencing |
| CK | Creatine kinase |
Appendix A

Figure A1.
Full-length model of calpain-3. The Alphafold model of the dimer (chain A in blue, and chain B in red) shows that Asp753 residue (green) locates in a region distant to the dimerization interface. Given the limitations of Alphafold in the modeling of domain organization, the model was checked against the crystallographic dimers (4OKH) and the location of Asp753 outside the dimerization interface was confirmed.
Table A1.
Reported cases of the CAPN3 p.Asp753Asn variant in the literature. Summary of reported cases across 16 studies published between 2004 and 2024. Clinical phenotypes, CK levels, muscle biopsy findings, and Western blot results are reported as originally stated by authors. No. PZ indicates the number of individuals with the p.Asp753Asn substitution reported in the referenced study. No. in Study refers to the specific patient numbering adopted within the original publication. CK: creatine kinase; WB: Western Blot; LGMD: limb-girdle muscular dystrophy; NGS: next-generation sequencing; WES: whole-exome sequencing; MLPA: multiplex ligation-dependent probe amplification; SSCP: single-strand conformation polymorphism; ARMS-PCR: amplification-refractory mutation system PCR; HT-DHPLC: high-throughput denaturing high-performance liquid chromatography. Empty fields “/” indicate unavailable data.
Table A1.
Reported cases of the CAPN3 p.Asp753Asn variant in the literature. Summary of reported cases across 16 studies published between 2004 and 2024. Clinical phenotypes, CK levels, muscle biopsy findings, and Western blot results are reported as originally stated by authors. No. PZ indicates the number of individuals with the p.Asp753Asn substitution reported in the referenced study. No. in Study refers to the specific patient numbering adopted within the original publication. CK: creatine kinase; WB: Western Blot; LGMD: limb-girdle muscular dystrophy; NGS: next-generation sequencing; WES: whole-exome sequencing; MLPA: multiplex ligation-dependent probe amplification; SSCP: single-strand conformation polymorphism; ARMS-PCR: amplification-refractory mutation system PCR; HT-DHPLC: high-throughput denaturing high-performance liquid chromatography. Empty fields “/” indicate unavailable data.
| Ref | No. PZ | No. in Study | Sex | Family History | Onset (Years) | Phenotype | Clinical Features | Severity | CK | WB Calpain-3 | Additional Mutations | Genetic Analysis |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [,,] | 4 | 52 | F | N | 3 | LGMD | LGMD | Moderate | / | Absent | No | SSCP, ARMS-PCR, SANGER HT-DHPLC |
| 71 | / | / | / | / | / | / | / | Normal | c.1355-6G4T | |||
| 48 | / | / | / | / | / | / | / | Reduced 5% | c.2242C4T (p.R748X) | |||
| 88 | / | / | / | LGMD | HyperCKmia | Mild | High | Normal | No | |||
| [] | 1 | / | / | / | 40 | Not suggestive of LGMD | / | Benign evolution | / | / | No | SSCP, Sanger |
| [] | 1 | 18 | F | N | 25 | Mild proximal weakness | Difficulty climbing stairs | Moderate | 140 U/L | / | No | Sanger |
| [] | 1 | XXXV | F | N | 37 | LGMD | / | Slow progression | 10–13× | 94 kDa Reduced 45.1%, 30 kDa Normal | No | Sanger |
| [] | 2 | 19 | / | N | 40 | LGMD | Pelvic girdle onset, Mild facial involvement | Mild, slow/moderate progression | 2× | Normal | No | Sanger |
| 20 | / | N | 55 | LGMD | Pelvic girdle onset | Mild, slow/moderate progression | 1.5× | Normal | No | |||
| [] | 2 | C21 | / | / | / | / | / | / | / | / | No | NGS panel |
| C36 | / | / | / | / | / | / | / | / | No | |||
| [] | 1 | P19 | M | / | 39 | Asymptomatic | HyperCKmia | Asymptomatic | 2000 U/L | / | c.1395-1397del (p.Leu465_Glu466del); c.1453A>G (p.Met485Val) | NGS panel |
| [] | 2 | / | / | / | / | / | / | / | / | / | N/A | WES |
| [] | 10 | / | / | / | / | Proximal weakness | / | / | / | / | N/A | NGS panel |
| [] | 1 | P8 | M | / | / | / | / | / | Normal | / | No | NGS panel, Sanger, MLPA |
| [] | 1 | A13 | F | N | 30 | Pseudometabolic | Paternal inheritance, Myalgia, fatigability | Mild | 6× | Normal | g.42350479A>G; g.42354194C>T | WES |
| [] | 3 | 29 | M | / | 21 | ataxia | / | / | / | / | No, carrying other causative variants in other genes | Custom Target Capture NGS panel |
| 32 | M | / | 2 | / | Difficulty climbing stairs | / | High | / | No, carrying causative variants in other genes | |||
| 35 | M | / | 1 | / | Hypotonia | / | 400 U/L | / | No | |||
| [] | 1 | U12 | M | / | 3 | Asymptomatic | HyperCKmia | Asymptomatic | High | / | Not found | NGS panel, Sanger |
| [] | 1 | 4 | F | / | 40 | LGMD | Scapular winging, waddling gait, severe upper and lower girlde weakness | Severe | 1169 UI/L | / | c.967G>T (p.Glu323*), c.1401_1403delGGA (p.Glu467del) | Sanger |
References
- Sorimachi, H.; Imajoh-Ohmi, S.; Emori, Y.; Kawasaki, H.; Ohno, S.; Minami, Y.; Suzuki, K. Molecular cloning of a novel mammalian calcium-dependent protease distinct from both m- and mu-types. Specific expression of the mRNA in skeletal muscle. J. Biol. Chem. 1989, 264, 20106–20111. [Google Scholar] [CrossRef]
- Beckmann, J.S.; Richard, I.; Hillaire, D.; Broux, O.; Antignac, C.; Bois, E.; Cann, H.; Cottingham, R.W., Jr.; Feingold, N.; Feingold, J.; et al. A gene for limb-girdle muscular dystrophy maps to chromosome 15 by linkage. Comptes Rendus Acad. Sci. III 1991, 312, 141–148. [Google Scholar]
- Richard, I.; Broux, O.; Allamand, V.; Fougerousse, F.; Chiannilkulchai, N.; Bourg, N.; Brenguier, L.; Devaud, C.; Pasturaud, P.; Roudaut, C.; et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995, 81, 27–40. [Google Scholar] [CrossRef]
- Sorimachi, H.; Ohmi, S.; Emori, Y.; Kawasaki, H.; Saido, T.C.; Ohno, S.; Minami, Y.; Suzuki, K. A novel member of the calcium-dependent cysteine protease family. Biol. Chem. Hoppe Seyler 1990, 371, 171–176. [Google Scholar]
- Partha, S.K.; Ravulapalli, R.; Allingham, J.S.; Campbell, R.L.; Davies, P.L. Crystal structure of calpain-3 penta-EF-hand (PEF) domain—A homodimerized PEF family member with calcium bound at the fifth EF-hand. FEBS J. 2014, 281, 3138–3149. [Google Scholar] [CrossRef]
- Sorimachi, H.; Toyama-Sorimachi, N.; Saido, T.C.; Kawasaki, H.; Sugita, H.; Miyasaka, M.; Arahata, K.; Ishiura, S.; Suzuki, K. Muscle-specific calpain, p94, is degraded by autolysis immediately after translation, resulting in disappearance from muscle. J. Biol. Chem. 1993, 268, 10593–10605. [Google Scholar] [CrossRef]
- Kinbara, K.; Sorimachi, H.; Ishiura, S.; Suzuki, K. Skeletal muscle-specific calpain, p49: Structure and physiological function. Biochem. Pharmacol. 1998, 56, 415–420. [Google Scholar]
- Chen, L.; Tang, F.; Gao, H.; Zhang, X.; Li, X.; Xiao, D. CAPN3: A muscle-specific calpain with an important role in the pathogenesis of diseases (Review). Int. J. Mol. Med. 2021, 48, 203. [Google Scholar] [CrossRef]
- Hata, S.; Doi, N.; Shinkai-Ouchi, F.; Ono, Y. A muscle-specific calpain, CAPN3, forms a homotrimer. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140411. [Google Scholar] [CrossRef]
- Ye, Q.; Henrickson, A.; Demeler, B.; Serrão, V.H.B.; Davies, P.L. Human calpain-3 and its structural plasticity: Dissociation of a homohexamer into dimers on binding titin. bioRxiv 2024. [Google Scholar] [CrossRef]
- Zhong, H.; Zheng, Y.; Zhao, Z.; Lin, P.; Xi, J.; Zhu, W.; Lin, J.; Lu, J.; Yu, M.; Zhang, W.; et al. Molecular landscape of CAPN3 mutations in limb-girdle muscular dystrophy type R1: From a Chinese multicentre analysis to a worldwide perspective. J. Med. Genet. 2021, 58, 729–736. [Google Scholar] [CrossRef]
- Anderson, L.V.; Davison, K.; Moss, J.A.; Richard, I.; Fardeau, M.; Tomé, F.M.; Hübner, C.; Lasa, A.; Colomer, J.; Beckmann, J.S. Characterization of monoclonal antibodies to calpain 3 and protein expression in muscle from patients with limb-girdle muscular dystrophy type 2A. Am. J. Pathol. 1998, 153, 1169–1179. [Google Scholar] [CrossRef]
- Fanin, M.; Nascimbeni, A.C.; Angelini, C. Screening of calpain-3 autolytic activity in LGMD muscle: A functional map of CAPN3 gene mutations. J. Med. Genet. 2007, 44, 38–43. [Google Scholar] [CrossRef]
- Groen, E.J.; Charlton, R.; Barresi, R.; Anderson, L.V.; Eagle, M.; Hudson, J.; Koref, M.S.; Straub, V.; Bushby, K.M. Analysis of the UK diagnostic strategy for limb girdle muscular dystrophy 2A. Brain 2007, 130 Pt 12, 3237–3249. [Google Scholar] [CrossRef]
- Vissing, J.; Barresi, R.; Witting, N.; Van Ghelue, M.; Gammelgaard, L.; Bindoff, L.A.; Straub, V.; Lochmüller, H.; Hudson, J.; Wahl, C.M.; et al. A heterozygous 21-bp deletion in CAPN3 causes dominantly inherited limb girdle muscular dystrophy. Brain 2016, 139 Pt 8, 2154–2163. [Google Scholar] [CrossRef]
- Martinez-Thompson, J.M.; Niu, Z.; Tracy, J.A.; Moore, S.A.; Swenson, A.; Wieben, E.D.; Milone, M. Autosomal dominant calpainopathy due to heterozygous CAPN3 C.643_663del21. Muscle Nerve 2018, 57, 679–683. [Google Scholar]
- Nallamilli, B.R.R.; Chakravorty, S.; Kesari, A.; Tanner, A.; Ankala, A.; Schneider, T.; da Silva, C.; Beadling, R.; Alexander, J.J.; Askree, S.H.; et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann. Clin. Transl. Neurol. 2018, 5, 1574–1587. [Google Scholar] [CrossRef]
- Cerino, M.; Campana-Salort, E.; Salvi, A.; Cintas, P.; Renard, D.; Juntas Morales, R.; Tard, C.; Leturcq, F.; Stojkovic, T.; Bonello-Palot, N.; et al. Novel CAPN3 variant associated with an autosomal dominant calpainopathy. Neuropathol. Appl. Neurobiol. 2020, 46, 564–578. [Google Scholar] [CrossRef]
- Vissing, J.; Dahlqvist, J.R.; Roudaut, C.; Poupiot, J.; Richard, I.; Duno, M.; Krag, T. A single c.1715G>C calpain 3 gene variant causes dominant calpainopathy with loss of calpain 3 expression and activity. Hum. Mutat. 2020, 41, 1507–1513. [Google Scholar] [CrossRef]
- González-Mera, L.; Ravenscroft, G.; Cabrera-Serrano, M.; Ermolova, N.; Domínguez-González, C.; Arteche-López, A.; Soltanzadeh, P.; Evesson, F.; Navas, C.; Mavillard, F.; et al. Heterozygous CAPN3 missense variants causing autosomal-dominant calpainopathy in seven unrelated families. Neuropathol. Appl. Neurobiol. 2021, 47, 283–296. [Google Scholar] [CrossRef]
- Mao, B.; Yang, J.; Zhao, X.; Jia, X.; Shi, X.; Zhao, L.; Banerjee, S.; Zhang, L.; Ma, X. Identification and functional characterization of a novel heterozygous splice-site mutation in the calpain 3 gene causes rare autosomal dominant limb-girdle muscular dystrophy. Exp. Ther. Med. 2024, 27, 97. [Google Scholar] [CrossRef]
- Krag, T.; Nasho, E.; Brady, L.; Verebi, C.; Leturcq, F.; Malfatti, E.; Duno, M.; Tarnopolsky, M.; Vissing, J. Variants in CAPN3 Causing Autosomal Dominant Limb-Girdle Muscular Dystrophy Combined With Calpain-3 Deficiency. Hum. Mutat. 2025, 2025, 9301465. [Google Scholar] [CrossRef]
- Massucco, S.; Fossa, P.; Fiorillo, C.; Faedo, E.; Gemelli, C.; Barresi, R.; Ripolone, M.; Patrone, S.; Gaudio, A.; Mandich, P.; et al. Case report: A single novel calpain 3 gene variant associated with mild myopathy. Front. Genet. 2024, 15, 1437859. [Google Scholar] [CrossRef]
- Ono, Y.; Ojima, K.; Shinkai-Ouchi, F.; Hata, S.; Sorimachi, H. An eccentric calpain, CAPN3/p94/calpain-3. Biochimie 2016, 122, 169–187. [Google Scholar] [CrossRef]
- Sáenz, A.; Leturcq, F.; Cobo, A.M.; Poza, J.J.; Ferrer, X.; Otaegui, D.; Camaño, P.; Urtasun, M.; Vílchez, J.; Gutiérrez-Rivas, E.; et al. LGMD2A: Genotype-phenotype correlations based on a large mutational survey on the calpain 3 gene. Brain 2005, 128, 732–742. [Google Scholar] [CrossRef]
- Fanin, M.; Fulizio, L.; Nascimbeni, A.C.; Spinazzi, M.; Piluso, G.; Ventriglia, V.M.; Ruzza, G.; Siciliano, G.; Trevisan, C.; Politano, L.; et al. Molecular diagnosis in LGMD2A: Mutation analysis or protein testing? Hum. Mutat. 2004, 24, 52–62. [Google Scholar] [CrossRef]
- Todorova, A.; Georgieva, B.; Tournev, I.; Todorov, T.; Bogdanova, N.; Mitev, V.; Mueller, C.R.; Kremensky, I.; Horst, J. A large deletion and novel point mutations in the calpain 3 gene (CAPN3) in Bulgarian LGMD2A patients. Neurogenetics 2007, 8, 225–229. [Google Scholar] [CrossRef]
- Guglieri, M.; Magri, F.; D’Angelo, M.G.; Prelle, A.; Morandi, L.; Rodolico, C.; Cagliani, R.; Mora, M.; Fortunato, F.; Bordoni, A.; et al. Clinical, molecular, and protein correlations in a large sample of genetically diagnosed Italian limb girdle muscular dystrophy patients. Hum. Mutat. 2008, 29, 258–266. [Google Scholar] [CrossRef]
- Perez, F.; Vital, A.; Martin-Negrier, M.L.; Ferrer, X.; Sole, G. Diagnostic procedure of limb girdle muscular dystrophies 2A or calpainopathies: French cohort from a neuromuscular center (Bordeaux). Rev. Neurol. 2010, 166, 502–508. [Google Scholar] [CrossRef]
- Rubegni, A.; Malandrini, A.; Dosi, C.; Astrea, G.; Baldacci, J.; Battisti, C.; Bertocci, G.; Donati, M.A.; Dotti, M.T.; Federico, A.; et al. Next-generation sequencing approach to hyperCKemia: A 2-year cohort study. Neurol. Genet. 2019, 5, e352. [Google Scholar] [CrossRef]
- Bevilacqua, J.A.; Guecaimburu Ehuletche, M.D.R.; Perna, A.; Dubrovsky, A.; Franca, M.C.; Vargas, S.; Hegde, M.; Claeys, K.G.; Straub, V.; Daba, N.; et al. The Latin American experience with a next generation sequencing genetic panel for recessive limb-girdle muscular weakness and Pompe disease. Orphanet J. Rare Dis. 2020, 15, 11. [Google Scholar] [CrossRef]
- Gonzalez-Quereda, L.; Rodriguez, M.J.; Diaz-Manera, J.; Alonso-Perez, J.; Gallardo, E.; Nascimento, A.; Ortez, C.; Benito, D.N.-D.; Olive, M.; Gonzalez-Mera, L.; et al. Targeted Next-Generation Sequencing in a Large Cohort of Genetically Undiagnosed Patients with Neuromuscular Disorders in Spain. Genes 2020, 11, 539. [Google Scholar] [CrossRef]
- Töpf, A.; Johnson, K.; Bates, A.; Phillips, L.; Chao, K.R.; England, E.M.; Laricchia, K.M.; Mullen, T.; Valkanas, E.; Xu, L.; et al. Sequential targeted exome sequencing of 1001 patients affected by unexplained limb-girdle weakness. Genet. Med. 2020, 22, 1478–1488. [Google Scholar]
- Macias, A.; Fichna, J.P.; Topolewska, M.; Rȩdowicz, M.J.; Kaminska, A.M.; Kostera-Pruszczyk, A. Targeted Next-Generation Sequencing Reveals Mutations in Non-coding Regions and Potential Regulatory Sequences of Calpain-3 Gene in Polish Limb-Girdle Muscular Dystrophy Patients. Front. Neurosci. 2021, 15, 692482. [Google Scholar] [CrossRef]
- Ozyilmaz, B.; Kirbiyik, O.; Ozdemir, T.R.; Ozer, O.K.; Kutbay, Y.B.; Erdogan, K.M.; Guvenc, M.S.; Arıkan, Ş.; Turk, T.S.; Kale, M.Y.; et al. Experiences in the molecular genetic and histopathological evaluation of calpainopathies. Neurogenetics 2022, 23, 103–114. [Google Scholar] [CrossRef]
- Çavdarlı, B.; Köken, Ö.; Satılmış, S.B.A.; Bilen, Ş.; Ardıçlı, D.; Ceylan, A.C.; Gündüz, C.N.S.; Topaloğlu, H. High diagnostic yield of targeted next-generation sequencing panel as a first-tier molecular test for the patients with myopathy or muscular dystrophy. Ann. Hum. Genet. 2023, 87, 104–114. [Google Scholar] [CrossRef]
- Hanna, R.A.; Campbell, R.L.; Davies, P.L. Calcium-bound structure of calpain and its mechanism of inhibition by calpastatin. Nature 2008, 456, 409–412. [Google Scholar] [CrossRef]
- Fanin, M.; Nascimbeni, A.C.; Aurino, S.; Tasca, E.; Pegoraro, E.; Nigro, V.; Angelini, C. Frequency of LGMD gene mutations in Italian patients with distinct clinical phenotypes. Neurology 2009, 72, 1432–1435. [Google Scholar] [CrossRef]
- Aguti, S.; Gallus, G.N.; Bianchi, S.; Salvatore, S.; Rubegni, A.; Berti, G.; Formichi, P.; De Stefano, N.; Malandrini, A.; Lopergolo, D. Novel Biomarkers for Limb Girdle Muscular Dystrophy (LGMD). Cells 2024, 13, 329. [Google Scholar] [CrossRef]
- Piluso, G.; Politano, L.; Aurino, S.; Fanin, M.; Ricci, E.; Ventriglia, V.M.; Belsito, A.; Totaro, A.; Saccone, V.; Topaloglu, H.; et al. Extensive scanning of the calpain-3 gene broadens the spectrum of LGMD2A phenotypes. J. Med. Genet. 2005, 42, 686–693. [Google Scholar] [CrossRef]
- Fanin, M.; Nascimbeni, A.C.; Fulizio, L.; Trevisan, C.P.; Meznaric-Petrusa, M.; Angelini, C. Loss of calpain-3 autocatalytic activity in LGMD2A patients with normal protein expression. Am. J. Pathol. 2003, 163, 1929–1936. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Bruno, C.; Cassandrini, D.; Martinuzzi, A.; Toscano, A.; Moggio, M.; Morandi, L.; Servidei, S.; Mongini, T.; Angelini, C.; Musumeci, O.; et al. McArdle disease: The mutation spectrum of PYGM in a large Italian cohort. Hum. Mutat. 2006, 27, 718. [Google Scholar] [CrossRef]
- Anderson, L.V.; Davison, K. Multiplex Western blotting system for the analysis of muscular dystrophy proteins. Am. J. Pathol. 1999, 154, 1017–1022. [Google Scholar] [CrossRef]
- Ermolova, N.; Kramerova, I.; Spencer, M.J. Autolytic activation of calpain 3 proteinase is facilitated by calmodulin protein. J. Biol. Chem. 2015, 290, 996–1004. [Google Scholar] [CrossRef]
- Bordoli, L.; Kiefer, F.; Arnold, K.; Benkert, P.; Battey, J.; Schwede, T. Protein structure homology modeling using SWISS-MODEL workspace. Nat. Protoc. 2009, 4, 1–13. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Krebs, W.G.; Gerstein, M. The morph server: A standardized system for analyzing and visualizing macromolecular motions in a database framework. Nucleic Acids Res. 2000, 28, 1665–1675. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).