
Non-Canonical Amino Acids in Analyses of Protease Structure and Function




Abstract
1. Introduction


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
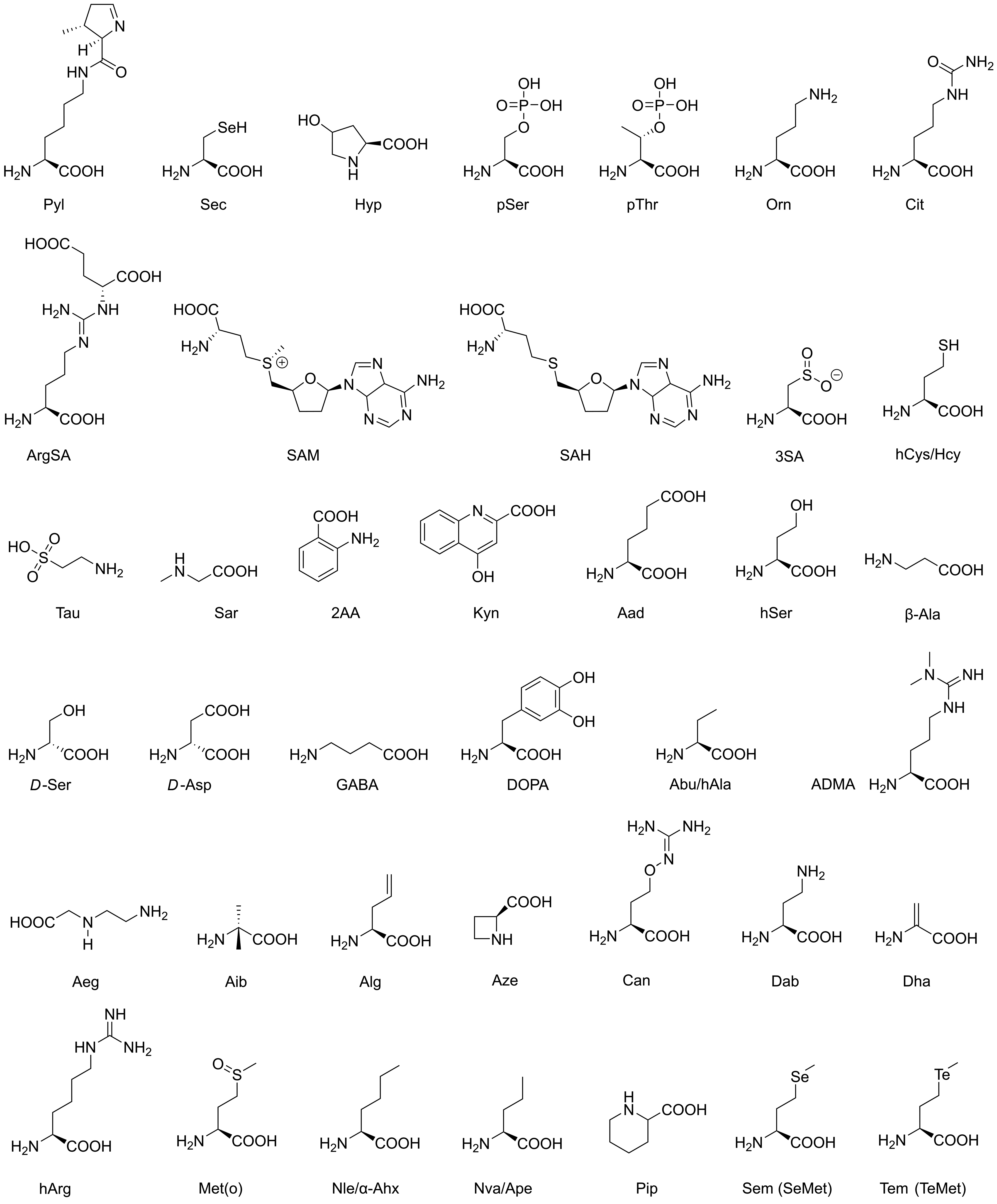
| AA | Standard Name | Type and Occurrence | Functions and Usage |
|---|---|---|---|
| Sec | selenocysteine | all organisms (ao) | redox processes |
| Pyl | pyrrolysine | archaea and bacteria | methyltransferase catalysis |
| Hyp | hydroxyproline | vertebrates (ve) | PTM, collagen stability |
| pSer | phosphoserine | eukaryotes (eu) | PTM, signaling/cancer |
| pThr | phosphothreonine | eu | PTM, signaling/cancer |
| Orn | ornithine | ao | urea cycle |
| Cit | citrulline | ao | urea cycle |
| ArgSA | argininosuccinic acid | ao | urea cycle |
| SAM | S-adenosylmethionine | ao | DNA methylation (eukaryotes) |
| SAH | S-adenosylhomocysteine | ao | sulfur metabolism |
| 3SA | 3-sulfinoalanine | ao | sulfur metabolism |
| hCys/Hcy | homocysteine | ao | sulfur metabolism |
| Tau | taurine | ao | sulfur metabolism |
| Sar | sarcosine | ao | glycine biosynthesis |
| 2AA | anthranilic acid | ao | tryptophan biosynthesis |
| Kyn | kynurenic acid | ao | tryptophan degradation |
| Aad | aminoadipic acid | ao | lysine biosynthesis |
| hSer | homoserine | ao | methionine metabolism |
| β-Ala | β-alanine | eu | vitamin B5 component |
| D-Ser | D-serine | ve | neurotransmitter, coagonist NMDA receptor |
| D-Asp | D-aspartic acid | ve | neurotransmitter, agonist NMDA receptors |
| GABA | γ-aminobutyric acid | ve | neurotransmitter, GABA receptors |
| DOPA | 3-hydroxytyrosine | ve | neurotransmitter, dopamine precursor |
| Abu/hAla | α-aminobutyric acid | ve | metabolite |
| ADMA | dimethylarginine | ve | nitric oxide synthase regulator |
| Aeg | N-2-aminoethylglycine | cyanobacteria | toxin |
| Aib | α-aminoisobutyric acid | ao | metabolite |
| Alg | allylglycine | rare metabolite | |
| Aze | azetidine-2-carboxylic acid | plants, Convallaria majalis | toxin |
| Can | canavanine | plants, Leguminosae | toxin |
| Dab | 2,4-diaminobutyric acid | cyanobacteria | toxin |
| Dha | dehydroalanine | bacteria, Lactococcus lactis | lantibiotics |
| hArg | homoarginine | ao | bacterial growth inhibitor |
| Met(o) | methionine sulfoxide | ao | aging proteins |
| Nle/α-Ahx | norleucine | bacteria | |
| Nva/Ape | norvaline | bacteria | |
| Pip | pipecolic acid | ao | immunity regulator |
| Sem/SeMet | selenomethionine | Met analog in proteins | X-ray crystallography |
| Tem/TeMet | telluromethionine | Met analog in proteins | X-ray crystallography |

| AA | Standard Name | Characteristics and Usage |
|---|---|---|
| Aca | trans-4-aminocyclohexylalanine | substrate, inhibitor |
| ACCA | cis-3-aminomethylcyclobutane carboxylic acid | inhibitor |
| ε-Ahx | aminocaproic acid/6-aminohexaonic acid | substrate, inhibitor, ABP |
| Ama | trans-4-aminomethylcyclohexylalanine | substrate, inhibitor |
| Amf | 4-aminomethylphenylalanine | substrate, inhibitor |
| Anb | 5-amino-2-nitrobenzoic acid | substrate, inhibitor, ABP |
| Cha | cyclohexylalanine | substrate, inhibitor |
| Chg | cyclohexylglycine | inhibitor |
| Cpa | 3-(2-cyano-4-pyridyl)alanine | inhibitor |
| CycL | cyclo-leucine/cyclo-Leu | inhibitor |
| DfeGly | difluoroethylgycine | inhibitor, ABP |
| DFM | difluoromethionine | protein modification |
| DfpGly | difluoropropylgycine | inhibitor |
| Dnp-Lys | N(6)-(2,4-dinitrophenyl)lysine | substrate (fluorophore) |
| 4F-Phe | 4-fluorophenylalanine (19F) | NMR |
| 5F-Trp | 5-fluorotryptophan (19F) | NMR |
| F(gua) | 4-guanidinophenylalanine | substrate, inhibitor, ABP |
| hCha | homocyclohexylalanine | substrate, inhibitor |
| Igl | 2-indanylglycine | substrate, ABP |
| MfeGly | monofluoroethylgycine | inhibitor |
| MLeu | α-methyl-leucine | inhibitor |
| 1-Nal | 1-naphthylalanine | inhibitor |
| 2-Nal | 2-naphthylalanine | inhibitor |
| Oic | octahydroindole-2-carboxylic acid | substrate, inhibitor, ABP |
| Phg | phenylglycine | substrate, inhibitor |
| Phi | perhydroindol-2-carboxylic acid | inhibitor |
| Sep | selenotryptophans (selenolo[3,2-b]pyrrole/[2,3-b]pyrrole) | X-ray cystallography |
| TfeGly | trifluoroethylgycine | inhibitor, ABP |
| Thfg | tetrahydrofuranylglycine | inhibitor |
| Thi | 3-(2-thienyl)alanine/β-thienylalanine or 3-(3-thienyl)alanine | substrate, inhibitor |
| Thp | thioproline | inhibitor |
| Tic | 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid | substrate, inhibitor |
| Tle | tert-leucine/tert-butyl-glycine | substrate, inhibitor, ABP |
| CN-Phe | 4-cyanophenylalanine | fluorophore |
| 3CY | 3-chlorotyrosine | protein modification |
| 3NY | 3-nitrotyrosine | protein modification, fluorophore |
| Bpa | 4-benzoylphenylalanine | photo-crosslinker |
| Dap | 2,3-diaminopropionic acid | ABP for Ser/Cys proteases |
| DiZPK | 3-(3-methyl-3H-diazirine-3-yl)-propamino(carbonyl-Nε-lysine) | protein modification/photo-crosslinker |
| Hco | 7-(hydroxy-coumarin-4-yl)-ethylglycine | protein modification/fluorophore |
| NPY | nitropiperonyltyrosine | caging and decaging |
| PABK | Nε-4-azidobenzyloxycarbonyllysine | click reactant, caging and decaging |
| Aha | azidohomoalanine | click reaction, 1,3-dipolar cycloaddition |
| AzF | 4-azidophenylalanine | 1,3-dipolar cycloaddition |
| Hpg | homopropargylglycine | 1,3-dipolar cycloaddition |
| Plk | N-propargyllysine | 1,3-dipolar cycloaddition |
| Sac | S-allylcysteine | photo-click reaction |
2. Methods for the Incorporation of ncAAs in Peptides and Proteins
2.1. Chemical Modification of Standard Amino Acids
2.2. Substitution of Specific cAAs by ncAAs in Auxotrophy-Based Methods and Its Relevance for Structural Biology

2.3. Site-Directed Insertion of ncAAs Using Orthogonal Pairs
3. Protease Substrates, Inhibitors and Activity-Based Probes with ncAAs
3.1. Serine Proteases
3.1.1. Digestive Trypsin-like Serine Proteases
3.1.2. Thrombin and Other Blood Coagulation Factors
3.1.3. Kallikrein-Related Peptidases, Cathepsins, Neutrophil Serine Proteases and Tryptases

3.1.4. Viral Serine Proteases
3.1.5. Subtilisin-like Serine Proteases and α/β-Hydrolases
3.2. ATP-Dependent Proteases

3.3. Cysteine Proteases

3.4. Aspartic Proteases

3.5. Metalloproteases


4. Modified Proteases
4.1. Incorporation of ncAAs into Proteases for Structure-Functional Studies
4.2. Cross-Linking with ncAAs
4.3. Click Reactions with Proteases and Their Inhibitors

5. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
| AA | IUPAC Name | CAS Number |
|---|---|---|
| Sec | (2R)-2-amino-3-selanylpropanoic acid | 10236-58-5 |
| Pyl | N6-[[(2R,3R)-3-methyl-3,4-dihydro-2H-pyrrol-2-yl]carbonyl]-L-lysine | 448235-52-7 |
| Hyp | (2S,3S)-3-hydroxypyrrolidine-2-carboxylic acid (2S,4R)-4-hydroxypyrrolidine-2-carboxylic acid | 4298-08-2 51-35-4 |
| pSer | (2S)-2-amino-3-phosphonooxypropanoic acid | 407-41-0 |
| pThr | (2S,3R)-2-amino-3-phosphonooxybutanoic acid | 27530-80-9 |
| Orn | (2S)-2,5-diaminopentanoic acid | 70-26-8 |
| Cit | (2S)-2-amino-5-(carbamoylamino)pentanoic acid | 372-75-8 |
| ArgSA | (2S)-2-[[N′-[(4S)-4-amino-4-carboxybutyl]carbamimidoyl]amino]butanedioic acid | 2387-71-5 |
| SAM | (2S)-2-amino-4-[[(2S,3S,4R,5R)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl-methylsulfonio]butanoate | 29908-03-0 |
| SAH | (2S)-2-amino-4-[[(2S,3S,4R,5R)-5-(6-aminopurin-9-yl)-3,4-dihydroxyoxolan-2-yl]methyl-sulfanyl]butanoate | 979-92-0 |
| 3SA | (2R)-2-amino-3-hydroxy-3-oxopropane-1-sulfinate | 1115-65-7 |
| hCys/Hcy | (2S)-2-amino-4-sulfanylbutanoic acid | 6027-13-0 |
| Tau | 2-aminoethanesulfonic acid | 107-35-7 |
| Sar | 2-(methylamino)acetic acid | 107-97-1 |
| 2AA | 2-aminobenzoic acid | 118-92-3 |
| Kyn | 4-oxo-1H-quinoline-2-carboxylic acid | 492-27-3 |
| Aad | (2S)-2-aminohexanedioic acid | 1118-90-7 |
| hSer | (2S)-2-amino-4-hydroxybutanoic acid | 672-15-1 |
| β-Ala | 3-aminopropanoic acid | 107-95-9 |
| D-Ser | (2R)-2-amino-3-hydroxypropanoic acid | 312-84-5 |
| D-Asp | (2R)-2-aminobutanedioic acid | 1783-96-6 |
| GABA | 4-aminobutanoic acid | 56-12-2 |
| DOPA | (2S)-2-amino-3-(3,4-dihydroxyphenyl)propanoic acid | 59-92-7 |
| Abu/hAla | (2S)-2-aminobutanoic acid | 1492-24-6 |
| ADMA | (2S)-5-(diaminomethylideneamino)-2-(dimethylamino)pentanoic acid | 30315-93-6 |
| Aeg | 2-(2-aminoethylamino)acetic acid | 24123-14-6 |
| Aib | 2-amino-2-methylpropanoic acid | 62-57-7 |
| Alg | (2S)-2-aminopent-4-enoic acid | 16338-48-0 |
| Aze | (2S)-azetidine-2-carboxylic acid | 2133-34-8 |
| Can | (2S)-2-amino-4-(diaminomethylideneamino)oxybutanoic acid | 543-38-4 |
| Dab | (2S)-2,4-diaminobutanoic acid | 1758-80-1 |
| Dha | 2-aminoprop-2-enoic acid | 1948-56-7 |
| hArg | (2S)-2-amino-6-(diaminomethylideneamino)hexanoic acid | 156-86-5 |
| Met(o) | (2S)-2-amino-4-methylsulfinylbutanoic acid | 3226-65-1 |
| Nle/α-Ahx | (2S)-2-aminohexanoic acid | 327-57-1 |
| Nva/Ape | (2S)-2-aminopentanoic acid | 6600-40-4 |
| Pip | (2S)-piperidine-2-carboxylic acid | 3105-95-1 |
| Sem/SeMet | (2S)-2-amino-4-methylselanylbutanoic acid | 3211-76-5 |
| Tem/TeMet | (2(2S)-2-amino-4-methyltellanylbutanoic acid | J663.934H * |
Appendix B
| AA | IUPAC Name | CAS Number |
|---|---|---|
| Aca | (2S)-2-amino-3-(4-aminocyclohexyl)propanoic acid | J1.610.797B * |
| ACCA | cis-(1S,3S)-3-(aminomethyl)cyclobutane-1-carboxylic acid | 1400744-20-8 (HCl) |
| ε-Ahx | 6-aminohexaonic acid | 60-32-2 |
| Ama | (2S)-2-amino-3-(4-aminomethylcyclohexyl)propanoic acid | - |
| Amf | (2S)-2-amino-3-[4-(aminomethyl)phenyl]propanoic acid | 1991-96-4 |
| Anb | 5-amino-2-nitrobenzoic acid | 13280-60-9 |
| Cha | (2S)-2-amino-3-cyclohexylpropanoic acid | 27527-05-5 |
| Chg | (2S)-2-amino-2-cyclohexylacetic acid | 14328-51-9 |
| Cpa | (2S)-2-amino-3-(2-cyanopyridin-4-yl)propanoic acid | 169949-53-5 |
| CycL | 1-aminocyclopentane-1-carboxylic acid | 52-52-8 |
| DfeGly | (2S)-2-amino-4,4-difluoro butanoic acid | J786.797B * |
| DFM | (2S)-2-amino-4-(difluoromethylsulfanyl)butanoic acid | J1.943.889I * |
| DfpGly | (2S)-2-amino-4,4-difluoro pentanoic acid | - |
| Dnp-Lys | (2S)-2-amino-6-(2,4-dinitroanilino)hexanoic acid | 14401-10-6 |
| 4F-Phe | (2S)-2-amino-3-(4-fluorophenyl)propanoic acid (19F) | 51-65-0 |
| 5F-Trp | (2S)-2-amino-3-(5-fluoro-1H-indol-3-yl)propanoic acid (19F) | 16626-02-1 |
| F(gua) | (2S)-2-amino-3-[4-(diaminomethylideneamino)phenyl]propanoic acid | 2776-36-5 |
| hCha | (2S)-2-amino-4-cyclohexylbutanoic acid | 116622-38-9 |
| Igl | (2S)-2-amino-2-(2,3-dihydro-1H-inden-2-yl)acetic acid | 155239-51-3 |
| MfeGly | (2S)-2-amino-4-fluorobutanoic acid | J1.434.018A * |
| MLeu | methyl (2S)-2-amino-2,4-dimethylpentanoic acid | 90104-02-2 |
| 1-Nal | (2S)-2-amino-3-naphthalen-1-ylpropanoic acid | 55516-54-6 |
| 2-Nal | (2S)-2-amino-3-naphthalen-2-ylpropanoic acid | 6960-34-5 |
| Oic | (2S,3aS,7aR)-2,3,3a,4,5,6,7,7a-octahydro-1H-indole-2-carboxylic acid | J532.164F * |
| Phg | (2S)-2-amino-2-phenylacetic acid | 2935-35-5 |
| Phi | (2S,3aS,7aS)-2,3,3a,4,5,6,7,7a-octahydro-1H-indole-2-carboxylic acid | 80875-98-5 |
| Sep | (2S)-2-amino-3-(4H-selenopheno[3,2-b]pyrrol-6-yl)propanoic acid (selenolo[3,2-b]pyrrole) | J1.076.917E * |
| TfeGly | (2S)-2-amino-4,4,4-trifluorobutanoic acid | 15960-05-1 |
| Thfg | (2S)-2-amino-2-(oxolan-3-yl)acetic acid | J1.827.275J * |
| Thi | (2S)-2-amino-3-thiophen-2-ylpropanoic acid/(2S)-2-amino-3-thiophen-3-ylpropanoic acid | 22574-47-6/3685-51-6 |
| Thp | (4R)-1,3-thiazolidine-4-carboxylic acid | 34592-47-7 |
| Tic | 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid | 67123-97-1 |
| Tle | (2S)-2-amino-3,3-dimethylbutanoic acid | 20859-02-3 |
| CN-Phe | (2S)-2-amino-3-(4-cyanophenyl)propanoic acid | 167479-78-9 |
| 3CY | (2S)-2-amino-3-(3-chloro-4-hydroxyphenyl)propanoic acid | 7423-93-0 |
| 3NY | (2S)-2-amino-3-(4-hydroxy-3-nitrophenyl)propanoic acid | 621-44-3 |
| Bpa | (2S)-2-amino-3-(4-benzoylphenyl)propanoic acid | 104504-45-2 |
| Dap | (2S)-2,3-diaminopropanoic acid | 4033-39-0 |
| DiZPK | (2S)-2-amino-6-[3-(3-methyldiazirin-3-yl)propylcarbamoylamino]hexanoic acid | 1337883-32-5 |
| Hco | (2R)-2-amino-4-(7-hydroxy-2-oxochromen-4-yl)butanoic acid | 905442-42-4 |
| NPY | (2S)-2-amino-3-(4-(1-(6-nitrobenzo[d][1,3]dioxol-5-yl)ethoxy)phenyl)propanoic acid | 207727-86-4 |
| PABK | (2S)-2-amino-6-(4-azido-benzyloxycarbonylamino)hexanoic acid | 2084913-49-3 |
| Aha | (2S)-2-amino-4-azidobutanoic acid | 120042-14-0 |
| AzF | (2S)-2-amino-3-(4-azidophenyl)propanoic acid | 33173-53-4 |
| Hpg | (2S)-2-aminohex-5-ynoic acid | 98841-36-2 |
| Plk | (2S)-2-amino-6-{[(prop-2-yn-1-yloxy)carbonyl]amino}hexanoic acid | 1215204-46-8 |
| Sac | (2R)-2-amino-3-prop-2-enylsulfanylpropanoic acid | 21593-77-1 |
References
- Rother, M.; Quitzke, V. Selenoprotein synthesis and regulation in Archaea. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2451–2462. [Google Scholar] [CrossRef]
- Meng, K.; Chung, C.Z.; Söll, D.; Krahn, N. Unconventional genetic code systems in archaea. Front. Microbiol. 2022, 13, 1007832. [Google Scholar] [CrossRef]
- Zhang, Y.; Gladyshev, V.N. High content of proteins containing 21st and 22nd amino acids, selenocysteine and pyrrolysine, in a symbiotic deltaproteobacterium of gutless worm Olavius algarvensis. Nucleic Acids Res. 2007, 35, 4952–4963. [Google Scholar] [CrossRef]
- Böck, A.; Forchhammer, K.; Heider, J.; Leinfelder, W.; Sawers, G.; Veprek, B.; Zinoni, F. Selenocysteine: The 21st amino acid. Mol. Microbiol. 1991, 5, 515–520. [Google Scholar] [CrossRef]
- Gong, P.; Lei, P.; Wang, S.; Zeng, A.; Lou, H. Post-Translational Modifications Aid Archaeal Survival. Biomolecules 2020, 10, 584. [Google Scholar] [CrossRef]
- Macek, B.; Forchhammer, K.; Hardouin, J.; Weber-Ban, E.; Grangeasse, C.; Mijakovic, I. Protein post-translational modifications in bacteria. Nat. Rev. Microbiol. 2019, 17, 651–664. [Google Scholar] [CrossRef]
- Prus, G.; Hoegl, A.; Weinert, B.T.; Choudhary, C. Analysis and Interpretation of Protein Post-Translational Modification Site Stoichiometry. Trends Biochem. Sci. 2019, 44, 943–960. [Google Scholar] [CrossRef]
- Kvenvolden, K.; Lawless, J.; Pering, K.; Peterson, E.; Flores, J.; Ponnamperuma, C.; Kaplan, I.R.; Moore, C. Evidence for Extraterrestrial Amino-acids and Hydrocarbons in the Murchison Meteorite. Nature 1970, 228, 923–926. [Google Scholar] [CrossRef]
- Elsila, J.E.; Aponte, J.C.; Blackmond, D.G.; Burton, A.S.; Dworkin, J.P.; Glavin, D.P. Meteoritic Amino Acids: Diversity in Compositions Reflects Parent Body Histories. ACS Cent. Sci. 2016, 2, 370–379. [Google Scholar] [CrossRef]
- Koga, T.; Naraoka, H. A new family of extraterrestrial amino acids in the Murchison meteorite. Sci. Rep. 2017, 7, 636. [Google Scholar] [CrossRef]
- Zou, H.; Li, L.; Zhang, T.; Shi, M.; Zhang, N.; Huang, J.; Xian, M. Biosynthesis and biotechnological application of non-canonical amino acids: Complex and unclear. Biotechnol. Adv. 2018, 36, 1917–1927. [Google Scholar] [CrossRef]
- Bastings, J.J.A.J.; van Eijk, H.M.; Olde Damink, S.W.; Rensen, S.S. d-amino Acids in Health and Disease: A Focus on Cancer. Nutrients 2019, 11, 2205. [Google Scholar] [CrossRef]
- Genchi, G. An overview on d-amino acids. Amino Acids 2017, 49, 1521–1533. [Google Scholar] [CrossRef]
- Fujii, N.; Takata, T.; Fujii, N.; Aki, K.; Sakaue, H. D-Amino acids in protein: The mirror of life as a molecular index of aging. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 840–847. [Google Scholar] [CrossRef]
- Kudo, F.; Miyanaga, A.; Eguchi, T. Biosynthesis of natural products containing β-amino acids. Nat. Prod. Rep. 2014, 31, 1056–1073. [Google Scholar] [CrossRef]
- von Nussbaum, F.; Spiteller, P. β-Amino Acids in Nature. In Highlights in Bioorganic Chemistry: Methods and Applications; Schmuck, C., Wennemers, H., Eds.; Wiley-VCH Verlag: Weinheim, Germany, 2004; Volume 1, pp. 63–89. [Google Scholar]
- Hedges, J.B.; Ryan, K.S. Biosynthetic Pathways to Nonproteinogenic α-Amino Acids. Chem. Rev. 2020, 120, 3161–3209. [Google Scholar] [CrossRef]
- Papageorgiou, N.; Androulakis, E.; Papaioannou, S.; Antoniades, C.; Tousoulis, D. Homoarginine in the shadow of asymmetric dimethylarginine: From nitric oxide to cardiovascular disease. Amino Acids 2015, 47, 1741–1750. [Google Scholar] [CrossRef]
- Hrncic, D.; Rasic-Markovic, A.; Macut, D.; Mladenovic, D.; Susic, V.; Djuric, D.; Stanojlovic, O. Sulfur–Containing Amino Acids in Seizures: Current State of the Art. Curr. Med. Chem. 2018, 25, 378–390. [Google Scholar] [CrossRef]
- Stipanuk, M.H. Metabolism of Sulfur-Containing Amino Acids: How the Body Copes with Excess Methionine, Cysteine, and Sulfide. J. Nutr. 2020, 150, 2494S–2505S. [Google Scholar] [CrossRef]
- Manolidi, K.; Triantis, T.M.; Kaloudis, T.; Hiskia, A. Neurotoxin BMAA and its isomeric amino acids in cyanobacteria and cyanobacteria-based food supplements. J. Hazard. Mater. 2019, 365, 346–365. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Barrett, A.J.; Thomas, P.D.; Huang, X.; Bateman, A.; Finn, R.D. The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and a comparison with peptidases in the PANTHER database. Nucleic Acids Res. 2018, 46, D624–D632. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Salvesen, G. Handbook of Proteolytic Enzymes, 3rd ed.; Academic Press: London, UK; Oxford, UK; Boston, MA, USA; New York, NY, USA; San Diego, CA, USA, 2013; pp. 1–4094. [Google Scholar]
- Elsässer, B.; Goettig, P. Mechanisms of Proteolytic Enzymes and Their Inhibition in QM/MM Studies. Int. J. Mol. Sci. 2021, 22, 3232. [Google Scholar] [CrossRef] [PubMed]
- Kubyshkin, V.; Davis, R.; Budisa, N. Biochemistry of fluoroprolines: The prospect of making fluorine a bioelement. Beilstein J. Org. Chem. 2021, 17, 439–460. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-M.; Yang, M.-Y.; Huang, Y.-C.; Li, Y.-T.; Chen, P.R.; Liu, L. Ligation of Expressed Protein α-Hydrazides via Genetic Incorporation of an α-Hydroxy Acid. ACS Chem. Biol. 2012, 7, 1015–1022. [Google Scholar] [CrossRef] [PubMed]
- Agostini, F.; Völler, J.-S.; Koksch, B.; Acevedo-Rocha, C.G.; Kubyshkin, V.; Budisa, N. Biocatalysis with Unnatural Amino Acids: Enzymology Meets Xenobiology. Angew. Chem. Int. Ed. Engl. 2017, 56, 9680–9703. [Google Scholar] [CrossRef]
- Chalker, J.M.; Bernardes, G.J.L.; Lin, Y.A.; Davis, B.G. Chemical Modification of Proteins at Cysteine: Opportunities in Chemistry and Biology. Chem. Asian J. 2009, 4, 630–640. [Google Scholar] [CrossRef]
- Dickens, F. Interaction of halogenacetates and SH compounds: The reaction of halogenacetic acids with glutathione and cysteine. The mechanism of iodoacetate poisoning of glyoxalase. Biochem. J. 1933, 27, 1141–1151. [Google Scholar] [CrossRef]
- Tsuchiya, K.; Numata, K. Chemoenzymatic synthesis of polypeptides containing the unnatural amino acid 2-aminoisobutyric acid. Chem. Commun. 2017, 53, 7318–7321. [Google Scholar] [CrossRef]
- Goettig, P. Reversed Proteolysis—Proteases as Peptide Ligases. Catalysts 2021, 11, 33. [Google Scholar] [CrossRef]
- Paetzel, M.; Strynadka, N.C.J.; Tschantz, W.R.; Casareno, R.; Bullinger, P.R.; Dalbey, R.E. Use of Site-directed Chemical Modification to Study an Essential Lysine in Escherichia coli Leader Peptidase. J. Biol. Chem. 1997, 272, 9994–10003. [Google Scholar] [CrossRef]
- DeSantis, G.; Berglund, P.; Stabile, M.R.; Gold, M.; Jones, J.B. Site-Directed Mutagenesis Combined with Chemical Modification as a Strategy for Altering the Specificity of the S1 and S1′ Pockets of Subtilisin Bacillus lentus. Biochemistry 1998, 37, 5968–5973. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.G.; Khumtaveeporn, K.; Bott, R.R.; Jones, J.B. Altering the specificity of subtilisin Bacillus lentus through the introduction of positive charge at single amino acid sites. Bioorg. Med. Chem. 1999, 7, 2303–2311. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.G.; Shang, X.; DeSantis, G.; Bott, R.R.; Jones, J.B. The controlled introduction of multiple negative charge at single amino acid sites in subtilisin Bacillus lentus. Bioorg. Med. Chem. 1999, 7, 2293–2301. [Google Scholar] [CrossRef] [PubMed]
- Cowie, D.B.; Cohen, G.N. Biosynthesis by Escherichia coli of active altered proteins containing selenium instead of sulfur. Biochim. Biophys. Acta 1957, 26, 252–261. [Google Scholar] [CrossRef]
- Richmond, H.M. The Effect of Amino Acid Analogues on Growth and Protein Synthesis in Microorganisms. Bacteriol. Rev. 1962, 26, 398–420. [Google Scholar] [CrossRef]
- Boles, J.O.; Cisneros, R.J.; Weir, M.S.; Odom, J.D.; Villafranca, J.E.; Dunlap, R.B. Purification and characterization of selenomethionyl thymidylate synthase from Escherichia coli: Comparison with the wild-type enzyme. Biochemistry 1991, 30, 11073–11080. [Google Scholar] [CrossRef]
- Yang, W.; Hendrickson, W.A.; Crouch, R.J.; Satow, Y. Structure of Ribonuclease H Phased at 2 Å Resolution by MAD Analysis of the Selenomethionyl Protein. Science 1990, 249, 1398–1405. [Google Scholar] [CrossRef]
- Boles, J.O.; Yu, H.N.; Patti, J.M. The biosynthetic incorporation of selenomethionine and telluromethionine into pyrrolidone carboxyl peptidase (PYRase) from S. aureus. SAAS Bull. Biochem. Biotechnol. 1997, 10, 13–17. [Google Scholar] [PubMed]
- Budisa, N.; Steipe, B.; Demange, P.; Eckerskorn, C.; Kellermann, J.; Huber, R. High-level Biosynthetic Substitution of Methionine in Proteins by its Analogs 2-Aminohexanoic Acid, Selenomethionine, Telluromethionine and Ethionine in Escherichia coli. Eur. J. Biochem. 1995, 230, 788–796. [Google Scholar] [CrossRef]
- Bae, J.H.; Alefelder, S.; Kaiser, J.T.; Friedrich, R.; Moroder, L.; Huber, R.; Budisa, N. Incorporation of β-selenolo[3,2-b]pyrrolyl-alanine into proteins for phase determination in protein X-ray crystallography. J. Mol. Biol. 2001, 309, 925–936. [Google Scholar] [CrossRef]
- Hendrickson, W.A.; Smith, J.L.; Sheriff, S. Direct phase determination based on anomalous scattering. Methods Enzymol. 1985, 115, 41–55. [Google Scholar] [CrossRef]
- Hendrickson, W.A.; Horton, J.R.; LeMaster, D.M. Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): A vehicle for direct determination of three-dimensional structure. EMBO J. 1990, 9, 1665–1672. [Google Scholar] [CrossRef]
- Both, E.B.; Shao, S.; Xiang, J.; Jókai, Z.; Yin, H.; Liu, Y.; Magyar, A.; Dernovics, M. Selenolanthionine is the major water-soluble selenium compound in the selenium tolerant plant Cardamine violifolia. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 2354–2362. [Google Scholar] [CrossRef]
- Templeton, L.K.; Templeton, D.H. Biaxial tensors for anomalous scattering of X-rays in selenolanthionine. Acta Crystallogr. Sect. A 1988, 44, 1045–1051. [Google Scholar] [CrossRef]
- Ferrer, J.L.; Roth, M.; Antoniadis, A. Data compression for diffraction patterns. Acta Crystallogr. Sect. D Biol. Crystallogr. 1998, 54, 184–199. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, W.A.; Pähler, A.; Smith, J.L.; Satow, Y.; Merritt, E.A.; Phizackerley, R.P. Crystal structure of core streptavidin determined from multiwavelength anomalous diffraction of synchrotron radiation. Proc. Natl. Acad. Sci. USA 1989, 86, 2190–2194. [Google Scholar] [CrossRef] [PubMed]
- Boles, J.O.; Lewinski, K.; Kunkle, M.; Odom, J.D.; Dunlap, R.B.; Lebioda, L.; Hatada, M. Bio–incorporation of telluromethionine into buried residues of dihydrofolate reductase. Nat. Struct. Biol. 1994, 1, 283–284. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, S.E.; Razak, A.A.; Ragab, A.M.; El-Meleigy, M. Incorporation of tellurium into amino acids and proteins in a tellurium-tolerant fungi. Biol. Trace Elem. Res. 1989, 20, 225–232. [Google Scholar] [CrossRef]
- Budisa, N.; Karnbrock, W.; Steinbacher, S.; Humm, A.; Prade, L.; Neuefeind, T.; Moroder, L.; Huber, R. Bioincorporation of telluromethionine into proteins: A promising new approach for X-ray structure analysis of proteins. J. Mol. Biol. 1997, 270, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Moroder, L.; Musiol, H.-J. Amino acid chalcogen analogues as tools in peptide and protein research. J. Pept. Sci. 2020, 26, e3232. [Google Scholar] [CrossRef]
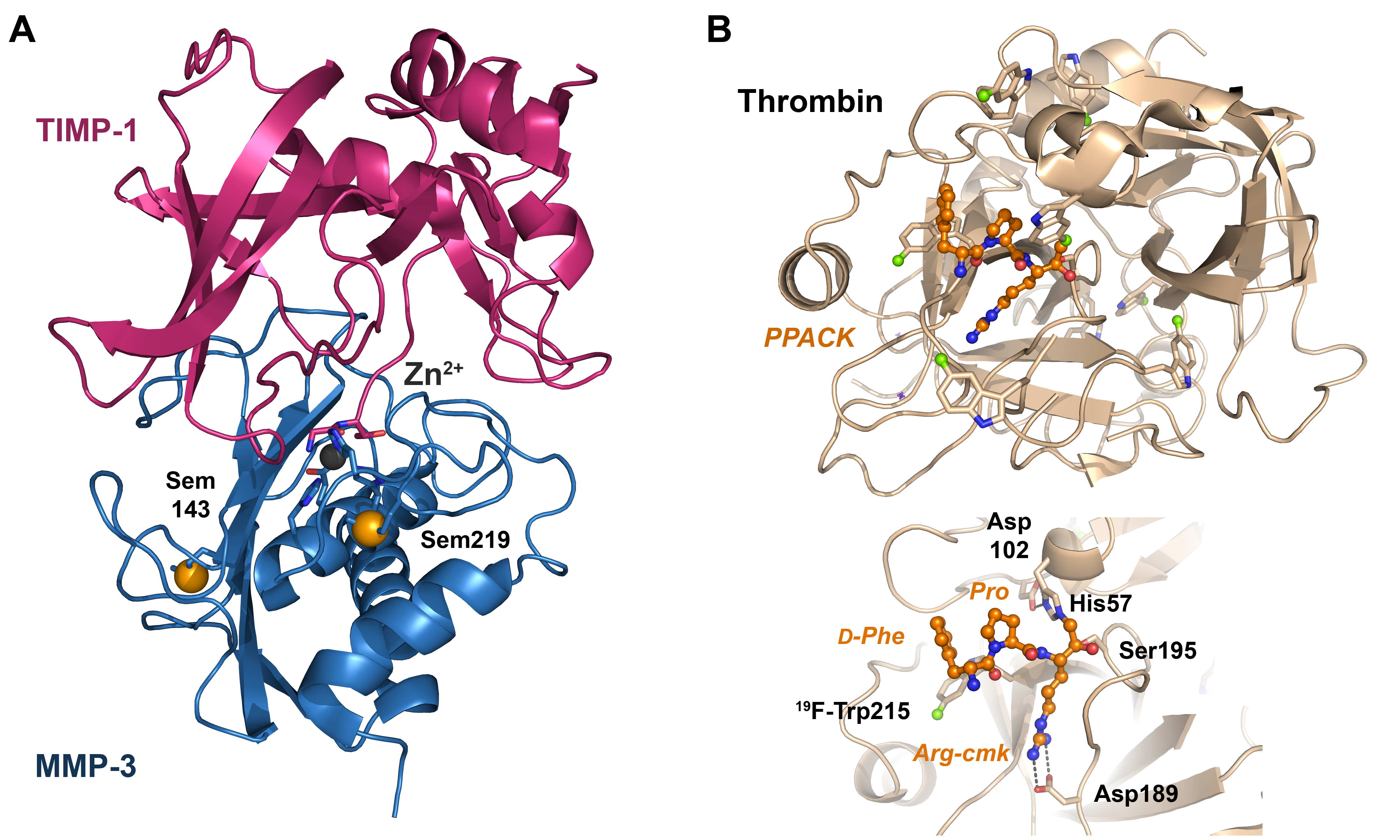
- Gomis-Rüth, F.-X.; Maskos, K.; Betz, M.; Bergner, A.; Huber, R.; Suzuki, K.; Yoshida, N.; Nagase, H.; Brew, K.; Bourenkov, G.P.; et al. Mechanism of inhibition of the human matrix metalloproteinase stromelysin-1 by TIMP-1. Nature 1997, 389, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Hendrickson, W.A. Contemporary Use of Anomalous Diffraction in Biomolecular Structure Analysis. In Protein Crystallography: Methods and Protocols; Wlodawer, A., Dauter, Z., Jaskolski, M., Eds.; Springer: New York, NY, USA, 2017; pp. 377–399. [Google Scholar]
- Rose, J.P.; Wang, B.-C. SAD phasing: History, current impact and future opportunities. Arch. Biochem. Biophys. 2016, 602, 80–94. [Google Scholar] [CrossRef] [PubMed]
- Barton, W.A.; Tzvetkova-Robev, D.; Erdjument-Bromage, H.; Tempst, P.; Nikolov, D.B. Highly efficient selenomethionine labeling of recombinant proteins produced in mammalian cells. Protein Sci. 2006, 15, 2008–2013. [Google Scholar] [CrossRef] [PubMed]
- Ouerdane, L.; Mester, Z. Production and Characterization of Fully Selenomethionine-Labeled Saccharomyces cerevisiae. J. Agric. Food Chem. 2008, 56, 11792–11799. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.; Imasaki, T.; Takagi, Y. A practical method for efficient and optimal production of Seleno-methionine-labeled recombinant protein complexes in the insect cells. Protein Sci. 2019, 28, 808–822. [Google Scholar] [CrossRef]
- Brewster, A.S.; Bhowmick, A.; Bolotovsky, R.; Mendez, D.; Zwart, P.H.; Sauter, N.K. SAD phasing of XFEL data depends critically on the error model. Acta Crystallogr. Sect. D 2019, 75, 959–968. [Google Scholar] [CrossRef]
- Rius, J.; Torrelles, X. Extending the novel |[rho]|-based phasing algorithm to the solution of anomalous scattering substructures from SAD data of protein crystals. Acta Crystallogr. Sect. A 2022, 78, 473–481. [Google Scholar] [CrossRef]
- Barbarin-Bocahu, I.; Graille, M. The X-ray crystallography phase problem solved thanks to AlphaFold and RoseTTAFold models: A case-study report. Acta Crystallogr. Sect. D 2022, 78, 517–531. [Google Scholar] [CrossRef]
- Hoesl, M.G.; Budisa, N. In vivo Incorporation of Multiple Noncanonical Amino Acids into Proteins. Angew. Chem. Int. Ed. Engl. 2011, 50, 2896–2902. [Google Scholar] [CrossRef]
- Kwon, I.; Kirshenbaum, K.; Tirrell, D.A. Breaking the Degeneracy of the Genetic Code. J. Am. Chem. Soc. 2003, 125, 7512–7513. [Google Scholar] [CrossRef]
- Bohlke, N.; Budisa, N. Sense codon emancipation for proteome-wide incorporation of noncanonical amino acids: Rare isoleucine codon AUA as a target for genetic code expansion. FEMS Microbiol. Lett. 2014, 351, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Budisa, N. Prolegomena to Future Experimental Efforts on Genetic Code Engineering by Expanding Its Amino Acid Repertoire. Angew. Chem. Int. Ed. Engl. 2004, 43, 6426–6463. [Google Scholar] [CrossRef] [PubMed]
- Budisa, N.; Minks, C.; Alefelder, S.; Wenger, W.; Dong, F.; Moroder, L.; Huber, R. Toward the experimental codon reassignment in vivo: Protein building with an expanded amino acid repertoire. FASEB J. 1999, 13, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Wong, J.T. Future of the Genetic Code. Life 2017, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Takai, K.; Hohsaka, T.; Sisido, M.; Takaku, H. Sense Codon-Dependent Introduction of Unnatural Amino Acids into Multiple Sites of a Protein. Biochem. Biophys. Res. Commun. 2000, 270, 1136–1139. [Google Scholar] [CrossRef]
- Baumann, T.; Nickling, J.H.; Bartholomae, M.; Buivydas, A.; Kuipers, O.P.; Budisa, N. Prospects of in vivo Incorporation of Non-canonical Amino Acids for the Chemical Diversification of Antimicrobial Peptides. Front. Microbiol. 2017, 8, 124. [Google Scholar] [CrossRef]
- Foster, M.P.; McElroy, C.A.; Amero, C.D. Solution NMR of Large Molecules and Assemblies. Biochemistry 2007, 46, 331–340. [Google Scholar] [CrossRef]
- Merkel, L.; Budisa, N. Organic fluorine as a polypeptide building element: In vivo expression of fluorinated peptides, proteins and proteomes. Org. Biomol. Chem. 2012, 10, 7241–7261. [Google Scholar] [CrossRef]
- Sugiki, T.; Furuita, K.; Fujiwara, T.; Kojima, C. Current NMR Techniques for Structure-Based Drug Discovery. Molecules 2018, 23, 148. [Google Scholar] [CrossRef]
- Ruben, E.A.; Gandhi, P.S.; Chen, Z.; Koester, S.K.; DeKoster, G.T.; Frieden, C.; Di Cera, E. 19F NMR reveals the conformational properties of free thrombin and its zymogen precursor prethrombin-2. J. Biol. Chem. 2020, 295, 8227–8235. [Google Scholar] [CrossRef]
- Gowda, G.A.N.; Shanaiah, N.; Raftery, D. Isotope enhanced approaches in metabolomics. Adv. Exp. Med. Biol. 2012, 992, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Verardi, R.; Traaseth, N.J.; Masterson, L.R.; Vostrikov, V.V.; Veglia, G. Isotope Labeling for Solution and Solid-State NMR Spectroscopy of Membrane Proteins BT—Isotope labeling in Biomolecular NMR. In Advances in Experimental Medicine and Biology; Atreya, H.S., Ed.; Springer: Dordrecht, The Netherlands, 2012; Volume 992, pp. 35–62. [Google Scholar]
- Marshall, D.D.; Powers, R. Beyond the paradigm: Combining mass spectrometry and nuclear magnetic resonance for metabolomics. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 100, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Permentier, H.P.; Bischoff, R. Chemical isotope labeling for quantitative proteomics. Mass Spectrom. Rev. 2023, 42, 546–576. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.Y.; Lieblich, S.A.; Tirrell, D.A. Incorporation of Non-Canonical Amino Acids into Proteins by Global Reassignment of Sense Codons. Methods Mol. Biol. 2018, 1798, 173–186. [Google Scholar] [CrossRef]
- Hoesl, M.G.; Acevedo-Rocha, C.G.; Nehring, S.; Royter, M.; Wolschner, C.; Wiltschi, B.; Budisa, N.; Antranikian, G. Lipase Congeners Designed by Genetic Code Engineering. ChemCatChem 2011, 3, 213–221. [Google Scholar] [CrossRef]
- Hoesl, M.G.; Budisa, N. Recent advances in genetic code engineering in Escherichia coli. Curr. Opin. Biotechnol. 2012, 23, 751–757. [Google Scholar] [CrossRef]
- Wang, L.; Schultz, P.G. Expanding the genetic code. Chem. Commun. 2002, 1, 1–11. [Google Scholar] [CrossRef]
- Wan, W.; Tharp, J.M.; Liu, W.R. Pyrrolysyl-tRNA synthetase: An ordinary enzyme but an outstanding genetic code expansion tool. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 1059–1070. [Google Scholar] [CrossRef]
- Wang, L.; Brock, A.; Herberich, B.; Schultz, P.G. Expanding the Genetic Code of Escherichia coli. Science 2001, 292, 498–500. [Google Scholar] [CrossRef]
- Baumann, T.; Exner, M.; Budisa, N. Orthogonal Protein Translation Using Pyrrolysyl-tRNA Synthetases for Single- and Multiple-Noncanonical Amino Acid Mutagenesis BT. In Synthetic Biology—Metabolic Engineering; Zhao, H., Zeng, A.-P., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 1–19. [Google Scholar]
- Wang, L. Genetically encoding new bioreactivity. New Biotechnol. 2017, 38, 16–25. [Google Scholar] [CrossRef]
- de la Torre, D.; Chin, J.W. Reprogramming the genetic code. Nat. Rev. Genet. 2021, 22, 169–184. [Google Scholar] [CrossRef] [PubMed]
- Ros, E.; Torres, A.G.; Ribas de Pouplana, L. Learning from Nature to Expand the Genetic Code. Trends Biotechnol. 2021, 39, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Exner, M.P.; Köhling, S.; Rivollier, J.; Gosling, S.; Srivastava, P.; Palyancheva, Z.I.; Herdewijn, P.; Heck, M.-P.; Rademann, J.; Budisa, N. Incorporation of Amino Acids with Long-Chain Terminal Olefins into Proteins. Molecules 2016, 21, 287. [Google Scholar] [CrossRef]
- Koch, N.G.; Goettig, P.; Rappsilber, J.; Budisa, N. Engineering Pyrrolysyl-tRNA Synthetase for the Incorporation of Non-Canonical Amino Acids with Smaller Side Chains. Int. J. Mol. Sci. 2021, 22, 11194. [Google Scholar] [CrossRef]
- Tseng, H.-W.; Baumann, T.; Sun, H.; Wang, Y.-S.; Ignatova, Z.; Budisa, N. Expanding the Scope of Orthogonal Translation with Pyrrolysyl-tRNA Synthetases Dedicated to Aromatic Amino Acids. Molecules 2020, 25, 4418. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Krzycki, J.A. PylSn and the Homologous N-terminal Domain of Pyrrolysyl-tRNA Synthetase Bind the tRNA That Is Essential for the Genetic Encoding of Pyrrolysine. J. Biol. Chem. 2012, 287, 32738–32746. [Google Scholar] [CrossRef]
- Dumas, A.; Lercher, L.; Spicer, C.D.; Davis, B.G. Designing logical codon reassignment—Expanding the chemistry in biology. Chem. Sci. 2015, 6, 50–69. [Google Scholar] [CrossRef]
- Reinkemeier, C.D.; Lemke, E.A. Synthetic biomolecular condensates to engineer eukaryotic cells. Curr. Opin. Chem. Biol. 2021, 64, 174–181. [Google Scholar] [CrossRef]
- Merkel, L.; Beckmann, H.S.G.; Wittmann, V.; Budisa, N. Efficient N-Terminal Glycoconjugation of Proteins by the N-End Rule. ChemBioChem 2008, 9, 1220–1224. [Google Scholar] [CrossRef]
- Arranz-Gibert, P.; Vanderschuren, K.; Isaacs, F.J. Next-generation genetic code expansion. Curr. Opin. Chem. Biol. 2018, 46, 203–211. [Google Scholar] [CrossRef]
- Brown, W.; Liu, J.; Deiters, A. Genetic Code Expansion in Animals. ACS Chem. Biol. 2018, 13, 2375–2386. [Google Scholar] [CrossRef]
- Brabham, R.; Fascione, M.A. Pyrrolysine Amber Stop-Codon Suppression: Development and Applications. ChemBioChem 2017, 18, 1973–1983. [Google Scholar] [CrossRef]
- Pirman, N.L.; Barber, K.W.; Aerni, H.R.; Ma, N.J.; Haimovich, A.D.; Rogulina, S.; Isaacs, F.J.; Rinehart, J. A flexible codon in genomically recoded Escherichia coli permits programmable protein phosphorylation. Nat. Commun. 2015, 6, 8130. [Google Scholar] [CrossRef] [PubMed]
- Rogerson, D.T.; Sachdeva, A.; Wang, K.; Haq, T.; Kazlauskaite, A.; Hancock, S.M.; Huguenin-Dezot, N.; Muqit, M.M.K.; Fry, A.M.; Bayliss, R.; et al. Efficient genetic encoding of phosphoserine and its nonhydrolyzable analog. Nat. Chem. Biol. 2015, 11, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Wiltschi, B. Incorporation of non-canonical amino acids into proteins in yeast. Fungal Genet. Biol. 2016, 89, 137–156. [Google Scholar] [CrossRef] [PubMed]
- Wissner, R.F.; Batjargal, S.; Fadzen, C.M.; Petersson, E.J. Labeling Proteins with Fluorophore/Thioamide Förster Resonant Energy Transfer Pairs by Combining Unnatural Amino Acid Mutagenesis and Native Chemical Ligation. J. Am. Chem. Soc. 2013, 135, 6529–6540. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Johnston, W.A.; Alexandrov, K. Cell-Free Approach for Non-canonical Amino Acids Incorporation Into Polypeptides. Front. Bioeng. Biotechnol. 2020, 8, 1031. [Google Scholar] [CrossRef]
- Hui, A.; de Boer, H.A. Specialized ribosome system: Preferential translation of a single mRNA species by a subpopulation of mutated ribosomes in Escherichia coli. Proc. Natl. Acad. Sci. USA 1987, 84, 4762–4766. [Google Scholar] [CrossRef]
- Rackham, O.; Chin, J.W. Cellular logic with orthogonal ribosomes. J. Am. Chem. Soc. 2005, 127, 17584–17585. [Google Scholar] [CrossRef]
- Dedkova, L.M.; Fahmi, N.E.; Golovine, S.Y.; Hecht, S.M. Construction of modified ribosomes for incorporation of D-amino acids into proteins. Biochemistry 2006, 45, 15541–15551. [Google Scholar] [CrossRef]
- Maini, R.; Nguyen, D.T.; Chen, S.; Dedkova, L.M.; Chowdhury, S.R.; Alcala-Torano, R.; Hecht, S.M. Incorporation of β-amino acids into dihydrofolate reductase by ribosomes having modifications in the peptidyltransferase center. Bioorg. Med. Chem. 2013, 21, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Ji, X.; Gao, M.; Dedkova, L.M.; Hecht, S.M. In Cellulo Synthesis of Proteins Containing a Fluorescent Oxazole Amino Acid. J. Am. Chem. Soc. 2019, 141, 5597–5601. [Google Scholar] [CrossRef]
- Shakya, B.; Joyner, O.G.; Hartman, M.C.T. Hyperaccurate Ribosomes for Improved Genetic Code Reprogramming. ACS Synth. Biol. 2022, 11, 2193–2201. [Google Scholar] [CrossRef] [PubMed]
- Mukai, T.; Lajoie, M.J.; Englert, M.; Söll, D. Rewriting the Genetic Code. Annu. Rev. Microbiol. 2017, 71, 557–577. [Google Scholar] [CrossRef]
- Fiacco, S.V.; Kelderhouse, L.E.; Hardy, A.; Peleg, Y.; Hu, B.; Ornelas, A.; Yang, P.; Gammon, S.T.; Howell, S.M.; Wang, P.; et al. Directed Evolution of Scanning Unnatural-Protease-Resistant (SUPR) Peptides for in vivo Applications. ChemBioChem 2016, 17, 1643–1651. [Google Scholar] [CrossRef]
- Otvos Jr, L.; Bokonyi, K.; Varga, I.; Ertl, H.C.J.; Hoffmann, R.; Bulet, P.; Otvos, B.I.; Wade, J.D.; McManus, A.M.; Craik, D.J. Insect peptides with improved protease-resistance protect mice against bacterial infection. Protein Sci. 2000, 9, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.M.; Lee, S.; Mak, J.S.W.; Helmy, A.S.; Deber, C.M. Differential binding of L- vs. D-isomers of cationic antimicrobial peptides to the biofilm exopolysaccharide alginate. Protein Pept. Lett. 2013, 20, 843–847. [Google Scholar] [CrossRef]
- Khara, J.S.; Priestman, M.; Uhía, I.; Hamilton, M.S.; Krishnan, N.; Wang, Y.; Yang, Y.Y.; Langford, P.R.; Newton, S.M.; Robertson, B.D.; et al. Unnatural amino acid analogues of membrane-active helical peptides with anti-mycobacterial activity and improved stability. J. Antimicrob. Chemother. 2016, 71, 2181–2191. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.E.; Lee, K.H. Synthesis of novel unnatural amino acid as a building block and its incorporation into an antimicrobial peptide. Bioorg. Med. Chem. 1999, 7, 2985–2990. [Google Scholar] [CrossRef]
- Sarkar, T.; Chetia, M.; Chatterjee, S. Antimicrobial Peptides and Proteins: From Nature’s Reservoir to the Laboratory and Beyond. Front. Chem. 2021, 9, 691532. [Google Scholar] [CrossRef]
- Lu, J.; Xu, H.; Xia, J.; Ma, J.; Xu, J.; Li, Y.; Feng, J. D- and Unnatural Amino Acid Substituted Antimicrobial Peptides With Improved Proteolytic Resistance and Their Proteolytic Degradation Characteristics. Front. Microbiol. 2020, 11, 563030. [Google Scholar] [CrossRef] [PubMed]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Maluch, I.; Czarna, J.; Drag, M. Applications of Unnatural Amino Acids in Protease Probes. Chem. Asian J. 2019, 14, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Poreba, M.; Salvesen, G.S.; Drag, M. Synthesis of a HyCoSuL peptide substrate library to dissect protease substrate specificity. Nat. Protoc. 2017, 12, 2189–2214. [Google Scholar] [CrossRef] [PubMed]
- Vizovišek, M.; Vidmar, R.; Drag, M.; Fonović, M.; Salvesen, G.S.; Turk, B. Protease Specificity: Towards in vivo Imaging Applications and Biomarker Discovery. Trends Biochem. Sci. 2018, 43, 829–844. [Google Scholar] [CrossRef]
- Liu, Y.; Patricelli, M.P.; Cravatt, B.F. Activity-based protein profiling: The serine hydrolases. Proc. Natl. Acad. Sci. USA 1999, 96, 14694–14699. [Google Scholar] [CrossRef]
- Smolewski, P.; Grabarek, J.; Halicka, H.D.; Darzynkiewicz, Z. Assay of caspase activation in situ combined with probing plasma membrane integrity to detect three distinct stages of apoptosis. J. Immunol. Methods 2002, 265, 111–121. [Google Scholar] [CrossRef]
- Lai, Z.; Tan, P.; Zhu, Y.; Shao, C.; Shan, A.; Li, L. Highly Stabilized α-Helical Coiled Coils Kill Gram-Negative Bacteria by Multicomplementary Mechanisms under Acidic Condition. ACS Appl. Mater. Interfaces 2019, 11, 22113–22128. [Google Scholar] [CrossRef]
- Sarwar Gilani, G.; Wu Xiao, C.; Cockell, K.A. Impact of Antinutritional Factors in Food Proteins on the Digestibility of Protein and the Bioavailability of Amino Acids and on Protein Quality. Br. J. Nutr. 2012, 108, S315–S332. [Google Scholar] [CrossRef]
- Kiso, Y.; Matsumoto, H.; Mizumoto, S.; Kimura, T.; Fujiwara, Y.; Akaji, K. Small dipeptide-based HIV protease inhibitors containing the hydroxymethylcarbonyl isostere as an ideal transition-state mimic. Pept. Sci. 1999, 51, 59–68. [Google Scholar] [CrossRef]
- Böhm, H.-J. Towards the automatic design of synthetically accessible protein ligands: Peptides, amides and peptidomimetics. J. Comput.-Aided Mol. Des. 1996, 10, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical modifications designed to improve peptide stability: Incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef] [PubMed]
- Quast, U.; Engel, J.; Steffen, E.; Mair, G.; Tschesche, H.; Jering, H. Kinetics of Binding of Bovine Trypsin-Kallikrein Inhibitor (Kunitz) in which the Reactive-Site Peptide Bond Lys-15-Ala-16 is Cleaved, to α-Chymotrypsin and β-Trypsin. Eur. J. Biochem. 1975, 52, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.V.; Elmore, D.T. Kinetics and mechanism of catalysis by proteolytic enzymes. A comparison of the kinetics of hydrolysis of synthetic substrates by bovine α- and β-trypsin. Biochem. J. 1974, 141, 545–554. [Google Scholar] [CrossRef] [PubMed]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef] [PubMed]
- Goettig, P.; Brandstetter, H.; Magdolen, V. Surface loops of trypsin-like serine proteases as determinants of function. Biochimie 2019, 166, 52–76. [Google Scholar] [CrossRef]
- Lu, W.; Apostol, I.; Qasim, M.A.; Warne, N.; Wynn, R.; Zhang, W.L.; Anderson, S.; Chiang, Y.W.; Ogin, E.; Rothberg, I.; et al. Binding of amino acid side-chains to S1 cavities of serine proteinases. J. Mol. Biol. 1997, 266, 441–461. [Google Scholar] [CrossRef]
- Schellenberger, V.; Braune, K.; Hofmann, H.-J.; Jakubke, H.-D. The specificity of chymotrypsin. Eur. J. Biochem. 1991, 199, 623–636. [Google Scholar] [CrossRef]
- Wysocka, M.; Lesner, A.; Legowska, A.; Jaśkiewicz, A.; Miecznikowska, H.; Rolka, K. Designing of substrates and inhibitors of bovine alpha-chymotrypsin with synthetic phenylalanine analogues in position P(1). Protein Pept. Lett. 2008, 15, 260–264. [Google Scholar] [CrossRef]
- Huang, K.; James, M.N.G.; Lu, W.; Laskowski, M., Jr.; Anderson, S. Water molecules participate in proteinase-inhibitor interactions: Crystal structures of Leu18, Ala18, and Gly18 variants of turkey ovomucoid inhibitor third domain complexed with Streptomyces griseus proteinase B. Protein Sci. 1995, 4, 1985–1997. [Google Scholar] [CrossRef]
- Lee, T.-W.; Qasim, M.A.; Laskowski, M.; James, M.N.G. Structural Insights into the Non-additivity Effects in the Sequence-to-Reactivity Algorithm for Serine Peptidases and their Inhibitors. J. Mol. Biol. 2007, 367, 527–546. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Loll, B.; Berger, A.A.; Mülow, U.; Alings, C.; Wahl, M.C.; Koksch, B. Fluorine teams up with water to restore inhibitor activity to mutant BPTI. Chem. Sci. 2015, 6, 5246–5254. [Google Scholar] [CrossRef] [PubMed]
- Leppkes, J.; Dimos, N.; Loll, B.; Hohmann, T.; Dyrks, M.; Wieseke, A.; Keller, B.G.; Koksch, B. Fluorine-induced polarity increases inhibitory activity of BPTI towards chymotrypsin. RSC Chem. Biol. 2022, 3, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Bark, S.J.; Kent, S.B.H. Engineering an unnatural Nα-anchored disulfide into BPTI by total chemical synthesis: Structural and functional consequences. FEBS Lett. 1999, 460, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Brown, K.C.; Lodder, M.; Craik, C.S.; Hecht, S.M. Chemically mediated site-specific proteolysis. Alteration of protein-protein interaction. Biochemistry 2002, 41, 2805–2813. [Google Scholar] [CrossRef] [PubMed]
- Debowski, D.; Łukajtis, R.; Filipowicz, M.; Strzelecka, P.; Wysocka, M.; Łęgowska, A.; Lesner, A.; Rolka, K. Hybrid analogues of SFTI-1 modified in P1 position by β- and γ-amino acids and N-substituted β-alanines. Pept. Sci. 2013, 100, 154–159. [Google Scholar] [CrossRef]
- Lukajtis, R.; Legowska, A.; Wysocka, M.; Debowski, D.; Lesner, A.; Rolka, K. Analogues of trypsin inhibitor SFTI-1 modified in the conserved P1′ position by synthetic or non-proteinogenic amino acids retain their inhibitory activity. J. Pept. Sci. 2011, 17, 281–287. [Google Scholar] [CrossRef]
- Fewer, D.P.; Jokela, J.; Rouhiainen, L.; Wahlsten, M.; Koskenniemi, K.; Stal, L.J.; Sivonen, K. The non-ribosomal assembly and frequent occurrence of the protease inhibitors spumigins in the bloom-forming cyanobacterium Nodularia spumigena. Mol. Microbiol. 2009, 73, 924–937. [Google Scholar] [CrossRef]
- Ouyang, X.; D’Agostino, P.M.; Wahlsten, M.; Delbaje, E.; Jokela, J.; Permi, P.; Gaiani, G.; Poso, A.; Bartos, P.; Gulder, T.A.M.; et al. Direct pathway cloning and expression of the radiosumin biosynthetic gene cluster. Org. Biomol. Chem. 2023, 21, 4893–4908. [Google Scholar] [CrossRef]
- Issac, M.; Aknin, M.; Gauvin-Bialecki, A.; De Voogd, N.; Ledoux, A.; Frederich, M.; Kashman, Y.; Carmeli, S. Cyclotheonellazoles A–C, Potent Protease Inhibitors from the Marine Sponge Theonella aff. swinhoei. J. Nat. Prod. 2017, 80, 1110–1116. [Google Scholar] [CrossRef]
- Matthew, S.; Ross, C.; Rocca, J.R.; Paul, V.J.; Luesch, H. Lyngbyastatin 4, a Dolastatin 13 Analogue with Elastase and Chymotrypsin Inhibitory Activity from the Marine Cyanobacterium Lyngbya confervoides. J. Nat. Prod. 2007, 70, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Talukder, P.; Dedkova, L.M.; Ellington, A.D.; Yakovchuk, P.; Lim, J.; Anslyn, E.V.; Hecht, S.M. Synthesis of alanyl nucleobase amino acids and their incorporation into proteins. Bioorg. Med. Chem. 2016, 24, 4177–4187. [Google Scholar] [CrossRef] [PubMed]
- Avrutina, O.; Schmoldt, H.-U.; Gabrijelcic-Geiger, D.; Le Nguyen, D.; Sommerhoff, C.P.; Diederichsen, U.; Kolmar, H. Trypsin inhibition by macrocyclic and open-chain variants of the squash inhibitor MCoTI-II. Biol. Chem. 2005, 386, 1301–1306. [Google Scholar] [CrossRef]
- Di Cera, E. Thrombin. Mol. Asp. Med. 2008, 29, 203–254. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, A.C.; Clement, C.C.; Zakia, S.; Gingold, J.; Philipp, M.; Pereira, P.J.B. Rational Design and Characterization of D-Phe-Pro-D-Arg-Derived Direct Thrombin Inhibitors. PLoS ONE 2012, 7, e34354. [Google Scholar] [CrossRef] [PubMed]
- De Marco, R.; Di Gioia, M.L.; Leggio, A.; Liguori, A.; Perri, F.; Siciliano, C.; Viscomi, M.C. A new non-natural arginine-like amino acid derivative with a sulfamoyl group in the side-chain. Amino Acids 2010, 38, 691–700. [Google Scholar] [CrossRef]
- De Nanteuil, G.; Gloanec, P.; Lila, C.; Portevin, B.; Boudon, A.; Rupin, A.; Verbeuren, T.J. New tripeptidic thrombin inhibitors. Influence of P2 and P3 residues on activity and selectivity. Bioorg. Med. Chem. 1995, 3, 1019–1024. [Google Scholar] [CrossRef]
- Portevin, B.; Lonchampt, M.; Canet, E.; De Nanteuil, G. Dual Inhibition of Human Leukocyte Elastase and Lipid Peroxidation: In vitro and in vivo Activities of Azabicyclo[2.2.2]octane and Perhydroindole Derivatives. J. Med. Chem. 1997, 40, 1906–1918. [Google Scholar] [CrossRef]
- Deadman, J.; Claeson, G.; Scully, M.F. Structure/Function Aspects of Neutral P1 Residue Peptide Inhibitors of Thrombin. J. Enzym. Inhib. 1995, 9, 29–41. [Google Scholar] [CrossRef]
- Tsuda, Y.; Szewczuk, Z.; Wang, J.; Yue, S.Y.; Purisima, E.; Konishi, Y. Interactions of Hirudin-Based Inhibitor with Thrombin: Critical Role of the IleH59 Side Chain of the Inhibitor. Biochemistry 1995, 34, 8708–8714. [Google Scholar] [CrossRef]
- Lombardi, A.; Nastri, F.; Della Morte, R.; Rossi, A.; De Rosa, A.; Staiano, N.; Pedone, C.; Pavone, V. Rational Design of True Hirudin Mimetics: Synthesis and Characterization of Multisite-Directed α-Thrombin Inhibitors. J. Med. Chem. 1996, 39, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- De Simone, G.; Lombardi, A.; Galdiero, S.; Nastri, F.; Morte, R.D.; Staiano, N.; Pedone, C.; Bolognesi, M.; Pavone, V. Hirunorms are true hirudin mimetics. The crystal structure of human α-thrombin-hirunorm V complex. Protein Sci. 1998, 7, 243–253. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, V.; Quarzago, D.; Vindigni, A.; Di Cera, E.; Fontana, A. Synthesis and Characterization of More Potent Analogues of Hirudin Fragment 1−47 Containing Non-Natural Amino Acids. Biochemistry 1998, 37, 13507–13515. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, V.; Russo, I.; Vindigni, A.; Cera, E.D.; Salmaso, S.; Fontana, A. Incorporation of noncoded amino acids into the N-terminal domain 1-47 of hirudin yields a highly potent and selective thrombin inhibitor. Protein Sci. 1999, 8, 2213–2217. [Google Scholar] [CrossRef]
- De Filippis, V.; De Boni, S.; De Dea, E.; Dalzoppo, D.; Grandi, C.; Fontana, A. Incorporation of the fluorescent amino acid 7-azatryptophan into the core domain 1-47 of hirudin as a probe of hirudin folding and thrombin recognition. Protein Sci. A Publ. Protein Soc. 2004, 13, 1489–1502. [Google Scholar] [CrossRef]
- De Filippis, V.; Frasson, R.; Fontana, A. 3-Nitrotyrosine as a spectroscopic probe for investigating protein–protein interactions. Protein Sci. 2006, 15, 976–986. [Google Scholar] [CrossRef]
- Steinmetzer, T.; Zhu, B.Y.; Konishi, Y. Potent Bivalent Thrombin Inhibitors: Replacement of the Scissile Peptide Bond at P1−P1‘ with Arginyl Ketomethylene Isosteres. J. Med. Chem. 1999, 42, 3109–3115. [Google Scholar] [CrossRef]
- Brandstetter, H.; Turk, D.; Hoeffken, H.W.; Grosse, D.; Stürzebecher, J.; Martin, P.D.; Edwards, B.F.P.; Bode, W. Refined 2·3ÅX-ray crystal structure of bovine thrombin complexes formed with the benzamidine and arginine-based thrombin inhibitors NAPAP, 4-TAPAP and MQPA: A starting point for improving antithrombotics. J. Mol. Biol. 1992, 226, 1085–1099. [Google Scholar] [CrossRef]
- Bergner, A.; Bauer, M.; Brandstetter, H.; Stürzebecher, J.; Bode, W. The X-ray crystal structure of thrombin in complex with N alpha-2-naphthylsulfonyl-L-3-amidino-phenylalanyl-4-methylpiperidide: The beneficial effect of filling out an empty cavity. J. Enzym. Inhib. 1995, 9, 101–110. [Google Scholar] [CrossRef]
- Steinmetzer, T.; Schweinitz, A.; Künzel, S.; Wikström, P.; Hauptmann, J.; Stürzebecher, J. Structure-activity relationships of new NAPAP-analogs. J. Enzym. Inhib. 2001, 16, 241–249. [Google Scholar] [CrossRef]
- Hauptmann, J.; Steinmetzer, T.; Vieweg, H.; Wikström, P.; Stürzebecher, J. Influence of Structural Variations in Peptidomimetic 4-Amidinophenylalanine-Derived Thrombin Inhibitors on Plasma Clearance and Biliary Excretion in Rats. Pharm. Res. 2002, 19, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Féthière, J.; Tsuda, Y.; Coulombe, R.; Konishi, Y.; Cygler, M. Crystal structure of two new bifunctional nonsubstrate type thrombin inhibitors complexed with human α-thrombin. Protein Sci. 1996, 5, 1174–1183. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, Y.; Cygler, M.; Gibbs, B.F.; Pedyczak, A.; Féthière, J.; Yue, S.Y.; Konishi, Y. Design of potent bivalent thrombin inhibitors based on hirudin sequence: Incorporation of nonsubstrate-type active site inhibitors. Biochemistry 1994, 33, 14443–14451. [Google Scholar] [CrossRef] [PubMed]
- Grütter, M.G.; Priestle, J.P.; Rahuel, J.; Grossenbacher, H.; Bode, W.; Hofsteenge, J.; Stone, S.R. Crystal structure of the thrombin-hirudin complex: A novel mode of serine protease inhibition. EMBO J. 1990, 9, 2361–2365. [Google Scholar] [CrossRef]
- Slon-Usakiewicz, J.J.; Purisima, E.; Tsuda, Y.; Sulea, T.; Pedyczak, A.; Féthière, J.; Cygler, M.; Konishi, Y. Nonpolar interactions of thrombin S’ subsites with its bivalent inhibitor: Methyl scan of the inhibitor linker. Biochemistry 1997, 36, 13494–13502. [Google Scholar] [CrossRef]
- Nieman, M.T.; Burke, F.; Warnock, M.; Zhou, Y.; Sweigart, J.; Chen, A.; Ricketts, D.; Lucchesi, B.R.; Chen, Z.; Di Cera, E.; et al. Thrombostatin FM compounds: Direct thrombin inhibitors—Mechanism of action in vitro and in vivo. J. Thromb. Haemost. JTH 2008, 6, 837–845. [Google Scholar] [CrossRef]
- Hanessian, S.; Ersmark, K.; Wang, X.; Del Valle, J.R.; Blomberg, N.; Xue, Y.; Fjellström, O. Structure-based organic synthesis of unnatural aeruginosin hybrids as potent inhibitors of thrombin. Bioorg. Med. Chem. Lett. 2007, 17, 3480–3485. [Google Scholar] [CrossRef]
- Calisto, B.M.; Ripoll-Rozada, J.; Dowman, L.J.; Franck, C.; Agten, S.M.; Parker, B.L.; Veloso, R.C.; Vale, N.; Gomes, P.; de Sanctis, D.; et al. Sulfotyrosine-Mediated Recognition of Human Thrombin by a Tsetse Fly Anticoagulant Mimics Physiological Substrates. Cell Chem. Biol. 2021, 28, 26–33.e28. [Google Scholar] [CrossRef]
- Hosseini, M.; Jiang, L.; Sørensen, H.P.; Jensen, J.K.; Christensen, A.; Fogh, S.; Yuan, C.; Andersen, L.M.; Huang, M.; Andreasen, P.A.; et al. Elucidation of the Contribution of Active Site and Exosite Interactions to Affinity and Specificity of Peptidylic Serine Protease Inhibitors Using Non-Natural Arginine Analogs. Mol. Pharmacol. 2011, 80, 585–597. [Google Scholar] [CrossRef]
- Zhao, B.; Xu, P.; Jiang, L.; Paaske, B.; Kromann-Hansen, T.; Jensen, J.K.; Sørensen, H.P.; Liu, Z.; Nielsen, J.T.; Christensen, A.; et al. A Cyclic Peptidic Serine Protease Inhibitor: Increasing Affinity by Increasing Peptide Flexibility. PLoS ONE 2014, 9, e115872. [Google Scholar] [CrossRef]
- Modrzycka, S.; Kołt, S.; Polderdijk, S.G.I.; Adams, T.E.; Potoczek, S.; Huntington, J.A.; Kasperkiewicz, P.; Drąg, M. Parallel imaging of coagulation pathway proteases activated protein C, thrombin, and factor Xa in human plasma. Chem. Sci. 2022, 13, 6813–6829. [Google Scholar] [CrossRef]
- Modrzycka, S.; Kołt, S.; Adams, T.E.; Potoczek, S.; Huntington, J.A.; Kasperkiewicz, P.; Drąg, M. Fluorescent Activity-Based Probe To Image and Inhibit Factor XIa Activity in Human Plasma. J. Med. Chem. 2023, 66, 3785–3797. [Google Scholar] [CrossRef] [PubMed]
- Guillen Schlippe, Y.V.; Hartman, M.C.T.; Josephson, K.; Szostak, J.W. In vitro Selection of Highly Modified Cyclic Peptides That Act as Tight Binding Inhibitors. J. Am. Chem. Soc. 2012, 134, 10469–10477. [Google Scholar] [CrossRef] [PubMed]
- Goettig, P.; Magdolen, V.; Brandstetter, H. Natural and synthetic inhibitors of kallikrein-related peptidases (KLKs). Biochimie 2010, 92, 1546–1567. [Google Scholar] [CrossRef] [PubMed]
- Melo, R.L.; Barbosa Pozzo, R.C.; Pimenta, D.C.; Perissutti, E.; Caliendo, G.; Santagada, V.; Juliano, L.; Juliano, M.A. Human Tissue Kallikrein S1 Subsite Recognition of Non-Natural Basic Amino Acids. Biochemistry 2001, 40, 5226–5232. [Google Scholar] [CrossRef]
- Pimenta, D.C.; Melo, R.L.; Caliendo, G.; Santagada, V.; Fiorino, F.; Severino, B.; de Nucci, G.; Juliano, L.; Juliano, M.A. Design of inhibitors for human tissue kallikrein using non-natural aromatic and basic amino acids. Biol. Chem. 2002, 383, 853–857. [Google Scholar] [CrossRef]
- Guo, S.; Skala, W.; Magdolen, V.; Briza, P.; Biniossek, M.L.; Schilling, O.; Kellermann, J.; Brandstetter, H.; Goettig, P. A Single Glycan at the 99-Loop of Human Kallikrein-related Peptidase 2 Regulates Activation and Enzymatic Activity. J. Biol. Chem. 2016, 291, 593–604. [Google Scholar] [CrossRef]
- LeBeau, A.M.; Banerjee, S.R.; Pomper, M.G.; Mease, R.C.; Denmeade, S.R. Optimization of peptide-based inhibitors of prostate-specific antigen (PSA) as targeted imaging agents for prostate cancer. Bioorg. Med. Chem. 2009, 17, 4888–4893. [Google Scholar] [CrossRef]
- Teufel, D.P.; Bennett, G.; Harrison, H.; van Rietschoten, K.; Pavan, S.; Stace, C.; Le Floch, F.; Van Bergen, T.; Vermassen, E.; Barbeaux, P.; et al. Stable and Long-Lasting, Novel Bicyclic Peptide Plasma Kallikrein Inhibitors for the Treatment of Diabetic Macular Edema. J. Med. Chem. 2018, 61, 2823–2836. [Google Scholar] [CrossRef]
- Patel, S.; Homaei, A.; El-Seedi, H.R.; Akhtar, N. Cathepsins: Proteases that are vital for survival but can also be fatal. Biomed. Pharmacother. 2018, 105, 526–532. [Google Scholar] [CrossRef]
- Wysocka, M.; Łȩgowska, A.; Bulak, E.; Jaśkiewicz, A.; Miecznikowska, H.; Lesner, A.; Rolka, K. New chromogenic substrates of human neutrophil cathepsin G containing non-natural aromatic amino acid residues in position P1 selected by combinatorial chemistry methods. Mol. Divers. 2007, 11, 93–99. [Google Scholar] [CrossRef]
- de Veer, S.J.; White, A.M.; Craik, D.J. Sunflower Trypsin Inhibitor-1 (SFTI-1): Sowing Seeds in the Fields of Chemistry and Biology. Angew. Chem. Int. Ed. Engl. 2021, 60, 8050–8071. [Google Scholar] [CrossRef] [PubMed]
- Łęgowska, A.; Dębowski, D.; Lesner, A.; Wysocka, M.; Rolka, K. Introduction of non-natural amino acid residues into the substrate-specific P1 position of trypsin inhibitor SFTI-1 yields potent chymotrypsin and cathepsin G inhibitors. Bioorg. Med. Chem. 2009, 17, 3302–3307. [Google Scholar] [CrossRef] [PubMed]
- St-Georges, C.; Désilets, A.; Béliveau, F.; Ghinet, M.; Dion, S.P.; Colombo, É.; Boudreault, P.-L.; Najmanovich, R.J.; Leduc, R.; Marsault, É. Modulating the selectivity of matriptase-2 inhibitors with unnatural amino acids. Eur. J. Med. Chem. 2017, 129, 110–123. [Google Scholar] [CrossRef]
- Groborz, K.; Kołt, S.; Kasperkiewicz, P.; Drag, M. Internally quenched fluorogenic substrates with unnatural amino acids for cathepsin G investigation. Biochimie 2019, 166, 103–111. [Google Scholar] [CrossRef]
- Grzywa, R.; Burchacka, E.; Łęcka, M.; Winiarski, Ł.; Walczak, M.; Łupicka-Słowik, A.; Wysocka, M.; Burster, T.; Bobrek, K.; Csencsits-Smith, K.; et al. Synthesis of Novel Phosphonic-Type Activity-Based Probes for Neutrophil Serine Proteases and Their Application in Spleen Lysates of Different Organisms. ChemBioChem 2014, 15, 2605–2612. [Google Scholar] [CrossRef]
- Asante, V.; Mortier, J.; Schlüter, H.; Koksch, B. Impact of fluorination on proteolytic stability of peptides in human blood plasma. Bioorg. Med. Chem. 2013, 21, 3542–3546. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, S.C.; MacGregor, I.; Zamani, A.; Gordon, M.W.; Robertson, C.E.; Steedman, D.J.; Little, K.; Haslett, C. Plasma elastase levels and the development of the adult respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1995, 151, 1428–1433. [Google Scholar] [CrossRef]
- Asante, V.; Mortier, J.; Wolber, G.; Koksch, B. Impact of fluorination on proteolytic stability of peptides: A case study with α-chymotrypsin and pepsin. Amino Acids 2014, 46, 2733–2744. [Google Scholar] [CrossRef]
- Kasperkiewicz, P.; Poreba, M.; Snipas, S.J.; Parker, H.; Winterbourn, C.C.; Salvesen, G.S.; Drag, M. Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling. Proc. Natl. Acad. Sci. USA 2014, 111, 2518–2523. [Google Scholar] [CrossRef]
- Lechtenberg, B.C.; Kasperkiewicz, P.; Robinson, H.; Drag, M.; Riedl, S.J. The Elastase-PK101 Structure: Mechanism of an Ultrasensitive Activity-based Probe Revealed. ACS Chem. Biol. 2015, 10, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, I.; Kinoshita, T.; Sato, A.; Tada, T. Structure of porcine pancreatic elastase complexed with FR901277, a novel macrocyclic inhibitor of elastases, at 1.6 Å resolution. Biopolymers 2000, 53, 434–445. [Google Scholar] [CrossRef]
- Kasperkiewicz, P.; Poreba, M.; Snipas, S.J.; Lin, S.J.; Kirchhofer, D.; Salvesen, G.S.; Drag, M. Design of a Selective Substrate and Activity Based Probe for Human Neutrophil Serine Protease 4. PLoS ONE 2015, 10, e0132818. [Google Scholar] [CrossRef] [PubMed]
- Kasperkiewicz, P.; Altman, Y.; D’Angelo, M.; Salvesen, G.S.; Drag, M. Toolbox of Fluorescent Probes for Parallel Imaging Reveals Uneven Location of Serine Proteases in Neutrophils. J. Am. Chem. Soc. 2017, 139, 10115–10125. [Google Scholar] [CrossRef]
- Janiszewski, T.; Kołt, S.; Kaiserman, D.; Snipas, S.J.; Li, S.; Kulbacka, J.; Saczko, J.; Bovenschen, N.; Salvesen, G.; Drąg, M.; et al. Noninvasive optical detection of granzyme B from natural killer cells with enzyme-activated fluorogenic probes. J. Biol. Chem. 2020, 295, 9567–9582. [Google Scholar] [CrossRef]
- Vitte, J. Human mast cell tryptase in biology and medicine. Mol. Immunol. 2015, 63, 18–24. [Google Scholar] [CrossRef]
- Murakami, Y.; Takei, M.; Shindo, K.; Kitazume, C.; Tanaka, J.; Higa, T.; Fukamachi, H. Cyclotheonamide E4 and E5, New Potent Tryptase Inhibitors from an Ircinia Species of Sponge. J. Nat. Prod. 2002, 65, 259–261. [Google Scholar] [CrossRef]
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef]
- Gouvea, I.E.; Izidoro, M.A.; Judice, W.A.S.; Cezari, M.H.S.; Caliendo, G.; Santagada, V.; dos Santos, C.N.D.; Queiroz, M.H.; Juliano, M.A.; Young, P.R.; et al. Substrate specificity of recombinant dengue 2 virus NS2B-NS3 protease: Influence of natural and unnatural basic amino acids on hydrolysis of synthetic fluorescent substrates. Arch. Biochem. Biophys. 2007, 457, 187–196. [Google Scholar] [CrossRef]
- Behnam, M.A.M.; Nitsche, C.; Vechi, S.M.; Klein, C.D. C-Terminal Residue Optimization and Fragment Merging: Discovery of a Potent Peptide-Hybrid Inhibitor of Dengue Protease. ACS Med. Chem. Lett. 2014, 5, 1037–1042. [Google Scholar] [CrossRef]
- Weigel, L.F.; Nitsche, C.; Graf, D.; Bartenschlager, R.; Klein, C.D. Phenylalanine and Phenylglycine Analogues as Arginine Mimetics in Dengue Protease Inhibitors. J. Med. Chem. 2015, 58, 7719–7733. [Google Scholar] [CrossRef] [PubMed]
- Behnam, M.A.M.; Graf, D.; Bartenschlager, R.; Zlotos, D.P.; Klein, C.D. Discovery of Nanomolar Dengue and West Nile Virus Protease Inhibitors Containing a 4-Benzyloxyphenylglycine Residue. J. Med. Chem. 2015, 58, 9354–9370. [Google Scholar] [CrossRef] [PubMed]
- Rut, W.; Groborz, K.; Zhang, L.; Modrzycka, S.; Poreba, M.; Hilgenfeld, R.; Drag, M. Profiling of flaviviral NS2B-NS3 protease specificity provides a structural basis for the development of selective chemical tools that differentiate Dengue from Zika and West Nile viruses. Antivir. Res. 2020, 175, 104731. [Google Scholar] [CrossRef] [PubMed]
- Rut, W.; Zhang, L.; Kasperkiewicz, P.; Poreba, M.; Hilgenfeld, R.; Drąg, M. Extended substrate specificity and first potent irreversible inhibitor/activity-based probe design for Zika virus NS2B-NS3 protease. Antivir. Res. 2017, 139, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Nitsche, C.; Onagi, H.; Quek, J.-P.; Otting, G.; Luo, D.; Huber, T. Biocompatible Macrocyclization between Cysteine and 2-Cyanopyridine Generates Stable Peptide Inhibitors. Org. Lett. 2019, 21, 4709–4712. [Google Scholar] [CrossRef]
- Wu, Z.; Yao, N.; Le, H.V.; Weber, P.C. Mechanism of autoproteolysis at the NS2–NS3 junction of the hepatitis C virus polyprotein. Trends Biochem. Sci. 1998, 23, 92–94. [Google Scholar] [CrossRef]
- Urbani, A.; Biasiol, G.; Brunetti, M.; Volpari, C.; Di Marco, S.; Sollazzo, M.; Orrú, S.; Piaz, F.D.; Casbarra, A.; Pucci, P.; et al. Multiple Determinants Influence Complex Formation of the Hepatitis C Virus NS3 Protease Domain with Its NS4A Cofactor Peptide. Biochemistry 1999, 38, 5206–5215. [Google Scholar] [CrossRef]
- Landro, J.A.; Raybuck, S.A.; Luong, Y.P.C.; O’Malley, E.T.; Harbeson, S.L.; Morgenstern, K.A.; Rao, G.; Livingston, D.J. Mechanistic Role of an NS4A Peptide Cofactor with the Truncated NS3 Protease of Hepatitis C Virus: Elucidation of the NS4A Stimulatory Effect via Kinetic Analysis and Inhibitor Mapping. Biochemistry 1997, 36, 9340–9348. [Google Scholar] [CrossRef]
- Frecer, V.r.; Kabeláč, M.; De Nardi, P.; Pricl, S.; Miertuš, S. Structure-based design of inhibitors of NS3 serine protease of hepatitis C virus. J. Mol. Graph. Model. 2004, 22, 209–220. [Google Scholar] [CrossRef]
- Arasappan, A.; Njoroge, F.G.; Chan, T.Y.; Bennett, F.; Bogen, S.L.; Chen, K.; Gu, H.; Hong, L.; Jao, E.; Liu, Y.T.; et al. Hepatitis C virus NS3-4A serine protease inhibitors: SAR of P2′ moiety with improved potency. Bioorg. Med. Chem. Lett. 2005, 15, 4180–4184. [Google Scholar] [CrossRef]
- Hagel, M.; Niu, D.; St Martin, T.; Sheets, M.P.; Qiao, L.; Bernard, H.; Karp, R.M.; Zhu, Z.; Labenski, M.T.; Chaturvedi, P.; et al. Selective irreversible inhibition of a protease by targeting a noncatalytic cysteine. Nat. Chem. Biol. 2011, 7, 22–24. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Ohe, T.; Takahashi, K.; Nakamura, S.; Mashino, T. Novel fullerene derivatives as dual inhibitors of Hepatitis C virus NS5B polymerase and NS3/4A protease. Bioorg. Med. Chem. Lett. 2016, 26, 4565–4567. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.G.; Zipfel, S.; Ramey, K.; Vivian, R.; Schrier, A.; Karki, K.K.; Katana, A.; Kato, D.; Kobayashi, T.; Martinez, R.; et al. Discovery of the pan-genotypic hepatitis C virus NS3/4A protease inhibitor voxilaprevir (GS-9857): A component of Vosevi®. Bioorg. Med. Chem. Lett. 2019, 29, 2428–2436. [Google Scholar] [CrossRef]
- Basak, A.; Jean, F.; Seidah, N.G.; Lazure, C. Design and synthesis of novel inhibitors of prohormone convertases. Int. J. Pept. Protein Res. 1994, 44, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Basak, A.; Schmidt, C.; Ismail, A.A.; Seidah, N.G.; Chrétien, M.; Lazure, C. Peptidyl substrates containing unnatural amino acid at the P’1 position are potent inhibitors of prohormone convertases. Int. J. Pept. Protein Res. 1995, 46, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Hardes, K.; Becker, G.L.; Lu, Y.; Dahms, S.O.; Köhler, S.; Beyer, W.; Sandvig, K.; Yamamoto, H.; Lindberg, I.; Walz, L.; et al. Novel Furin Inhibitors with Potent Anti-infectious Activity. ChemMedChem 2015, 10, 1218–1231. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Mohottalage, D.; Basak, A. Multibranch and Pseudopeptide Approach for Design of Novel Inhibitors of Subtilisin Kexin Isozyme-1. Protein Pept. Lett. 2006, 13, 863–876. [Google Scholar] [CrossRef]
- Dwyer, M.A.; Lu, W.; Dwyer, J.J.; Kossiakoff, A.A. Biosynthetic phage display: A novel protein engineering tool combining chemical and genetic diversity. Chem. Biol. 2000, 7, 263–274. [Google Scholar] [CrossRef]
- Burchacka, E.; Sieńczyk, M.; Frick, I.-M.; Wysocka, M.; Lesner, A.; Oleksyszyn, J. Substrate profiling of Finegoldia magna SufA protease, inhibitor screening and application to prevent human fibrinogen degradation and bacteria growth in vitro. Biochimie 2014, 103, 137–143. [Google Scholar] [CrossRef]
- Babkova, K.; Korabecny, J.; Soukup, O.; Nepovimova, E.; Jun, D.; Kuca, K. Prolyl oligopeptidase and its role in the organism: Attention to the most promising and clinically relevant inhibitors. Future Med. Chem. 2017, 9, 1015–1038. [Google Scholar] [CrossRef]
- Portevin, B.; Benoist, A.; Rémond, G.; Hervé, Y.; Vincent, M.; Lepagnol, J.; De Nanteuil, G. New Prolyl Endopeptidase Inhibitors: In vitro and in vivo Activities of Azabicyclo[2.2.2]octane, Azabicyclo[2.2.1]heptane, and Perhydroindole Derivatives. J. Med. Chem. 1996, 39, 2379–2391. [Google Scholar] [CrossRef] [PubMed]
- Racys, D.T.; Rea, D.; Fülöp, V.; Wills, M. Inhibition of prolyl oligopeptidase with a synthetic unnatural dipeptide. Bioorg. Med. Chem. 2010, 18, 4775–4782. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.; Katane, M.; Saitoh, Y.; Sekine, M.; Homma, H. Involvement of penicillin-binding proteins in the metabolism of a bacterial peptidoglycan containing a non-canonical d-amino acid. Amino Acids 2020, 52, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Lucana, M.C.; Arruga, Y.; Petrachi, E.; Roig, A.; Lucchi, R.; Oller-Salvia, B. Protease-Resistant Peptides for Targeting and Intracellular Delivery of Therapeutics. Pharmaceutics 2021, 13, 2065. [Google Scholar] [CrossRef]
- Massucci, M.T.; Giansanti, F.; Di Nino, G.; Turacchio, M.; Giardi, M.F.; Botti, D.; Ippoliti, R.; De Giulio, B.; Siciliano, R.A.; Donnarumma, G.; et al. Proteolytic activity of bovine lactoferrin. Biometals Int. J. Role Met. Ions Biol. Biochem. Med. 2004, 17, 249–255. [Google Scholar] [CrossRef]
- Kish-Trier, E.; Hill, C.P. Structural Biology of the Proteasome. Annu. Rev. Biophys. 2013, 42, 29–49. [Google Scholar] [CrossRef]
- Gersch, M.; Stahl, M.; Poreba, M.; Dahmen, M.; Dziedzic, A.; Drag, M.; Sieber, S.A. Barrel-shaped ClpP Proteases Display Attenuated Cleavage Specificities. ACS Chem. Biol. 2016, 11, 389–399. [Google Scholar] [CrossRef]
- Ju, Y.; He, L.; Zhou, Y.; Yang, T.; Sun, K.; Song, R.; Yang, Y.; Li, C.; Sang, Z.; Bao, R.; et al. Discovery of Novel Peptidomimetic Boronate ClpP Inhibitors with Noncanonical Enzyme Mechanism as Potent Virulence Blockers in vitro and in vivo. J. Med. Chem. 2020, 63, 3104–3119. [Google Scholar] [CrossRef]
- Kazmaier, U.; Junk, L. Recent Developments on the Synthesis and Bioactivity of Ilamycins/Rufomycins and Cyclomarins, Marine Cyclopeptides That Demonstrate Anti-Malaria and Anti-Tuberculosis Activity. Mar. Drugs 2021, 19, 446. [Google Scholar] [CrossRef]
- Shin, M.; Watson, E.R.; Song, A.S.; Mindrebo, J.T.; Novick, S.J.; Griffin, P.R.; Wiseman, R.L.; Lander, G.C. Structures of the human LONP1 protease reveal regulatory steps involved in protease activation. Nat. Commun. 2021, 12, 3239. [Google Scholar] [CrossRef]
- Babin, B.M.; Kasperkiewicz, P.; Janiszewski, T.; Yoo, E.; Dra̧g, M.; Bogyo, M. Leveraging Peptide Substrate Libraries to Design Inhibitors of Bacterial Lon Protease. ACS Chem. Biol. 2019, 14, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, T.A.; Smith, D.M. A Practical Review of Proteasome Pharmacology. Pharmacol Rev 2019, 71, 170–197. [Google Scholar] [CrossRef] [PubMed]
- Gladysz, R.; Malek, N.; Rut, W.; Drag, M. Investigation of the P1′ and P2′ sites of IQF substrates and their selectivity toward 20S proteasome subunits. Biol. Chem. 2023, 404, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Zerfas, B.L.; Coleman, R.A.; Salazar-Chaparro, A.F.; Macatangay, N.J.; Trader, D.J. Fluorescent Probes with Unnatural Amino Acids to Monitor Proteasome Activity in Real-Time. ACS Chem. Biol. 2020, 15, 2588–2596. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Adwal, A.; Turner, A.G.; Callen, D.F.; Abell, A.D. New Peptidomimetic Boronates for Selective Inhibition of the Chymotrypsin-like Activity of the 26S Proteasome. ACS Med. Chem. Lett. 2016, 7, 1039–1043. [Google Scholar] [CrossRef]
- Geurink, P.P.; van der Linden, W.A.; Mirabella, A.C.; Gallastegui, N.; de Bruin, G.; Blom, A.E.M.; Voges, M.J.; Mock, E.D.; Florea, B.I.; van der Marel, G.A.; et al. Incorporation of non-natural amino acids improves cell permeability and potency of specific inhibitors of proteasome trypsin-like sites. J. Med. Chem. 2013, 56, 1262–1275. [Google Scholar] [CrossRef]
- Merz, T.; Heck, T.; Geueke, B.; Mittl, P.R.; Briand, C.; Seebach, D.; Kohler, H.-P.E.; Grütter, M.G. Autoproteolytic and Catalytic Mechanisms for the β-Aminopeptidase BapA—A Member of the Ntn Hydrolase Family. Structure 2012, 20, 1850–1860. [Google Scholar] [CrossRef]
- Heck, T.; Reimer, A.; Seebach, D.; Gardiner, J.; Deniau, G.; Lukaszuk, A.; Kohler, H.-P.E.; Geueke, B. β-Aminopeptidase-Catalyzed Biotransformations of β2-Dipeptides: Kinetic Resolution and Enzymatic Coupling. ChemBioChem 2010, 11, 1129–1136. [Google Scholar] [CrossRef]
- Poreba, M.; Groborz, K.; Vizovisek, M.; Maruggi, M.; Turk, D.; Turk, B.; Powis, G.; Drag, M.; Salvesen, G.S. Fluorescent probes towards selective cathepsin B detection and visualization in cancer cells and patient samples. Chem. Sci. 2019, 10, 8461–8477. [Google Scholar] [CrossRef]
- Tholen, M.; Yim, J.J.; Groborz, K.; Yoo, E.; Martin, B.A.; van den Berg, N.S.; Drag, M.; Bogyo, M. Design of Optical-Imaging Probes by Screening of Diverse Substrate Libraries Directly in Disease-Tissue Extracts. Angew. Chem. Int. Ed. Engl. 2020, 59, 19143–19152. [Google Scholar] [CrossRef]
- Poreba, M.; Mihelic, M.; Krai, P.; Rajkovic, J.; Krezel, A.; Pawelczak, M.; Klemba, M.; Turk, D.; Turk, B.; Latajka, R.; et al. Unnatural amino acids increase activity and specificity of synthetic substrates for human and malarial cathepsin C. Amino Acids 2014, 46, 931–943. [Google Scholar] [CrossRef] [PubMed]
- Alves, L.C.; Melo, R.L.; Cezari, M.H.S.; Sanderson, S.J.; Mottram, J.C.; Coombs, G.H.; Juliano, L.; Juliano, M.A. Analysis of the S2 subsite specificities of the recombinant cysteine proteinases CPB of Leishmania mexicana, and cruzain of Trypanosoma cruzi, using fluorescent substrates containing non-natural basic amino acids. Mol. Biochem. Parasitol. 2001, 117, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Alves, L.C.; Melo, R.L.; Sanderson, S.J.; Mottram, J.C.; Coombs, G.H.; Caliendo, G.; Santagada, V.; Juliano, L.; Juliano, M.A. S1 subsite specificity of a recombinant cysteine proteinase, CPB, of Leishmania mexicana compared with cruzain, human cathepsin L and papain using substrates containing non-natural basic amino acids. Eur. J. Biochem. 2001, 268, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Del Nery, E.; Alves, L.C.; Melo, R.L.; Cesari, M.H.; Juliano, L.; Juliano, M.A. Specificity of cathepsin B to fluorescent substrates containing benzyl side-chain-substituted amino acids at P1 subsite. J. Protein Chem. 2000, 19, 33–38. [Google Scholar] [CrossRef]
- van der Linden, W.A.; Segal, E.; Child, M.A.; Byzia, A.; Drąg, M.; Bogyo, M. Design and Synthesis of Activity-Based Probes and Inhibitors for Bleomycin Hydrolase. Chem. Biol. 2015, 22, 995–1001. [Google Scholar] [CrossRef][Green Version]
- Cuerrier, D.; Moldoveanu, T.; Campbell, R.L.; Kelly, J.; Yoruk, B.; Verhelst, S.H.L.; Greenbaum, D.; Bogyo, M.; Davies, P.L. Development of Calpain-specific Inactivators by Screening of Positional Scanning Epoxide Libraries. J. Biol. Chem. 2007, 282, 9600–9611. [Google Scholar] [CrossRef]
- Renfrew, P.D.; Choi, E.J.; Bonneau, R.; Kuhlman, B. Incorporation of Noncanonical Amino Acids into Rosetta and Use in Computational Protein-Peptide Interface Design. PLoS ONE 2012, 7, e32637. [Google Scholar] [CrossRef]
- Wang, Y.; Jia, S.; Tseng, B.; Drewe, J.; Cai, S.X. Dipeptidyl aspartyl fluoromethylketones as potent caspase inhibitors: Peptidomimetic replacement of the P2 amino acid by 2-aminoaryl acids and other non-natural amino acids. Bioorg. Med. Chem. Lett. 2007, 17, 6178–6182. [Google Scholar] [CrossRef]
- Vickers, C.J.; González-Páez, G.E.; Wolan, D.W. Selective Detection of Caspase-3 versus Caspase-7 Using Activity-Based Probes with Key Unnatural Amino Acids. ACS Chem. Biol. 2013, 8, 1558–1566. [Google Scholar] [CrossRef]
- Vickers, C.J.; González-Páez, G.E.; Litwin, K.M.; Umotoy, J.C.; Coutsias, E.A.; Wolan, D.W. Selective Inhibition of Initiator versus Executioner Caspases Using Small Peptides Containing Unnatural Amino Acids. ACS Chem. Biol. 2014, 9, 2194–2198. [Google Scholar] [CrossRef]
- Poreba, M.; Kasperkiewicz, P.; Snipas, S.J.; Fasci, D.; Salvesen, G.S.; Drag, M. Unnatural amino acids increase sensitivity and provide for the design of highly selective caspase substrates. Cell Death Differ. 2014, 21, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Poreba, M.; Szalek, A.; Rut, W.; Kasperkiewicz, P.; Rutkowska-Wlodarczyk, I.; Snipas, S.J.; Itoh, Y.; Turk, D.; Turk, B.; Overall, C.M.; et al. Highly sensitive and adaptable fluorescence-quenched pair discloses the substrate specificity profiles in diverse protease families. Sci. Rep. 2017, 7, 43135. [Google Scholar] [CrossRef] [PubMed]
- Kasperkiewicz, P.; Kołt, S.; Janiszewski, T.; Groborz, K.; Poręba, M.; Snipas, S.J.; Salvesen, G.S.; Drąg, M. Determination of extended substrate specificity of the MALT1 as a strategy for the design of potent substrates and activity-based probes. Sci. Rep. 2018, 8, 15998. [Google Scholar] [CrossRef] [PubMed]
- van de Plassche, M.A.T.; O’Neill, T.J.; Seeholzer, T.; Turk, B.; Krappmann, D.; Verhelst, S.H.L. Use of Non-Natural Amino Acids for the Design and Synthesis of a Selective, Cell-Permeable MALT1 Activity-Based Probe. J. Med. Chem. 2020, 63, 3996–4004. [Google Scholar] [CrossRef]
- Lee, J.; Bogyo, M. Synthesis and evaluation of aza-peptidyl inhibitors of the lysosomal asparaginyl endopeptidase, legumain. Bioorg. Med. Chem. Lett. 2012, 22, 1340–1343. [Google Scholar] [CrossRef]
- Rut, W.; Zmudzinski, M.; Snipas, S.J.; Bekes, M.; Huang, T.T.; Drag, M. Engineered unnatural ubiquitin for optimal detection of deubiquitinating enzymes. Chem. Sci. 2020, 11, 6058–6069. [Google Scholar] [CrossRef]
- Rajković, J.; Poreba, M.; Caglič, D.; Vidmar, R.; Wilk, A.; Borowik, A.; Salvesen, G.; Turk, V.; Drag, M.; Turk, B. Biochemical Characterization and Substrate Specificity of Autophagin-2 from the Parasite Trypanosoma cruzi. J. Biol. Chem. 2015, 290, 28231–28244. [Google Scholar] [CrossRef]
- Breuning, A.; Degel, B.; Schulz, F.; Büchold, C.; Stempka, M.; Machon, U.; Heppner, S.; Gelhaus, C.; Leippe, M.; Leyh, M.; et al. Michael Acceptor Based Antiplasmodial and Antitrypanosomal Cysteine Protease Inhibitors with Unusual Amino Acids. J. Med. Chem. 2010, 53, 1951–1963. [Google Scholar] [CrossRef]
- Pérez, B.C.; Teixeira, C.; Figueiras, M.; Gut, J.; Rosenthal, P.J.; Gomes, J.R.B.; Gomes, P. Novel cinnamic acid/4-aminoquinoline conjugates bearing non-proteinogenic amino acids: Towards the development of potential dual action antimalarials. Eur. J. Med. Chem. 2012, 54, 887–899. [Google Scholar] [CrossRef]
- Joshi, R.P.; Schultz, K.J.; Wilson, J.W.; Kruel, A.; Varikoti, R.A.; Kombala, C.J.; Kneller, D.W.; Galanie, S.; Phillips, G.; Zhang, Q.; et al. AI-Accelerated Design of Targeted Covalent Inhibitors for SARS-CoV-2. J. Chem. Inf. Model. 2023, 63, 1438–1453. [Google Scholar] [CrossRef]
- Kronenberger, T.; Laufer, S.A.; Pillaiyar, T. COVID-19 therapeutics: Small-molecule drug development targeting SARS-CoV-2 main protease. Drug Discov. Today 2023, 28, 103579. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, K.S.; Geng, Z.Z.; Alugubelli, Y.R.; Shaabani, N.; Vatansever, E.C.; Ma, X.R.; Cho, C.-C.; Khatua, K.; Blankenship, L.; et al. The P3 O-Tert-Butyl-Threonine is Key to High Cellular and Antiviral Potency for Aldehyde-Based SARS-CoV-2 Main Protease Inhibitors. bioRxiv 2021, preprint. [Google Scholar] [CrossRef]
- Ma, Y.; Yang, K.S.; Geng, Z.Z.; Alugubelli, Y.R.; Shaabani, N.; Vatansever, E.C.; Ma, X.R.; Cho, C.-C.; Khatua, K.; Xiao, J.; et al. A multi-pronged evaluation of aldehyde-based tripeptidyl main protease inhibitors as SARS-CoV-2 antivirals. Eur. J. Med. Chem. 2022, 240, 114570. [Google Scholar] [CrossRef] [PubMed]
- Katagishi, D.; Yasuda, D.; Takahashi, K.; Nakamura, S.; Mashino, T.; Ohe, T. Fullerene derivatives as inhibitors of the SARS-CoV-2 main protease. Bioorg. Med. Chem. Lett. 2023, 80, 129121. [Google Scholar] [CrossRef] [PubMed]
- van de Plassche, M.A.T.; Barniol-Xicota, M.; Verhelst, S.H.L. Peptidyl Acyloxymethyl Ketones as Activity-Based Probes for the Main Protease of SARS-CoV-2. ChemBioChem Eur. J. Chem. Biol. 2020, 21, 3383–3388. [Google Scholar] [CrossRef]
- Rut, W.; Groborz, K.; Zhang, L.; Sun, X.; Zmudzinski, M.; Pawlik, B.; Wang, X.; Jochmans, D.; Neyts, J.; Młynarski, W.; et al. SARS-CoV-2 Mpro inhibitors and activity-based probes for patient-sample imaging. Nat. Chem. Biol. 2021, 17, 222–228. [Google Scholar] [CrossRef]
- Goto, Y.; Katoh, T.; Suga, H. Flexizymes for genetic code reprogramming. Nat. Protoc. 2011, 6, 779–790. [Google Scholar] [CrossRef]
- Miura, T.; Malla, T.R.; Owen, C.D.; Tumber, A.; Brewitz, L.; McDonough, M.A.; Salah, E.; Terasaka, N.; Katoh, T.; Lukacik, P.; et al. In vitro selection of macrocyclic peptide inhibitors containing cyclic γ2,4-amino acids targeting the SARS-CoV-2 main protease. Nat. Chem. 2023, 15, 998–1005. [Google Scholar] [CrossRef]
- Lorenz, I.C.; Marcotrigiano, J.; Dentzer, T.G.; Rice, C.M. Structure of the catalytic domain of the hepatitis C virus NS2-3 protease. Nature 2006, 442, 831–835. [Google Scholar] [CrossRef]
- Shaw, J.; Fishwick, C.W.G.; Harris, M. Epoxide based inhibitors of the hepatitis C virus non-structural 2 autoprotease. Antivir. Res. 2015, 117, 20–26. [Google Scholar] [CrossRef]
- Fruton, J.S. A history of pepsin and related enzymes. Q. Rev. Biol. 2002, 77, 127–147. [Google Scholar] [CrossRef] [PubMed]
- Marciniszyn, J.; Hartsuck, J.A.; Tang, J. Mode of inhibition of acid proteases by pepstatin. J. Biol. Chem. 1976, 251, 7088–7094. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, M.; Chernaia, M.M.; Mosimann, S.C.; James, M.N.G.; Tarasova, N.I. Crystal structure of human pepsin and its complex with pepstatin. Protein Sci. 1995, 4, 960–972. [Google Scholar] [CrossRef] [PubMed]
- Kakizawa, T.; Sanjoh, A.; Kobayashi, A.; Hattori, Y.; Teruya, K.; Akaji, K. Evaluation of superior BACE1 cleavage sequences containing unnatural amino acids. Bioorg. Med. Chem. 2011, 19, 2785–2789. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.-F.; Wang, B.-J.; Hsu, W.-M.; Lee, H.; Liao, C.-Y.; Wu, S.-Y.; Cheng, H.-T.; Hu, M.-K. Unnatural Amino Acid-Substituted (Hydroxyethyl)urea Peptidomimetics Inhibit γ-Secretase and Promote the Neuronal Differentiation of Neuroblastoma Cells. Mol. Pharmacol. 2007, 71, 588–601. [Google Scholar] [CrossRef]
- Iizuka, K.; Kamijo, T.; Kubota, T.; Akahane, K.; Umeyama, H.; Kiso, Y. New human renin inhibitors containing an unnatural amino acid, norstatine. J. Med. Chem. 1988, 31, 701–704. [Google Scholar] [CrossRef]
- Paruszewski, R.; Jaworski, P.; Winiecka, I.; Tautt, J.; Dudkiewicz, J. New Renin Inhibitors with Pseudodipeptidic Units in P1–P1′ and P2′–P3′ Positions. Chem. Pharm. Bull. 2002, 50, 850–853. [Google Scholar] [CrossRef][Green Version]
- Winiecka, I.; Jaworski, P.; Mazurek, A.P.; Marszałek, D.; Goldnik, A.; Sokulski, D. Novel renin inhibitors containing derivatives of N-alkylleucyl-β-hydroxy-γ-amino acids. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 2016, 22, 106–115. [Google Scholar] [CrossRef]
- Winiecka, I.; Marszałek, D.; Goldnik, A.; Jaworski, P.; Mazurek, A.P. Novel renin inhibitors containing a non-peptide aminoalkanoyl moiety at P1–P1′ position. Die Pharm. 2014, 69, 263–270. [Google Scholar] [CrossRef]
- Kurt Yilmaz, N.; Swanstrom, R.; Schiffer, C.A. Improving Viral Protease Inhibitors to Counter Drug Resistance. Trends Microbiol. 2016, 24, 547–557. [Google Scholar] [CrossRef]
- Thompson, W.J.; Ghosh, A.K.; Holloway, M.K.; Lee, H.Y.; Munson, P.M.; Schwering, J.E.; Wai, J.; Darke, P.L.; Zugay, J.; Emini, E.A.; et al. 3′-Tetrahydrofuranylglycine as a Novel, Unnatural Amino Acid Surrogate for Asparagine in the Design of Inhibitors of the HIV Protease. J. Am. Chem. Soc. 1993, 115, 801–803. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.K.; Thompson, W.J.; Holloway, M.K.; McKee, S.P.; Duong, T.T.; Lee, H.Y.; Munson, P.M.; Smith, A.M.; Wai, J.M.; Darke, P.L.; et al. Potent HIV protease inhibitors: The development of tetrahydrofuranylglycines as novel P2-ligands and pyrazine amides as P3-ligands. J. Med. Chem. 1993, 36, 2300–2310. [Google Scholar] [CrossRef] [PubMed]
- Mimoto, T.; Kato, R.; Takaku, H.; Nojima, S.; Terashima, K.; Misawa, S.; Fukazawa, T.; Ueno, T.; Sato, H.; Shintani, M.; et al. Structure−Activity Relationship of Small-Sized HIV Protease Inhibitors Containing Allophenylnorstatine. J. Med. Chem. 1999, 42, 1789–1802. [Google Scholar] [CrossRef] [PubMed]
- Mimoto, T.; Hattori, N.; Takaku, H.; Kisanuki, S.; Fukazawa, T.; Terashima, K.; Kato, R.; Nojima, S.; Misawa, S.; Ueno, T.; et al. Structure-Activity Relationship of Orally Potent Tripeptide-Based HIV Protease Inhibitors Containing Hydroxymethylcarbonyl Isostere. Chem. Pharm. Bull. 2000, 48, 1310–1326. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nakatani, S.; Hidaka, K.; Ami, E.I.; Nakahara, K.; Sato, A.; Nguyen, J.-T.; Hamada, Y.; Hori, Y.; Ohnishi, N.; Nagai, A.; et al. Combination of Non-natural d-Amino Acid Derivatives and Allophenylnorstatine−Dimethylthioproline Scaffold in HIV Protease Inhibitors Have High Efficacy in Mutant HIV. J. Med. Chem. 2008, 51, 2992–3004. [Google Scholar] [CrossRef]
- Baldwin, E.T.; Bhat, T.N.; Gulnik, S.; Liu, B.; Topol, I.A.; Kiso, Y.; Mimoto, T.; Mitsuya, H.; Erickson, J.W. Structure of HIV-1 protease with KNI-272, a tight-binding transition-state analog containing allophenylnorstatine. Structure 1995, 3, 581–590. [Google Scholar] [CrossRef]
- Adachi, M.; Ohhara, T.; Kurihara, K.; Tamada, T.; Honjo, E.; Okazaki, N.; Arai, S.; Shoyama, Y.; Kimura, K.; Matsumura, H.; et al. Structure of HIV-1 protease in complex with potent inhibitor KNI-272 determined by high-resolution X-ray and neutron crystallography. Proc. Natl. Acad. Sci. USA 2009, 106, 4641–4646. [Google Scholar] [CrossRef]
- Churchill, D.R.; Slade, P.M.; Youle, M.; Gazzard, B.G.; Weber, J.N. A phase I/II study of the safety and activity of a microsphere formulation of KNI-272 in patients with HIV-1 infection. J. Antimicrob. Chemother. 2001, 47, 353–355. [Google Scholar] [CrossRef][Green Version]
- Chen, M.-H.; Chang, S.-S.; Dong, B.; Yu, L.-Y.; Wu, Y.-X.; Wang, R.-Z.; Jiang, W.; Gao, Z.-P.; Si, S.-Y. Ahmpatinin iBu, a new HIV-1 protease inhibitor, from Streptomyces sp. CPCC 202950. RSC Adv. 2018, 8, 5138–5144. [Google Scholar] [CrossRef]
- Schramm, H.J.; de Rosny, E.; Reboud-Ravaux, M.; Büttner, J.; Dick, A.; Schramm, W. Lipopeptides as dimerization inhibitors of HIV-1 protease. Biol. Chem. 1999, 380, 593–596. [Google Scholar] [CrossRef]
- Mak, C.C.; Brik, A.; Lerner, D.L.; Elder, J.H.; Morris, G.M.; Olson, A.J.; Wong, C.-H. Design and synthesis of broad-Based mono- and bi-cyclic inhibitors of FIV and HIV proteases. Bioorg. Med. Chem. 2003, 11, 2025–2040. [Google Scholar] [CrossRef] [PubMed]
- Strom, T.A.; Durdagi, S.; Ersoz, S.S.; Salmas, R.E.; Supuran, C.T.; Barron, A.R. Fullerene-based inhibitors of HIV-1 protease. J. Pept. Sci. 2015, 21, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Skwarecki, A.S.; Nowak, M.G.; Milewska, M.J. Amino Acid and Peptide-Based Antiviral Agents. ChemMedChem 2021, 16, 3106–3135. [Google Scholar] [CrossRef]
- Kiso, Y. Design and synthesis of substrate-based peptidomimetic human immunodeficiency virus protease inhibitors containing the hydroxymethylcarbonyl isostere. Pept. Sci. 1996, 40, 235–244. [Google Scholar] [CrossRef]
- Abdel-Rahman, H.M.; Kimura, T.; Hidaka, K.; Kiso, A.; Nezami, A.; Freire, E.; Hayashi, Y.; Kiso, Y. Design of inhibitors against HIV, HTLV-I, and Plasmodium falciparum aspartic proteases. Biol. Chem. 2004, 385, 1035–1039. [Google Scholar] [CrossRef]
- Nguyen, J.-T.; Zhang, M.; Kumada, H.-O.; Itami, A.; Nishiyama, K.; Kimura, T.; Cheng, M.; Hayashi, Y.; Kiso, Y. Truncation and non-natural amino acid substitution studies on HTLV-I protease hexapeptidic inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 366–370. [Google Scholar] [CrossRef]
- Hodder, A.N.; Sleebs, B.E.; Czabotar, P.E.; Gazdik, M.; Xu, Y.; O’Neill, M.T.; Lopaticki, S.; Nebl, T.; Triglia, T.; Smith, B.J.; et al. Structural basis for plasmepsin V inhibition that blocks export of malaria proteins to human erythrocytes. Nat. Struct. Mol. Biol. 2015, 22, 590–596. [Google Scholar] [CrossRef]
- Worst, E.G.; Exner, M.P.; De Simone, A.; Schenkelberger, M.; Noireaux, V.; Budisa, N.; Ott, A. Residue-specific Incorporation of Noncanonical Amino Acids into Model Proteins Using an Escherichia coli Cell-free Transcription-translation System. J. Vis. Exp. JoVE 2016, 114, e54273. [Google Scholar] [CrossRef]
- Chang, M. Matrix metalloproteinase profiling and their roles in disease. RSC Adv. 2023, 13, 6304–6316. [Google Scholar] [CrossRef]
- Leipoldt, F.; Santos-Aberturas, J.; Stegmann, D.P.; Wolf, F.; Kulik, A.; Lacret, R.; Popadić, D.; Keinhörster, D.; Kirchner, N.; Bekiesch, P.; et al. Warhead biosynthesis and the origin of structural diversity in hydroxamate metalloproteinase inhibitors. Nat. Commun. 2017, 8, 1965. [Google Scholar] [CrossRef]
- Mustafa, S.; Koran, S.; Al Omair, L. Insights Into the Role of Matrix Metalloproteinases in Cancer and its Various Therapeutic Aspects: A Review. Front. Mol. Biosci. 2022, 9, 896099. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Tang, J.; Guo, F. Identification of Inhibitors of MMPS Enzymes via a Novel Computational Approach. Int. J. Biol. Sci. 2018, 14, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.A.; Venkataraman, S.; Ghaffari, M.A.; Libby, A.; Mookhtiar, K.A.; Mallya, S.K.; Birkedal-Hansen, H.; Van Wart, H.E. Inhibition of human collagenases by sulfur-based substrate analogs. Biochem. Biophys. Res. Commun. 1991, 176, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Ferry, G.; Boutin, J.A.; Atassi, G.; Fauchère, J.L.; Tucker, G.C. Selection of a histidine-containing inhibitor of gelatinases through deconvolution of combinatorial tetrapeptide libraries. Mol. Divers. 1997, 2, 135–146. [Google Scholar] [CrossRef]
- Levy, D.E.; Lapierre, F.; Liang, W.; Ye, W.; Lange, C.W.; Li, X.; Grobelny, D.; Casabonne, M.; Tyrrell, D.; Holme, K.; et al. Matrix Metalloproteinase Inhibitors: A Structure−Activity Study. J. Med. Chem. 1998, 41, 199–223. [Google Scholar] [CrossRef]
- Rowsell, S.; Hawtin, P.; Minshull, C.A.; Jepson, H.; Brockbank, S.M.V.; Barratt, D.G.; Slater, A.M.; McPheat, W.L.; Waterson, D.; Henney, A.M.; et al. Crystal Structure of Human MMP9 in Complex with a Reverse Hydroxamate Inhibitor. J. Mol. Biol. 2002, 319, 173–181. [Google Scholar] [CrossRef]
- Uttamchandani, M.; Wang, J.; Li, J.; Hu, M.; Sun, H.; Chen, K.Y.T.; Liu, K.; Yao, S.Q. Inhibitor Fingerprinting of Matrix Metalloproteases Using a Combinatorial Peptide Hydroxamate Library. J. Am. Chem. Soc. 2007, 129, 7848–7858. [Google Scholar] [CrossRef]
- Hayun, H.; Coban, M.; Bhagat, A.K.; Ozer, E.; Alfonta, L.; Caulfield, T.R.; Radisky, E.S.; Papo, N. Utilizing genetic code expansion to modify N-TIMP2 specificity towards MMP-2, MMP-9, and MMP-14. Sci. Rep. 2023, 13, 5186. [Google Scholar] [CrossRef]
- Tranchant, I.; Vera, L.; Czarny, B.; Amoura, M.; Cassar, E.; Beau, F.; Stura, E.A.; Dive, V. Halogen Bonding Controls Selectivity of FRET Substrate Probes for MMP-9. Chem. Biol. 2014, 21, 408–413. [Google Scholar] [CrossRef]
- Takeuchi, T.; Hayashi, M.; Tamita, T.; Nomura, Y.; Kojima, N.; Mitani, A.; Takeda, T.; Hitaka, K.; Kato, Y.; Kamitani, M.; et al. Discovery of Aryloxyphenyl–Heptapeptide Hybrids as Potent and Selective Matrix Metalloproteinase-2 Inhibitors for the Treatment of Idiopathic Pulmonary Fibrosis. J. Med. Chem. 2022, 65, 8493–8510. [Google Scholar] [CrossRef]
- Mucha, A.; Cuniasse, P.; Kannan, R.; Beau, F.; Yiotakis, A.; Basset, P.; Dive, V. Membrane Type-1 Matrix Metalloprotease and Stromelysin-3 Cleave More Efficiently Synthetic Substrates Containing Unusual Amino Acids in Their P1′ Positions. J. Biol. Chem. 1998, 273, 2763–2768. [Google Scholar] [CrossRef] [PubMed]
- Holtz, B.; Cuniasse, P.; Boulay, A.; Kannan, R.; Mucha, A.; Beau, F.; Basset, P.; Dive, V. Role of the S1‘ Subsite Glutamine 215 in Activity and Specificity of Stromelysin-3 by Site-Directed Mutagenesis. Biochemistry 1999, 38, 12174–12179. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.S.; Rademann, J. Extra- and Intracellular Imaging of Human Matrix Metalloprotease 11 (hMMP-11) with a Cell-penetrating FRET Substrate. J. Biol. Chem. 2012, 287, 37857–37867. [Google Scholar] [CrossRef] [PubMed]
- Gall, F.M.; Hohl, D.; Frasson, D.; Wermelinger, T.; Mittl, P.R.E.; Sievers, M.; Riedl, R. Drug Design Inspired by Nature: Crystallographic Detection of an Auto-Tailored Protease Inhibitor Template. Angew. Chem. Int. Ed. Engl. 2019, 58, 4051–4055. [Google Scholar] [CrossRef] [PubMed]
- Cowan, J.D.; Moss, L.M.; Andrews, C.R.; Musso, L.D.; McDougald, L.D.; Becherer, D.J.; Wiethe, W.R.; Rabinowitz, H.M.; Petrov, G.K.; Gaul, D.M.; et al. Substrate Specificity and Novel Selective Inhibitors of TNF-alpha Converting Enzyme (TACE) from Two-Dimensional Substrate Mapping. Comb. Chem. High Throughput Screen. 2005, 8, 327–339. [Google Scholar] [CrossRef]
- Mallari, J.P.; Choy, C.J.; Hu, Y.; Martinez, A.R.; Hosaka, M.; Toriyabe, Y.; Maung, J.; Blecha, J.E.; Pavkovic, S.F.; Berkman, C.E. Stereoselective inhibition of glutamate carboxypeptidase by organophosphorus derivatives of glutamic acid. Bioorg. Med. Chem. 2004, 12, 6011–6020. [Google Scholar] [CrossRef]
- Turner, A.J. Chapter 79—Aminopeptidase N. In Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings, N.D., Salvesen, G., Eds.; Academic Press: Cambridge, MA, USA, 2013; pp. 397–403. [Google Scholar]
- Drag, M.; Bogyo, M.; Ellman, J.A.; Salvesen, G.S. Aminopeptidase Fingerprints, an Integrated Approach for Identification of Good Substrates and Optimal Inhibitors. J. Biol. Chem. 2010, 285, 3310–3318. [Google Scholar] [CrossRef]
- Kiehstaller, S.; Ottmann, C.; Hennig, S. MMP activation–associated aminopeptidase N reveals a bivalent 14-3-3 binding motif. J. Biol. Chem. 2020, 295, 18266–18275. [Google Scholar] [CrossRef]
- Węglarz-Tomczak, E.; Poręba, M.; Byzia, A.; Berlicki, Ł.; Nocek, B.; Mulligan, R.; Joachimiak, A.; Drąg, M.; Mucha, A. An integrated approach to the ligand binding specificity of Neisseria meningitidis M1 alanine aminopeptidase by fluorogenic substrate profiling, inhibitory studies and molecular modeling. Biochimie 2013, 95, 419–428. [Google Scholar] [CrossRef]
- Gajda, A.D.; Pawełczak, M.; Drag, M. Substrate specificity screening of oat (Avena sativa) seeds aminopeptidase demonstrate unusually broad tolerance in S1 pocket. Plant Physiol. Biochem. 2012, 54, 6–9. [Google Scholar] [CrossRef]
- Velmourougane, G.; Harbut, M.B.; Dalal, S.; McGowan, S.; Oellig, C.A.; Meinhardt, N.; Whisstock, J.C.; Klemba, M.; Greenbaum, D.C. Synthesis of New (−)-Bestatin-Based Inhibitor Libraries Reveals a Novel Binding Mode in the S1 Pocket of the Essential Malaria M1 Metalloaminopeptidase. J. Med. Chem. 2011, 54, 1655–1666. [Google Scholar] [CrossRef] [PubMed]
- Poreba, M.; McGowan, S.; Skinner-Adams, T.S.; Trenholme, K.R.; Gardiner, D.L.; Whisstock, J.C.; To, J.; Salvesen, G.S.; Dalton, J.P.; Drag, M. Fingerprinting the Substrate Specificity of M1 and M17 Aminopeptidases of Human Malaria, Plasmodium falciparum. PLoS ONE 2012, 7, e31938. [Google Scholar] [CrossRef] [PubMed]
- Poreba, M.; Gajda, A.; Picha, J.; Jiracek, J.; Marschner, A.; Klein, C.D.; Salvesen, G.S.; Drag, M. S1 pocket fingerprints of human and bacterial methionine aminopeptidases determined using fluorogenic libraries of substrates and phosphorus based inhibitors. Biochimie 2012, 94, 704–710. [Google Scholar] [CrossRef] [PubMed]
- Byzia, A.; Szeffler, A.; Kalinowski, L.; Drag, M. Activity profiling of aminopeptidases in cell lysates using a fluorogenic substrate library. Biochimie 2016, 122, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Babaie, F.; Hosseinzadeh, R.; Ebrazeh, M.; Seyfizadeh, N.; Aslani, S.; Salimi, S.; Hemmatzadeh, M.; Azizi, G.; Jadidi-Niaragh, F.; Mohammadi, H. The roles of ERAP1 and ERAP2 in autoimmunity and cancer immunity: New insights and perspective. Mol. Immunol. 2020, 121, 7–19. [Google Scholar] [CrossRef]
- Węglarz-Tomczak, E.; Vassiliou, S.; Mucha, A. Discovery of potent and selective inhibitors of human aminopeptidases ERAP1 and ERAP2 by screening libraries of phosphorus-containing amino acid and dipeptide analogues. Bioorg. Med. Chem. Lett. 2016, 26, 4122–4126. [Google Scholar] [CrossRef]
- Fink, C.A.; Qiao, Y.; Berry, C.J.; Sakane, Y.; Ghai, R.D.; Trapani, A.J. New alpha-thiol dipeptide dual inhibitors of angiotensin-I converting enzyme and neutral endopeptidase EC 3.4.24.11. J. Med. Chem. 1995, 38, 5023–5030. [Google Scholar] [CrossRef]
- Fink, C.A.; Carlson, J.E.; McTaggart, P.A.; Qiao, Y.; Webb, R.; Chatelain, R.; Jeng, A.Y.; Trapani, A.J. Mercaptoacyl Dipeptides as Orally Active Dual Inhibitors of Angiotensin-Converting Enzyme and Neutral Endopeptidase. J. Med. Chem. 1996, 39, 3158–3168. [Google Scholar] [CrossRef]
- Vincent, M.; Marchand, B.; Rémond, G.; Jaguelin-Guinamant, S.; Damien, G.; Portevin, B.; Baumal, J.Y.; Volland, J.P.; Bouchet, J.P.; Lambert, P.H. Synthesis and ACE inhibitory activity of the stereoisomers of perindopril (S 9490) and perindoprilate (S 9780). Drug Des. Discov. 1992, 9, 11–28. [Google Scholar] [PubMed]
- Wester, A.; Devocelle, M.; Tallant, E.A.; Chappell, M.C.; Gallagher, P.E.; Paradisi, F. Stabilization of Angiotensin-(1-7) by key substitution with a cyclic non-natural amino acid. Amino Acids 2017, 49, 1733–1742. [Google Scholar] [CrossRef]
- Dunn, F.W.; Dittmer, K. Action of carboxypeptidase toward peptides containing unnatural aromatic amino acids. Science 1950, 111, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Dunn, F.W.; Smith, E.L. Action of Carboxypeptidase on Derivatives of Unnatural Amino Acids. J. Biol. Chem. 1950, 187, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Ogishima, T.; Ito, A. Analysis of Recognition Elements for Mitochondrial Processing Peptidase Using Artificial Amino Acids: Roles of the Intervening Portion and Proximal Arginine1. J. Biochem. 1999, 126, 874–878. [Google Scholar] [CrossRef]
- Martin, S.E.; Ganguly, T.; Munske, G.R.; Fulton, M.D.; Hopkins, M.R.; Berkman, C.E.; Black, M.E. Development of Inhibitor-Directed Enzyme Prodrug Therapy (IDEPT) for Prostate Cancer. Bioconjug. Chem. 2014, 25, 1752–1760. [Google Scholar] [CrossRef]
- Nigam, P.K.; Nigam, A. Botulinum toxin. Indian J. Dermatol. 2010, 55, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Krilich, J.; Baudys, J.; Barr, J.R.; Kalb, S.R. Enhanced detection of type C botulinum neurotoxin by the Endopep-MS assay through optimization of peptide substrates. Bioorg. Med. Chem. 2015, 23, 3667–3673. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Baudys, J.; Hoyt, K.; Barr, J.R.; Kalb, S.R. Sensitive detection of type G botulinum neurotoxin through Endopep-MS peptide substrate optimization. Anal. Bioanal. Chem. 2019, 411, 5489–5497. [Google Scholar] [CrossRef] [PubMed]
- Dadová, J.; Wu, K.-J.; Isenegger, P.G.; Errey, J.C.; Bernardes, G.J.L.; Chalker, J.M.; Raich, L.; Rovira, C.; Davis, B.G. Precise Probing of Residue Roles by Post-Translational β,γ-C,N Aza-Michael Mutagenesis in Enzyme Active Sites. ACS Cent. Sci. 2017, 3, 1168–1173. [Google Scholar] [CrossRef]
- Osamura, T.; Okuda, M.; Yamaguchi, A.; Ohtake, K.; Sakamoto, K.; Takimura, Y. Variants of the industrially relevant protease KP-43 with suppressed activity under alkaline conditions developed using expanded genetic codes. Biochem. Biophys. Rep. 2019, 17, 93–96. [Google Scholar] [CrossRef]
- Luo, J.; Torres-Kolbus, J.; Liu, J.; Deiters, A. Genetic Encoding of Photocaged Tyrosines with Improved Light-Activation Properties for the Optical Control of Protease Function. ChemBioChem 2017, 18, 1442–1447. [Google Scholar] [CrossRef]
- Short, G.F.; Golovine, S.Y.; Hecht, S.M. Effects of Release Factor 1 on in vitro Protein Translation and the Elaboration of Proteins Containing Unnatural Amino Acids. Biochemistry 1999, 38, 8808–8819. [Google Scholar] [CrossRef]
- Short, G.F.; Laikhter, A.L.; Lodder, M.; Shayo, Y.; Arslan, T.; Hecht, S.M. Probing the S1/S1‘ Substrate Binding Pocket Geometry of HIV-1 Protease with Modified Aspartic Acid Analogues. Biochemistry 2000, 39, 8768–8781. [Google Scholar] [CrossRef]
- Ugwumba, I.N.; Ozawa, K.; de la Cruz, L.; Xu, Z.-Q.; Herlt, A.J.; Hadler, K.S.; Coppin, C.; Brown, S.E.; Schenk, G.; Oakeshott, J.G.; et al. Using a Genetically Encoded Fluorescent Amino Acid as a Site-Specific Probe to Detect Binding of Low-Molecular-Weight Compounds. ASSAY Drug Dev. Technol. 2010, 9, 50–57. [Google Scholar] [CrossRef]
- Walasek, P.; Honek, J.F. Nonnatural amino acid incorporation into the methionine 214 position of the metzincin Pseudomonas aeruginosa alkaline protease. BMC Biochem. 2005, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Fan, X.; Chen, P.R. A genetically encoded multifunctional unnatural amino acid for versatile protein manipulations in living cells. Chem. Sci. 2016, 7, 7055–7060. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.; Wesalo, J.; Tsang, M.; Deiters, A. Engineering Small Molecule Switches of Protein Function in Zebrafish Embryos. J. Am. Chem. Soc. 2023, 145, 2395–2403. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Beattie, A.T.; Kafkova, L.; Petris, G.; Huguenin-Dezot, N.; Fiedler, M.; Freeman, M.; Chin, J.W. Mechanism-based traps enable protease and hydrolase substrate discovery. Nature 2022, 602, 701–707. [Google Scholar] [CrossRef]
- Fu, X.; Chang, Z. Biogenesis, quality control, and structural dynamics of proteins as explored in living cells via site-directed photocrosslinking. Protein Sci. 2019, 28, 1194–1209. [Google Scholar] [CrossRef]
- Sinz, A. The advancement of chemical cross-linking and mass spectrometry for structural proteomics: From single proteins to protein interaction networks. Expert Rev. Proteom. 2014, 11, 733–743. [Google Scholar] [CrossRef]
- Aydin, Y.; Coin, I. Genetically encoded crosslinkers to address protein–protein interactions. Protein Sci. 2023, 32, e4637. [Google Scholar] [CrossRef]
- Yi, D.; Xing, J.; Gao, Y.; Pan, X.; Xie, P.; Yang, J.; Wang, Q.; Gao, X. Enhancement of keratin-degradation ability of the keratinase KerBL from Bacillus licheniformis WHU by proximity-triggered chemical crosslinking. Int. J. Biol. Macromol. 2020, 163, 1458–1470. [Google Scholar] [CrossRef]
- Cheng, Y.; Wu, J.; Han, Y.; Xu, J.; Da, Y.; Zhao, Q.; Guo, G.; Zhou, Y.; Chen, Y.; Liu, J.; et al. A CDR-based approach to generate covalent inhibitory antibody for human rhinovirus protease. Bioorg. Med. Chem. 2021, 42, 116219. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Wang, R.; Ma, J.; Liu, Y.; Ezemaduka, A.N.; Chen, P.R.; Fu, X.; Chang, Z. DegP primarily functions as a protease for the biogenesis of β-barrel outer membrane proteins in the Gram-negative bacterium Escherichia coli. FEBS J. 2014, 281, 1226–1240. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Wang, Y.; Shao, H.; Ma, J.; Song, X.; Zhang, M.; Chang, Z. DegP functions as a critical protease for bacterial acid resistance. FEBS J. 2018, 285, 3525–3538. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Wang, Y.; Song, X.; Shi, X.; Shao, H.; Liu, Y.; Zhang, M.; Chang, Z. Subunit interactions as mediated by “non-interface” residues in living cells for multiple homo-oligomeric proteins. Biochem. Biophys. Res. Commun. 2019, 512, 100–105. [Google Scholar] [CrossRef]
- Liebsch, F.; Aurousseau, M.R.P.; Bethge, T.; McGuire, H.; Scolari, S.; Herrmann, A.; Blunck, R.; Bowie, D.; Multhaup, G. Full-length cellular β-secretase has a trimeric subunit stoichiometry, and its sulfur-rich transmembrane interaction site modulates cytosolic copper compartmentalization. J. Biol. Chem. 2017, 292, 13258–13270. [Google Scholar] [CrossRef]
- Nguyen, D.P.; Lusic, H.; Neumann, H.; Kapadnis, P.B.; Deiters, A.; Chin, J.W. Genetic Encoding and Labeling of Aliphatic Azides and Alkynes in Recombinant Proteins via a Pyrrolysyl-TRNA Synthetase/TRNACUA Pair and Click Chemistry. J. Am. Chem. Soc. 2009, 131, 8720. [Google Scholar] [CrossRef]
- Kawamoto, S.A.; Coleska, A.; Ran, X.; Yi, H.; Yang, C.-Y.; Wang, S. Design of Triazole-Stapled BCL9 α-Helical Peptides to Target the β-Catenin/B-Cell CLL/lymphoma 9 (BCL9) Protein–Protein Interaction. J. Med. Chem. 2012, 55, 1137–1146. [Google Scholar] [CrossRef]
- Schneider, T.; Schneider, D.; Rösner, D.; Malhotra, S.; Mortensen, F.; Mayer, T.U.; Scheffner, M.; Marx, A. Dissecting Ubiquitin Signaling with Linkage-Defined and Protease Resistant Ubiquitin Chains. Angew. Chem. Int. Ed. Engl. 2014, 53, 12925–12929. [Google Scholar] [CrossRef]
- Abdelkader, E.H.; Feintuch, A.; Yao, X.; Adams, L.A.; Aurelio, L.; Graham, B.; Goldfarb, D.; Otting, G. Protein conformation by EPR spectroscopy using gadolinium tags clicked to genetically encoded p-azido-l-phenylalanine. Chem. Commun. 2015, 51, 15898–15901. [Google Scholar] [CrossRef]
- Mahawaththa, M.C.; Lee, M.D.; Giannoulis, A.; Adams, L.A.; Feintuch, A.; Swarbrick, J.D.; Graham, B.; Nitsche, C.; Goldfarb, D.; Otting, G. Small neutral Gd(iii) tags for distance measurements in proteins by double electron–electron resonance experiments. Phys. Chem. Chem. Phys. 2018, 20, 23535–23545. [Google Scholar] [CrossRef] [PubMed]
- Penna, E.; Cerciello, A.; Chambery, A.; Russo, R.; Cernilogar, F.M.; Pedone, E.M.; Perrone-Capano, C.; Cappello, S.; Di Giaimo, R.; Crispino, M. Cystatin B Involvement in Synapse Physiology of Rodent Brains and Human Cerebral Organoids. Front. Mol. Neurosci. 2019, 12, 195. [Google Scholar] [CrossRef] [PubMed]
- Bard, J.A.M.; Martin, A. Recombinant Expression, Unnatural Amino Acid Incorporation, and Site-Specific Labeling of 26S Proteasomal Subcomplexes BT. In The Ubiquitin Proteasome System: Methods and Protocols; Mayor, T., Kleiger, G., Eds.; Springer: New York, NY, USA, 2018; pp. 219–236. [Google Scholar]
- Bard, J.A.M.; Bashore, C.; Dong, K.C.; Martin, A. The 26S Proteasome Utilizes a Kinetic Gateway to Prioritize Substrate Degradation. Cell 2019, 177, 286–298.e215. [Google Scholar] [CrossRef] [PubMed]
- Regev, O.; Linder, H.; Gur, E. Pup-Click—A New Chemoenzymatic Method for the Generation of Singly Pupylated Targets. Bioconjug. Chem. 2019, 30, 2909–2916. [Google Scholar] [CrossRef] [PubMed]
- Exner, M.P.; Kuenzl, T.; To, T.M.T.; Ouyang, Z.; Schwagerus, S.; Hoesl, M.G.; Hackenberger, C.P.R.; Lensen, M.C.; Panke, S.; Budisa, N. Design of S-Allylcysteine in Situ Production and Incorporation Based on a Novel Pyrrolysyl-tRNA Synthetase Variant. ChemBioChem 2017, 18, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Koch, N.G.; Goettig, P.; Rappsilber, J.; Budisa, N. “Cold” Orthogonal Translation: Psychrophilic Pyrrolysyl-tRNA Synthetase as Efficient Tool for Expanding the Genetic Code. bioRxiv 2023, preprint. [Google Scholar] [CrossRef]
- Turk, N.; Raza, A.; Wuytens, P.; Demol, H.; Daele, M.V.; Detavernier, C.; Skirtach, A.; Gevaert, K.; Baets, R. Waveguide-based surface-enhanced Raman spectroscopy detection of protease activity using non-natural aromatic amino acids. Biomed. Opt. Express 2020, 11, 4800–4816. [Google Scholar] [CrossRef]
- Hostetler, Z.M.; Cory, M.B.; Jones, C.M.; Petersson, E.J.; Kohli, R.M. The Kinetic and Molecular Basis for the Interaction of LexA and Activated RecA Revealed by a Fluorescent Amino Acid Probe. ACS Chem. Biol. 2020, 15, 1127–1133. [Google Scholar] [CrossRef]
- Poreba, M. Protease-activated prodrugs: Strategies, challenges, and future directions. FEBS J. 2020, 287, 1936–1969. [Google Scholar] [CrossRef]
- Garner, P.; Sherry, B.; Moilanen, S.; Huang, Y. In vitro stability of α-Helical peptide nucleic acids (αPNAs). Bioorg. Med. Chem. Lett. 2001, 11, 2315–2317. [Google Scholar] [CrossRef]
- Radchenko, T.; Brink, A.; Siegrist, Y.; Kochansky, C.; Bateman, A.; Fontaine, F.; Morettoni, L.; Zamora, I. Software-aided approach to investigate peptide structure and metabolic susceptibility of amide bonds in peptide drugs based on high resolution mass spectrometry. PLoS ONE 2017, 12, e0186461. [Google Scholar] [CrossRef] [PubMed]
- Radchenko, T.; Fontaine, F.; Morettoni, L.; Zamora, I. WebMetabase: Cleavage sites analysis tool for natural and unnatural substrates from diverse data source. Bioinformatics 2019, 35, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Radchenko, T.; Fontaine, F.; Morettoni, L.; Zamora, I. Software-aided workflow for predicting protease-specific cleavage sites using physicochemical properties of the natural and unnatural amino acids in peptide-based drug discovery. PLoS ONE 2019, 14, e0199270. [Google Scholar] [CrossRef] [PubMed]
- Courtney, T.; Deiters, A. Recent advances in the optical control of protein function through genetic code expansion. Curr. Opin. Chem. Biol. 2018, 46, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Fok, J.A.; Mayer, C. Genetic-Code-Expansion Strategies for Vaccine Development. ChemBioChem 2020, 21, 3291–3300. [Google Scholar] [CrossRef] [PubMed]
- Rezhdo, A.; Islam, M.; Huang, M.; Van Deventer, J.A. Future prospects for noncanonical amino acids in biological therapeutics. Curr. Opin. Biotechnol. 2019, 60, 168–178. [Google Scholar] [CrossRef]
- Zhu, C.; Xu, L.; Chen, L.; Zhang, Z.; Zhang, Y.; Wu, W.; Li, C.; Liu, S.; Xiang, S.; Dai, S.; et al. Epitope-Directed Antibody Elicitation by Genetically Encoded Chemical Cross-Linking Reactivity in the Antigen. ACS Cent. Sci. 2023, 9, 1229–1240. [Google Scholar] [CrossRef]
- Benner, S.A. Expanding the genetic lexicon: Incorporating non-standard amino acids into proteins by ribosome-based synthesis. Trends Biotechnol. 1994, 12, 158–163. [Google Scholar] [CrossRef]
- Chen, Y.; He, X.; Ma, B.; Liu, K.; Gao, T.; Niu, W.; Guo, J. Noncanonical amino acid mutagenesis in response to recoding signal-enhanced quadruplet codons. Nucleic Acids Res. 2022, 50, e94. [Google Scholar] [CrossRef]
- Anderson, J.C.; Magliery, T.J.; Schultz, P.G. Exploring the limits of codon and anticodon size. Chem. Biol. 2002, 9, 237–244. [Google Scholar] [CrossRef]
- Hartman, M.C.T.; Josephson, K.; Lin, C.-W.; Szostak, J.W. An Expanded Set of Amino Acid Analogs for the Ribosomal Translation of Unnatural Peptides. PLoS ONE 2007, 2, e972. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, F.; Krautwurst, S.; Salentin, S.; Haupt, V.J.; Leberecht, C.; Bittrich, S.; Labudde, D.; Schroeder, M. The structural basis of the genetic code: Amino acid recognition by aminoacyl-tRNA synthetases. Sci. Rep. 2020, 10, 12647. [Google Scholar] [CrossRef]
- Hartman, M.C.T.; Josephson, K.; Szostak, J.W. Enzymatic aminoacylation of tRNA with unnatural amino acids. Proc. Natl. Acad. Sci. USA 2006, 103, 4356–4361. [Google Scholar] [CrossRef] [PubMed]
- Krishnakumar, R.; Prat, L.; Aerni, H.R.; Ling, J.; Merryman, C.; Glass, J.I.; Rinehart, J.; Söll, D. Transfer RNA misidentification scrambles sense codon recoding. Chembiochem 2013, 14, 1967–1972. [Google Scholar] [CrossRef]
- Eremeeva, E.; Herdewijn, P. Non canonical genetic material. Curr Opin Biotechnol 2019, 57, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Völler, J.-S.; Budisa, N. Coupling genetic code expansion and metabolic engineering for synthetic cells. Curr. Opin. Biotechnol. 2017, 48, 1–7. [Google Scholar] [CrossRef]
- Agostini, F.; Sinn, L.; Petras, D.; Schipp, C.J.; Kubyshkin, V.; Berger, A.A.; Dorrestein, P.C.; Rappsilber, J.; Budisa, N.; Koksch, B. Multiomics Analysis Provides Insight into the Laboratory Evolution of Escherichia coli toward the Metabolic Usage of Fluorinated Indoles. ACS Cent Sci 2021, 7, 81–92. [Google Scholar] [CrossRef]
- Dragosits, M.; Mattanovich, D. Adaptive laboratory evolution—Principles and applications for biotechnology. Microb. Cell Factories 2013, 12, 64. [Google Scholar] [CrossRef] [PubMed]
- Hoesl, M.G.; Oehm, S.; Durkin, P.; Darmon, E.; Peil, L.; Aerni, H.-R.; Rappsilber, J.; Rinehart, J.; Leach, D.; Söll, D.; et al. Chemical Evolution of a Bacterial Proteome. Angew. Chem. Int. Ed. Engl. 2015, 54, 10030–10034. [Google Scholar] [CrossRef]
- Tolle, I.; Oehm, S.; Hoesl, M.G.; Treiber-Kleinke, C.; Peil, L.; Bozukova, M.; Albers, S.; Bukari, A.-R.A.; Semmler, T.; Rappsilber, J.; et al. Evolving a mitigation of the stress response pathway to change the basic chemistry of life. Front. Synth. Biol. 2021, 1, 1248065. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goettig, P.; Koch, N.G.; Budisa, N. Non-Canonical Amino Acids in Analyses of Protease Structure and Function. Int. J. Mol. Sci. 2023, 24, 14035. https://doi.org/10.3390/ijms241814035
Goettig P, Koch NG, Budisa N. Non-Canonical Amino Acids in Analyses of Protease Structure and Function. International Journal of Molecular Sciences. 2023; 24(18):14035. https://doi.org/10.3390/ijms241814035
Chicago/Turabian StyleGoettig, Peter, Nikolaj G. Koch, and Nediljko Budisa. 2023. "Non-Canonical Amino Acids in Analyses of Protease Structure and Function" International Journal of Molecular Sciences 24, no. 18: 14035. https://doi.org/10.3390/ijms241814035
APA StyleGoettig, P., Koch, N. G., & Budisa, N. (2023). Non-Canonical Amino Acids in Analyses of Protease Structure and Function. International Journal of Molecular Sciences, 24(18), 14035. https://doi.org/10.3390/ijms241814035