Abstract
The takin lungworm Varestrongylus eleguneniensis (Strongylida: Protostrongylidae) causes lethal bronchopneumonia and represents severe threats to captive and wild populations. However, until now there has been very limited information available concerning the molecular epidemiology and evolutionary biology of V. eleguneniensis. Mitochondrial genomes (mtDNAs) can provide resources for investigations in these areas and, therefore, can assist with the surveillance and control of this lungworm. Herein, the complete mtDNA of V. eleguneniensis was sequenced and characterized with Illumina pipeline analyses. This circular genome (13,625 bp) encoded twelve protein-coding genes (PCGs), two rRNAs, and twenty-two tRNAs, with notable levels of AT and GC skews. Comparative genomics revealed a purifying selection among PCGs, with cox1 and nad6 having the lowest and the highest evolutionary rate, respectively. Genome-wide phylogenies showed a close relationship between V. eleguneniensis and Protostrongylus rufescens in Strongylida. Single gene (PCGs or rRNAs)-based phylogenies indicated that cox1 and nad5 genes shared the same family-level topology with that inferred from genomic datasets, suggesting that both genes could be suitable genetic markers for evolutionary and phylogenetic studies of Strongylida species. This was the first mtDNA of any member of the genus Varestrongylus, and its comprehensive molecular characterization represents a new resource for systematic, population genetic and evolutionary biological studies of Varestrongylus lungworms in wildlife.
1. Introduction
The takin (Budorcas taxicolor) is considered endangered by the International Union for Conservation of Nature Criteria (IUCN) and endemic to high elevation regions ranging from the Eastern Himalayas to South-Central China [1]. The nematode Varestrongylus eleguneniensis (Strongylida: Protostrongylidae) is one of the commonest endoparasites found in the lung of the takin and represents a significant threat to captive and wild populations [2]. Like other lungworms, V. eleguneniensis adults parasitize in the alveoli and bronchus of the takin, where they mate and reproduce first-stage larvae (L1)-containing eggs. Then, the L1 larvae hatch, travel up the bronchus to the oral cavity, enter the intestinal tract and are finally shed into feces. The terrestrial snails ingest L1s and become their intermediate hosts, where the L1s further develop into infective third-stage larvae (L3s). Takins, as the definitive host, infected V. eleguneniensis by digestion of L3-containing snails when grazing [3]. Clinically, V. eleguneniensis infection causes pulmonary complications including lung collapse and bronchopneumonia, and in severe cases host death, especially when co-infections with dictyocaulidosis [4].
Current diagnosis of V. eleguneniensis infections typically relies on morphological examination [5,6,7], and key morphological features of V. eleguneniensis adults include the fine cuticle markings with one blunt and rounded tip and four papillae around the buccal foramen. However, the morphology is often unrecognized, even by experienced parasitologists, and mistaken identification, particularly of the eggs and larvae, is not uncommon [8]. Additionally, the diagnosis becomes more challenging when identification and differentiation of eggs or larvae are carried out within an environment cross-contaminated by other lungworm eggs and larvae, including morphologically similar Dictyocaulus spp. [9,10]. Therefore, obtaining a more efficient and reliable method to detect V. eleguneniensis eggs or larvae has become crucial for clinical diagnosis, epidemiological survey, and laboratory examination, and achieving this goal is foreseeable only through the application of molecular methodologies.
Recent methods using ribosomal or mitochondrial DNA have proven valuable complementary tools to overcome this problem and are widely utilized for species identification of many Strongylida species, including lungworms [11,12]. For example, Verocai et al. studied the prevalence of lungworms from muskoxen and caribous and molecularly identified V. eleguneniensis as a widespread, multi-host species of wild North American ungulates based on sequencing and genetically analyzing the ribosomal internal transcribed spacer 2 (ITS-2) [13]. Considering that the mitochondrial DNA (mtDNA) could be more suitable than the ribosomal sequences for species identification owing to its maternal inheritance, high mutation, and lack of recombination [14], Li and colleagues determined the mitochondrial cox1 gene of Tibetan pig lungworms and verified that all lungworms were Metastrongylus pudendotectus [15]. Furthermore, Jabbar et al. applied the mitochondrial cox1, cytb, and rrnS genes-based multiplex PCR to monitor the prevalence of Dictyoculus lungworms and demonstrated the presence of Dictyoculus cervi infection in the Polish moose population [16]. However, compared to single mitochondrial gene loci, the complete mtDNA data can provide the genome-level genetic information and, therefore, would yield novel insights into evolutionary and phylogenetic-based analyses of lungworms.
Until now, there has been no information available on the mtDNAs of any Varestrongylus species. In this study, we sequenced, de novo assembled and characterized the complete mtDNA of V. eleguneniensis, the first member of the genus Varestrongylus, using Illumina technology. Then, the structure and organization of V. eleguneniensis mtDNA were compared with those of other Strongylida species to explore the conservation and variability of the whole genome at the nucleotide- and amino acid-levels. Following comparative genomics, the phylogenetic relationships of V. eleguneniensis in the family Protostrongylidae and of the family Protostrongylidae within the order Strongylida were also studied by reconstruction of phylogenetic trees (maximum parsimony (MP), maximum likelihood (ML), and Bayesian inference (BI)) using either single or concatenated datasets of twelve PCGs and two rRNAs.
2. Results and Discussion
2.1. Genome Feature of the Takin Lungworm
The circular mtDNA of V. eleguneniensis was 13,625 bp in length, comparable to other sequenced Strongylida species (ranging from 13,310 to 15,221 bp) [17,18,19,20]. The entire genome encoded 36 genes, including 12 PCGs (atp6, cox1-3, cytb, nad1-6, and nad4L), two mitochondrial rRNA genes (small (rrnS) and large (rrnL) subunits) and 22 tRNA genes (Figure 1). All these genes were present on the same strand and transcribed in the same direction (5′ to 3′). The gene order of the mtDNA of V. eleguneniensis followed the gene arrangement (GA3) [21,22,23], similar to that of the other Strongylida members. Moreover, there were two non-coding regions, namely NCR1 and NCR2, presented in this circular genome. NCR1 was 192 bp in length and located between the nad5 and nad6 genes, flanking at the 5′-end by the tRNA-Ala gene and at the 3′-end by the tRNA-Pro. Because of its high AT content (81.3%), this region was also known as AT-region. NCR2 was 57 bp in length and located between nad4L and rrnS, flanking at the 5′-end by the tRNA-Trp gene and at the 3′-end by the tRNA-Glu. In addition, five overlapping regions, ranging from 1 bp to 21bp, were located between cytb and tRNA-Leu (UAG) (1 bp), cox2 and tRNA-His (2 bp), tRNA-His and rrnL (1 bp), tRNA-Glu and rrnS (21 bp), and tRNA-Ser (UGA) and tRNA-Asn (1 bp), respectively. 140 nucleotides were observed to be scattered in 11 intergenic spacers, ranging from 1 to 36 bp in length.
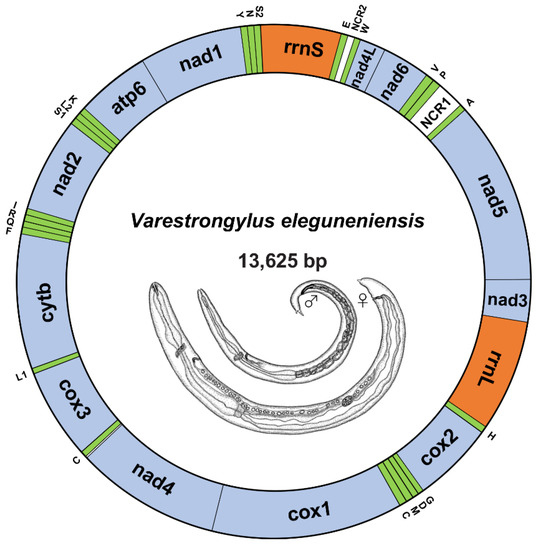
Figure 1.
The circular map of V. eleguneniensis mtDNA. Genes are represented by different colored blocks. PCGs are denoted in blue, rRNAs in orange, tRNAs in green and non-coding regions in white. Gene abbreviations: atp6, ATP synthase subunits 6; cox1-3, cytochrome oxidase subunits 1-3; cytb, cytochrome b; nad1-6/nad4L, NADH dehydrogenase subunits 1-6/4 L; rrnL, large rRNA subunit; rrnS, small rRNA subunits; NCR1, AT-rich region; NCR2, the non-coding region. The tRNAs are indicated with their one-letter corresponding amino acids. Line drawings of V. eleguneniensis is adapted with permission from Ref. [13]. 2014, Guilherme G. Verocai.
2.2. Nucleotide Content and Codon Usage
The nucleotide content of V. eleguneniensis mtDNA was 27.0% A, 48.8% T, 6.4% C, and 17.8% G, respectively, with a total of AT content of 75.9% (Table S1). This significant AT bias was similar to other Strongylida members and was within the range of AT contents described for other species in the same order (69.1 to 79.2%) [24,25]. And tRNAs and rRNAs also showed a strong AT bias with AT content of 79.17% and 78.16%, respectively. Generally, the AT and GC skews on the complementary strands of each mtDNA could be used to measure the compositional asymmetry [26]. For V. eleguneniensis mtDNA, the AT and GC skews were −0.287 and 0.471, respectively, and their comparisons with other 43 Strongylida species were shown in Figure 2. It was obvious that the nucleotide compositions of mtDNAs of these species consistently exhibited an extreme skew away from A and C in favor of T and G, with −0.48 to −0.145 of AT skew and 0.348 to 0.608 of GC skew. Specifically, the respective AT and GC skews were −0.198~0.043 and 0.368~0.577 in the rRNAs, −0.259~0.039 and 0.328~0.522 in the tRNAs, and −0.480~−0.004 and 0.337~0.608 in the concatenated 12 PCGs. Notably, among the PCGs, the TG bias was also significant at the first, second, and third positions of codons with AT and GC skews being −0.408~−0.099 and 0.363~0.603, −0.421~−0.122 and 0.260~0.548, and −0.459~−0.148 and 0.273~0.696, respectively.
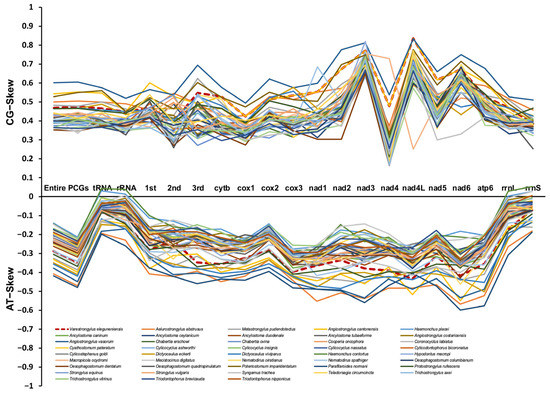
Figure 2.
The GC and AT skews for mtDNAs of 21 different regions (entire mtDNA, 12PCGs, rRNAs, tRNAs, the 1st site in codon, the 2nd site in codon, the 3rd site in codon, rrnL, rrnS, and each single PCG) within the 43 Strongylida mtDNAs. Different species are indicated by different colored lines graph.
Interestingly, the genome-wide AT bias was also reflected in the relative synonymous codon usage (RSCU) and codon usage patterns of PCGs. As shown in Figure 3, among 12 PCGs, the most frequently used codon was UUA (RSCU = 3.25; N = 243), followed by AGU (RSCU = 2.61; N = 99), GAU (RSCU = 2.29; N = 50), and GUU (RSCU = 2.55, N = 213). Correspondingly, the most frequently used amino acids were Phe (UUC & UUU; N = 566), Leu (UUG & UUA; N = 401), Val (GUG & GUA & GUC & GUU; N = 334) and Ile (AUA & AUC & AUU; N = 293). Further, a similar nucleotide bias was also observed in the choices of start and stop codons. Six of twelve PCGs were inferred to start with codon TTR (TTG was used for cytb, cox3, nad1, nad4, and nad6, and TTA was used for nad3) and the other six PCGs were initiated by the codon ATW as start codon (ATT was used for cox2, nad5, nad4L, and atp6, ATA was used for cox1 and nad2). In addition, six PCGs were terminated by an incomplete stop codon, and the incomplete stop codons were used for cox3, nad5, nad1, and nad4L (T-), for cox1 and atp6 (TA-) (Table 1). The incomplete stop codons may be converted into complete ones by post-transcriptional polyadenylation during mRNA maturation [27,28].
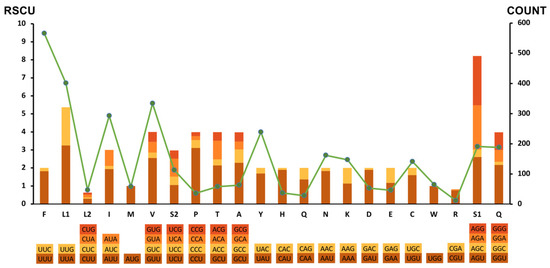
Figure 3.
Relative synonymous codon usage (RSCU) and codon numbers in V. eleguneniensis mtDNA. Codon families are provided on the X-axis, and the RSCU of each codon is represented by different color bars (left axis scale). The codon numbers are indicated by the green line graph (right axis scale).

Table 1.
Structures and organizations of V. eleguneniensis mtDNA.
2.3. tRNAs and rRNAs
Twenty-two tRNAs ranged from 52 to 64 bp in length, with a total length of 1263 bp in the mtDNA of V. eleguneniensis. Similar to other chromadorean nematodes sequenced so far [19,20,29], except for two tRNA-Ser, twenty tRNAs had a typical secondary structure with 4-bp DHU arms and 3~11-bp DHU loops, and its variable TψC arm and loop were replaced by a 6~12-bp TV loop (Figure 4). For these two tRNA-Ser, both were equipped with 4-bp TψC arms and 4~7-bp loops instead of the DHU arm and loop. Within two rRNAs, the rrnL gene was present between tRNA-His and nad3, and the rrnS was placed between tRNA-Glu and tRNA-Ser (UGA). The lengths of rrnL and rrnS were 995 bp and 722 bp, which were within the length ranges of other rRNAs published in Strongylida species with 946–983 bp for rrnL and 684–724 bp for rrnS, respectively.
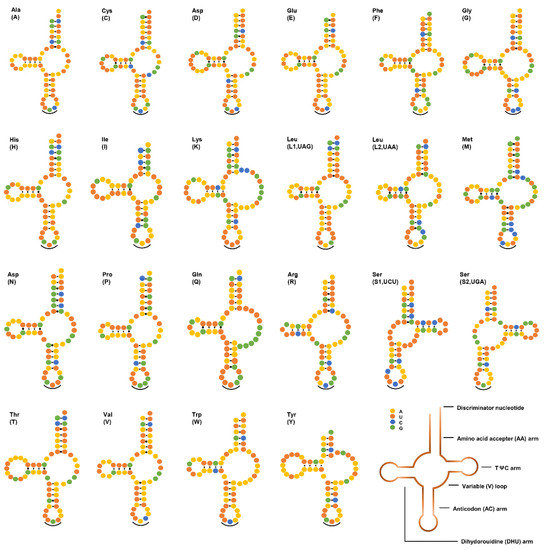
Figure 4.
Secondary structures of 22 tRNAs in V. eleguneniensis mtDNA. All tRNAs are differed with their corresponding amino acid abbreviations. Each base is represented by a different colored circle, yellow for adenine (A), orange for uracil (U), green for guanine (G), and blue for cytosine (C). Normal base pairings are illustrated by lines (-), whereas non-standard base pairs are illustrated by dots (·). Anticodons are marked by black curved lines (‿).
2.4. Comparison with Other Strongylida Genomes
Pairwise comparisons of the 12 PCGs of mtDNAs between V. eleguneniensis and other Strongylida species were shown in Figure 5 and Table S2. It was clear that the lengths of 12 PCGs in V. eleguneniensis were similar to those of most other Strongylida nematodes. Specifically, the atp6 gene among the PCGs was the most conserved with 600 bp in length followed by nad3 (336 bp) and nad4 (1230 bp). In contrast, the lengths of the cox1 and nad1 genes were the most variable, with 1572~1617 bp and 825~924 bp, respectively. Further sequence identity analyses showed that 74.7~78.7% nucleotide and 68.5~76.1% amino acid identities of 12 PCGs between V. eleguneniensis and other Strongylida species. It appeared that V. eleguneniensis shared relatively higher levels of the nucleotide and amino acid identities (in decreasing order) with Protostrongylus rufescens (78.7% and 75.6%)/Angiostrongylus cantonensis (78.3% and 76.1%), Aelurostrongylus abstrusus (77.5% and 73.5%), Ancylostoma tubaeforme (77.5% and 71.1%), and Ancylostoma caninum (77.4% and 71.0%). For each PCG, cox1 and cox3 were the most conserved genes because their high nucleotide (81.1~86.9% for cox1 and 79.6~84.1% for cox3) and amino acid (88.4~93.7% for cox1 and 80.0~86.3% for cox3) identities, whereas nad2 and nad6 were the most variable genes because their low nucleotide (64.4~72.4% for nad2 and 62.0~74.4% for nad6) and amino acid (44.1~61.3% for nad2 and 48.9~69.5% for nad6) identities.
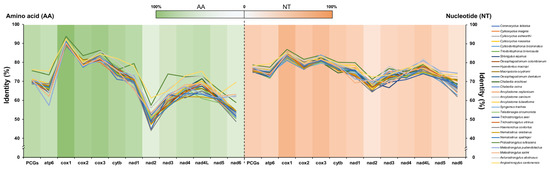
Figure 5.
Identity comparison of PCGs for 28 related Strongylida species with V. eleguneniensis. Different species are indicated by different colored broken lines. The heat maps in green and orange represent the average values of sequence identities of amino acid and nucleotide datasets, respectively.
To further explore the diversities within and between PCGs, the sliding window analysis was employed using the nucleotide alignments of V. eleguneniensis and other Strongylida species. As shown in Figure 6A, the values of the nucleotide diversity Pi of PCGs ranged from 0.123 to 0.321 among Strongylida species included here. Combined with the calculation of the number of variable positions per unit length of the gene, the nad6 was determined as the most variable gene (Pi = 0.2838), followed by nad2 (Pi = 0.2800), nad5 (Pi = 0.2473), and nad3 (Pi = 0.2311). However, the cox1 was determined as the most gene conserved with 0.1561 of Pi. This finding was consistent with the result of the identity analysis, which further demonstrates the accuracy of data from this study.
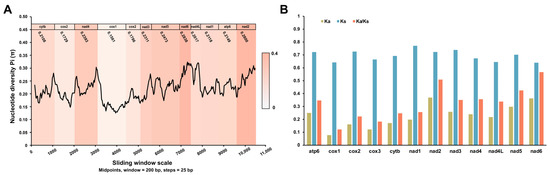
Figure 6.
Evolutionary diversity analyses of the mtDNA among 40 Strongylida species including V. eleguneniensis. (A), Sliding window analyses of 12 PCGs. The black line shows the value of nucleotide diversity (Pi) in a sliding window analysis (200-bp sliding window with 25-bp step size); the Pi values of each gene are shown under the gene name and reflected by the heat maps on the background; (B), The evolutionary rates of 12 PCGs in Strongylida mtDNAs. The Ka, Ks, and Ka/Ks are calculated for each PCG.
In parallel, Ka, Ks, and the Ka/Ks ratios were also calculated for all PCGs among the aforementioned Strongylida nematodes in order to estimate their evolutionary rates [30]. As shown in Figure 6B, the nad6 among the 12 PCGs showed the maximum ratio of Ka/Ks (0.566), followed by nad2 (0.509), nad5 (0.425), nad4 (0.356), and atp6 (0.375). Nevertheless, these ratios all were less than 1. In general, the ratio of Ka/Ks is often regarded as a measure of selective pressure of PCGs and indicates neutral mutation when Ka/Ks = 1, negative or purifying selection when Ka/Ks < 1 and positive or diversifying selection when Ka/Ks > 1 [31,32]. Therefore, it was reasonable that these PCGs in Strongylida were all subjected to a purifying selection during their evolutions.
2.5. Phylogenetic Analysis
The available mtDNA of V. eleguneniensis provided us an opportunity to study the evolutionary relationships of V. eleguneniensis in the family Protostrongylidae and of the family Protostrongylidae within the order Strongylida. As shown in Figure 7A,B, it was clear that three identical trees (MP/ML/BI) inferred from either the concatenated nucleotide or amino acid datasets consistently revealed that compared to other Strongylida species, V. eleguneniensis shared the closest relationship with P. rufescens, with high statistical support (all values ≥ 95 or = 1.00), consistent with previously proposed molecular phylogeny based on the ITS-2 [13]. Further, V. eleguneniensis and P. rufescens under the family Protostrongylidae clustered with species of the families Angiostrongylidae and Metastrongylidae, and together formed one branch and exhibited a paraphyletic relationship with species of the families Trichostrongylidae, Haemonchidae, Strongylidae, Chabertiidae, Ancylostomatidae, and Molineidae, supporting recent mtDNA-based phylogenetic conclusion [33,34,35,36].

Figure 7.
Phylogenies were inferred using three methods (MP, ML, and BI) based on mtDNA datasets containing 12 PCGs and two rRNAs. Dictyocaulus eckerti and Dictyocaulus viviparus are used as the outgroups. Different colored regions represent different families. (A), Phylogenetic tree based on the concatenated nucleotide sequences of the 12 PCGs; (B), Phylogenetic tree based on the concatenated amino acid sequences of the 12 PCGs; (C), Phylogenetic trees based on nucleotide sequences of 14 individual genes (12 PCGs and two rRNAs). The phylogenetic trees consistent with the trees in (A,B) are marked with red line boxes. The species sequenced in this study is shown in red fonts. The numbers along the branches indicate bootstrap values/posterior probabilities resulting from different analyses in the order MP/ML/BI.
In addition, single gene-based phylogenetic analyses were used to scoop out the optimal genetic marker candidates among the PCGs and rRNAs for phylogeny and species identification. As shown in Figure 7C, although most of PCGs and rRNAs showed different phylogenetic topologies, the positions of the family Protostrongylidae, Metastrongylidae, and Angiostrongylidae were steady in the cox1-, cytb-, nad1-, nad3-, nad5-, and nad6-based phylogenetic analyses. Notably, the cox1 and nad5 genes shared the same phylogenetic topology as that of the genome-based phylogenetic analysis, suggesting that both genes might be the most appropriate genetic markers and, therefore, could be used instead of mtDNA for evolutionary and phylogenetic studies of this parasite and other related species in Strongylida. Certainly, the marker validity of the cox1 and nad5 remains further validated when more additional Strongylida mtDNAs become available, especially those from the genus Varestrongylus, although the cox1 has been widely used as a DNA barcode for species identification and differentiation in parasitic nematodes [37,38].
3. Materials and Methods
3.1. Parasite Sampling
From September to October 2020, there were three dead wild B. taxicolor adults found in Tangjiahe National Nature Reserve, Sichuan, China (Figure 8); one of them was then brought to the Veterinary Medical Teaching Hospital, Sichuan Agricultural University (Ya’an, China), for postmortem examination. These takins died with the same clinical manifestations, including dyspnea, coughing and wheezing. After a standard necropsy under the Scientific Procedures Premises License for the College of Veterinary Medicine, Sichuan Agricultural University (SYXK 2014-187), diseased lungs were dissected, and about 56 nematode specimens were collected from the respiratory tracts of lungs. Through washing in physiological saline, all worms were preliminarily identified as V. eleguneniensis according to morphological keys [3]. Then, two specimens were further chosen for molecular identification by PCR amplifying the ITS-2 region, followed by sequence comparison with the previously reported corresponding sequence for V. eleguneniensis (GenBank accession number: JX115006) [13]. A result of 99.8% sequence identity of the ITS-2 between both worms and V. eleguneniensis confirmed their species identity.
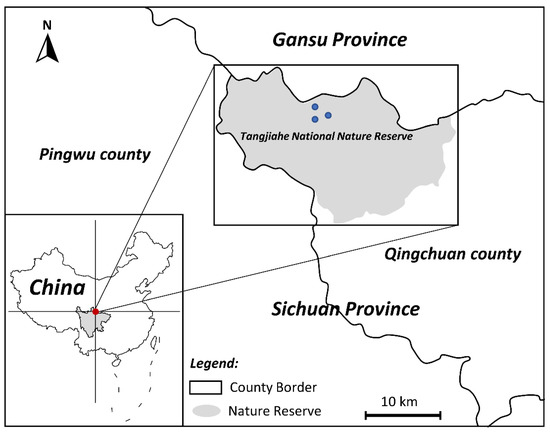
Figure 8.
Sampling plots of takin lungworms collected from Tangjiahe National Nature Reserve, China. The red and blue dots on the map are the location of the Tangjiahe National Nature Reserve and the sampling points, respectively.
3.2. V. eleguneniensis mtDNA Sequencing and Annotation
To decode the mtDNA of V. eleguneniensis, the total genomic DNA was extracted from pooled lungworm specimens (n = 20) using the Genomic DNA Extraction Kit ver. 3.0 (TaKaRa Biotech, Dalian, China). After DNA yield and integrity evaluation, a 0.25-μg aliquot of genomic DNA was fragmented, end-paired, and ligated to adaptor. The ligated fragments were isolated on agarose gels and purified by PCR amplification to produce the sequencing library. A 350-bp paired-end (PE) library was constructed and sequenced using the Illumina HiSeq TM 4000 platform (Illumina, San Diego, CA, USA). The raw reads were quality-trimmed by the following criteria: filtered reads with adapters, filtered reads with N bases >15 bp, and filtered reads with low-quality bases (≤3). In total, 2.5 Gb of clean data (250 bp each, paired-end reads) were obtained and assembled using MITObim v1.9 software [39] with default parameters, followed by manual inspection for searching the overlapping regions to circularize the complete mtDNA. This mtDNA was further verified by PCR amplifications using nine overlapping segments (ranging in length from 1.42 kp to 2.33 kb), which were designed on the basis of the alignments of the relatively conserved regions of the available Strongylida mtDNAs. The corresponding primer sequences are shown in Table S3. PCR reactions were achieved using a 50 μL reaction volume containing 3 μL genomic DNA (≥15 ng), 25 μL 2 × HiFi TransTaq PCR SuperMix, 1 μL sense primer (10 pmol), 1 μL anti-sense primer (10 pmol), and 20 μL nuclease-free water. PCR conditions were 5 min denaturation at 95 °C, followed by 30 cycles of 45 s at 95 °C, 40 s at 45~55 °C, and 1~3 min at 68 °C according to the Tm value and the product length, with a final extension at 68 °C for 10 min. After agarose gel detections, all target amplicons were sequenced either directly or following sub-cloning into the pMD19-T vector (TaKaRa, Biotech, Dalian, China). Each amplicon was sequenced three times to ensure maximum accuracy. This consensus circular genome was drawn with MacVector ver. 9.5 and annotated using MITOS and online BLAST tools available on the NCBI website [39]. The boundaries of PCGs and rRNA genes were verified by alignment with homologous genes of other related nematodes using DNAMAN ver. 8.0 (Lynnon Biosoft, Vaudreuil-Dorion, QC, Canada). In addition to MITOS, open Reading Frame (ORF) Finder and Primer software were also used to define the amino acid sequences of PCGs and to find the start and stop codons using the invertebrate mitochondrial genetic code [40]. tRNA genes were identified using ARWEN and tRNAscan-SE [41,42], and their secondary structures were predicted and manually plotted using the software Adobe Illustrator CS5 (www.adobe.com/products/illustrator.html (accessed on 17 June 2022)). The complete sequence of V. eleguneniensis mtDNA was deposited in GenBank under accession number: OP464906.
3.3. Sequence Analysis
Nucleotide content and codon usage of V. eleguneniensis mtDNA were measured using Geneious and PhyloSuite [43,44]. The nucleotide skewness of V. eleguneniensis and their comparisons with 42 other nematodes in the order Strongylida (Table 2) were performed using the formulas [26,34,35,36,45,46,47,48,49,50,51,52,53,54,55,56,57]: AT skew = (A − T)/(A + T) and GC skew = (G − C)/(G + C). Sequence lengths and nucleotide/amino acid identities of 12 PCGs among Strongylida species were determined using DNAstar [58]. Based on the multi-alignment of 12 PCGs for 43 Strongylida mtDNAs (including V. eleguneniensis), the sliding window analysis was used to compute the nucleotide diversity Pi (π) using 200-bp window and 25-bp steps in DnaSP ver.5.10 [59]. The Pi value was plotted against the midpoint position of each window. Additionally, the evolutionary rate and the ratio of the nonsynonymous substitutions (Ka) and synonymous substitutions (Ks) of each PCG were also calculated using KaKs_Calculator [60].
Table 2.
Summary mtDNA information included in this study.
3.4. Phylogenetic Analysis
To determine the classification positions of V. eleguneniensis in the family Protostrongylidae and of the family Protostrongylidae within the order Strongylida, the mtDNAs of twenty-nine selected Strongylida species were retrieved from GenBank, and their concatenated amino acid sequences of twelve PCGs and the nucleotide sequence of each PCG/rRNA were separately aligned using Clustal X ver. 2.0 for phylogenetic analyses. Dictyocaulus eckerti (GenBank accession no. NC_019809) and Dictyocaulus viviparus (GenBank accession no. NC_019810) were treated as the outgroups and included in each analysis. Phylogenetic analyses were performed with MP, ML, and BI. For the MP analysis, either the concatenated 12 PCGs dataset or multi-alignments of the nucleotide sequence for each PCG or rRNA were analyzed using equally weighted parsimony in PAUP* [61] and heuristic searches with a tree-bisection-reconnection (TBR) branch-swapping. 1000 replicates of Wagner trees were selected and five trees per replication were saved, followed by the harvest of the optimal topology using the Kishino–Hasegawa method. Bootstrap resampling with 1000 replications was calculated for each nodal support. For ML analyses, the optimal evolutionary model was selected with the “Auto” option on W-IQ-TREE web server (http://iqtree.cibiv.univie.ac.at (accessed on 20 June 2022)) and “TIM+F+I+G4” model was chosen for both matrices according to Bayesian Information Criterion (BIC). ML trees were constructed using an ultrafast bootstrap approximation approach with 100,000 replications. Within the BI trees, the optimal evolutionary model CAT+GTR+G was selected for mtDNA datasets using ModelFinder [62]. The BI computations were achieved using MrBayes 3.2.7 [63], with four independent Markov chains, running for 100,000 (concatenated dataset of 12 PCGs) and 10,000,000 (single PCG or rRNA dataset) metropolises coupled with Monte Carlo generations, sampling a tree every 100 and 10,000 generations. When the average standard deviation (SD) of the split frequencies was reduced to below 0.01, the first 25% trees were discarded as “burn-in” and the remaining were used to compute Bayesian posterior probabilities. The evolutionary distance was estimated using the MrBayes order (aamodelpr = mixed) with default parameters. A consensus tree was obtained and visualized using TreeviewX [64].
4. Conclusions
In this study, V. eleguneniensis mtDNA represented the first genome sequenced in the members of the genus Varestrongylus, and its comparison with the other Strongylida species revealed that cox1 and nad6 were the most and least conserved genes, respectively, across the order. Genome-wide phylogenies refined the phylogenetic relationship of V. eleguneniensis in the family Protostrongylidae of Strongylida and showed a close relationship between V. eleguneniensis and P. rufescens. Further single gene-based phylogenies suggested that the cox1 and nad5 genes could be suitable genetic markers for phylogeny and species identification in Strongylida because their topologies are the same as that of the whole genome. The results should contribute to a better understanding of systematics and evolutionary biology of Varestrongylus lungworms and, therefore, can assist in their prevention and control by providing resources of molecular markers for molecular diagnostic, epidemiology and population genetic studies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms232113597/s1.
Author Contributions
Conceptualization, Y.X. and X.Z.; methodology, Y.X.; software, Y.C. and L.W.; validation, Y.X. and X.Z.; formal analysis, Z.W., P.Z., Z.H., X.G., R.H., J.X., B.J., X.P., G.Y. and X.Z.; investigation, Y.X., Y.C. and L.W.; resources, Y.X.; data curation, Y.C., L.W., X.Z., Z.W. and P.Z.; writing—original draft preparation, Y.C. and Y.X.; writing—review and editing, Y.X.; visualization, Y.X., Y.C. and L.W.; supervision, Y.X.; funding acquisition, Y.X. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Natural Science Foundation of China program (32273028).
Institutional Review Board Statement
All sample collection and research processes conformed to the ethical guidelines, religious beliefs, and laws and regulations of the People’s Republic of China (PRC). And all procedures including the autopsy were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, Bethesda, MD, USA) and the recommendations in the ARRIVE guidelines (https://www.nc3rs.org.uk/arriveguidelines, (accessed on 11 September 2020)).
Informed Consent Statement
Not applicable.
Data Availability Statement
Molecular data have been deposited to GenBank with the following accession number: OP464906.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhou, M.; Yu, J.; Li, B.; Ouyang, B.; Yang, J. The complete mitochondrial genome of Budorcas taxicolor tibetana (Artiodactyla: Bovidae) and comparison with other Caprinae species: Insight into the phylogeny of the genus Budorcas. Int. J. Biol. Macromol. 2019, 121, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.X. A new species of protostrongylid lungworm from takin. Acta Zootax Sin. 1989, 14, 269–272. [Google Scholar]
- Kafle, P.; Sullivan, J.; Verocai, G.G.; Kutz, S.J. Experimental life-cycle of Varestrongylus eleguneniensis (Nematoda: Protostrongylidae) in a Captive Reindeer (Rangifer tarandus tarandus) and a Muskox (Ovibos moschatus moschatus). J. Parasitol. 2017, 103, 584–587. [Google Scholar] [CrossRef][Green Version]
- Yang, G.Y.; Zhang, Z.H. Parasitic Diseases of Wildlife; Science Press: Beijing, China, 2013. [Google Scholar]
- Blaxter, M.L.; De Ley, P.; Garey, J.R.; Liu, L.X.; Scheldeman, P.; Vierstraete, A.; Vanfleteren, J.R.; Mackey, L.Y.; Dorris, M.; Frisse, L.M.; et al. A molecular evolutionary framework for the phylum Nematoda. Nature 1998, 392, 71–75. [Google Scholar] [CrossRef]
- De Ley, P.; Blaxter, M.L. Systematic position and phylogeny. In The Biology of Nematodes; Lee, D.L., Ed.; CRC Press: New York, NY, USA, 2002; pp. 1–30. [Google Scholar]
- Kern, E.M.A.; Kim, T.; Park, J.K. The mitochondrial genome in nematode phylogenetics. Front. Ecol. Evol. 2020, 8, 250. [Google Scholar] [CrossRef]
- Bryan, H.M.; Sim, K.A.; Darimont, C.T.; Paquet, P.C.; Wagner, B.; Muñoz-Fuentes, V.; Smits, J.E.; Chilton, N.B. Identification of Parelaphostrongylus odocoilei (Nematoda: Protostrongylidae) first-stage larvae in the feces of gray wolves (Canis lupus) by molecular methods. J. Wildl. Dis. 2010, 46, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Bowman, D.D.; Lynn, R.C.; Eberhard, D.M.L. Georgis’ Parasitology for Veterinarians; Elsevier Science: Saint Louis, MI, USA, 2003. [Google Scholar]
- Chilton, N.B. The use of nuclear ribosomal DNA markers for the identification of bursate nematodes (order Strongylida) and for the diagnosis of infections. Anim. Health Res. Rev. 2004, 5, 173–187. [Google Scholar] [CrossRef]
- Hu, M.; Jex, A.R.; Campbell, B.E.; Gasser, R.B. Long PCR amplification of the entire mitochondrial genome from individual helminths for direct sequencing. Nat. Protoc. 2007, 2, 2339–2344. [Google Scholar] [CrossRef]
- Jex, A.R.; Littlewood, D.T.; Gasser, R.B. Toward next-generation sequencing of mitochondrial genome-focus on parasitic worms of animals and biotechnological implications. Biotechnol. Adv. 2010, 28, 151–159. [Google Scholar] [CrossRef]
- Verocai, G.G.; Kutz, S.J.; Simard, M.; Hoberg, E.P. Varestrongylus eleguneniensis sp. n. (Nematoda: Protostrongylidae): A widespread, multi-host lungworm of wild North American ungulates, with an emended diagnosis for the genus and explorations of biogeography. Parasit. Vectors 2014, 7, 566. [Google Scholar] [CrossRef]
- Park, J.K.; Sultana, T.; Lee, S.H.; Kang, S.; Kim, H.K.; Min, G.S.; Eom, K.S.; Nadler, S.A. Monophyly of clade III nematodes is not supported by phylogenetic analysis of complete mitochondrial genome sequences. BMC Genom. 2011, 12, 392. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Luo, H.; Zhang, H.; Lan, Y.; Han, Z.; Shahzad, M.; Wang, X.; Qiu, G.; Huang, S.; Jiang, W.; et al. First report of Metastrongylus pudendotectus by the genetic characterization of mitochondria genome of cox1 in pigs from Tibet, China. Vet. Parasitol. 2016, 223, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Filip-Hutsch, K.; Demiaszkiewicz, A.W.; Chęcińska, A.; Hutsch, T.; Czopowicz, M.; Pyziel, A.M. First report of a newly-described lungworm, Dictyocaulus cervi (Nematoda: Trichostrongyloidea), in moose (Alces alces) in central Europe. Int. J. Parasitol. Parasites Wildl. 2020, 13, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads–A baiting and iterative mapping approach. Nucleic Acids Res. 2013, 13, e129. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Chilton, N.; Gasser, R. The Mitochondrial Genomics of Parasitic Nematodes of Socio-Economic Importance: Recent Progress, and Implications for Population Genetics and Systematics. In Advances in Parasitology; Elsevier: Amsterdam, The Netherlands, 2003; Volume 56, pp. 133–212. ISBN 978-0-12-031756-1. [Google Scholar]
- Hu, M.; Gasser, R.B. Mitochondrial Genomes of Parasitic Nematodes–Progress and Perspectives. Trends Parasitol. 2006, 22, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Gasser, R.B.; Otranto, D.; Xu, M.J.; Shen, J.L.; Mohandas, N.; Zhou, D.H.; Zhu, X.Q. Mitochondrial Genome of the Eyeworm, Thelazia callipaeda (Nematoda: Spirurida), as the First Representative from the Family Thelaziidae. PLoS Negl. Trop. Dis. 2013, 7, e2029. [Google Scholar] [CrossRef]
- Xie, Y.; Niu, L.; Zhao, B.; Wang, Q.; Nong, X.; Chen, L.; Zhou, X.; Gu, X.; Wang, S.; Peng, X.; et al. Complete Mitochondrial Genomes of Chimpanzee- and Gibbon-Derived Ascaris Isolated from a Zoological Garden in Southwest China. PLoS ONE 2013, 8, e82795. [Google Scholar] [CrossRef]
- Yatawara, L.; Wickramasinghe, S.; Rajapakse, R.P.V.J.; Agatsuma, T. The Complete Mitochondrial Genome of Setaria digitata (Nematoda: Filarioidea): Mitochondrial Gene Content, Arrangement and Composition Compared with Other Nematodes. Mol. Biochem. Parasitol. 2010, 173, 32–38. [Google Scholar] [CrossRef]
- Duan, H. Analysis of Complete Mitochondrial Genomes and Genetic Relationships of Three Nematodes of Genus Triodontophorus; Heilongjiang Bayi Agricultural University: Heilongjiang, China, 2006. [Google Scholar]
- Liu, G.H.; Wang, Y.; Song, H.Q.; Li, M.W.; Ai, L.; Yu, X.L.; Zhu, X.Q. Characterization of the Complete Mitochondrial Genome of Spirocerca lupi: Sequence, Gene Organization and Phylogenetic Implications. Parasit. Vectors 2013, 6, 45. [Google Scholar] [CrossRef]
- Sun, L.; Zhuo, K.; Lin, B.; Wang, H.; Liao, J. The Complete Mitochondrial Genome of Meloidogyne graminicola (Tylenchina): A Unique Gene Arrangement and Its Phylogenetic Implications. PLoS ONE 2014, 9, e98558. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of Nucleotide Composition at Fourfold Degenerate Sites of Animal Mitochondrial Genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. TRNA Punctuation Model of RNA Processing in Human Mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Schuster, G.; Stern, D. Chapter 10 RNA Polyadenylation and Decay in Mitochondria and Chloroplasts. In Progress in Molecular Biology and Translational Science; Elsevier: Amsterdam, The Netherlands, 2009; Volume 85, pp. 393–422. ISBN 978-0-12-374761-7. [Google Scholar]
- Jacob, J.E.M.; Vanholme, B.; Van Leeuwen, T.; Gheysen, G. A Unique Genetic Code Change in the Mitochondrial Genome of the Parasitic Nematode Radopholus similis. BMC Res. Notes 2009, 2, 192. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Nielsen, R. Estimating Synonymous and Nonsynonymous Substitution Rates under Realistic Evolutionary Models. Mol. Biol. Evol. 2000, 17, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.D. The Ka/Ks Ratio: Diagnosing the Form of Sequence Evolution. Trends Genet. 2002, 18, 486. [Google Scholar] [CrossRef]
- Li, H.; Liu, H.; Shi, A.; Štys, P.; Zhou, X.; Cai, W. The Complete Mitochondrial Genome and Novel Gene Arrangement of the Unique-Headed Bug Stenopirates sp. (Hemiptera: Enicocephalidae). PLoS ONE 2012, 7, e29419. [Google Scholar] [CrossRef]
- Jabbar, A.; Jex, A.R.; Mohandas, N.; Hall, R.S.; Littlewood, D.T.; Gasser, R.B. The mitochondrial genome of Aelurostrongylus abstrusus-diagnostic, epidemiological and systematic implications. Gene 2013, 516, 294–300. [Google Scholar] [CrossRef]
- Jex, A.R.; Hall, R.S.; Littlewood, D.T.; Gasser, R.B. An integrated pipeline for next-generation sequencing and annotation of mitochondrial genomes. Nucleic Acids Res. 2010, 38, 522–533. [Google Scholar] [CrossRef]
- Jex, A.R.; Hu, M.; Littlewood, D.T.J.; Waeschenbach, A.; Gasser, R.B. Using 454 Technology for Long-PCR Based Sequencing of the Complete Mitochondrial Genome from Single Haemonchus Contortus (Nematoda). BMC Genom. 2008, 9, 11. [Google Scholar] [CrossRef]
- Jabbar, A.; Mohandas, N.; Jex, A.R.; Gasser, R.B. The Mitochondrial Genome of Protostrongylus rufescens–Implications for Population and Systematic Studies. Parasit. Vectors 2013, 6, 263. [Google Scholar] [CrossRef]
- Prosser, S.W.; Velarde-Aguilar, M.G.; León-Règagnon, V.; Hebert, P.D. Advancing nematode barcoding: A primer cocktail for the cytochrome c oxidase subunit I gene from vertebrate parasitic nematodes. Mol. Ecol. Resour. 2013, 13, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, L.T.; Bianchi, F.M.; Deprá, M.; Calegaro-Marques, C. Barcoding a can of worms: Testing cox1 performance as a DNA barcode of Nematoda. Genome 2021, 64, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Mangalam, A.K.; Dwivedi, S.; Naik, S. Primer premier: Program for design of degenerate primers from a protein sequence. Biotechniques 1998, 24, 318–319. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE on-line: Search and contextual analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.L.; Li, W.X.; Jakovlić, I.; Zou, H.; Zhang, J.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Jex, A.R.; Waeschenbach, A.; Hu, M.; van Wyk, J.A.; Beveridge, I.; Littlewood, D.T.; Gasser, R.B. The mitochondrial genomes of Ancylostoma caninum and Bunostomum phlebotomum- two hookworms of animal health and zoonotic importance. BMC Genom. 2009, 10, 79. [Google Scholar] [CrossRef]
- Hu, M.; Chilton, N.B.; Gasser, R.B. The mitochondrial genomes of the human hookworms, Ancylostoma duodenale and Necator americanus (Nematoda: Secernentea). Int. J. Parasitol. 2002, 32, 145–158. [Google Scholar] [CrossRef]
- Shi, X.L.; Fu, Y.Q.; Abdullahi, A.Y.; Wang, M.W.; Yang, F.; Yu, X.G.; Pan, W.D.; Yan, X.X.; Hang, J.X.; Zhang, P.; et al. The mitochondrial genome of Ancylostoma tubaeforme from cats in China. J. Helminthol. 2018, 92, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Gasser, R.B.; Jabbar, A.; Mohandas, N.; Schnyder, M.; Deplazes, P.; Littlewood, D.T.; Jex, A.R. Mitochondrial genome of Angiostrongylus vasorum: Comparison with congeners and implications for studying the population genetics and epidemiology of this parasite. Infect. Genet. Evol. 2012, 12, 1884–1891. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Zhao, L.; Song, H.Q.; Zhao, G.H.; Cai, J.Z.; Zhao, Q.; Zhu, X.Q. Chabertia erschowi (Nematoda) is a distinct species based on nuclear ribosomal DNA sequences and mitochondrial DNA sequences. Parasit. Vectors 2014, 7, 44. [Google Scholar] [CrossRef]
- Jabbar, A.; Beveridge, I.; Mohandas, N.; Chilton, N.B.; Littlewood, D.T.; Jex, A.R.; Gasser, R.B. Analyses of mitochondrial amino acid sequence datasets support the proposal that specimens of Hypodontus macropi from three species of macropodid hosts represent distinct species. BMC Evol. Biol. 2013, 13, 259. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.H.; Hu, B.; Cheng, W.Y.; Jia, Y.Q.; Li, H.M.; Yu, S.K.; Liu, G.H. The complete mitochondrial genomes of Oesophagostomum asperum and Oesophagostomum columbianum in small ruminants. Infect. Evol. 2013, 19, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.Q.; Liu, G.H.; Hu, M.; Song, H.Q.; Wu, X.Y.; Li, M.W.; Zhang, Y.; Zou, F.C.; Zhu, X.Q. Oesophagostomum dentatum and Oesophagostomum quadrispinulatum: Characterization of the complete mitochondrial genome sequences of the two pig nodule worms. Exp. Parasitol. 2012, 131, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Van der Veer, M.; de Vries, E. A single nucleotide polymorphism map of the mitochondrial genome of the parasitic nematode Cooperia oncophora. Parasitology 2004, 128, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Gasser, R.B.; Jabbar, A.; Mohandas, N.; Höglund, J.; Hall, R.S.; Littlewood, D.T.J.; Jex, A.R. Assessment of the Genetic Relationship between Dictyocaulus Species from Bos Taurus and Cervus Elaphus Using Complete Mitochondrial Genomic Datasets. Parasit. Vectors 2012, 5, 241. [Google Scholar] [CrossRef]
- Jabbar, A.; Mohandas, N.; Gasser, R.B. Characterisation of the Mitochondrial Genome of Parafilaroides normani (Lungworm) of Arctocephalus Pusillus Doriferus (Australian Fur Seal). Parasitol. Res. 2014, 113, 3049–3055. [Google Scholar] [CrossRef]
- Zhao, G.H.; Jia, Y.Q.; Cheng, W.Y.; Zhao, W.; Bian, Q.Q.; Liu, G.H. Characterization of the Complete Mitochondrial Genomes of Nematodirus oiratianus and Nematodirus spathiger of Small Ruminants. Parasit. Vectors 2014, 7, 319. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Li, P.; Sun, C.; Liu, W. The Complete Mitochondrial Genome of Coronocyclus Labratus (Rhabditida: Cyathostominae). Mitochondrial DNA Part B 2020, 5, 1044–1045. [Google Scholar] [CrossRef] [PubMed]
- Burland, T.G. DNASTAR’s Lasergene Sequence Analysis Software. In Bioinformatics Methods and Protocols; Humana Press: Totowa, NJ, USA, 1999; Volume 132, pp. 71–91. ISBN 978-1-59259-192-3. [Google Scholar]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, J.; Zhao, X.-Q.; Wang, J.; Wong, G.K.-S.; Yu, J. KaKs_Calculator: Calculating Ka and Ks Through Model Selection and Model Averaging. Genom. Proteom. Bioinform. 2006, 4, 259–263. [Google Scholar] [CrossRef]
- Goloboff, P.A.; Catalano, S.A.; Torres, A. Parsimony Analysis of Phylogenomic Datasets (II): Evaluation of PAUP*, MEGA and MPBoot. Cladistics 2022, 38, 126–146. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Page, R.D.M. Visualizing Phylogenetic Trees Using TreeView. Curr. Protoc. Bioinform. 2002, 6. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).