
Biological and Catalytic Properties of Selenoproteins

Abstract
1. Introduction
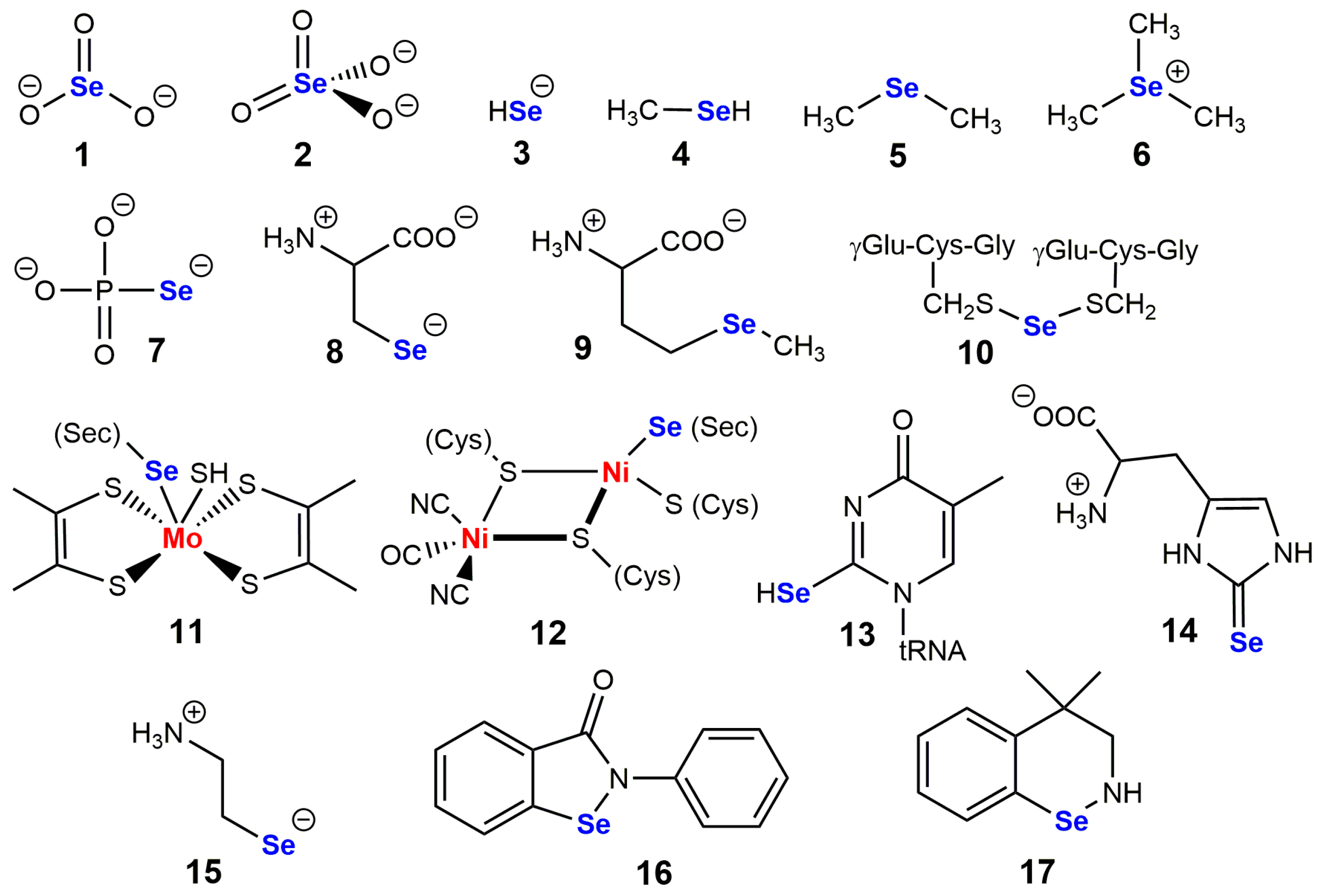
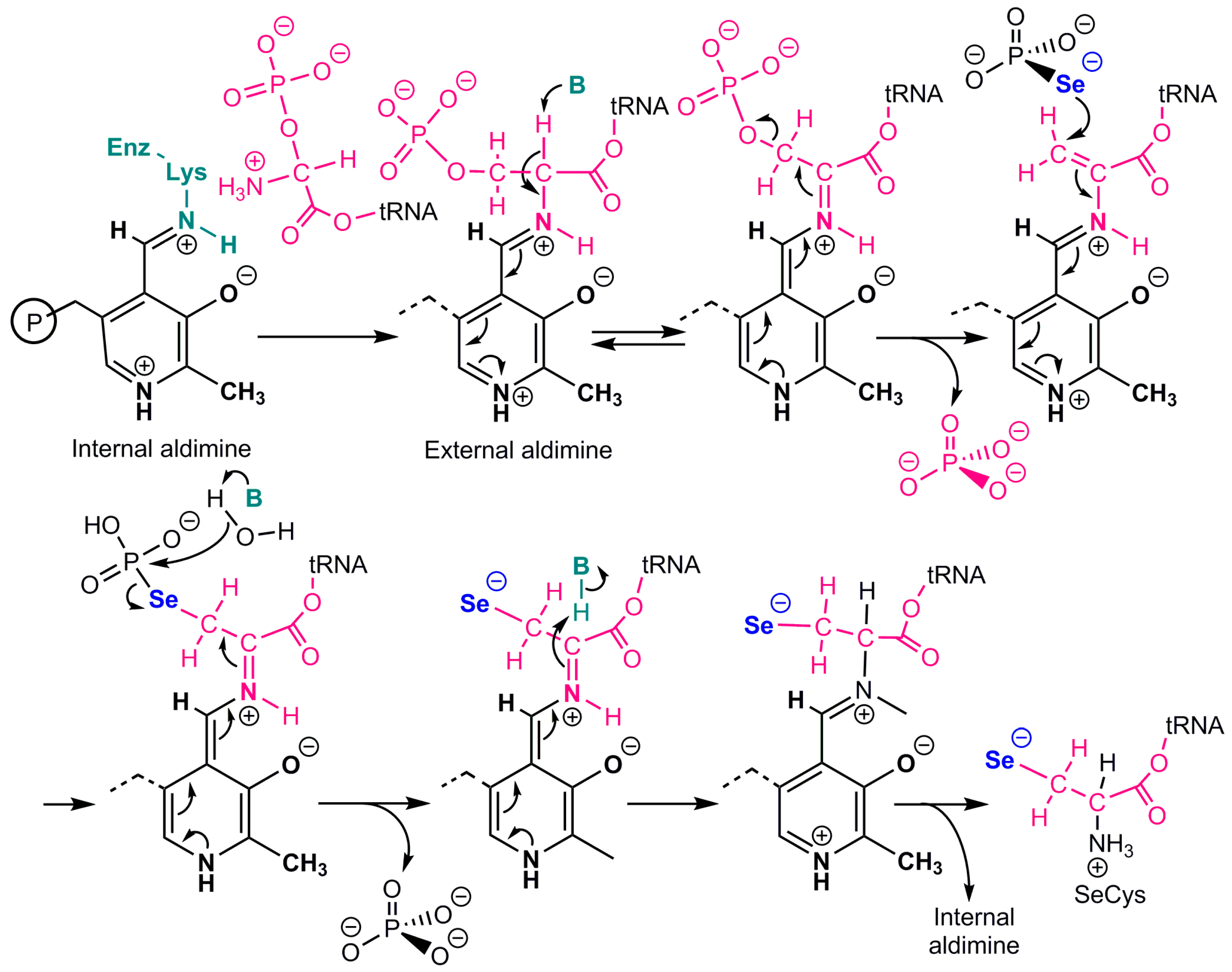
2. Co-Translational Incorporation of Selenocysteine
3. Mammalian Selenoproteins
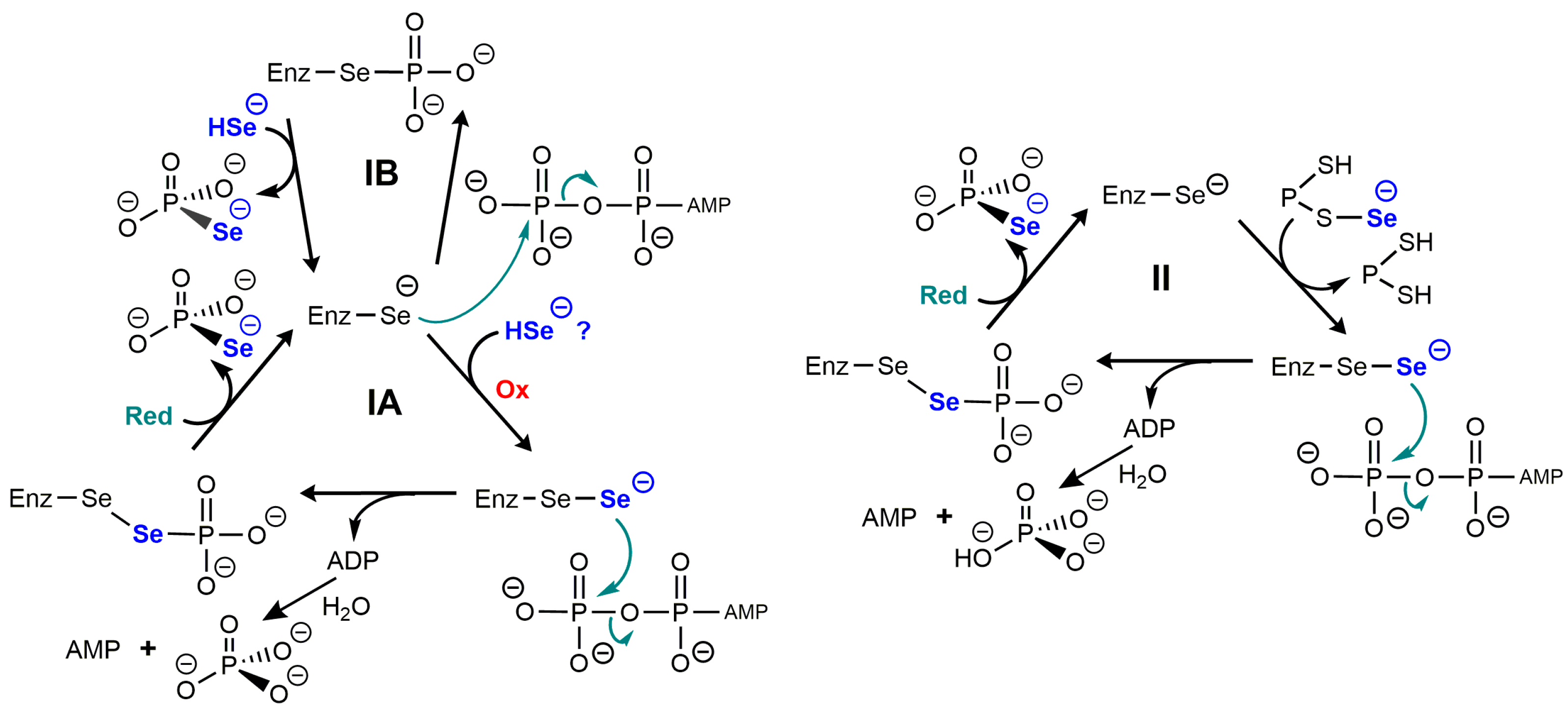
3.1. Selenophosphate Synthetase
3.2. Selenoprotein P (Selenop)
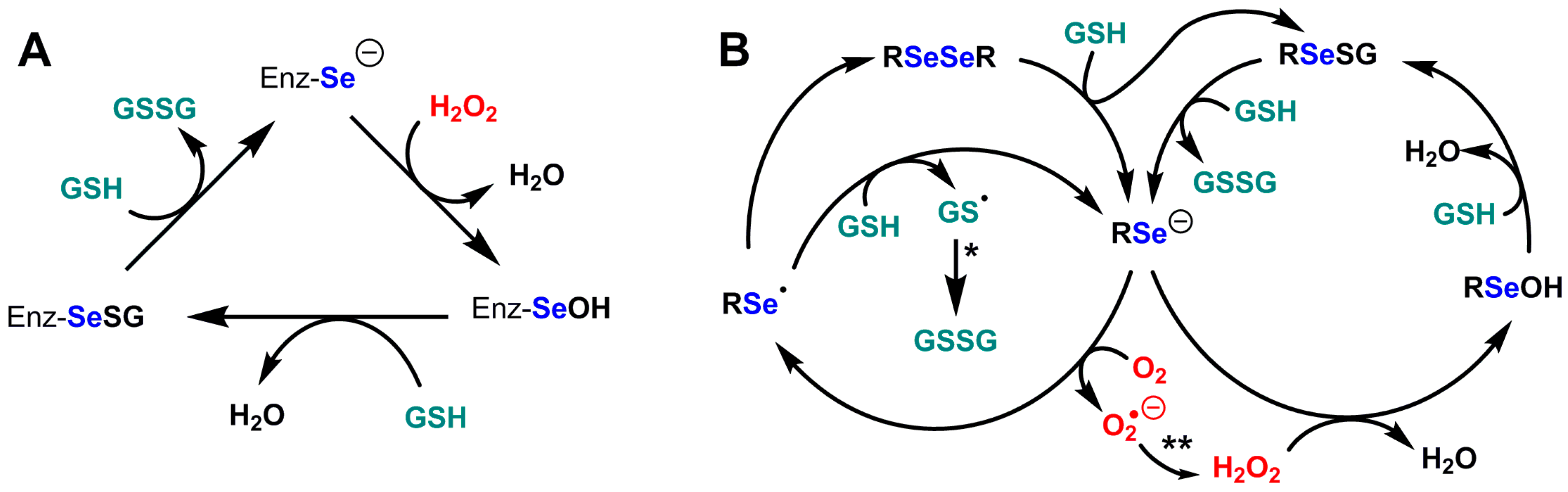
3.3. Selenium-Glutathione Peroxidases (Se-GPx)
3.3.1. GPx1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Localization | Structure | Reducing Cosubstrate | Hydroperoxide Substrates | Inhibitors Inactivators | Function 2 |
|---|---|---|---|---|---|---|
| GPx1 | Ubiquitous in cytosol and mitochondrial matrix | Homotetramer, five conserved residues at GSH binding site (four Arg, one Lys) | GSH | H2O2 and alkyl hydroperoxides | Mercaptosuccinate 3 O2.−, HOCl Heavy metals | Antioxidant Redox regulator? |
| GPx2 | Cytosol of epithelial cells | Homotetramer, extracellular glycoprotein | GSH | H2O2 and alkyl hydroperoxides | undocumented | Antioxidant Redox regulator? |
| GPx3 | Mostly extracell. Blood plasma, Secreted/kidney mammary gland | Homotetramer, Glycoprotein | Thioredoxin; GSH (nonphysiological) | H2O2 and alkyl hydroperoxides | undocumented | Antioxidant Redox regulator? |
| GPx4 | Membrane-bound Cytoplasm Mitochondria Nucleus (minor) | Monomer | Thioredoxin Protein thiols GSH (nonphysiological), | Phospholipid-OOH, Cholesterol-OOH, LDL-OOH | RSL3 ML162 | Antiox/Redox regul? Anti-ferroptosis Spermatozoid specific- protein crosslinker |
| GpX6 | Olfactory epithelium | Homotetramer, Strong sequence homology with GPx3 | Thioredoxin; GSH? | undocumented | undocumented | Antioxidant Redox regulator? |
3.3.2. GPx2
3.3.3. GPx3
3.3.4. GPx4
3.3.5. Other Members of the GPx Family

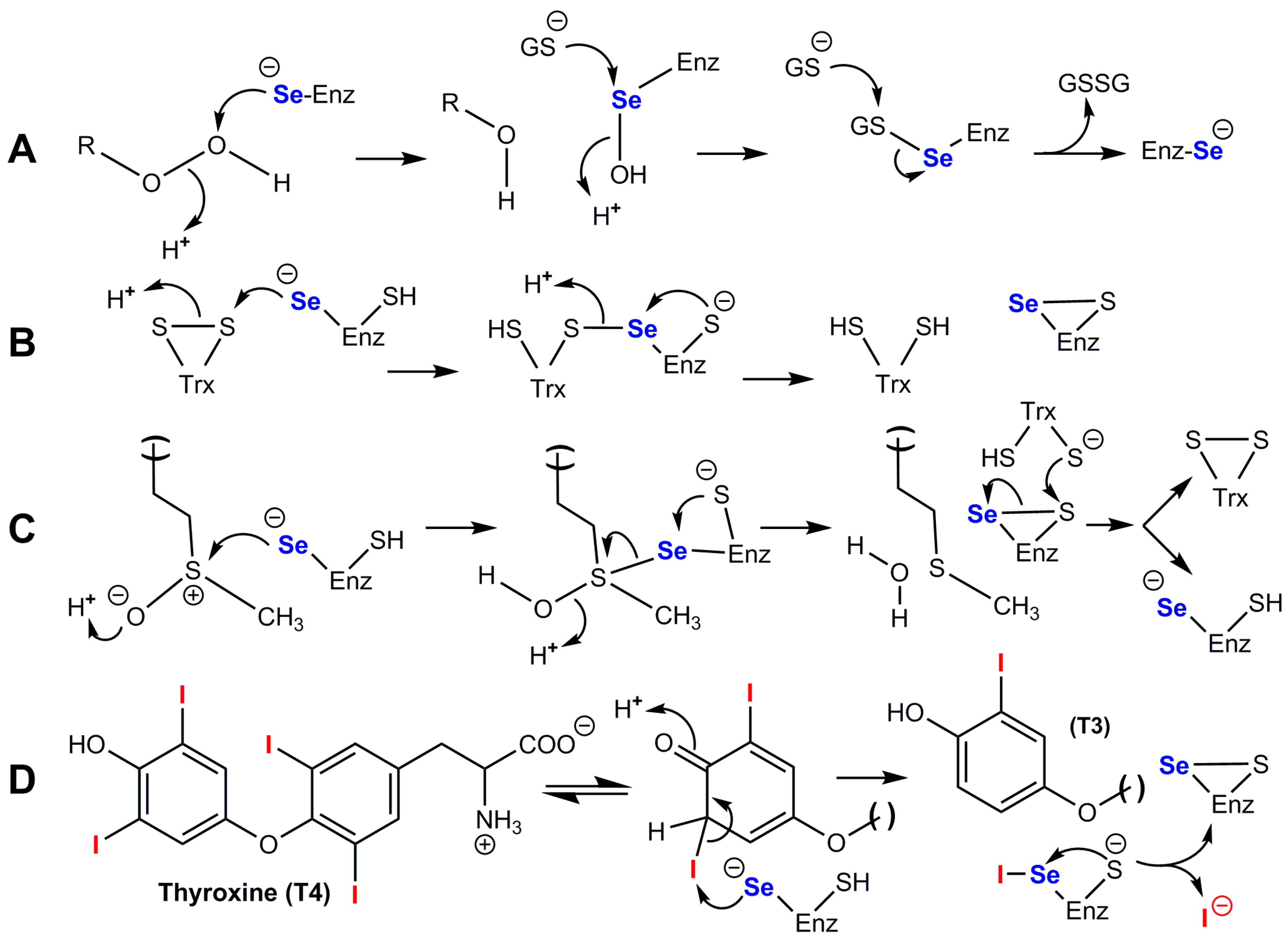
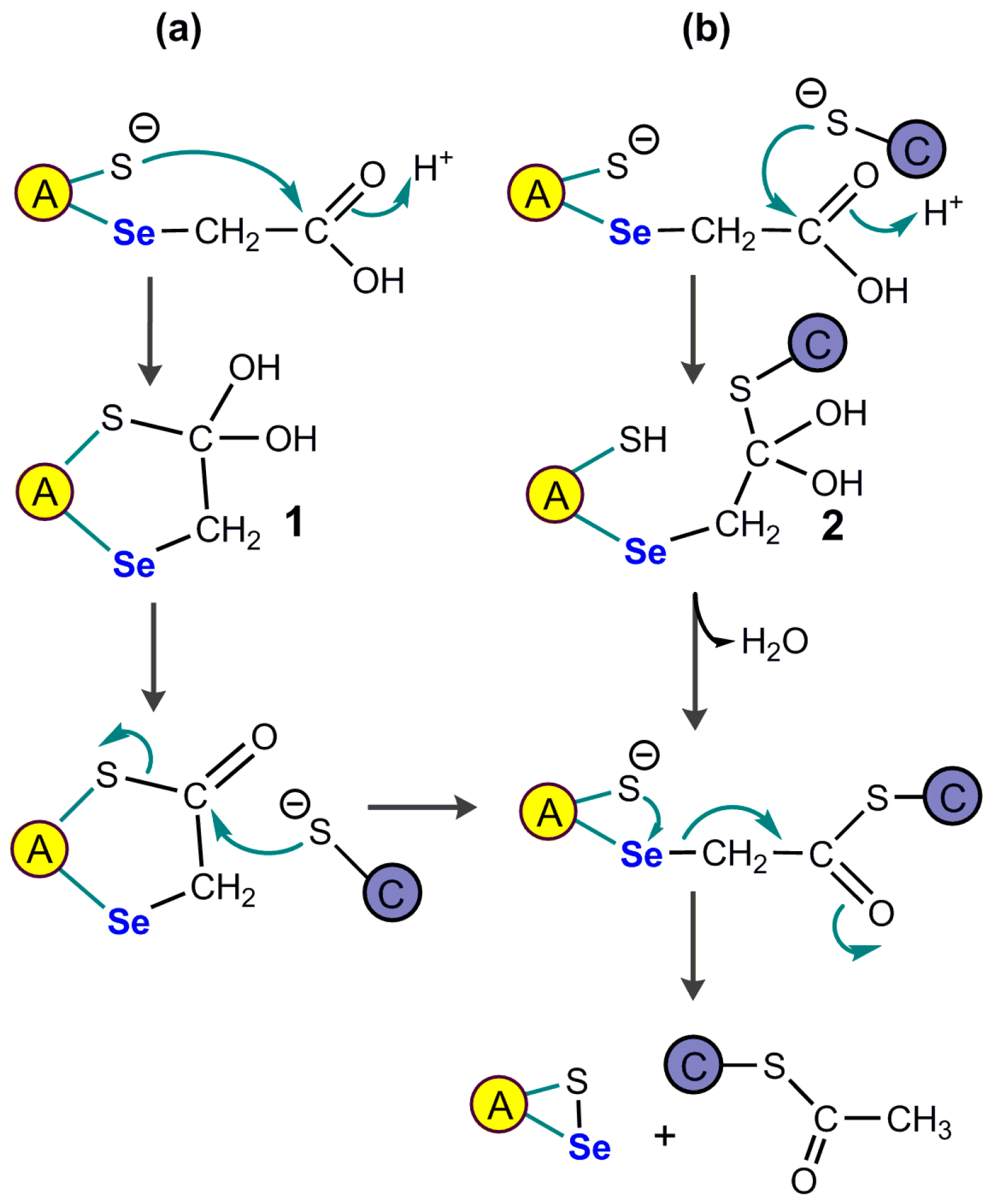
3.3.6. Mechanistic Complexity of Glutathione Peroxidases
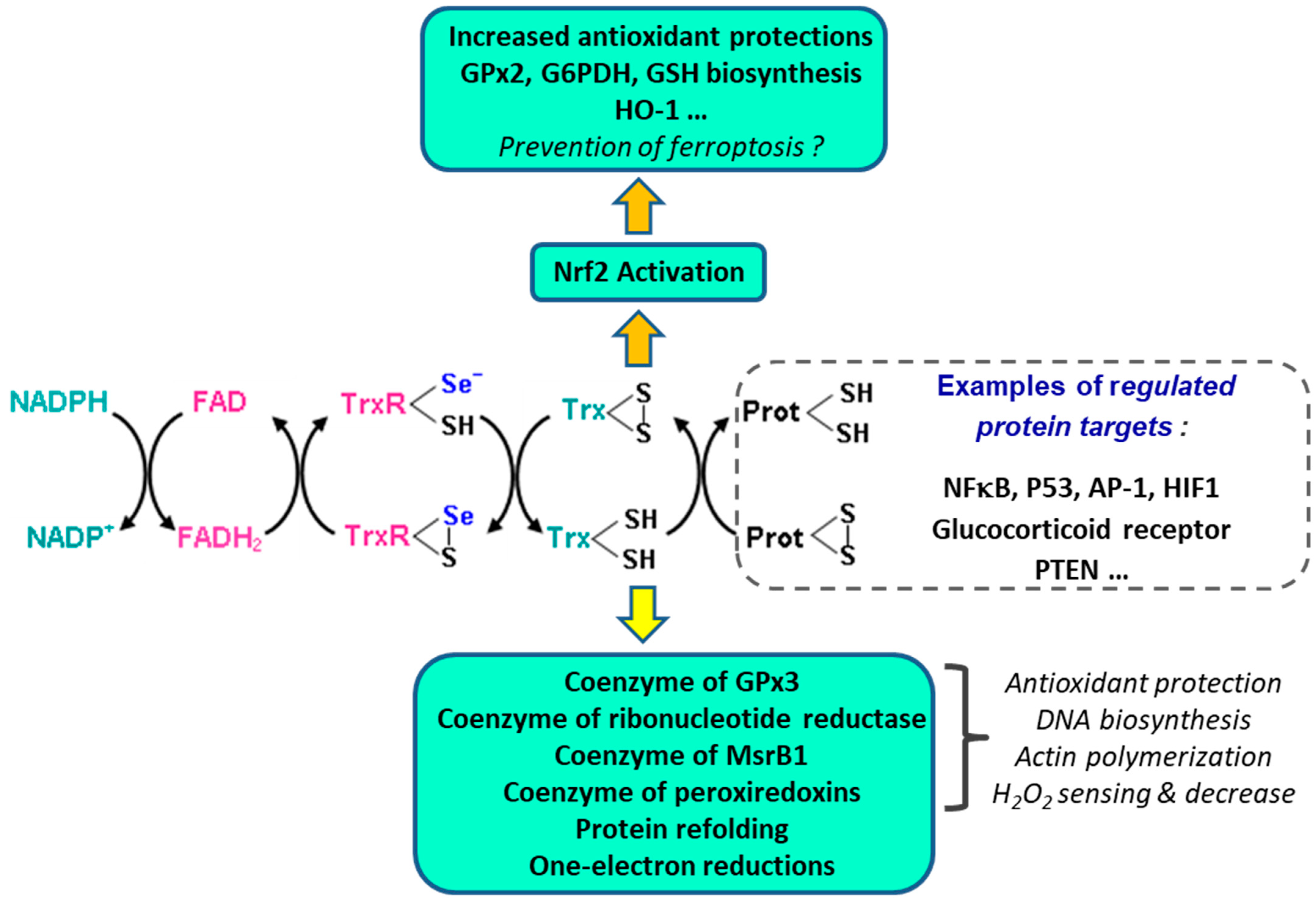
3.4. Thioredoxin Reductases (TrxR)
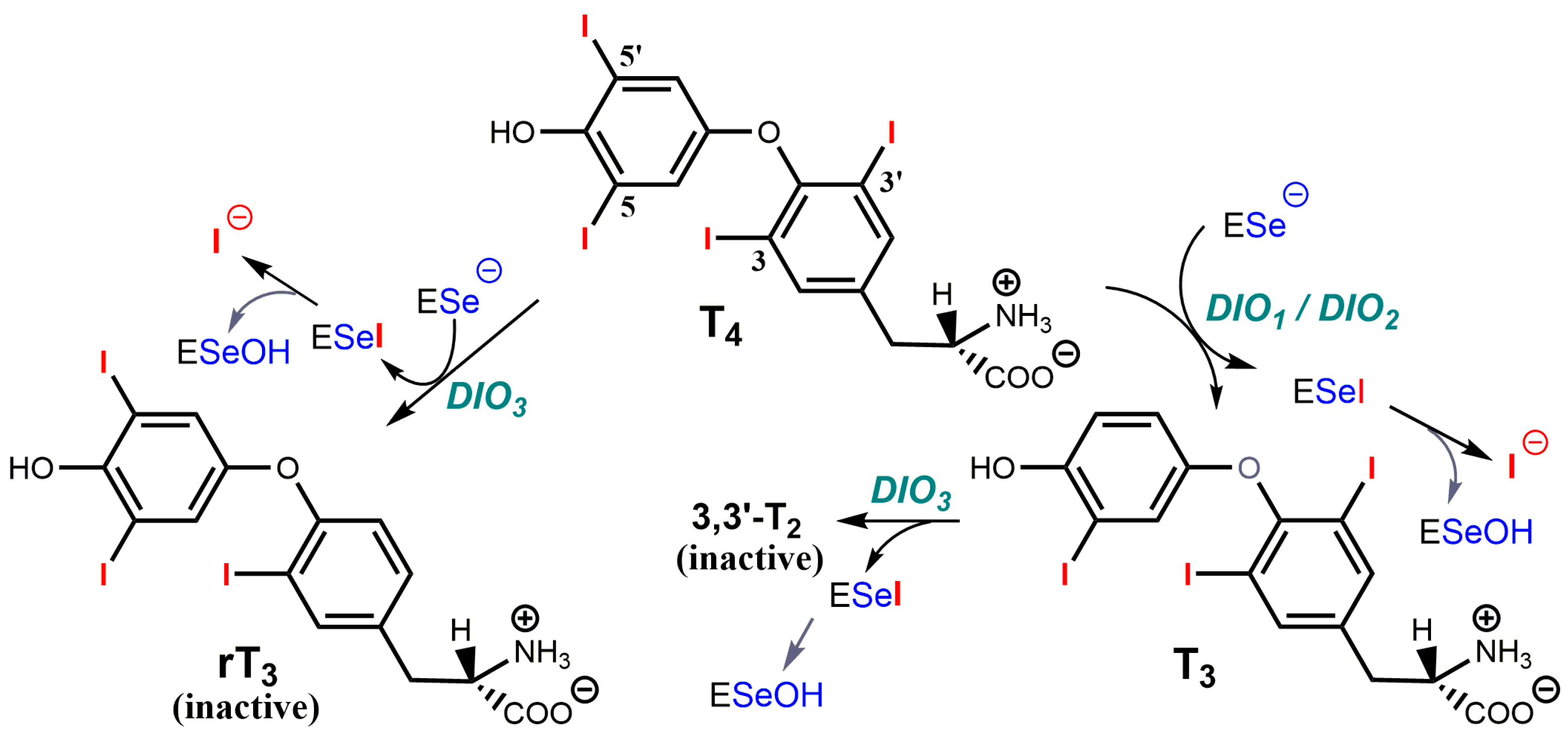
3.5. Selenium-Dependent Deiodinases
3.6. Methionine R-Sulfoxide Reductase B1 [MsrB1]
3.7. Other Mammalian Selenoproteins
3.7.1. Selenoprotein O and Protein AMPylation
3.7.2. Selenoprotein F and Endoplasmic Reticulum Glycoproteins
3.7.3. Selenoprotein N and Endoplasmic Reticulum Calcium Sensing
3.7.4. Selenoprotein I and Ethanolamine Phospholipids
3.7.5. Selenoprotein K, Palmitoylation Cofactor and Inhibitor of ER-Induced Apoptosis
3.7.6. Selenoprotein W and EGF Regulation
3.7.7. Selenoprotein T
3.7.8. Selenoprotein S
3.7.9. Selenoprotein-H
4. Bacterial Selenoenzymes
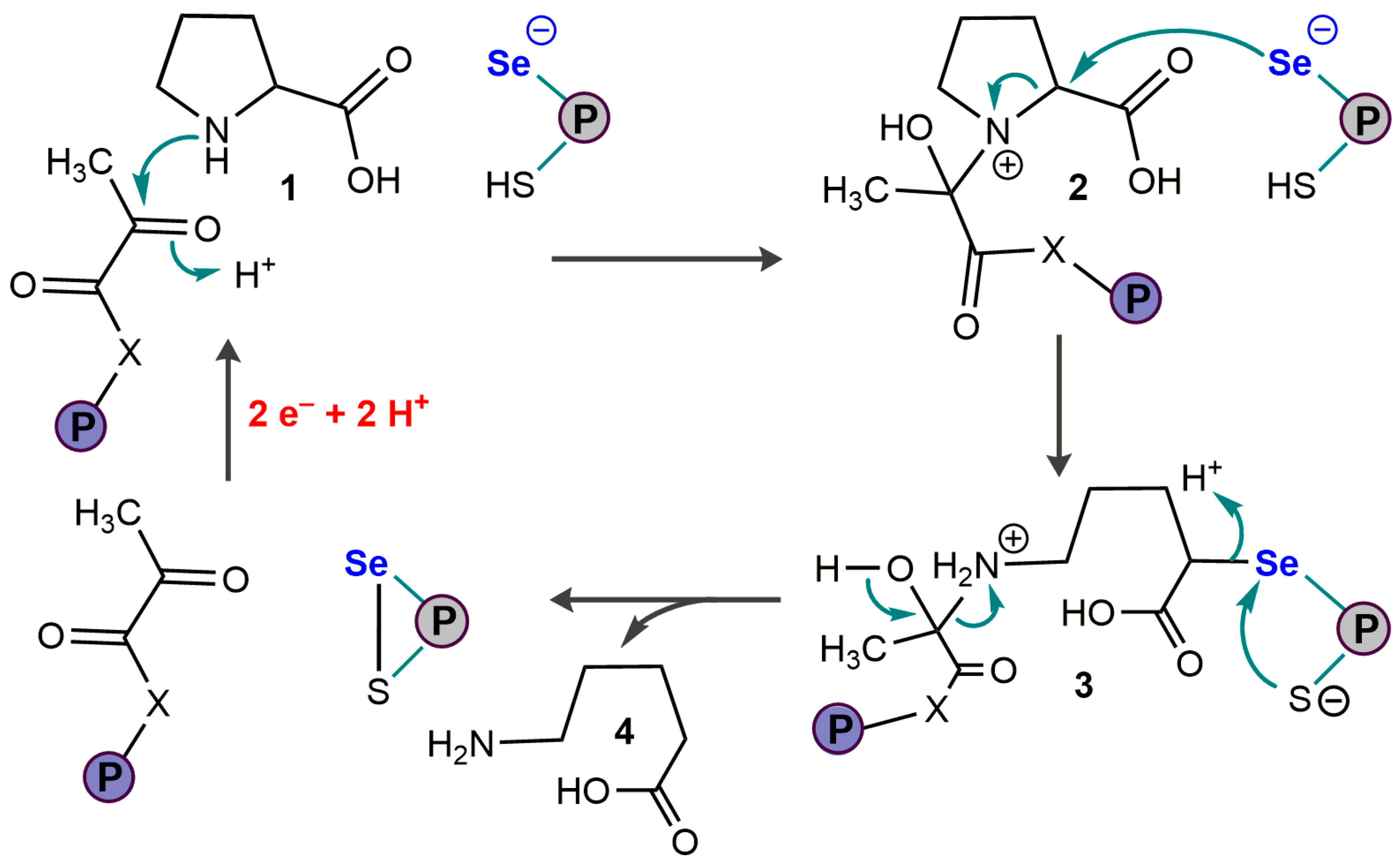
4.1. Glycine Reductase
4.2. D-Proline Reductase
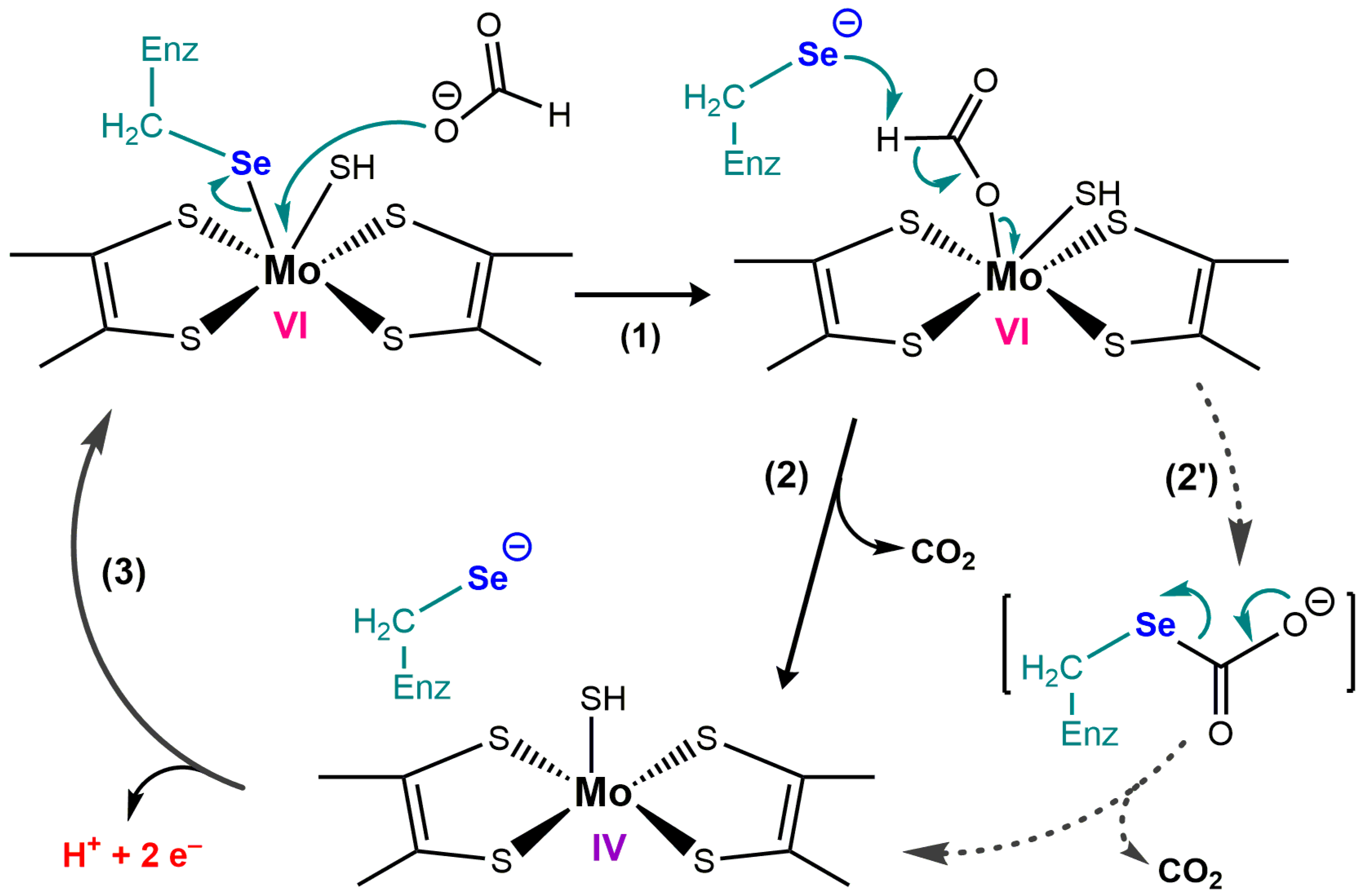
4.3. Formate Dehydrogenase
4.4. [NiFeSe] Hydrogenases
5. Is Redox Regulation an Essential Function of Most Mammalian Selenoproteins?
6. Advantages and Constraints Associated with the Choice of Selenocysteine at the Active Site of Selenoenzymes
7. Conclusions
- The co-translational incorporation of selenium in selenoproteins is probably unique among the elements situated below period 3 of the periodic table, and one of the major advantages of selenium compared with sulfur should be the better reversibility of its oxidation reactions in biological conditions.
- Selenoproteins are mostly involved in anaerobic metabolism in bacteria, whereas they are involved in antioxidant protection, protein repair, redox signaling, and regulation of cell proliferation/cell death, and energetic metabolism in mammals.
- Many functions of mammalian selenoproteins, especially those which are anti-inflammatory, anti-apoptotic, or anti-ferroptotic, or which interfere with energetic metabolism, require regulation in space and time. Specifically, most mammalian selenoenzymes should not only be regulating but also regulated, although, in this area, much information is probably still missing.
- With the recent development of new techniques of selenocysteine insertion into protein sequences, we can expect that new discoveries will shed light on the hidden face of the moon.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AIF, apoptosis inducing factor; Alkbh, mammalian alkylation repair homolog; Alox15 gene, gene encoding an arachidonate 15-lipoxygenase; AP-1, activator protein 1; ASK1, apoptosis signal kinase 1; CAMKII, Ca2+ calmodulin-dependent kinase; COX2, Type 2 cyclooxygenase; DIO, deiodinase; DUOX, dual oxidase; EFsec, selenocysteine dedicated elongation factor; EMT, epithelial-mesenchymal transition; GLS2, liver-type glutaminase; GPx, glutathione peroxidase; GR, glutathione reductase; HCV, hepatitis C virus; HIF, hypoxia inducible factor; Keap1, Kelch associated protein 1; LOX, Lipoxygenase; LPS, lipopolysaccharide; MAPK, mitogen-activated protein kinase; MICAL, molecules interacting with CasL; MsrB1, methionine sulfoxide reductase B1; NFκB, nuclear factor kB; Nox, NADPH oxidase; Nrf2, nuclear factor erythroid-2-related factor; NQQ1, NAD[P)H: Quinone oxidoreductase 1; PDI, protein disulfide isomerase; Prx, peroxiredoxin; PTEN, phosphatase and tensin homolog; SBP2, SECIS-binding protein 2; SecTRAPs, selenium compromised thioredoxin reductase-derived apoptotic proteins; SECYS, selenocysteine insertion sequence; Selenoprotein P, selenoprotein P; Sephs2, selenophosphate synthetase 2; TGS1, trimethylguanosine synthase 1; TNFα, tumor necrosis factor a; TPO, thyroid peroxidase; Trx, thioredoxin; TrxR, thioredoxin reductase; T4, thyroxine; T3 triiodothyronine; ZEB1, zinc-finger E-box binding protein 1. |
References
- Flohé, L. The labour pains of biochemical selenology: The history of selenoprotein biosynthesis. Biochim. Biophys. Acta 2009, 1790, 1389–1403. [Google Scholar] [CrossRef] [PubMed]
- Elhodaky, M.; Diamond, A.M. Selenium-Binding Protein 1 in Human Health and Disease. Int. J. Mol. Sci. 2018, 19, 3437. [Google Scholar] [CrossRef] [PubMed]
- Schrauzer, G.N.; Surai, P.F. Selenium in human and animal nutrition: Resolved and unresolved issues. Crit. Rev. Biotechnol. 2009, 29, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, J.K.; Power, R.; Toborek, M. Biological activity of selenium: Revisited. IUBMB Life 2015, 68, 97–105. [Google Scholar] [CrossRef]
- Genchi, G.; Lauria, G.; Catalano, A.; Sinicropi, M.S.; Carocci, A. Biological activity of selenium and its impact on human health. Int. J. Mol. Sci. 2023, 24, 2633. [Google Scholar] [CrossRef]
- Steinbrenner, H.; Sies, H. Selenium homeostasis and antioxidant selenoproteins in brain: Implications for disorders in the central nervous system. Arch. Biochem. Biophys. 2013, 536, 152–157. [Google Scholar] [CrossRef]
- Conrad, M.; Schweizer, U. Unveiling the molecular mechanisms behind selenium-related diseases through knockout mouse studies. Antioxid. Redox Signal. 2010, 12, 851–865. [Google Scholar] [CrossRef]
- Agamy, O.; Ben Zeev, B.; Lev, D.; Marcus, B.; Fine, D.; Su, D.; Narkis, G.; Ofir, R.; Hoffmann, C.; Leshinsky-Silver, E.; et al. Mutations disrupting selenocysteine formation cause progressive cerebello-cerebral atrophy. Am. J. Hum. Genet. 2010, 87, 538–544. [Google Scholar] [CrossRef]
- Wirth, E.K.; Bharathi, B.S.; Hatfield, D.; Conrad, M.; Brielmeier, M.; Schweizer, U. Cerebellar hypoplasia in mice lacking selenoprotein biosynthesis in neurons. Biol. Trace Elem. Res. 2014, 158, 203–210. [Google Scholar] [CrossRef]
- Chen, J. An original discovery: Selenium deficiency and Keshan disease (an endemic heart disease). Asia Pac. J. Clin. Nutr. 2012, 21, 320–326. [Google Scholar]
- Xie, D.; Liao, Y.; Yue, J.; Zhang, C.; Wang, Y.; Deng, C.; Chen, L. Effects of five types of selenium supplementation for treatment of Kashin-Beck disease in children: A systematic review and network meta-analysis. BMJ Open 2018, 8, e017883. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, T.; Li, Q.; Li, D. Prevention of Keshan Disease by Selenium Supplementation: A Systematic Review and Meta-analysis. Biol. Trace Elem. Res. 2018, 186, 98–105. [Google Scholar] [CrossRef]
- Koivistoinen, P.; Huttunen, J.K. Selenium in food and nutrition in Finland: An overview on research and action. Ann. Clin. Res. 1986, 18, 13–17. [Google Scholar]
- Jedrychowski, M.P.; Lu, G.Z.; Szpyt, J.; Mariotti, M.; Garrity, R.; Paulo, J.A.; Schweppe, D.K.; Laznik-Bogoslavski, D.; Kazak, L.; Murphy, M.P.; et al. Facultative protein selenation regulates redox sensitivity, adipose tissue thermogenesis, and obesity. Proc. Natl. Acad. Sci. USA 2020, 117, 10789–10796. [Google Scholar] [CrossRef]
- Hosnedlova, B.; Kepinska, M.; Skalickova, S.; Fernandez, C.; Ruttkay-Nedecky, B.; Malevu, T.D.; Sochor, J.; Baron, M.; Melcova, M.; Zidkova, J.; et al. A summary of new findings on the biological effects of selenium in selected animal species—A critical review. Int. J. Mol. Sci. 2017, 18, 2209. [Google Scholar] [CrossRef]
- Rahman, M.M.; Hossain, K.F.B.; Banik, S.; Sikder, M.T.; Akter, M.; Bondad, S.E.C.; Rahaman, M.S.; Hosokawa, T.; Saito, T.; Kurasaki, M. Selenium and zinc protections against metal-(loids)-induced toxicity and disease manifestations: A review. Ecotoxicol. Environ. Saf. 2019, 168, 146–163. [Google Scholar] [CrossRef]
- Chaudière, J.; Tappel, A.L. Interaction of gold(I) with the active site of selenium-glutathione peroxidase. J. Inorg. Biochem. 1984, 20, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Flohé, L.; Günzler, W.A.; Schock, H.H. Glutathione peroxidase: A selenoenzyme. FEBS Lett. 1973, 32, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Rotruck, J.T.; Pope, A.L.; Ganther, H.E.; Swanson, A.B.; Hafeman, D.G.; Hoekstra, W.G. Selenium: Biochemical role as a component of glutathione peroxidase. Science 1973, 179, 588–590. [Google Scholar] [CrossRef] [PubMed]
- Böck, A.; Forchhammer, K.; Heider, J.; Leinfelder, W.; Sawers, G.; Veprek, B.; Zinoni, F. Selenocysteine: The 21st amino acid. Mol. Microbiol. 1991, 5, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Chapple, C.E.; Guigó, R. Relaxation of selective constraints causes independent selenoprotein extinction in insect genomes. PLoS ONE 2008, 3, e2968. [Google Scholar] [CrossRef] [PubMed]
- Squires, J.E.; Berry, M.J. Eukaryotic selenoprotein synthesis: Mechanistic insight incorporating new factors and new functions for old factors. IUBMB Life 2008, 60, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Allmang, C.; Wurth, L.; Krol, A. The selenium to selenoprotein pathway in eukaryotes: More molecular partners than anticipated. Biochim. Biophys. Acta 2009, 1790, 1415–1423. [Google Scholar] [CrossRef]
- Vindry, C.; Ohlmann, T.; Chavatte, L. Translation regulation of mammalian selenoproteins. Biochim. Biophys. Acta 2018, 1862, 2480–2492. [Google Scholar] [CrossRef]
- Copeland, P.R.; Howard, M.T. Ribosome fate during decoding of UGA-Sec codons. Int. J. Mol. Sci. 2021, 22, 13204. [Google Scholar] [CrossRef]
- Ganichkin, O.M.; Xu, X.M.; Carlson, B.A.; Mix, H.; Hatfield, D.L.; Gladyshev, V.N.; Wahl, M.C. Structure and catalytic mechanism of eukaryotic selenocysteine synthase. J. Biol. Chem. 2008, 283, 5849–5865. [Google Scholar] [CrossRef]
- Yamada, T.; Komoto, J.; Takata, Y.; Ogawa, H.; Pitot, H.C.; Takusagawa, F. Crystal structure of serine dehydratase from rat liver. Biochemistry 2003, 42, 12854–12865. [Google Scholar] [CrossRef]
- Wurth, L.; Gribling-Burrer, A.S.; Verheggen, C.; Leichter, M.; Takeuchi, A.; Baudrey, S.; Martin, F.; Krol, A.; Bertrand, E.; Allmang, C. Hypermethylated-capped selenoprotein mRNAs in mammals. Nucleic Acids Res. 2014, 42, 8663–8677. [Google Scholar] [CrossRef]
- Schoenmakers, E.; Chatterjee, K. Human Genetic Disorders Resulting in Systemic Selenoprotein Deficiency. Int. J. Mol. Sci. 2021, 22, 12927. [Google Scholar] [CrossRef]
- Cheng, Q.; Arnér, E.S. Selenocysteine Insertion at a Predefined UAG Codon in a Release Factor 1 (RF1)-depleted Escherichia coli Host Strain Bypasses Species Barriers in Recombinant Selenoprotein Translation. J. Biol. Chem. 2017, 292, 5476–5487. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Arnér, E.S.J. Expressing recombinant selenoproteins using redefinition of a single UAG codon in an RF1-depleted E. coli host strain. Methods Enzymol. 2022, 662, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Z.; Miller, C.; Söll, D.; Krahn, N. Introducing selenocysteine into recombinant proteins in Escherichia coli. Curr. Protoc. 2021, 1, e54. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Liu, J.; Wang, L.; Forstner, M.B.; Rozovsky, S. Methods|Re-engineering the site-specific Incorporation of selenocysteine into proteins. In Encyclopedia of Biological Chemistry III; Elsevier: Amsterdam, The Netherlands, 2021; pp. 757–765. [Google Scholar] [CrossRef]
- Mukai, T.; Sevostyanova, A.; Suzuki, T.; Fu, X.; Söll, D. A facile method for producing selenocysteine-containing proteins. Angew. Chem. Int. Ed. Engl. 2018, 57, 7215–7219. [Google Scholar] [CrossRef] [PubMed]
- Rakauskaitė, R.; Urbanavičiūtė, G.; Rukšėnaitė, A.; Liutkevičiūtė, Z.; Juškėnas, R.; Masevičius, V.; Klimašauskas, S. Biosynthetic selenoproteins with genetically encoded photocaged selenocysteines. Chem. Commun. 2015, 51, 8245–8248. [Google Scholar] [CrossRef]
- Welegedara, A.P.; Adams, L.A.; Huber, T.; Graham, B.; Otting, G. Site-specific incorporation of selenocysteine by genetic encoding as a photocaged unnatural amino acid. Bioconjug. Chem. 2018, 29, 2257–2264. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zheng, F.; Cheng, R.; Li, S.; Rozovsky, S.; Wang, Q.; Wang, L. Site-specific incorporation of selenocysteine using an expanded genetic code and palladium-mediated chemical deprotection. J. Am. Chem. Soc. 2018, 140, 8807–8816. [Google Scholar] [CrossRef]
- Peeler, J.C.; Falco, J.A.; Kelemen, R.E.; Abo, M.; Chartier, B.V.; Edinger, L.C.; Chen, J.; Chatterjee, A.; Weerapana, E. Generation of recombinant mammalian selenoproteins through genetic code expansion with photocaged selenocysteine. ACS Chem. Biol. 2020, 15, 1535–1540. [Google Scholar] [CrossRef]
- Peeler, J.C.; Weerapana, E. Expression of selenoproteins via genetic code expansion in mammalian cells. Methods Enzymol. 2022, 662, 143–158. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Gladyshev, V.N. Mammalian selenoprotein gene signature: Identification and functional analysis of selenoprotein genes using bioinformatics methods. Methods Enzymol. 2002, 347, 84–100. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigó, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef]
- Wingler, K.; Böcher, M.; Flohé, L.; Kollmus, H.; Brigelius-Flohé, R. mRNA stability and selenocysteine insertion sequence efficiency rank gastrointestinal glutathione peroxidase high in the hierarchy of selenoproteins. Eur. J. Biochem. 1999, 259, 149–157. [Google Scholar] [CrossRef]
- Low, S.C.; Grundner-Culemann, E.; Harney, J.W.; Berry, M.J. SECIS-SBP2 interactions dictate selenocysteine incorporation efficiency and selenoprotein hierarchy. EMBO J. 2000, 19, 6882–6890. [Google Scholar] [CrossRef]
- Sonet, J.; Bulteau, A.L.; Touat-Hamici, Z.; Mosca, M.; Bierla, K.; Mounicou, S.; Lobinski, R.; Chavatte, L. Selenoproteome expression studied by non-radioactive isotopic selenium-labeling in human cell lines. Int. J. Mol. Sci. 2021, 22, 7308. [Google Scholar] [CrossRef]
- Ehrenreich, A.; Forchhammer, K.; Tormay, P.; Veprek, B.; Böck, A. Selenoprotein synthesis in E. coli Purification and characterisation of the enzyme catalysing selenium activation. Eur. J. Biochem. 1992, 206, 767–773. [Google Scholar] [CrossRef]
- Noinaj, N.; Wattanasak, R.; Lee, D.Y.; Wally, J.L.; Piszczek, G.; Chock, P.B.; Stadtman, T.C.; Buchanan, S.K. Structural insights into the catalytic mechanism of Escherichia coli selenophosphate synthetase. J. Bacteriol. 2012, 194, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Manta, B.; Makarova, N.E.; Mariotti, M. The selenophosphate synthetase family: A review. Free Radic. Biol. Med. 2022, 192, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Bang, J.; Kang, D.; Jung, J.; Yoo, T.J.; Shim, M.S.; Gladyshev, V.N.; Tsuji, P.A.; Hatfield, D.L.; Kim, J.H.; Lee, B.J. SEPHS1: Its evolution, function and roles in development and diseases. Arch. Biochem. Biophys. 2022, 730, 109426. [Google Scholar] [CrossRef]
- Lobanov, A.V.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteinless animals: Selenophosphate synthetase SPS1 functions in a pathway unrelated to selenocysteine biosynthesis. Protein Sci. 2008, 17, 176–182. [Google Scholar] [CrossRef]
- Itoh, Y.; Sekine, S.; Matsumoto, E.; Akasaka, R.; Takemoto, C.; Shirouzu, M.; Yokoyama, S. Structure of selenophosphate synthetase essential for selenium incorporation into proteins and RNAs. J. Mol. Biol. 2009, 385, 1456–1469. [Google Scholar] [CrossRef]
- Tobe, R.; Mihara, H. Delivery of selenium to selenophosphate synthetase for selenoprotein biosynthesis. Biochim. Biophys. Acta. Gen. Subj. 2018, 1862, 2433–2440. [Google Scholar] [CrossRef]
- Ogasawara, Y.; Lacourciere, G.; Stadtman, T.C. Formation of a selenium-substituted rhodanese by reaction with selenite and glutathione: Possible role of a protein perselenide in a selenium delivery system. Proc. Natl. Acad. Sci. USA 2001, 98, 9494–9498. [Google Scholar] [CrossRef] [PubMed]
- Motsenbocker, M.A.; Tappel, A.L. Selenocysteine-containing proteins from rat and monkey plasma. Biochim. Biophys. Acta 1982, 704, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Burk, R.F.; Hill, K.E. Selenoprotein P-expression, functions, and roles in mammals. Biochim. Biophys. Acta 2009, 1790, 1441–1447. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E. Regulation of selenium metabolism and transport. Annu. Rev. Nutr. 2015, 35, 109–134. [Google Scholar] [CrossRef]
- Shetty, S.; Copeland, P.R. Molecular mechanism of selenoprotein P synthesis. Biochim. Biophys. Acta 2018, 186, 22506–22510. [Google Scholar] [CrossRef]
- Olson, G.E.; Winfrey, V.P.; Hill, K.E.; Burk, R.F. Megalin mediates selenoprotein P uptake by kidney proximal tubule epithelial cells. J. Biol. Chem. 2008, 283, 6854–6860. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E.; Motley, A.K.; Winfrey, V.P.; Kurokawa, S.; Mitchell, S.L.; Zhang, W. Selenoprotein P and apolipoprotein E receptor-2 interact at the blood-brain barrier and also within the brain to maintain an essential selenium pool that protects against neurodegeneration. FASEB J. 2014, 28, 3579–3588. [Google Scholar] [CrossRef]
- Olson, G.E.; Winfrey, V.P.; Nagdas, S.K.; Hill, K.E.; Burk, R.F. Apolipoprotein E receptor-2 (ApoER2) mediates selenium uptake from selenoprotein P by the mouse testis. J. Biol. Chem. 2007, 282, 12290–12297. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Labunsk, V.M.; Hatfield, D.L.; Gladyshev, V.N. Selenoproteins: Molecular pathways and physiological roles. Physiol. Rev. 2014, 94, 739–777. [Google Scholar] [CrossRef]
- Esworthy, R.S.; Doroshow, J.H.; Chu, F.F. The beginning of GPX2 and 30 years later. Free Radic. Biol. Med. 2022, 188, 419–433. [Google Scholar] [CrossRef]
- Dalziel, K.; Dickinson, F.M. The kinetics and mechanism of liver alcohol dehydrogenase with primary and secondary alcohols as substrates. Biochem. J. 1966, 100, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Chaudière, J.; Tappel, A.L. Purification and characterization of selenium-glutathione peroxidase from hamster liver. Arch. Biochem. Biophys. 1983, 226, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Chaudière, J.; Wilhelmsen, E.C.; Tappel, A.L. Mechanism of selenium-glutathione peroxidase and its inhibition by mercaptocarboxylic acids and other mercaptans. J. Biol. Chem. 1984, 259, 1043–1050. [Google Scholar] [CrossRef]
- Hall, M.D.; Marshall, T.S.; Kwit, A.D.; Miller Jenkins, L.M.; Dulcey, A.E.; Madigan, J.P.; Pluchino, K.M.; Goldsborough, A.S.; Brimacombe, K.R.; Griffiths, G.L.; et al. Inhibition of glutathione peroxidase mediates the collateral sensitivity of multidrug-resistant cells to tiopronin. J. Biol. Chem. 2014, 289, 21473–21489. [Google Scholar] [CrossRef] [PubMed]
- Cheff, D.M.; Huang, C.; Scholzen, K.C.; Gencheva, R.; Ronzetti, M.H.; Cheng, Q.; Hall, M.D.; Arnér, E.S.J. The ferroptosis inducing compounds RSL3 and ML162 are not direct inhibitors of GPX4 but of TXNRD1. Redox Biol. 2023, 62, 102703. [Google Scholar] [CrossRef]
- Ho, Y.S.; Magnenat, J.L.; Bronson, R.T.; Cao, J.; Gargano, M.; Sugawara, M.; Funk, C.D. Mice deficient in cellular glutathione peroxidase develop normally and show no increased sensitivity to hyperoxia. J. Biol. Chem. 1997, 272, 16644–16651. [Google Scholar] [CrossRef] [PubMed]
- Klivenyi, P.; Andreassen, O.A.; Ferrante, R.J.; Dedeoglu, A.; Mueller, G.; Lancelot, E.; Bogdanov, M.; Andersen, J.K.; Jiang, D.; Beal, M.F. Mice deficient in cellular glutathione peroxidase show increased vulnerability to malonate, 3-nitropropionic acid, and 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. J. Neurosci. 2000, 20, 1–7. [Google Scholar] [CrossRef]
- Cheng, W.H.; Ho, Y.S.; Valentine, B.A.; Ross, D.A.; Combs, G.F.; Lei, X.G. Cellular glutathione peroxidase is the mediator of body selenium to protect against paraquat lethality in transgenic mice. J. Nutr. 1998, 128, 1070–1076. [Google Scholar] [CrossRef]
- McClung, J.P.; Roneker, C.A.; Mu, W.; Lisk, D.J.; Langlais, P.; Liu, F.; Lei, X.G. Development of insulin resistance and obesity in mice overexpressing cellular glutathione peroxidase. Proc. Natl. Acad. Sci. USA 2004, 101, 8852–8857. [Google Scholar] [CrossRef]
- Wang, X.D.; Vatamaniuk, M.Z.; Wang, S.K.; Roneker, C.A.; Simmons, R.A.; Lei, X.G. Molecular mechanisms for hyperinsulinaemia induced by overproduction of selenium-dependent glutathione peroxidase-1 in mice. Diabetologia 2008, 51, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.Q.; Zhou, J.C.; Wu, Y.Y.; Ren, F.Z.; Lei, X.G. Role of glutathione peroxidase 1 in glucose and lipid metabolism-related diseases. Free Radic. Biol. Med. 2018, 127, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Barquissau, V.; Capel, F.; Dardevet, D.; Feillet-Coudray, C.; Gallinier, A.; Chauvin, M.A.; Rieusset, J.; Morio, B. Reactive oxygen species enhance mitochondrial function, insulin sensitivity and glucose uptake in skeletal muscle of senescence accelerated prone mice SAMP8. Free Radic. Biol. Med. 2017, 113, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Harmon, J.S.; Bogdani, M.; Parazzoli, S.D.; Mak, S.S.; Oseid, E.A.; Berghmans, M.; Leboeuf, R.C.; Robertson, R.P. Beta-cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 2009, 150, 4855–4862. [Google Scholar] [CrossRef] [PubMed]
- Lein, X.G.; Vatamaniuk, M.Z. Two tales of antioxidant enzymes on beta cells and diabetes. Antioxid. Redox Signal. 2011, 14, 489–503. [Google Scholar]
- Zhang, Y.; Han, S.J.; Park, I.; Kim, I.; Chay, K.O.; Kim, S.M.; Jang, D.I.; Lee, T.H.; Lee, S.R. Redox regulation of the tumor suppressor PTEN by hydrogen peroxide and tert-butyl hydroperoxide. Int. J. Mol. Sci. 2017, 18, 982. [Google Scholar] [CrossRef]
- Han, S.J.; Zhang, Y.; Kim, I.; Chay, K.O.; Yoon, H.J.; Jang, D.I.; Yang, S.Y.; Park, J.; Woo, H.A.; Park, I.; et al. Redox regulation of the tumor suppressor PTEN by the thioredoxin system and cumene hydroperoxide. Free Radic. Biol. Med. 2017, 112, 277–286. [Google Scholar] [CrossRef]
- Xirouchaki, C.E.; Jia, Y.; McGrath, M.J.; Greatorex, S.; Tran, M.; Merry, T.L.; Hong, D.; Eramo, M.J.; Broome, S.C.; Woodhead, J.S.T.; et al. Skeletal muscle NOX4 is required for adaptive responses that prevent insulin resistance. Sci Adv. 2021, 7, eabl4988. [Google Scholar] [CrossRef]
- Eberhardt, R.T.; Forgione, M.A.; Cap, A.; Leopold, J.A.; Rudd, M.A.; Trolliet, M.; Heydrick, S.; Stark, R.; Klings, E.S.; Moldovan, N.I.; et al. Endothelial dysfunction in a murine model of mild hyperhomocyst(e)inemia. J. Clin. Investig. 2000, 106, 483–491. [Google Scholar] [CrossRef]
- Forgione, M.A.; Weiss, N.; Heydrick, S.; Cap, A.; Klings, E.S.; Bierl, C.; Eberhardt, R.T.; Farber, H.W.; Loscalzo, J. Cellular glutathione peroxidase deficiency and endothelial dysfunction. Am. J. Physiol. Heart Circ. Physiol. 2002, 282, H1255–H1261. [Google Scholar] [CrossRef]
- Weiss, N.; Zhang, Y.; Heydrick, S.; Bierl, C.; Loscalzo, J. Overexpression of cellular glutathione peroxidase rescues homocyst(e)ine-induced endothelial dysfunction. Proc. Natl. Acad. Sci. USA 2001, 98, 12503–12508. [Google Scholar] [CrossRef]
- Upchurch, G.R., Jr.; Welch, G.N.; Fabian, A.J.; Freedman, J.E.; Johnson, J.L.; Keaney, J.F., Jr.; Loscalzo, J. Homocyst(e)ine decreases bioavailable nitric oxide by a mechanism involving glutathione peroxidase. J. Biol. Chem. 1999, 272, 17012–17017. [Google Scholar] [CrossRef]
- Handy, D.E.; Zhang, Y.; Loscalzo, J. Homocysteine down-regulates cellular glutathione peroxidase (GPx1) by decreasing translation. J. Biol. Chem. 2005, 280, 15518–15525. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.F.; Doroshow, J.H.; Esworthy, R.S. Expression, characterization, and tissue distribution of a new cellular selenium-dependent glutathione peroxidase, GSHPx-GI. J. Biol. Chem. 1993, 268, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- Esworthy, R.S.; Swiderek, K.M.; Ho, Y.S.; Chu, F.F. Selenium-dependent glutathione peroxidase-GI is a major glutathione peroxidase activity in the mucosal epithelium of rodent intestine. Biochim. Biophys. Acta 1998, 1381, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Song, J.; Guan, T.; Wang, S.; Wang, Y.; Meng, Y.; Guo, J.; Li, T.; Ma, C.; Wei, J. Characterization of recombinant human gastrointestinal glutathione peroxidase mutant produced in Escherichia coli. Free Radic. Res. 2015, 49, 228–235. [Google Scholar] [CrossRef]
- Florian, S.; Wingler, K.; Schmehl, K.; Jacobasch, G.; Kreuzer, O.J.; Meyerhof, W.; Brigelius-Flohé, R. Cellular and subcellular localization of gastrointestinal glutathione peroxidase in normal and malignant human intestinal tissue. Free Radic. Res. 2001, 35, 655–663. [Google Scholar] [CrossRef]
- Esworthy, R.S.; Mann, J.R.; Sam, M.; Chu, F.F. Low glutathione peroxidase activity in Gpx1 knockout mice protects jejunum crypts from gamma-irradiation damage. Am. J. Physiol. Gastrointest. Liver Physiol. 2000, 279, G426–G436. [Google Scholar] [CrossRef]
- Florian, S.; Krehl, S.; Loewinger, M.; Kipp, A.; Banning, A.; Esworthy, S.; Chu, F.F.; Brigelius-Flohé, R. Loss of GPx2 increases apoptosis, mitosis, and GPx1 expression in the intestine of mice. Free Radic. Biol. Med. 2010, 49, 1694–1702. [Google Scholar] [CrossRef]
- Pinto, D.; Gregorieff, A.; Begthel, H.; Clevers, H. Canonical Wnt signals are essential for homeostasis of the intestinal epithelium. Genes Dev. 2003, 17, 1709–1713. [Google Scholar] [CrossRef]
- Kipp, A.; Banning, A.; Brigelius-Flohé, R. Activation of the glutathione peroxidase2 (GPx2) promoter by beta-catenin. Biol. Chem. 2007, 388, 1027–1033. [Google Scholar] [CrossRef]
- Kipp, A.P.; Müller, M.F.; Göken, E.M.; Deubel, S.; Brigelius-Flohé, R. The selenoproteins GPx2, TrxR2 and TrxR3 are regulated by Wnt signalling in the intestinal epithelium. Biochim. Biophys. Acta 2012, 1820, 1588–1596. [Google Scholar] [CrossRef]
- Pérez, S.; Taléns-Visconti, R.; Rius-Pérez, S.; Finamor, I.; Sastre, J. Redox signaling in the gastrointestinal tract. Free Radic. Biol. Med. 2017, 104, 75–103. [Google Scholar] [CrossRef]
- Wilson, C.L.; Ouellette, A.J.; Satchell, D.P.; Ayabe, T.; López-Boado, Y.S.; Stratman, J.L.; Hultgren, S.J.; Matrisian, L.M.; Parks, W.C. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science 1999, 286, 113–117. [Google Scholar] [CrossRef]
- Dittrich, A.M.; Meyer, H.A.; Krokowski, M.; Quarcoo, D.; Ahrens, B.; Kube, S.M.; Witzenrath, M.; Esworthy, R.S.; Chu, F.F.; Hamelmann, E. Glutathione peroxidase-2 protects from allergen-induced airway inflammation in mice. Eur. Respir. J. 2010, 35, 1148–1154. [Google Scholar] [CrossRef]
- Cho, H.Y.; Jedlicka, A.E.; Reddy, S.P.; Kensler, T.W.; Yamamoto, M.; Zhang, L.Y.; Kleeberger, S.R. Role of Nrf2 in protection against hyperoxic lung injury in mice. Am. J. Respir. Cell. Mol. Biol. 2002, 26, 175–182. [Google Scholar] [CrossRef]
- Singh, A.; Rangasamy, T.; Thimmulappa, R.K.; Lee, H.; Osburn, W.O.; Brigelius-Flohé, R.; Kensler, T.W.; Yamamoto, M.; Biswal, S. Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am. J. Respir. Cell. Mol. Biol. 2006, 35, 639–650. [Google Scholar] [CrossRef]
- Cho, H.Y.; Miller-DeGraff, L.; Blankenship-Paris, T.; Wang, X.; Bell, D.A.; Lih, F.; Deterding, L.; Panduri, V.; Morgan, D.L.; Yamamoto, M.; et al. Sulforaphane enriched transcriptome of lung mitochondrial energy metabolism and provided pulmonary injury protection via Nrf2 in mice. Toxicol. Appl. Pharmacol. 2019, 364, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Baek, I.J.; Yon, J.M.; Lee, S.R.; Kim, M.R.; Hong, J.T.; Lee, B.J.; Yun, Y.W.; Nam, S.Y. Differential expression of gastrointestinal glutathione peroxidase (GI-GPx) gene during mouse organogenesis. Anat. Histol. Embryol. 2011, 40, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Al-Taie, O.H.; Uceyler, N.; Eubner, U.; Jakob, F.; Mörk, H.; Scheurlen, M.; Brigelius-Flohé, R.; Schöttker, K.; Abel, J.; Thalheimer, A.; et al. Expression profiling and genetic alterations of the selenoproteins GI-GPx and SePP in colorectal carcinogenesis. Nutr. Cancer 2004, 48, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Chiu, S.T.; Hsieh, F.J.; Chen, S.W.; Chen, C.L.; Shu, H.F.; Li, H. Clinicopathologic correlation of up-regulated genes identified using cDNA microarray and real-time reverse transcription-PCR in human colorectal cancer. Cancer Epidemiol. Biomark. Prev. 2005, 14, 437–443. [Google Scholar] [CrossRef]
- Mörk, H.; al-Taie, O.H.; Bähr, K.; Zierer, A.; Beck, C.; Scheurlen, M.; Jakob, F.; Köhrle, J. Inverse mRNA expression of the selenocysteine-containing proteins GI-GPx and SeP in colorectal adenomas compared with adjacent normal mucosa. Nutr. Cancer 2000, 37, 108–116. [Google Scholar] [CrossRef]
- Mörk, H.; Scheurlen, M.; Al-Taie, O.; Zierer, A.; Kraus, M.; Schöttker, K.; Jakob, F.; Köhrle, J. Glutathione peroxidase isoforms as part of the local antioxidative defense system in normal and Barrett’s esophagus. Int. J. Cancer 2003, 105, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Serewko, M.M.; Popa, C.; Dahler, A.L.; Smith, L.; Strutton, G.M.; Coman, W.; Dicker, A.J.; Saunders, N.A. Alterations in gene expression and activity during squamous cell carcinoma development. Cancer Res. 2002, 62, 3759–3765. [Google Scholar] [PubMed]
- Woenckhaus, M.; Klein-Hitpass, L.; Grepmeier, U.; Merk, J.; Pfeifer, M.; Wild, P.; Bettstetter, M.; Wuensch, P.; Blaszyk, H.; Hartmann, A.; et al. Smoking and cancer-related gene expression in bronchial epithelium and non-small-cell lung cancers. J. Pathol. 2006, 210, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Dai, L.; Niu, J. GPX2 silencing relieves epithelial-mesenchymal transition, invasion, and metastasis in pancreatic cancer by downregulating Wnt pathway. J. Cell. Physiol. 2019, 235, 7780–7790. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cao, P.; Alshwmi, M.; Jiang, N.; Xiao, Z.; Jiang, F.; Gu, J.; Wang, X.; Sun, X.; Li, S. GPx2 suppression of H2O2 stress regulates cervical cancer metastasis and apoptosis via activation of the β-catenin-WNT pathway. Onco Targets Ther. 2019, 12, 6639–6651. [Google Scholar] [CrossRef] [PubMed]
- Emmink, B.L.; Laoukili, J.; Kipp, A.P.; Koster, J.; Govaert, K.M.; Fatrai, S.; Verheem, A.; Steller, E.J.; Brigelius-Flohé, R.; Jimenez, C.R.; et al. GPx2 suppression of H2O2 stress links the formation of differentiated tumor mass to metastatic capacity in colorectal cancer. Cancer Res. 2014, 74, 6717–6730. [Google Scholar] [CrossRef]
- Yan, W.; Chen, X. GPX2, a direct target of p63, inhibits oxidative stress-induced apoptosis in a p53-dependent manner. J. Biol. Chem. 2006, 281, 7856–7862. [Google Scholar] [CrossRef]
- Banning, A.; Deubel, S.; Kluth, D.; Zhou, Z.; Brigelius-Flohé, R. The GI-GPx gene is a target for Nrf2. Mol. Cell. Biol. 2005, 25, 4914–4923. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-Nrf2 System: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Müller, M.; Lippmann, D.; Kipp, A.P. The yin and yang of nrf2-regulated selenoproteins in carcinogenesis. Int. J. Cell Biol. 2012, 2012, 486147. [Google Scholar] [CrossRef]
- Menegon, S.; Columbano, A.; Giordano, S. The Dual Roles of Nrf2 in Cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Lu, H.; Bai, Y. Nrf2 in cancers: A double-edged sword. Cancer Med. 2019, 8, 2252–2267. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Zimta, A.A.; Cenariu, D.; Irimie, A.; Magdo, L.; Nabavi, S.M.; Atanasov, A.G.; Berindan-Neagoe, I. The Role of Nrf2 Activity in Cancer Development and Progression. Cancers 2019, 11, 1755. [Google Scholar] [CrossRef] [PubMed]
- Narayanankutty, A.; Job, J.T.; Narayanankutty, V. Glutathione, an Antioxidant Tripeptide: Dual Roles in Carcinogenesis and Chemoprevention. Curr. Protein Pept. Sci. 2019, 20, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Kan, X.F.; Ma, C.; Chen, L.L.; Cheng, T.T.; Zou, Z.W.; Li, Y.; Cao, F.J.; Zhang, W.J.; Yao, J.; et al. GPX2 overexpression indicates poor prognosis in patients with hepatocellular carcinoma. Tumour Biol. 2017, 39, 1010428317700410. [Google Scholar] [CrossRef]
- Weitzel, F.; Wendel, A. Selenoenzymes regulate the activity of leukocyte 5-lipoxygenase via the peroxide tone. J. Biol. Chem. 1993, 268, 6288–6292. [Google Scholar] [CrossRef]
- Bürkert, E.; Arnold, C.; Hammarberg, T.; Rådmark, O.; Steinhilber, D.; Werz, O. The C2-like beta-barrel domain mediates the Ca2+-dependent resistance of 5-lipoxygenase activity against inhibition by glutathione peroxidase-1. J. Biol. Chem. 2003, 278, 42846–42853. [Google Scholar] [CrossRef]
- Imai, H.; Narashima, K.; Arai, M.; Sakamoto, H.; Chiba, N.; Nakagawa, Y. Suppression of leukotriene formation in RBL-2H3 cells that overexpressed phospholipid hydroperoxide glutathione peroxidase. J. Biol. Chem. 1998, 273, 1990–1997. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.H.; Zhuang, M.K.; Xie, W.H.; Du, F.; Huang, Y.H.; Chen, Z.X.; Chen, F.L.; Wang, X.Z. 12-Lipoxygenase promotes epithelial-mesenchymal transition via the Wnt/β-catenin signaling pathway in gastric cancer cells. Onco Targets Ther. 2019, 12, 5551–5561. [Google Scholar] [CrossRef] [PubMed]
- Banning, A.; Florian, S.; Deubel, S.; Thalmann, S.; Müller-Schmehl, K.; Jacobasch, G.; Brigelius-Flohé, R. GPx2 counteracts PGE2 production by dampening COX-2 and mPGES-1 expression in human colon cancer cells. Antioxid Redox Signal. 2008, 10, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Te Velde, A.A.; Pronk, I.; de Kort, F.; Stokkers, P.C. Glutathione peroxidase 2 and aquaporin 8 as new markers for colonic inflammation in experimental colitis and inflammatory bowel diseases: An important role for H2O2? Eur. J Gastroenterol. Hepatol. 2008, 20, 555–560. [Google Scholar] [CrossRef]
- Esworthy, R.S.; Aranda, R.; Martín, M.G.; Doroshow, J.H.; Binder, S.W.; Chu, F.F. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G848–G855. [Google Scholar] [CrossRef]
- Chu, F.F.; Esworthy, R.S.; Chu, P.G.; Longmate, J.A.; Huycke, M.M.; Wilczynski, S.; Doroshow, J.H. Bacteria-induced intestinal cancer in mice with disrupted Gpx1 and Gpx2 genes. Cancer Res. 2004, 64, 962–968. [Google Scholar] [CrossRef]
- Esworthy, R.S.; Kim, B.W.; Chow, J.; Shen, B.; Doroshow, J.H.; Chu, F.F. Nox1 causes ileocolitis in mice deficient in glutathione peroxidase-1 and -2. Free Radic. Biol. Med. 2014, 68, 315–325. [Google Scholar] [CrossRef]
- Esworthy, R.S.; Yang, L.; Frankel, P.H.; Chu, F.F. Epithelium-specific glutathione peroxidase, Gpx2, is involved in the prevention of intestinal inflammation in selenium-deficient mice. J. Nutr. 2005, 135, 740–745. [Google Scholar] [CrossRef]
- Choi, J.; Ou, J.H. Mechanisms of liver injury. III. Oxidative stress in the pathogenesis of hepatitis C virus. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G847–G851. [Google Scholar] [CrossRef]
- Choi, J. Oxidative stress, endogenous antioxidants, alcohol, and hepatitis C: Pathogenic interactions and therapeutic considerations. Free Radic. Biol. Med. 2012, 52, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, R.; Ploen, D.; Hildt, E. HCV and Oxidative Stress: Implications for HCV Life Cycle and HCV-Associated Pathogenesis. Oxid. Med. Cell. Longev. 2016, 2016, 9012580. [Google Scholar] [CrossRef]
- Morbitzer, M.; Herget, T. Expression of gastrointestinal glutathione peroxidase is inversely correlated to the presence of hepatitis C virus subgenomic RNA in human liver cells. J. Biol. Chem. 2005, 280, 8831–8841. [Google Scholar] [CrossRef]
- MacCallum, P.R.; Jack, S.C.; Egan, P.A.; McDermott, B.T.; Elliott, R.M.; Chan, S.W. Cap-dependent and hepatitis C virus internal ribosome entry site-mediated translation are modulated by phosphorylation of eIF2alpha under oxidative stress. J. Gen. Virol. 2006, 87 Pt 11, 3251–3262. [Google Scholar] [CrossRef]
- Jack, S.C.; Chan, S.W. The role of PERK and GCN2 in basal and hydrogen peroxide-regulated translation from the hepatitis C virus internal ribosome entry site. Virus Genes 2011, 43, 208–214. [Google Scholar] [CrossRef]
- Nguyen, B.N.; Okuno, Y.; Ajiro, M.; Iida, K.; Denawa, M.; Yamamoto, M.; Sakamoto, N.; Kagechika, H.; Hagiwara, M. Retinoid derivative Tp80 exhibits anti-hepatitis C virus activity through restoration of GI-GPx expression. J. Med. Virol. 2017, 89, 1224–1234. [Google Scholar] [CrossRef]
- Whitin, J.C.; Bhamre, S.; Tham, D.M.; Cohen, H.J. Extracellular glutathione peroxidase is secreted basolaterally by human renal proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2002, 283, F20–F28. [Google Scholar] [CrossRef]
- Avissar, N.; Kerl, E.A.; Baker, S.S.; Cohen, H.J. Extracellular glutathione peroxidase mRNA and protein in human cell lines. Arch. Biochem. Biophys. 1994, 309, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Björnstedt, M.; Xue, J.; Huang, W.; Akesson, B.; Holmgren, A. The thioredoxin and glutaredoxin systems are efficient electron donors to human plasma glutathione peroxidase. J. Biol. Chem. 1994, 269, 29382–29384. [Google Scholar] [CrossRef]
- Freedman, J.E.; Loscalzo, J.; Benoit, S.E.; Valeri, C.R.; Barnard, M.R.; Michelson, A.D. Decreased platelet inhibition by nitric oxide in two brothers with a history of arterial thrombosis. J. Clin. Investig. 1996, 97, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Kenet, G.; Freedman, J.; Shenkman, B.; Regina, E.; Brok-Simoni, F.; Holzman, F.; Vavva, F.; Brand, N.; Michelson, A.; Trolliet, M.; et al. Plasma glutathione peroxidase deficiency and platelet insensitivity to nitric oxide in children with familial stroke. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2017–2023. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.C.; Mahoney, C.E.; Anderson, L.; Ottaviano, F.; Croce, K.; Leopold, J.A.; Zhang, Y.Y.; Tang, S.S.; Handy, D.E.; Loscalzo, J. Glutathione peroxidase-3 deficiency promotes platelet-dependent thrombosis in vivo. Circulation 2011, 123, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Barrett, C.W.; Ning, W.; Chen, X.; Smith, J.J.; Washington, M.K.; Hill, K.E.; Coburn, L.A.; Peek, R.M.; Chaturvedi, R.; Wilson, K.T.; et al. Tumor suppressor function of the plasma glutathione peroxidase GPx3 in colitis-associated carcinoma. Cancer Res. 2013, 73, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhu, J.; Liu, Y.; Wang, Z.; Cao, X.; Gu, Y. Tumor-polarized GPX3+ AT2 lung epithelial cells promote premetastatic niche formation. Proc. Natl. Acad. Sci. USA 2022, 119, e2201899119. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Valente, M.; Ferri, L.; Gregolin, C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochim. Biophys. Acta 1982, 710, 197–211. [Google Scholar] [CrossRef]
- Ursini, F.; Bosello Travain, V.; Cozza, G.; Miotto, G.; Roveri, A.; Toppo, S.; Maiorino, M. A white paper on phospholipid Hydroperoxide Glutathione Peroxidase (GPx4) forty years later. Free Radic. Biol. Med. 2022, 188, 117–133. [Google Scholar] [CrossRef]
- Maiorino, M.; Chu, F.F.; Ursini, F.; Davies, K.J.; Doroshow, J.H.; Esworthy, R.S. Phospholipid hydroperoxide glutathione peroxidase is the 18-kDa selenoprotein expressed in human tumor cell lines. J. Biol. Chem. 1991, 266, 7728–7732. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Aumann, K.D.; Blöcker, H.; Gross, G.; Kiess, M.; Klöppel, K.D.; Maiorino, M.; Roveri, A.; Schuckelt, R.; Usani, F.; et al. Phospholipid-hydroperoxide glutathione peroxidase. Genomic DNA, cDNA, and deduced amino acid sequence. J. Biol. Chem. 1994, 269, 7342–7348. [Google Scholar] [CrossRef]
- Pushpa-Rekha, T.R.; Burdsall, A.L.; Oleksa, L.M.; Chisolm, G.M.; Driscoll, D.M. Rat phospholipid-hydroperoxide glutathione peroxidase. cDNA cloning and identification of multiple transcription and translation start sites. J. Biol. Chem. 1995, 270, 26993–26999. [Google Scholar] [CrossRef]
- Thomas, J.P.; Geiger, P.G.; Maiorino, M.; Ursini, F.; Girotti, A.W. Enzymatic reduction of phospholipid and cholesterol hydroperoxides in artificial bilayers and lipoproteins. Biochim. Biophys. Acta 1990, 1045, 252–260. [Google Scholar] [CrossRef]
- Cozza, G.; Rossetto, M.; Bosello-Travain, V.; Maiorino, M.; Roveri, A.; Toppo, S.; Zaccarin, M.; Zennaro, L.; Ursini, F. Glutathione peroxidase 4-catalyzed reduction of lipid hydroperoxides in membranes: The polar head of membrane phospholipids binds the enzyme and addresses the fatty acid hydroperoxide group toward the redox center. Free Radic. Biol. Med. 2017, 112, 1–11. [Google Scholar] [CrossRef]
- Stolwijk, J.M.; Stefely, J.A.; Veling, M.T.; van‘t Erve, T.J.; Wagner, B.A.; Raife, T.J.; Buettner, G.R. Red blood cells contain enzymatically active GPx4 whose abundance anticorrelates with hemolysis during blood bank storage. Redox Biol. 2021, 46, 102073. [Google Scholar] [CrossRef]
- Pfeifer, H.; Conrad, M.; Roethlein, D.; Kyriakopoulos, A.; Brielmeier, M.; Bornkamm, G.W.; Behne, D. Identification of a specific sperm nuclei selenoenzyme necessary for protamine thiol cross-linking during sperm maturation. FASEB J. 2001, 15, 1236–1238. [Google Scholar] [CrossRef]
- Maiorino, M.; Roveri, A.; Benazzi, L.; Bosello, V.; Mauri, P.; Toppo, S.; Tosatto, S.C.; Ursini, F. Functional interaction of phospholipid hydroperoxide glutathione peroxidase with sperm mitochondrion-associated cysteine-rich protein discloses the adjacent cysteine motif as a new substrate of the selenoperoxidase. J. Biol. Chem. 2005, 280, 38395–38402. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Förster, H.; Boersma, A.; Seiler, A.; Wehnes, H.; Sinowatz, F.; Neumüller, C.; Deutsch, M.J.; Walch, A.; Hrabé de Angelis, M.; et al. Mitochondrial glutathione peroxidase 4 disruption causes male infertility. FASEB J. 2009, 23, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Brütsch, S.H.; Rademacher, M.; Roth, S.R.; Müller, K.; Eder, S.; Viertel, D.; Franz, C.; Kuhn, H.; Borchert, A. Male subfertility induced by heterozygous expression of catalytically inactive glutathione peroxidase 4 is rescued in vivo by systemic inactivation of the Alox15 gene. J. Biol. Chem. 2016, 291, 23578–23588. [Google Scholar] [CrossRef]
- Brütsch, S.H.; Wang, C.C.; Li, L.; Stender, H.; Neziroglu, N.; Richter, C.; Kuhn, H.; Borchert, A. Expression of inactive glutathione peroxidase 4 leads to embryonic lethality, and inactivation of the Alox15 gene does not rescue such knock-in mice. Antioxid. Redox Signal. 2015, 22, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Borchert, A.; Wang, C.C.; Ufer, C.; Schiebel, H.; Savaskan, N.E.; Kuhn, H. The role of phospholipid hydroperoxide glutathione peroxidase isoforms in murine embryogenesis. J. Biol. Chem. 2006, 281, 19655–19664. [Google Scholar] [CrossRef]
- Ufer, C.; Wang, C.C. The Roles of Glutathione Peroxidases during Embryo Development. Front. Mol. Neurosci. 2011, 4, 1–13. [Google Scholar] [CrossRef]
- Ingold, I.; Berndt, C.; Schmitt, S.; Doll, S.; Poschmann, G.; Buday, K.; Roveri, A.; Peng, X.; Porto Freitas, F.; Seibt, T.; et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 2018, 172, 409–422. [Google Scholar] [CrossRef]
- Wenk, J.; Schüller, J.; Hinrichs, C.; Syrovets, T.; Azoitei, N.; Podda, M.; Wlaschek, M.; Brenneisen, P.; Schneider, L.A.; Sabiwalsky, A.; et al. Overexpression of phospholipid-hydroperoxide glutathione peroxidase in human dermal fibroblasts abrogates UVA irradiation-induced expression of interstitial collagenase/matrix metalloproteinase-1 by suppression of phosphatidylcholine hydroperoxide-mediated NFkappaB activation and interleukin-6 release. J. Biol. Chem. 2004, 279, 45634–45642. [Google Scholar] [PubMed]
- Wu, J.; Sun, L.; Chen, X.; Du, F.; Shi, H.; Chen, C.; Chen, Z.J. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013, 339, 826–830. [Google Scholar] [CrossRef]
- Jia, M.; Qin, D.; Zhao, C.; Chai, L.; Yu, Z.; Wang, W.; Tong, L.; Lv, L.; Wang, Y.; Rehwinkel, J.; et al. Redox homeostasis maintained by GPX4 facilitates STING activation. Nat. Immunol. 2020, 21, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Hirschhorn, T.; Stockwell, B.R. The development of the concept of ferroptosis. Free Radic. Biol. Med. 2019, 133, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of ferroptosis and relations with regulated cell death: A review. Front. Physiol. 2019, 10, 139. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef]
- Weaver, K.; Skouta, R. The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities. Biomedicines 2022, 10, 891. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Deng, X.; Zhang, W.; Xie, X.; Conrad, M.; Liu, Y.; Angeli, J.P.F.; Lai, L. Novel allosteric activators for ferroptosis regulator glutathione peroxidase 4. J. Med. Chem. 2019, 62, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell. Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn DEOrman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; Stockwell, B.R. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Madungwe, N.B.; Imam Aliagan, A.D.; Tombo, N.; Bopassa, J.C. Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels. Biochem. Biophys. Res. Commun. 2019, 520, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Miotto, G.; Rossetto, M.; Di Paolo, M.L.; Orian, L.; Venerando, R.; Roveri, A.; Vučković, A.M.; Bosello Travain, V.; Zaccarin, M.; Zennaro, L.; et al. Insight into the mechanism of ferroptosis inhibition by ferrostatin-1. Redox Biol. 2020, 28, 101328. [Google Scholar] [CrossRef]
- Gao, M.; Jiang, X. To eat or not to eat—The metabolic flavor of ferroptosis. Curr. Opin. Cell Biol. 2018, 51, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohé, R.; Flohé, L. Regulatory phenomena in the glutathione peroxidase superfamily. Antioxid. Redox Signal. 2020, 33, 498–516. [Google Scholar] [CrossRef] [PubMed]
- Randolph, J.T.; O’Connor, M.J.; Han, F.; Hutchins, C.W.; Siu, Y.A.; Cho, M.; Zheng, Y.; Hickson, J.A.; Markley, J.L.; Manaves, V.; et al. Discovery of a Potent Chloroacetamide GPX4 Inhibitor with Bioavailability to Enable Target Engagement in Mice, a Potential Tool Compound for Inducing Ferroptosis In Vivo. J. Med. Chem. 2023, 66, 3852–3865. [Google Scholar] [CrossRef]
- Moosmayer, D.; Hilpmann, A.; Hoffmann, J.; Schnirch, L.; Zimmermann, K.; Badock, V.; Furst, L.; Eaton, J.K.; Viswanathan, V.S.; Schreiber, S.L.; et al. Crystal structures of the selenoprotein glutathione peroxidase 4 in its apo form and in complex with the covalently bound inhibitor ML162. Acta Crystallogr. D Struct. Biol. 2021, 77, 237–248. [Google Scholar] [CrossRef]
- Eaton, J.K.; Furst, L.; Cai, L.L.; Viswanathan, V.S.; Schreiber, S.L. Structure-activity relationships of GPx4 inhibitor warheads. Bioorg. Med. Chem. Lett. 2020, 30, 127538. [Google Scholar] [CrossRef]
- Vučković, A.M.; Bosello Travain, V.; Bordin, L.; Cozza, G.; Miotto, G.; Rossetto, M.; Toppo, S.; Venerando, R.; Zaccarin, M.; Maiorino, M.; et al. Inactivation of the glutathione peroxidase GPx4 by the ferroptosis-inducing molecule RSL3 requires the adaptor protein 14-3-3ε. FEBS Lett. 2020, 594, 611–624. [Google Scholar] [CrossRef]
- Xi, J.; Tian, L.L.; Xi, J.; Girimpuhwe, D.; Huang, C.; Ma, R.; Yao, X.; Shi, D.; Bai, Z.; Wu, Q.X.; et al. Alterperylenol as a novel thioredoxin reductase inhibitor induces liver cancer cell apoptosis and ferroptosis. J. Agric. Food Chem. 2022, 70, 15763–15775. [Google Scholar] [CrossRef]
- Carlisle, A.E.; Lee, N.; Matthew-Onabanjo, A.N.; Spears, M.E.; Park, S.J.; Youkana, D.; Doshi, M.B.; Peppers, A.; Li, R.; Joseph, A.B.; et al. Selenium detoxification is required for cancer-cell survival. Nat. Metab. 2020, 2, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, V.S.; Ryan, M.J.; Dhruv, H.D.; Gill, S.; Eichhoff, O.M.; Seashore-Ludlow, B.; Kaffenberger, S.D.; Eaton, J.K.; Shimada, K.; Aguirre, A.J.; et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017, 547, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Schreiber, S.L.; Stockwell, B.R. Targeting Dependency on the GPX4 Lipid Peroxide Repair Pathway for Cancer Therapy. Biochemistry 2018, 57, 2059–2060. [Google Scholar] [CrossRef]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Mathow, D.; Chessa, F.; Rabionet, M.; Kaden, S.; Jennemann, R.; Sandhoff, R.; Gröne, H.J.; Feuerborn, A. Zeb1 affects epithelial cell adhesion by diverting glycosphingolipid metabolism. EMBO Rep. 2015, 16, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Palte, M.J.; Deik, A.A.; Li, H.; Eaton, J.K.; Wang, W.; Tseng, Y.Y.; Deasy, R.; Kost-Alimova, M.; Dančík, V.; et al. A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat. Commun. 2019, 10, 1617. [Google Scholar] [CrossRef]
- Toppo, S.; Flohé, L.; Ursini, F.; Vanin, S.; Maiorino, M. Catalytic mechanisms and specificities of glutathione peroxidases: Variations of a basic scheme. Biochim. Biophys. Acta 2009, 1790, 1486–1500. [Google Scholar] [CrossRef]
- Passaia, G.; Margis-Pinheiro, M. Glutathione peroxidases as redox sensor proteins in plant cells. Plant Sci. 2015, 234, 22–26. [Google Scholar] [CrossRef]
- Maiorino, M.; Bosello-Travain, V.; Cozza, G.; Miotto, G.; Roveri, A.; Toppo, S.; Zaccarin, M.; Ursini, F. Understanding mammalian glutathione peroxidase 7 in the light of its homologs. Free Radic. Biol. Med. 2015, 83, 352–360. [Google Scholar] [CrossRef]
- Bosello-Travain, V.; Conrad, M.; Cozza, G.; Negro, A.; Quartesan, S.; Rossetto, M.; Roveri, A.; Toppo, S.; Ursini, F.; Zaccarin, M.; et al. Protein disulfide isomerase and glutathione are alternative substrates in the one Cys catalytic cycle of glutathione peroxidase 7. Biochim. Biophys. Acta 2013, 1830, 3846–3857. [Google Scholar] [CrossRef]
- Chaudière, J.; Courtin, O.; Leclaire, J. Glutathione oxidase activity of selenocystamine: A mechanistic study. Arch. Biochem. Biophys. 1992, 296, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Cadenas, E.; Graf, P.; Sies, H. A novel biologically active seleno-organic compound—I. Glutathione peroxidase-like activity in vitro and antioxidant capacity of PZ 51 (Ebselen). Biochem. Pharmacol. 1984, 33, 3235–3239. [Google Scholar] [CrossRef] [PubMed]
- Wendel, A.; Fausel, M.; Safayhi, H.; Tiegs, G.; Otter, R. A novel biologically active seleno-organic compound—II. Activity of PZ 51 in relation to glutathione peroxidase. Biochem. Pharmacol. 1984, 33, 3241–3245. [Google Scholar] [CrossRef]
- Chaudière, J.; Yadan, J.C.; Erdelmeier, I.; Tailhan-Lomont, C.; Moutet, M. Design of new selenium-containing mimics of glutathione peroxidase. In Oxidative Stress and Antioxidants; Raven Press, Ltd.: New York, NY, USA, 1994; pp. 165–184. [Google Scholar]
- d’Alessio, P.; Moutet, M.; Coudrier, E.; Darquenne, S.; Chaudière, J. ICAM-1 and VCAM-1 expression induced by TNF-alpha are inhibited by a glutathione peroxidase mimic. Free Radic. Biol. Med. 1998, 24, 979–987. [Google Scholar] [CrossRef]
- Moutet, M.; d’Alessio, P.; Malette, P.; Devaux, V.; Chaudière, J. Glutathione peroxidase mimics prevent TNF-alpha- and neutrophil-induced endothelial alterations. Free Radic. Biol. Med. 1998, 25, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Chaudière, J.; Erdelmeier, I.; Moutet, M.; Yadan, J.C. One- versus two-electron transfers: Cytotoxic and cytoprotective effects of seleno-organic catalysts. In Phosphorus, Sulfur, and Silicon and the Related Elements; Taylor & Francis: Oxfordshire, UK, 1998; Volume 136, pp. 467–470. [Google Scholar]
- Payne, N.C.; Barber, D.R.; Ruggles, E.L.; Hondal, R.J. Can dimedone be used to study selenoproteins? An investigation into the reactivity of dimedone toward oxidized forms of selenocysteine. Protein Sci. 2018, 28, 41–55. [Google Scholar] [CrossRef]
- Liu, J.; Rozovsky, S. Contribution of selenocysteine to the peroxidase activity of selenoprotein S. Biochemistry 2013, 52, 5514–5516. [Google Scholar] [CrossRef]
- Masuda, R.; Kimura, R.; Karasaki, T.; Sase, S.; Goto, K. Modeling the catalytic cycle of glutathione peroxidase by nuclear magnetic resonance spectroscopic analysis of selenocysteine selenenic acids. J. Am. Chem. Soc. 2021, 143, 6345–6350. [Google Scholar] [CrossRef]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef]
- Dagnell, M.; Schmidt, E.E.; Arnér, E.S.J. The A to Z of modulated cell patterning by mammalian thioredoxin reductases. Free Radic. Biol. Med. 2018, 115, 484–496. [Google Scholar] [CrossRef]
- Cebula, M.; Schmidt, E.E.; Arnér, E.S. TrxR1 as a potent regulator of the Nrf2-Keap1 response system. Antioxid. Redox Signal. 2015, 23, 823–853. [Google Scholar] [CrossRef]
- Lee, S.; Kim, S.M.; Lee, R.T. Thioredoxin and thioredoxin target proteins: From molecular mechanisms to functional significance. Antioxid. Redox Signal. 2013, 18, 1165–1207. [Google Scholar] [CrossRef]
- Lillig, C.H.; Holmgren, A. Thioredoxin and related molecules—From biology to health and disease. Antioxid. Redox Signal. 2007, 9, 25–47. [Google Scholar] [CrossRef]
- Collet, J.F.; Messens, J. Structure, function, and mechanism of thioredoxin proteins. Antioxid. Redox Signal. 2010, 13, 1205–1216. [Google Scholar] [CrossRef]
- Laurent, T.C.; Moore, E.C.; Reichard, P. Enzymatic synthesis of deoxyribonucleotides. IV: Isolation and characterization of thioredoxin, the hydrogen donor from Escherichia coli B. J. Biol. Chem. 1964, 239, 3436–3444. [Google Scholar] [CrossRef]
- Holmgren, A.; Sengupta, R. The use of thiols by ribonucleotide reductase. Free Radic. Biol. Med. 2010, 49, 1617–1628. [Google Scholar] [CrossRef]
- Nakao, L.S.; Everley, R.A.; Marino, S.M.; Lo, S.M.; de Souza, L.E.; Gygi, S.P.; Gladyshev, V.N. Mechanism-based proteomic screening identifies targets of thioredoxin-like proteins. J. Biol. Chem. 2015, 290, 5685–5695. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhang, H.; Lu, J.; Holmgren, A. Glutathione and glutaredoxin act as a backup of human thioredoxin reductase 1 to reduce thioredoxin 1 preventing cell death by aurothioglucose. J. Biol. Chem. 2012, 287, 38210–38219. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Du, Y.; Zhang, X.; Lu, J.; Holmgren, A. Glutaredoxin 2 reduces both thioredoxin 2 and thioredoxin 1 and protects cells from apoptosis induced by auranofin and 4-hydroxynonenal. Antioxid. Redox Signal. 2014, 21, 669–681. [Google Scholar] [CrossRef] [PubMed]
- López-Grueso, M.J.; González-Ojeda, R.; Requejo-Aguilar, R.; McDonagh, B.; Fuentes-Almagro, C.A.; Muntané, J.; Bárcena, J.A.; Padilla, C.A. Thioredoxin and glutaredoxin regulate metabolism through different multiplex thiol switches. Redox Biol. 2019, 21, 101049. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Mandal, P.K.; Kaminsk, V.O.; Lindqvist, A.; Conrad, M.; Arnér, E.S. Sec-containing TrxR1 is essential for self-sufficiency of cells by control of glucose-derived H2O2. Cell Death Dis. 2014, 5, e1235. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Arnér, E.S.; Holmgren, A. Structure and mechanism of mammalian thioredoxin reductase: The active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc. Natl. Acad. Sci. USA 2000, 97, 5854–5859. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Sandalova, T.; Lindqvist, Y.; Arnér, E.S. Crystal structure and catalysis of the selenoprotein thioredoxin reductase 1. J. Biol. Chem. 2009, 284, 3998–4008. [Google Scholar] [CrossRef] [PubMed]
- Lacey, B.M.; Eckenroth, B.E.; Flemer, S.; Hondal, R.J. Selenium in thioredoxin reductase: A mechanistic perspective. Biochemistry 2008, 47, 12810–12821. [Google Scholar] [CrossRef]
- Hondal, R.J. Using chemical approaches to study selenoproteins-focus on thioredoxin reductases. Biochim. Biophys. Acta 2009, 1790, 1501–1512. [Google Scholar] [CrossRef]
- Arnér, E.S. Focus on mammalian thioredoxin reductases--important selenoproteins with versatile functions. Biochim. Biophys. Acta 2009, 1790, 495–526. [Google Scholar] [CrossRef]
- Zhong, L.; Holmgren, A. Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J. Biol. Chem. 2000, 275, 18121–18128. [Google Scholar] [CrossRef]
- Barber, D.R.; Hondal, R.J. Gain of function conferred by selenocysteine: Catalytic enhancement of one-electron transfer reactions by thioredoxin reductase. Protein Sci. 2019, 28, 79–89. [Google Scholar] [CrossRef]
- Xu, J.; Cheng, Q.; Arnér, E.S. Details in the catalytic mechanism of mammalian thioredoxin reductase 1 revealed using point mutations and juglone-coupled enzyme activities. Free Radic. Biol. Med. 2016, 94, 110–120. [Google Scholar] [CrossRef]
- Turanov, A.A.; Hatfield, D.L.; Gladyshev, V.N. Characterization of protein targets of mammalian thioredoxin reductases. Methods Enzymol. 2010, 474, 245–254. [Google Scholar]
- Lu, J.; Berndt, C.; Holmgren, A. Metabolism of selenium compounds catalyzed by the mammalian selenoprotein thioredoxin reductase. Biochim. Biophys. Acta 2009, 1790, 1513–1519. [Google Scholar] [CrossRef]
- Lee, B.C.; Dikiy, A.; Kim, H.Y.; Gladyshev, V.N. Functions and evolution of selenoprotein methionine sulfoxide reductases. Biochim. Biophys. Acta 2009, 1790, 1471–1477. [Google Scholar] [CrossRef]
- Kim, H.Y.; Gladyshev, V.N. Different catalytic mechanisms in mammalian selenocysteine- and cysteine-containing methionine-R-sulfoxide reductases. PLoS Biol. 2005, 3, e375. [Google Scholar] [CrossRef]
- Lourenço Dos Santos, S.; Petropoulos, I.; Friguet, B. The oxidized protein repair enzymes methionine sulfoxide reductases and their roles in protecting against oxidative stress, in ageing and in regulating protein function. Antioxidants 2018, 7, 191. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Woo, H.A.; Kang, D. The role of peroxiredoxins in the transduction of H2O2 signals. Antioxid. Redox Signal. 2018, 28, 537–557. [Google Scholar] [CrossRef]
- Rhee, S.G.; Woo, H.A. Multiple functions of peroxiredoxins: Peroxidases, sensors and regulators of the intracellular messenger H2O2, and protein chaperones. Antioxid. Redox Signal. 2011, 15, 781–794. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.S.; Kang, S.W.; Woo, H.A.; Hwang, S.C.; Chae, H.Z.; Kim, K.; Rhee, S.G. Inactivation of human peroxiredoxin I during catalysis as the result of the oxidation of the catalytic site cysteine to cysteine-sulfinic acid. J. Biol. Chem. 2002, 277, 38029–38036. [Google Scholar] [CrossRef]
- Biteau, B.; Labarre, J.; Toledano, M.B. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature 2003, 425, 980–984. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.H.; Lee, K.O.; Chi, Y.H.; Jung, B.G.; Park, S.K.; Park, J.H.; Lee, J.R.; Lee, S.S.; Moon, J.C.; Yun, J.W.; et al. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell 2004, 117, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Weissbach, H.; Etienne, F.; Hoshi, T.; Heinemann, S.H.; Lowther, W.T.; Matthews, B.; St John, G.; Nathan, C.; Brot, N. Peptide methionine sulfoxide reductase: Structure, mechanism of action, and biological function. Arch. Biochem. Biophys. 2002, 397, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Kaya, A.; Lee, B.C.; Gladyshev, V.N. Regulation of protein function by reversible methionine oxidation and the role of selenoprotein MsrB1. Antioxid. Redox Signal. 2015, 23, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Giménez-Cassina, A.; Petrus, P.; Conrad, M.; Rydén, M.; Arnér, E.S. Thioredoxin reductase 1 suppresses adipocyte differentiation and insulin responsiveness. Sci. Rep. 2016, 6, 28080. [Google Scholar] [CrossRef]
- Dagnell, M.; Pace, P.E.; Cheng, Q.; Frijhoff, J.; Östman, A.; Arnér, E.S.J.; Hampton, M.B.; Winterbourn, C.C. Thioredoxin reductase 1 and NADPH directly protect protein tyrosine phosphatase 1B from inactivation during H2O2 exposure. J. Biol. Chem. 2017, 292, 14371–14380. [Google Scholar] [CrossRef]
- Cenas, N.; Nivinskas, H.; Anusevicius, Z.; Sarlauskas, J.; Lederer, F.; Arner, E.S. Interactions of quinones with thioredoxin reductase: A challenge to the antioxidant role of the mammalian selenoprotein. J. Biol. Chem. 2004, 279, 2583–2592. [Google Scholar] [CrossRef]
- Anestal, K.; Prast-Nielsen, S.; Cenas, N.; Arner, E.S. Cell death by SecTRAPs: Thioredoxin reductase as a prooxidant killer of cells. PLoS ONE 2008, 3, e1846. [Google Scholar] [CrossRef]
- Hansen, J.M.; Watson, W.H.; Jones, D.P. Compartmentation of Nrf-2 redox control: Regulation of cytoplasmic activation by glutathione and DNA binding by thioredoxin-1. Toxicol. Sci. 2004, 82, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Murata, M.; Sachi, Y.; Nakamura, H.; Takeuchi, J.; Mori, K.; Yodoi, J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J. Biol. Chem. 1999, 274, 27891–27897. [Google Scholar] [CrossRef] [PubMed]
- Ueno, M.; Masutani, H.; Arai, R.J.; Yamauchi, A.; Hirota, K.; Sakai, T.; Inamoto, T.; Yamaoka, Y.; Yodoi, J.; Nikaido, T. Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J. Biol. Chem. 1999, 274, 35809–35815. [Google Scholar] [CrossRef]
- Hirota, K.; Matsui, M.; Iwata, S.; Nishiyama, A.; Mori, K.; Yodoi, J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc. Natl. Acad. Sci. USA 1997, 94, 3633–3638. [Google Scholar] [CrossRef]
- Go, Y.M.; Jones, D.P. Redox control systems in the nucleus: Mechanisms and functions. Antioxid. Redox Signal. 2010, 13, 489–509. [Google Scholar] [CrossRef]
- Grippo, J.F.; Holmgren, A.; Pratt, W.B. Proof that the endogenous, heat-stable glucocorticoid receptor-activating factor is thioredoxin. J. Biol. Chem. 1985, 260, 93–97. [Google Scholar] [CrossRef]
- Meuillet, E.J.; Mahadevan, D.; Berggren, M.; Coon, A.; Powis, G. Thioredoxin-1 binds to the C2 domain of PTEN inhibiting PTEN’s lipid phosphatase activity and membrane binding: A mechanism for the functional loss of PTEN’s tumor suppressor activity. Arch. Biochem. Biophys. 2004, 429, 123–133. [Google Scholar] [CrossRef]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef]
- Gencheva, R.; Arnér, E.S.J. Thioredoxin Reductase Inhibition for Cancer Therapy. Annu. Rev. Pharmacol. Toxicol. 2022, 62, 177–196. [Google Scholar] [CrossRef]
- Eriksson, S.E.; Prast-Nielsen, S.; Flaberg, E.; Szekely, L.; Arnér, E.S. High levels of thioredoxin reductase 1 modulate drug-specific cytotoxic efficacy. Free Radic. Biol. Med. 2009, 47, 1661–1671. [Google Scholar] [CrossRef]
- Watson, W.H.; Heilman, J.M.; Hughes, L.L.; Spielberger, J.C. Thioredoxin reductase-1 knock down does not result in thioredoxin-1 oxidation. Biochem. Biophys. Res. Commun. 2008, 368, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, X.; Han, X.; Liu, R.; Fang, J. Targeting the thioredoxin system for cancer therapy. Trends Pharmacol. Sci. 2017, 38, 794–808. [Google Scholar] [CrossRef] [PubMed]
- Bian, M.; Fan, R.; Zhao, S.; Liu, W. Targeting the thioredoxin system as a strategy for cancer therapy. J. Med. Chem. 2019, 62, 7309–7321. [Google Scholar] [CrossRef]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef]
- Yoshihara, E.; Chen, Z.; Matsuo, Y.; Masutani, H.; Yodoi, J. Thiol redox transitions by thioredoxin and thioredoxin binding protein-2 in cell signaling. Methods Enzymol. 2010, 474, 67–82. [Google Scholar] [PubMed]
- Kuiper, G.G.; Kester, M.H.; Peeters, R.P.; Visser, T.J. Biochemical mechanisms of thyroid hormone deiodination. Thyroid 2005, 15, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Callebaut, I.; Curcio-Morelli, C.; Mornon, J.P.; Gereben, B.; Buettner, C.; Huang, S.; Castro, B.; Fonseca, T.L.; Harney, J.W.; Larsen, P.R.; et al. The iodothyronine selenodeiodinases are thioredoxin-fold family proteins containing a glycoside hydrolase clan GH-A-like structure. J. Biol. Chem. 2003, 278, 36887–36896. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, U.; Schlicker, C.; Braun, D.; Köhrle, J.; Steegborn, C. Crystal structure of mammalian selenocysteine-dependent iodothyronine deiodinase suggests a peroxiredoxin-like catalytic mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, 10526–10531. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, U.; Steegborn, C. New insights into the structure and mechanism of iodothyronine deiodinases. J. Mol. Endocrinol. 2015, 55, R37–R52. [Google Scholar] [CrossRef]
- Bianco, A.C.; da Conceição, R.R. The Deiodinase Trio and Thyroid Hormone Signaling. Methods Mol. Biol. 2018, 1801, 67–83. [Google Scholar]
- Köhrle, J.; Frädrich, C. Deiodinases control local cellular and systemic thyroid hormone availability. Free Radic. Biol. Med. 2022, 193 Pt 1, 59–79. [Google Scholar] [CrossRef]
- Cicatiello, A.G.; Di Girolamo, D.; Dentice, M. Metabolic Effects of the Intracellular Regulation of Thyroid Hormone: Old Players, New Concepts. Front. Endocrinol. 2018, 9, 474. [Google Scholar] [CrossRef]
- Zavacki, A.M.; Arrojo EDrigo, R.; Freitas, B.C.; Chung, M.; Harney, J.W.; Egri, P.; Wittmann, G.; Fekete, C.; Gereben, B.; Bianco, A.C. The E3 ubiquitin ligase TEB4 mediates degradation of type 2 iodothyronine deiodinase. Mol. Cell. Biol. 2009, 29, 5339–5347. [Google Scholar] [CrossRef]
- Sase, S.; Kakimoto, R.; Kimura, R.; Goto, K. Synthesis of a stable primary-alkyl-substituted selenenyl iodide and its hydrolytic conversion to the corresponding selenenic acid. Molecules 2015, 20, 21415–21420. [Google Scholar] [CrossRef]
- Steegborn, C.; Schweizer, U. Structure and Mechanism of Iodothyronine Deiodinases—What we know, what we don’t know, and what would be nice to know. Exp. Clin. Endocrinol. Diabetes 2019, 128, 375–378. [Google Scholar] [CrossRef]
- Di Jeso, B.; Arvan, P. Thyroglobulin from molecular and cellular biology to clinical endocrinology. Endocr. Rev. 2016, 37, 2–36. [Google Scholar] [CrossRef]
- Ruf, J.; Carayon, P. Structural and functional aspects of thyroid peroxidase. Arch. Biochem. Biophys. 2006, 445, 269–277. [Google Scholar] [CrossRef]
- Dunn, J.T.; Dunn, A.D. Update on intrathyroidal iodine metabolism. Thyroid 2001, 11, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Dumitrescu, A.M. Inherited defects in thyroid hormone cell-membrane transport and metabolism. Best Pract. Res. Clin. Endocrinol. Metab. 2014, 28, 189–201. [Google Scholar] [CrossRef]
- Fugazzola, L.; Muzza, M.; Weber, G.; Beck-Peccoz, P.; Persani, L. DUOXS defects: Genotype-phenotype correlations. Ann. Endocrinol. 2011, 72, 82–86. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Kumar, R.A.; Koc, A.; Sun, Z.; Gladyshev, V.N. Selenoprotein R is a zinc-containing stereo-specific methionine sulfoxide reductase. Proc. Natl. Acad. Sci. USA 2002, 99, 4245–4250. [Google Scholar] [CrossRef] [PubMed]
- Aachmann, F.L.; Sal, L.S.; Kim, H.Y.; Marino, S.M.; Gladyshev, V.N.; Dikiy, A. Insights into function, catalytic mechanism, and fold evolution of selenoprotein methionine sulfoxide reductase B1 through structural analysis. J. Biol. Chem. 2010, 285, 33315–33323. [Google Scholar] [CrossRef]
- Kim, H.Y. The methionine sulfoxide reduction system: Selenium utilization and methionine sulfoxide reductase enzymes and their functions. Antioxid. Redox Signal. 2013, 19, 958–969. [Google Scholar] [CrossRef]
- Tarrago, L.; Kaya, A.; Kim, H.Y.; Manta, B.; Lee, B.C.; Gladyshev, V.N. The selenoprotein methionine sulfoxide reductase B1 (MSRB1). Free Radic. Biol. Med. 2022, 191, 228–240. [Google Scholar] [CrossRef]
- Fomenko, D.E.; Novoselov, S.V.; Natarajan, S.K.; Lee, B.C.; Koc, A.; Carlson, B.A.; Lee, T.H.; Kim, H.Y.; Hatfield, D.L.; Gladyshev, V.N. MsrB1 (methionine-R-sulfoxide reductase 1) knock-out mice: Roles of MsrB1 in redox regulation and identification of a novel selenoprotein form. J. Biol. Chem. 2009, 284, 5986–5993. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Yesilyurt, H.G.; Yoon, J.; Terman, J.R. The MICALs are a Family of F-actin Dismantling oxidoreductases conserved from drosophila to humans. Sci. Rep. 2018, 8, 937. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Péterfi, Z.; Hoffmann, F.W.; Moore, R.E.; Kaya, A.; Avanesov, A.; Tarrago, L.; Zhou, Y.; Weerapana, E.; Fomenko, D.E.; et al. MsrB1 and MICALs regulate actin assembly and macrophage function via reversible stereoselective methionine oxidation. Mol. Cell. 2013, 51, 397–404. [Google Scholar] [CrossRef]
- Lee, B.C.; Lee, S.G.; Choo, M.K.; Kim, J.H.; Lee, H.M.; Kim, S.; Fomenko, D.E.; Kim, H.Y.; Park, J.M.; Gladyshev, V.N. Selenoprotein MsrB1 promotes anti-inflammatory cytokine gene expression in macrophages and controls immune response in vivo. Sci. Rep. 2017, 7, 5119. [Google Scholar] [CrossRef]
- Konstantinidis, K.; Bezzerides, V.J.; Lai, L.; Isbell, H.M.; Wei, A.C.; Wu, Y.; Viswanathan, M.C.; Blum, I.D.; Granger, J.M.; Heims-Waldron, D.; et al. MICAL1 constrains cardiac stress responses and protects against disease by oxidizing CaMKII. J. Clin. Investig. 2020, 130, 4663–4678. [Google Scholar] [CrossRef]
- Lai, L.; Sun, J.; Tarafdar, S.; Liu, C.; Murphy, E.; Kim, G.; Levine, R.L. Loss of methionine sulfoxide reductases increases resistance to oxidative stress. Free Radic. Biol. Med. 2019, 145, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Dudkiewicz, M.; Szczepińska, T.; Grynberg, M.; Pawłowski, K. A novel protein kinase-like domain in a selenoprotein, widespread in the tree of life. PLoS ONE 2012, 7, e32138. [Google Scholar] [CrossRef]
- Sreelatha, A.; Yee, S.S.; Lopez, V.A.; Park, B.C.; Kinch, L.N.; Pilch, S.; Servage, K.A.; Zhang, J.; Jiou, J.; Karasiewicz-Urbańska, M.; et al. Protein AMPylation by an evolutionarily conserved pseudokinase. Cell 2018, 175, 809–821.e19. [Google Scholar] [CrossRef]
- Mieyal, J.J.; Gallogly, M.M.; Qanungo, S.; Sabens, E.A.; Shelton, M.D. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 2008, 10, 1941–1988. [Google Scholar] [CrossRef]
- Labunskyy, V.M.; Ferguson, A.D.; Fomenko, D.E.; Chelliah, Y.; Hatfield, D.L.; Gladyshev, V.N. A novel cysteine-rich domain of Sep15 mediates the interaction with UDP-glucose:glycoprotein glucosyltransferase. J. Biol. Chem. 2005, 280, 37839–37845. [Google Scholar] [CrossRef]
- Ferguson, A.D.; Labunskyy, V.M.; Fomenko, D.E.; Araç, D.; Chelliah, Y.; Amezcua, C.A.; Rizo, J.; Gladyshev, V.N.; Deisenhofer, J. NMR structures of the selenoproteins Sep15 and SelM reveal redox activity of a new thioredoxin-like family. J. Biol. Chem. 2006, 281, 3536–3543. [Google Scholar] [CrossRef] [PubMed]
- Yim, S.H.; Everley, R.A.; Schildberg, F.A.; Lee, S.G.; Orsi, A.; Barbati, Z.R.; Karatepe, K.; Fomenko, D.E.; Tsuji, P.A.; Luo, H.R.; et al. Role of selenoprotein F as a gatekeeper of secreted disulfide-rich glycoproteins. Cell Rep. 2018, 23, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Kasaikina, M.V.; Fomenko, D.E.; Labunskyy, V.M.; Lachke, S.A.; Qiu, W.; Moncaster, J.A.; Zhang, J.; Wojnarowicz MWJr Natarajan, S.K.; Malinouski, M.; Schweizer, U.; et al. Roles of the 15-kDa selenoprotein (Sep15) in redox homeostasis and cataract development revealed by the analysis of Sep 15 knockout mice. J. Biol. Chem. 2011, 286, 33203–33212. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Ren, B.; Li, X.; Yan, H.; Xie, Q.; Liu, H.; Zhou, J.; Tian, J.; Huang, K. Selenoprotein F knockout leads to glucose and lipid metabolism disorders in mice. J. Biol. Inorg. Chem. 2020, 25, 1009–1022. [Google Scholar] [CrossRef] [PubMed]
- Petit, N.; Lescure, A.; Rederstorff, M.; Krol, A.; Moghadaszadeh, B.; Wewer, U.M.; Guicheney, P. Selenoprotein N: An endoplasmic reticulum glycoprotein with an early developmental expression pattern. Hum. Mol. Genet. 2003, 12, 1045–1053. [Google Scholar] [CrossRef]
- Li, Y.; Camacho, P. Ca2+-dependent redox modulation of SERCA 2b by ERp57. J. Cell Biol. 2004, 164, 35–46. [Google Scholar] [CrossRef]
- Chernorudskiy, A.; Varone, E.; Colombo, S.F.; Fumagalli, S.; Cagnotto, A.; Cattaneo, A.; Briens, M.; Baltzinger, M.; Kuhn, L.; Bachi, A.; et al. Selenoprotein N is an endoplasmic reticulum calcium sensor that links luminal calcium levels to a redox activity. Proc. Natl. Acad. Sci. USA 2020, 117, 21288–21298. [Google Scholar] [CrossRef]
- Horibata, Y.; Hirabayashi, Y. Identification and characterization of human ethanolaminephosphotransferase1. J. Lipid Res. 2007, 48, 503–508. [Google Scholar] [CrossRef]
- Horibata, Y.; Elpeleg, O.; Eran, A.; Hirabayashi, Y.; Savitzki, D.; Tal, G.; Mandel, H.; Sugimoto, H. EPT1 (selenoprotein I) is critical for the neural development and maintenance of plasmalogen in humans. J. Lipid Res. 2018, 59, 1015–1026. [Google Scholar] [CrossRef]
- Nunes, L.G.A.; Pitts, M.W.; Hoffmann, P.R. Selenoprotein I (selenoi) as a critical enzyme in the central nervous system. Arch. Biochem. Biophys. 2022, 729, 109376. [Google Scholar] [CrossRef]
- Ma, C.; Hoffmann, F.W.; Marciel, M.P.; Page, K.E.; Williams-Aduja, M.A.; Akana, E.N.L.; Gojanovich, G.S.; Gerschenson, M.; Urschitz, J.; Moisyadi, S.; et al. Upregulated ethanolamine phospholipid synthesis via selenoprotein I is required for effective metabolic reprogramming during T cell activation. Mol. Metab. 2021, 47, 101170. [Google Scholar] [CrossRef]
- Fredericks, G.J.; Hoffmann, P.R. Selenoprotein K and protein palmitoylation. Antioxid. Redox Signal. 2015, 23, 854–862. [Google Scholar] [CrossRef]
- Fredericks, G.J.; Hoffmann, F.W.; Rose, A.H.; Osterheld, H.J.; Hess, F.M.; Mercier, F.; Hoffmann, P.R. Stable expression and function of the inositol 1,4,5-triphosphate receptor requires palmitoylation by a DHHC6/selenoprotein K complex. Proc. Natl. Acad. Sci. USA 2014, 111, 16478–16483. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.Z.; Xu, X.W.; Zhang, Z.H.; Chen, C.; Chen, Y.B.; Huang, S.L.; Liu, Q.; Hoffmann, P.R.; Song, G.L. Selenoprotein K deficiency-induced apoptosis: A role for calpain and the ERS pathway. Redox Biol. 2021, 47, 102154. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, X.; Liu, Q.; Wang, Y.; Li, S.; Xu, S. Selenoprotein K protects skeletal muscle from damage and is required for satellite cells-mediated myogenic differentiation. Redox Biol. 2022, 50, 102255. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, W.C.; Alkan, Z. Delayed cell cycle progression from S.E.PW1 depletion is p53- and p21-dependent in MCF-7 breast cancer cells. Biochem. Biophys. Res. Commun. 2011, 413, 36–40. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, W.C.; Printsev, I.; Alkan, Z. Selenoprotein W depletion induces a p53- and p21-dependent delay in cell cycle progression in RWPE-1 prostate epithelial cells. J. Cell. Biochem. 2012, 113, 61–69. [Google Scholar] [CrossRef]
- Hawkes, W.C.; Alkan, Z. Delayed cell cycle progression in selenoprotein W depleted cells is regulated by a Mitogen-Activated Protein Kinase Kinase 4 (MKK4)-p38/c-Jun NH2-terminal kinase (JNK)-p53 pathway. J. Biol. Chem. 2012, 287, 27371–27379. [Google Scholar] [CrossRef]
- Alkan, Z.; Duong, F.L.; Hawkes, W.C. Selenoprotein W controls epidermal growth factor receptor surface expression, activation and degradation via receptor ubiquitination. Biochim. Biophys. Acta 2015, 1853, 1087–1095. [Google Scholar] [CrossRef]
- Jeon, Y.H.; Ko, K.Y.; Lee, J.H.; Park, K.J.; Jang, J.K.; Kim, I.Y. Identification of a redox-modulatory interaction between selenoprotein W and 14-3-3 protein. Biochim. Biophys. Acta 2016, 1863, 10–18. [Google Scholar] [CrossRef]
- Anouar, Y.; Lihrmann, I.; Falluel-Morel, A.; Boukhzar, L. Selenoprotein T is a key player in ER proteostasis, endocrine homeostasis and neuroprotection. Free Radic. Biol. Med. 2018, 127, 145–152. [Google Scholar] [CrossRef]
- Shao, Z.Q.; Zhang, X.; Fan, H.H.; Wang, X.S.; Wu, H.M.; Zhang, L.; Cheng, W.H.; Zhu, J.H. Selenoprotein T Promotes Proliferation and G1-to-S Transition in SK-N-SH Cells: Implications in Parkinson’s Disease. J. Nutr. 2019, 149, 2110–2119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, R.P.; Cheng, W.H.; Zhu, J.H. Prioritized brain selenium retention and selenoprotein expression: Nutritional insights into Parkinson’s disease. Mech. Ageing Dev. 2019, 180, 89–96. [Google Scholar] [CrossRef]
- Liu, J.; Li, F.; Rozovsky, S. The intrinsically disordered membrane protein selenoprotein S is a reductase in vitro. Biochemistry 2013, 52, 3051–3061. [Google Scholar] [CrossRef]
- Novoselov, S.V.; Kryukov, G.V.; Xu, X.M.; Carlson, B.A.; Hatfield, D.L.; Gladyshev, V.N. Selenoprotein H is a nucleolar thioredoxin-like protein with a unique expression pattern. J. Biol. Chem. 2007, 282, 11960–11968. [Google Scholar] [CrossRef]
- Wu, R.T.; Cao, L.; Chen, B.P.; Cheng, W.H. Selenoprotein H suppresses cellular senescence through genome maintenance and redox regulation. J. Biol. Chem. 2014, 289, 34378–34388. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, L.; Zhu, J.H.; Cheng, W.H. Nuclear selenoproteins and genome maintenance. IUBMB Life 2016, 68, 5–12. [Google Scholar] [CrossRef]
- Stadtman, T. Selenium biochemistry. Annu. Rev. Biochem. 1990, 59, 111–127. [Google Scholar] [CrossRef]
- Stadtman, T.C. A gold mine of fascinating enzymes: Those remarkable, strictly anaerobic bacteria, Methanococcus vannielii and Clostridium sticklandii. J. Biol. Chem. 2002, 277, 49091–49100. [Google Scholar] [CrossRef]
- Cone, J.E.; Del Rio, R.M.; Davis, J.N.; Stadtman, T.C. Chemical characterization of the selenoprotein component of clostridial glycine reductase: Identification of selenocysteine as the organoselenium moiety. Proc. Natl. Acad. Sci. USA 1976, 73, 2659–2663. [Google Scholar] [CrossRef] [PubMed]
- Arkowitz, R.A.; Abeles, R.H. Identification of acetyl phosphate as the product of clostridial glycine reductase: Evidence for an acyl enzyme intermediate. Biochemistry 1989, 28, 4639–4644. [Google Scholar] [CrossRef] [PubMed]
- Andreesen, J.R. Glycine reductase mechanism. Curr. Opin. Chem. Biol. 2004, 8, 454–461. [Google Scholar] [CrossRef]
- Arkowitz, R.A.; Abeles, R.H. Mechanism of action of clostridial glycine reductase: Isolation and characterization of a covalent acetyl enzyme intermediate. Biochemistry 1991, 30, 4090–4097. [Google Scholar] [CrossRef]
- Kabisch, U.C.; Gräntzdörffer, A.; Schierhorn, A.; Rücknagel, K.P.; Andreesen, J.R.; Pich, A. Identification of D-proline reductase from Clostridium sticklandii as a selenoenzyme and indications for a catalytically active pyruvoyl group derived from a cysteine residue by cleavage of a proprotein. J. Biol. Chem. 1999, 274, 8445–8454. [Google Scholar] [CrossRef] [PubMed]
- Arkowitz, R.A.; Dhe-Paganon, S.; Abeles, R.H. The fate of the carboxyl oxygens during D-proline reduction by clostridial proline reductase. Arch. Biochem. Biophys. 1994, 311, 457–459. [Google Scholar] [CrossRef]
- Bednarski, B.; Andreesen, J.R.; Pich, A. In vitro processing of the proproteins GrdE of protein B of glycine reductase and PrdA of D-proline reductase from Clostridium sticklandii: Formation of a pyruvoyl group from a cysteine residue. Eur. J. Biochem. 2001, 268, 3538–3544. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.B.; Stadtman, T.C. Selenium-dependent and selenium-independent formate dehydrogenases of Methanococcus vannielii. Separation of the two forms and characterization of the purified selenium-independent form. J. Biol. Chem. 1981, 256, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Axley, M.J.; Böck, A.; Stadtman, T.C. Catalytic properties of an Escherichia coli formate dehydrogenase mutant in which sulfur replaces selenium. Proc. Natl. Acad. Sci. USA 1991, 88, 8450–8454. [Google Scholar] [CrossRef]
- Boyington, J.C.; Gladyshev, V.N.; Khangulov, S.V.; Stadtman, T.C.; Sun, P.D. Crystal structure of formate dehydrogenase H: Catalysis involving Mo, molybdopterin, selenocysteine, and a Fe4S4 cluster. Science 1997, 275, 1305–1308. [Google Scholar] [CrossRef]
- Khangulov, S.V.; Gladyshev, V.N.; Dismukes, G.C.; Stadtman, T.C. Selenium-containing formate dehydrogenase H from Escherichia coli: A molybdopterin enzyme that catalyzes formate oxidation without oxygen transfer. Biochemistry 1998, 37, 3518–3528. [Google Scholar] [CrossRef]
- Raaijmakers, H.C.; Romão, M.J. Formate-reduced E. coli formate dehydrogenase H: The reinterpretation of the crystal structure suggests a new reaction mechanism. J. Biol. Inorg. Chem. 2006, 11, 849–854. [Google Scholar] [CrossRef]
- Leopoldini, M.; Chiodo, S.G.; Toscano, M.; Russo, N. Reaction mechanism of molybdoenzyme formate dehydrogenase. Chemistry 2008, 14, 8674–8681. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Ryde, U. Reaction mechanism of formate dehydrogenase studied by computational methods. J. Biol. Inorg. Chem. 2018, 23, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Baltazar, C.S.A.; Marques, M.C.; Soares, C.M.; DeLacey, A.M.; Pereira, I.A.C.; Matias, P.M. Nickel-iron-selenium hydrogenases—An Overview. Eur. J. Inorg. Chem. 2011, 2011, 948–962. [Google Scholar] [CrossRef]
- Wombwell, C.; Caputo, C.A.; Reisner, E. [NiFeSe]-hydrogenase chemistry. Acc. Chem. Res. 2015, 48, 2858–2865. [Google Scholar] [CrossRef]
- Mersch, D.; Lee, C.Y.; Zhang, J.Z.; Brinkert, K.; Fontecilla-Camps, J.C.; Rutherford, A.W.; Reisner, E. Wiring of photosystem II to hydrogenase for photoelectrochemical water splitting. J. Am. Chem. Soc. 2015, 137, 8541–8549. [Google Scholar] [CrossRef]
- Nam, D.H.; Zhang, J.Z.; Andrei, V.; Kornienko, N.; Heidary, N.; Wagner, A.; Nakanishi, K.; Sokol, K.P.; Slater, B.; Zebger, I.; et al. Solar water splitting with a hydrogenase integrated in photoelectrochemical tandem cells. Angew. Chem. Int. Ed. Engl. 2018, 57, 10595–10599. [Google Scholar] [CrossRef]
- Parkin, A.; Goldet, G.; Cavazza, C.; Fontecilla-Camps, J.C.; Armstrong, F.A. The difference a Se makes? Oxygen-tolerant hydrogen production by the [NiFeSe]-hydrogenase from Desulfomicrobium baculatum. J. Am. Chem. Soc. 2008, 130, 13410–13416. [Google Scholar] [CrossRef]
- Marques, M.C.; Tapia, C.; Gutiérrez-Sanz, O.; Ramos, A.R.; Keller, K.L.; Wall, J.D.; De Lacey, A.L.; Matias, P.M.; Pereira, I.A.C. The direct role of selenocysteine in [NiFeSe] hydrogenase maturation and catalysis. Nat. Chem. Biol. 2017, 13, 544–550. [Google Scholar] [CrossRef]
- Yang, X.; Elrod, L.C.; Le, T.; Vega, V.S.; Naumann, H.; Rezenom, Y.; Reibenspies, J.H.; Hall, M.B.; Darensbourg, M.Y. Controlling O2 reactivity in synthetic analogues of [NiFeS]- and [NiFeSe]-hydrogenase active sites. J. Am. Chem. Soc. 2019, 141, 15338–15347. [Google Scholar] [CrossRef]
- Hawkes, W.C.; Alkan, Z. Regulation of redox signaling by selenoproteins. Biol. Trace Elem. Res. 2010, 134, 235–251. [Google Scholar] [CrossRef]
- Flohé, L. The impact of thiol peroxidases on redox regulation. Free Radic. Res. 2016, 50, 126–142. [Google Scholar] [CrossRef]
- Chaudière, J.; Gérard, D.; Clément, M.; Bourre, J.M. Induction of selenium-glutathione peroxidase by stimulation of metabolic hydrogen peroxide production in vivo. Bioelectrochem. Bioenerg. 1987, 18, 247–256. [Google Scholar] [CrossRef]
- Touat-Hamici, Z.; Legrain, Y.; Bulteau, A.L.; Chavatte, L. Selective up-regulation of human selenoproteins in response to oxidative stress. J. Biol. Chem. 2014, 289, 14750–14761. [Google Scholar] [CrossRef] [PubMed]
- Endres, L.; Begley, U.; Clark, R.; Gu, C.; Dziergowska, A.; Małkiewicz, A.; Melendez, J.A.; Dedon, P.C.; Begley, T.J. Alkbh8 regulates selenocysteine-protein expression to protect against reactive oxygen species damage. PLoS ONE 2015, 10, e0131335. [Google Scholar] [CrossRef]
- Schwarz, M.; Löser, A.; Cheng, Q.; Wichmann-Costaganna, M.; Schädel, P.; Werz, O.; Arnér, E.S.; Kipp, A.P. Side-by-side comparison of recombinant human glutathione peroxidases identifies overlapping substrate specificities for soluble hydroperoxides. Redox Biol. 2023, 59, 102593. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.F.; Esworthy, R.S. The expression of an intestinal form of glutathione peroxidase (GSHPx-GI) in rat intestinal epithelium. Arch. Biochem. Biophys. 1995, 323, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Marinho, H.S.; Real, C.; Cyrne, L.; Soares, H.; Antunes, F. Hydrogen peroxide sensing, signaling and regulation of transcription factors. Redox Biol. 2014, 2, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef]
- Espinosa-Diez, C.; Miguel, V.; Mennerich, D.; Kietzmann, T.; Sánchez-Pérez, P.; Cadenas, S.; Lamas, S. Antioxidant responses and cellular adjustments to oxidative stress. Redox Biol. 2015, 6, 183–197. [Google Scholar] [CrossRef]
- Moldogazieva, N.T.; Mokhosoev, I.M.; Feldman, N.B.; Lutsenko, S.V. ROS and RNS signalling: Adaptive redox switches through oxidative/nitrosative protein modifications. Free Radic. Res. 2018, 52, 507–543. [Google Scholar] [CrossRef]
- Oliveira-Marques, V.; Marinho, H.S.; Cyrne, L.; Antunes, F. Role of hydrogen peroxide in NF-kappaB activation: From inducer to modulator. Antioxid. Redox Signal. 2009, 11, 2223–2243. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, S.; Kitamura, M. Bidirectional regulation of NF-κB by reactive oxygen species: A role of unfolded protein response. Free Radic. Biol. Med. 2013, 65, 162–174. [Google Scholar] [CrossRef]
- Kulmacz, R.J. Regulation of cyclooxygenase catalysis by hydroperoxides. Biochem. Biophys. Res. Commun. 2005, 338, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Netto, L.E.S.; Machado, L.E.S.F. Preferential redox regulation of cysteine-based protein tyrosine phosphatases: Structural and biochemical diversity. FEBS J. 2022, 289, 5480–5504. [Google Scholar] [CrossRef]
- Dagnell, M.; Cheng, Q.; Rizvi, S.H.M.; Pace, P.E.; Boivin, B.; Winterbourn, C.C.; Arnér, E.S.J. Bicarbonate is essential for protein-tyrosine phosphatase 1B (PTP1B) oxidation and cellular signaling through EGF-triggered phosphorylation cascades. J. Biol. Chem. 2019, 294, 12330–12338. [Google Scholar] [CrossRef] [PubMed]
- Kretz-Remy, C.; Mehlen, P.; Mirault, M.E.; Arrigo, A.P. Inhibition of I kappa B-alpha phosphorylation and degradation and subsequent NF-kappa B activation by glutathione peroxidase overexpression. J. Cell Biol. 1996, 133, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Agbor, T.A.; Demma, Z.; Mrsny, R.J.; Castillo, A.; Boll, E.J.; McCormick, B.A. The oxido-reductase enzyme glutathione peroxidase 4 (GPX4) governs Salmonella typhimurium-induced neutrophil transepithelial migration. Cell. Microbiol. 2014, 16, 1339–1353. [Google Scholar] [CrossRef]
- Cao, C.; Leng, Y.; Huang, W.; Liu, X.; Kufe, D. Glutathione peroxidase 1 is regulated by the c-Abl and Arg tyrosine kinases. J. Biol. Chem. 2003, 278, 39609–39614. [Google Scholar] [CrossRef] [PubMed]
- Arjunan, P.; Lin, X.; Tang, Z.; Du, Y.; Kumar, A.; Liu, L.; Yin, X.; Huang, L.; Chen, W.; Chen, Q.; et al. VEGF-B is a potent antioxidant. Proc. Natl. Acad. Sci. USA 2018, 115, 10351–10356. [Google Scholar] [CrossRef]
- Chaudière, J. Selenium-Glutathione Peroxidase: Properties of the Active Site and Distribution in Mammals. Ph.D. Thesis, University of California, Davis, CA, USA, 1983. Available online: https://hdl.handle.net/2027/uc1.x15939 (accessed on 7 June 2023).
- Wessjohann, L.A.; Schneider, A.; Abbas, M.; Brandt, W. Selenium in chemistry and biochemistry in comparison to sulfur. Biol. Chem. 2007, 388, 997–1006. [Google Scholar] [CrossRef]
- Arnér, E.S. Selenoproteins-What unique properties can arise with selenocysteine in place of cysteine? Exp. Cell Res. 2010, 316, 1296–1303. [Google Scholar] [CrossRef]
- Reich, H.J.; Hondal, R.J. Why nature chose selenium. ACS Chem. Biol. 2016, 11, 821–841. [Google Scholar] [CrossRef]
- Maroney, M.J.; Hondal, R.J. Selenium versus sulfur: Reversibility of chemical reactions and resistance to permanent oxidation in proteins and nucleic acids. Free Radic. Biol. Med. 2018, 127, 228–237. [Google Scholar] [CrossRef]
- Dalla Tiezza, M.; Bickelhaupt, F.M.; Flohé, L.; Maiorino, M.; Ursini, F.; Orian, L. A dual attack on the peroxide bond. The common principle of peroxidatic cysteine or selenocysteine residues. Redox Biol. 2020, 34, 101540. [Google Scholar] [CrossRef] [PubMed]
- Flohé, L.; Toppo, S.; Orian, L. The glutathione peroxidase family: Discoveries and mechanism. Free Radic. Biol. Med. 2022, 187, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Rocher, C.; Lalanne, J.L.; Chaudière, J. Purification and properties of a recombinant sulfur analog of murine selenium-glutathione peroxidase. Eur. J. Biochem. 1992, 205, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Toscano, M.D.; Woycechowsky, K.J.; Hilvert, D. Minimalist active-site redesign: Teaching old enzymes new tricks. Angew. Chem. Int. Ed. Engl. 2007, 46, 3212–3236. [Google Scholar] [CrossRef] [PubMed]
- Metanis, N.; Hilvert, D. Natural and synthetic selenoproteins. Curr. Opin. Chem. Biol. 2014, 22, 27–34. [Google Scholar] [CrossRef]
- Bell, I.M.; Fisher, M.L.; Wu, Z.P.; Hilvert, D. Kinetic studies on the peroxidase activity of selenosubtilisin. Biochemistry 1993, 32, 3754–3762, Erratum in Biochemistry 1993, 32, 8980. [Google Scholar] [CrossRef]
- Wang, T.; Li, J.; Xu, J.; Fan, X.; Zhao l Qiao, S.; Pan, T.; Liu, J. Rational redesign of the active site of selenosubtilisin with strongly enhanced glutathione peroxidase activity. J. Catal. 2018, 359, 27–35. [Google Scholar] [CrossRef]
- Yu, H.J.; Liu, J.Q.; Bock, A.; Li, J.; Luo, G.M.; Shen, J.C. Engineering glutathione transferase to a novel glutathione peroxidase mimic with high catalytic efficiency: Incorporation of selenocysteine into a glutathione-binding scaffold using an auxotrophic expression system. J. Biol. Chem. 2005, 280, 11930–11935. [Google Scholar] [CrossRef]
- Casi, G.; Roelfes, G.; Hilvert, D. Selenoglutaredoxin as a glutathione peroxidase mimic. Chembiochem 2008, 9, 1623–1631. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Qi, Z.; Wang, Y.; Liu, X.; Li, J.; Xu, J.; Liu, J.; Shen, J. Engineered selenium-containing glutaredoxin displays strong glutathione peroxidase activity rivaling natural enzyme. Int. J. Biochem. Cell Biol. 2009, 41, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Boschi-Muller, S.; Muller, S.; Van Dorsselaer, A.; Böck, A.; Branlant, G. Substituting selenocysteine for active site cysteine of phosphorylating glyceraldehyde 3-phosphate dehydrogenase reveals a peroxidase activity. FEBS Lett. 1998, 439, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Huber, R.E.; Criddle, R.S. Comparison of the chemical properties of selenocysteine and selenocystine with their sulfur analogs. Arch. Biochem. Biophys. 1967, 122, 164–173. [Google Scholar] [CrossRef]
- Rhee, S.G.; Cho, C.S. Blot-based detection of dehydroalanine-containing glutathione peroxidase with the use of biotin-conjugated cysteamine. Methods Enzymol. 2010, 474, 23–34. [Google Scholar] [CrossRef]
- Dimastrogiovanni, D.; Anselmi, M.; Miele, A.E.; Boumis, G.; Petersson, L.; Angelucci, F.; Nola, A.D.; Brunori, M.; Bellelli, A. Combining crystallography and molecular dynamics: The case of Schistosoma mansoni phospholipid glutathione peroxidase. Proteins Struct. Funct. Genet. 2010, 78, 259–270. [Google Scholar] [CrossRef]
- Orian, L.; Mauri, P.; Roveri, A.; Toppo, S.; Benazzi, L.; Bosello-Travain, V.; De Palma, A.; Maiorino, M.; Miotto, G.; Zaccarin, M.; et al. Selenocysteine oxidation in glutathione peroxidase catalysis: An MS-supported quantum mechanics study. Free Radic. Biol. Med. 2015, 87, 1–14. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaudière, J. Biological and Catalytic Properties of Selenoproteins. Int. J. Mol. Sci. 2023, 24, 10109. https://doi.org/10.3390/ijms241210109
Chaudière J. Biological and Catalytic Properties of Selenoproteins. International Journal of Molecular Sciences. 2023; 24(12):10109. https://doi.org/10.3390/ijms241210109
Chicago/Turabian StyleChaudière, Jean. 2023. "Biological and Catalytic Properties of Selenoproteins" International Journal of Molecular Sciences 24, no. 12: 10109. https://doi.org/10.3390/ijms241210109
APA StyleChaudière, J. (2023). Biological and Catalytic Properties of Selenoproteins. International Journal of Molecular Sciences, 24(12), 10109. https://doi.org/10.3390/ijms241210109