
The Cytoplasmic Actins in the Regulation of Endothelial Cell Function


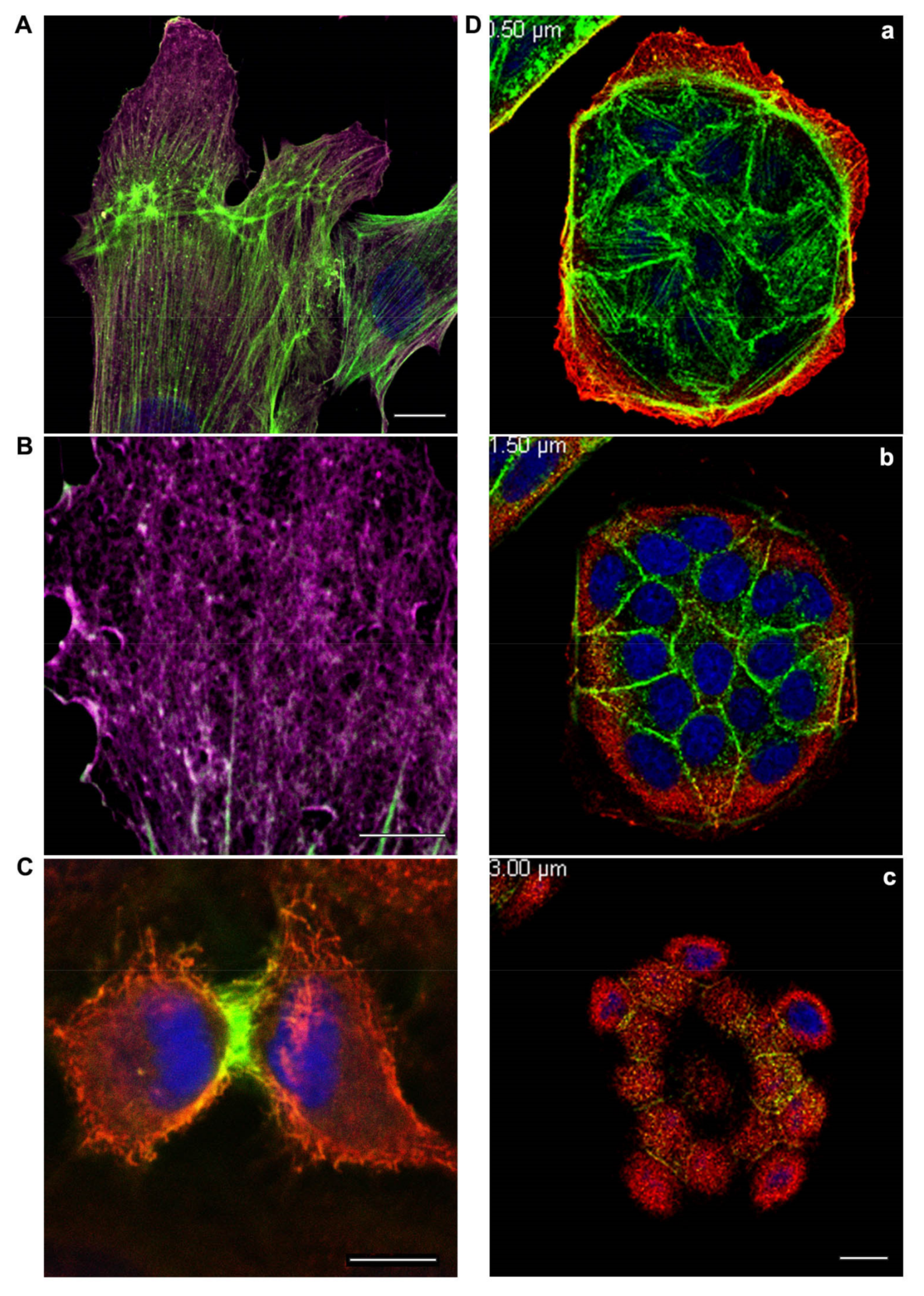
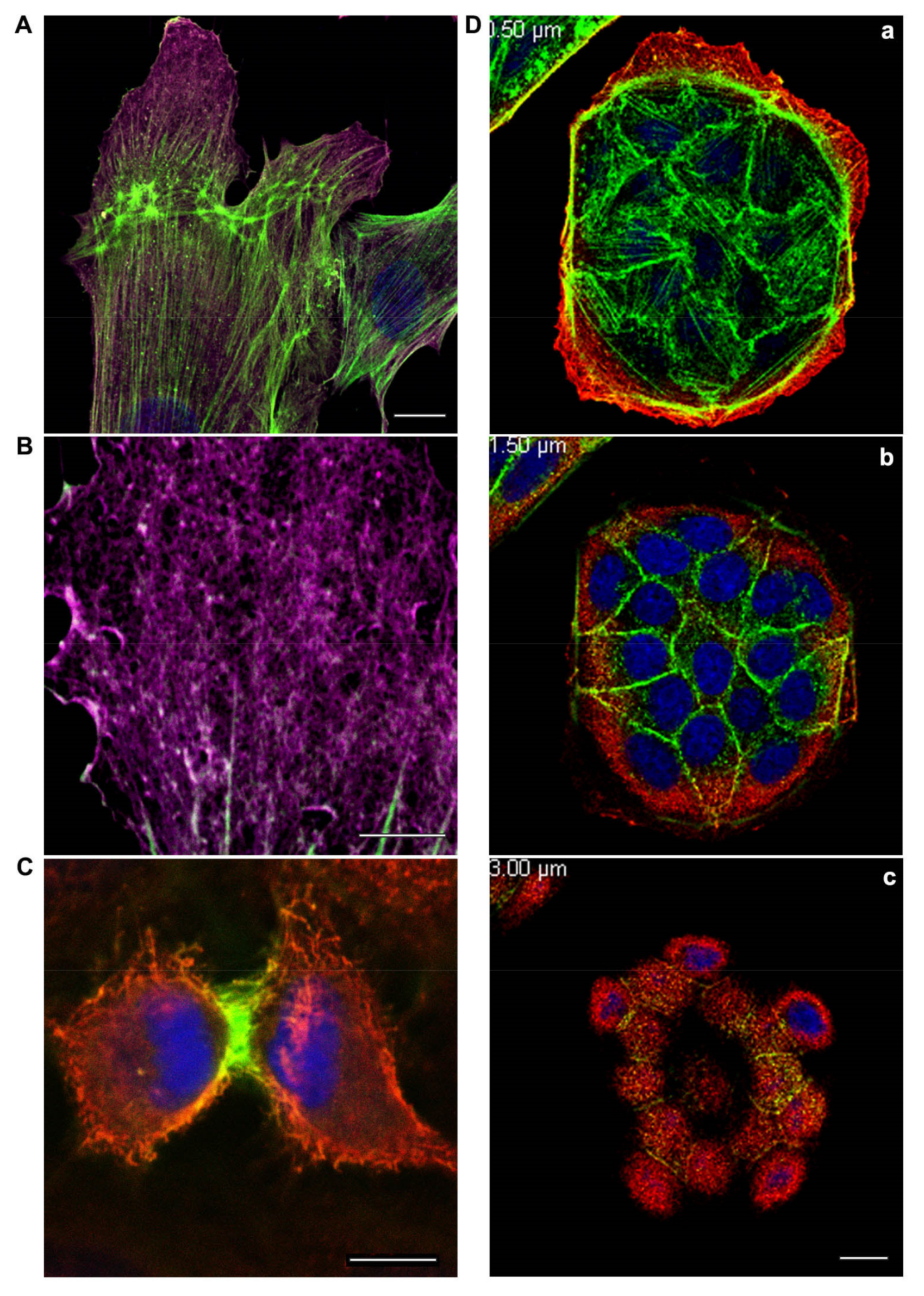{kind=link}
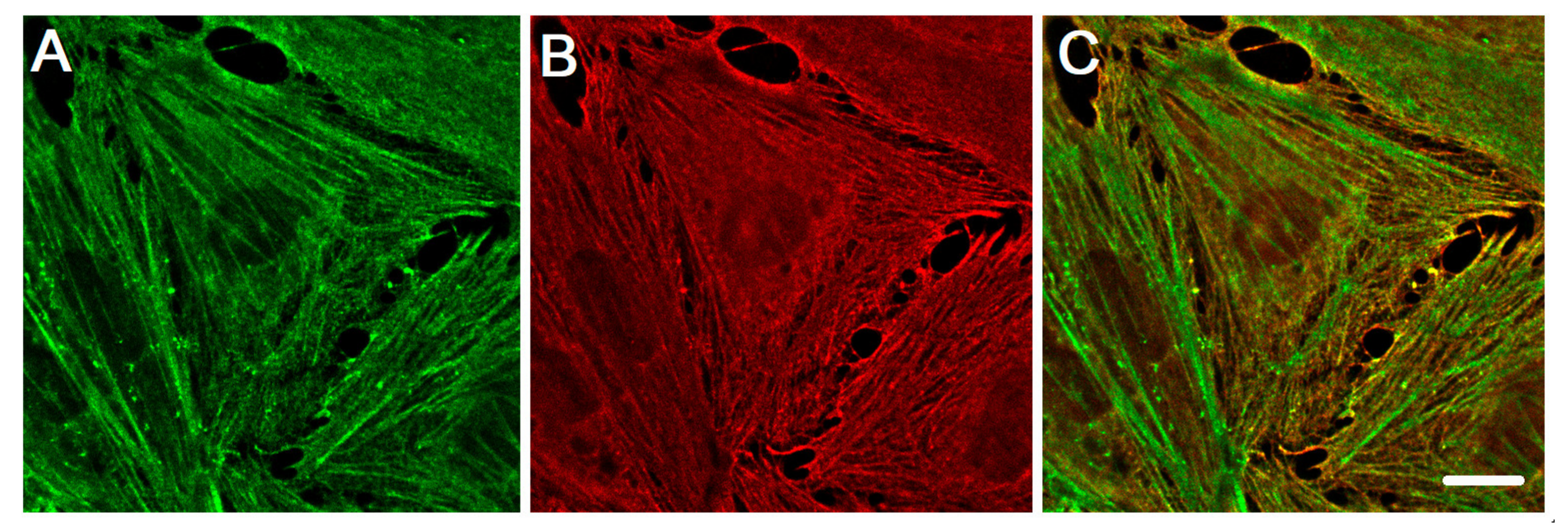{kind=link}
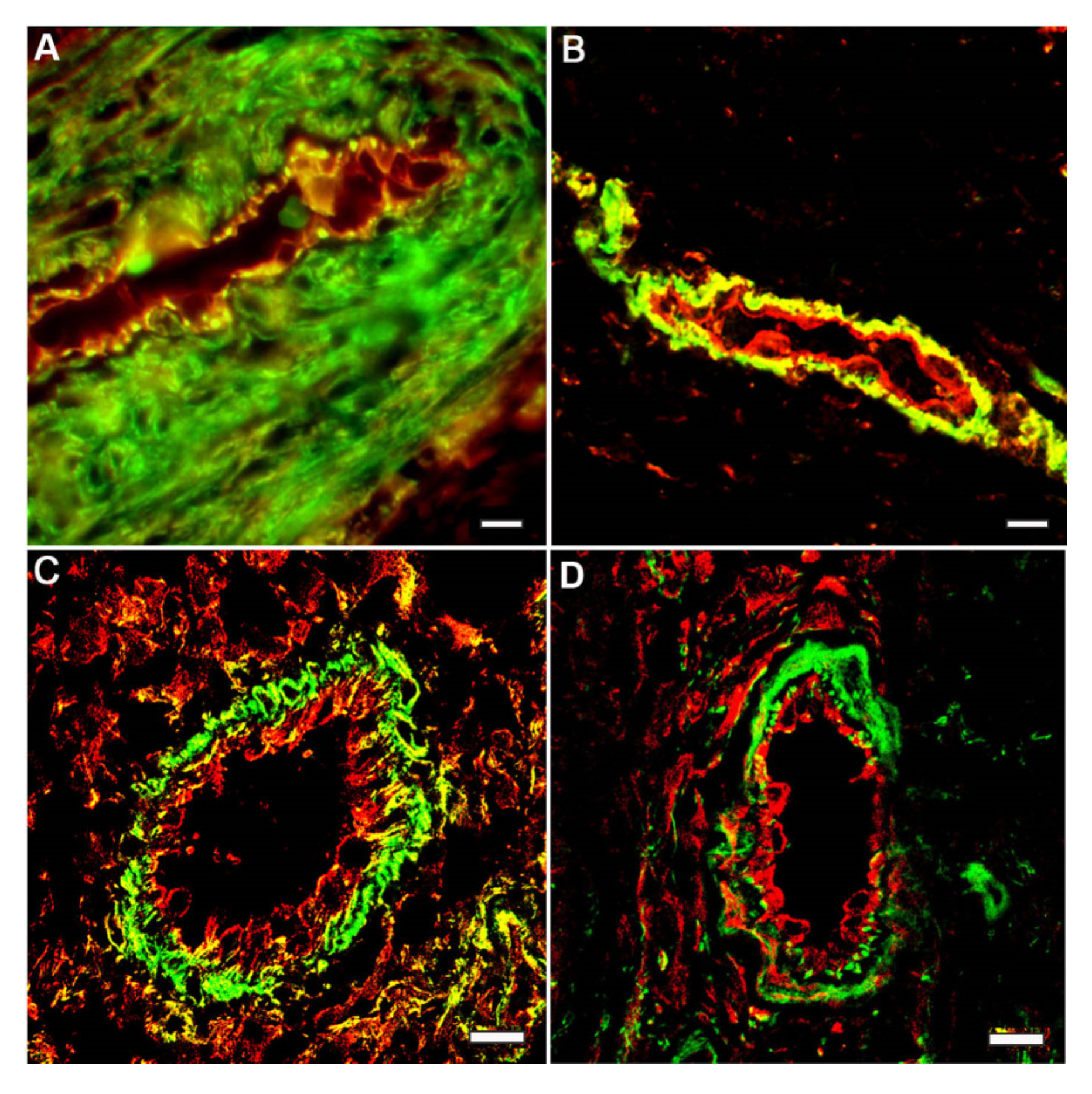{kind=link}
Abstract
:1. Introduction
2. Cytoplasmic Actin Isoforms: Structures and Functions in the Interphase Non-Muscle Cells
3. Different Impact of Actin Isoforms on the Process of Cell Division
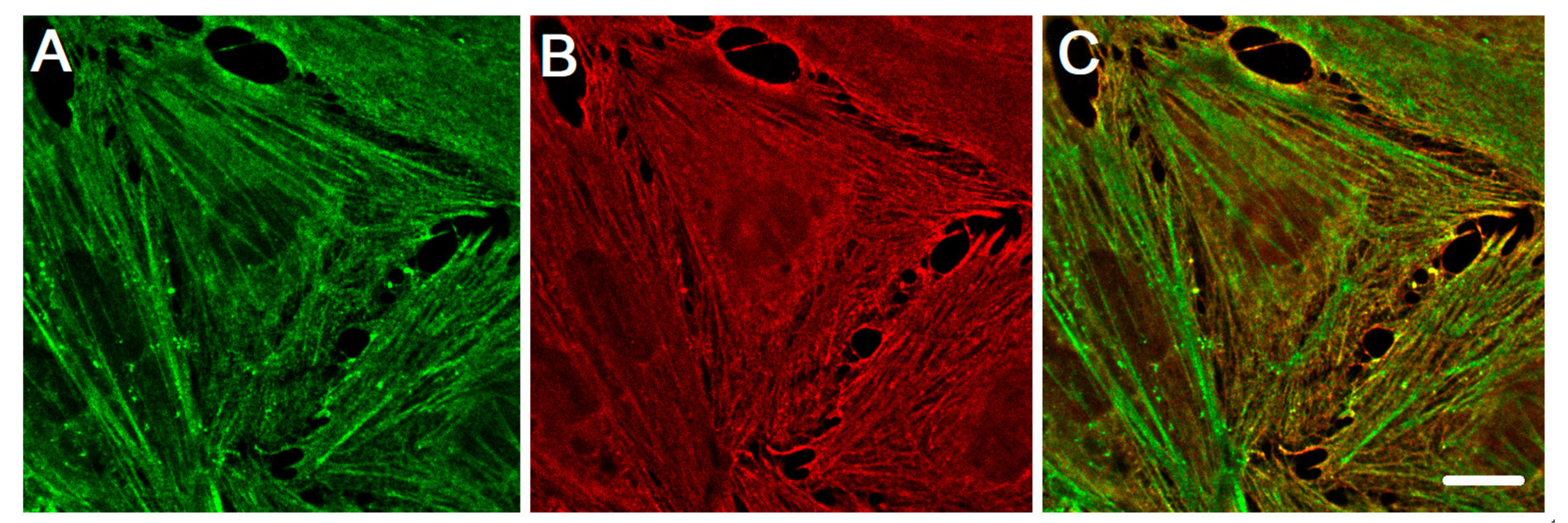
4. Crucial Involvement of Cytoplasmic Actins in the Endothelial Cell Motility and Angiogenesis
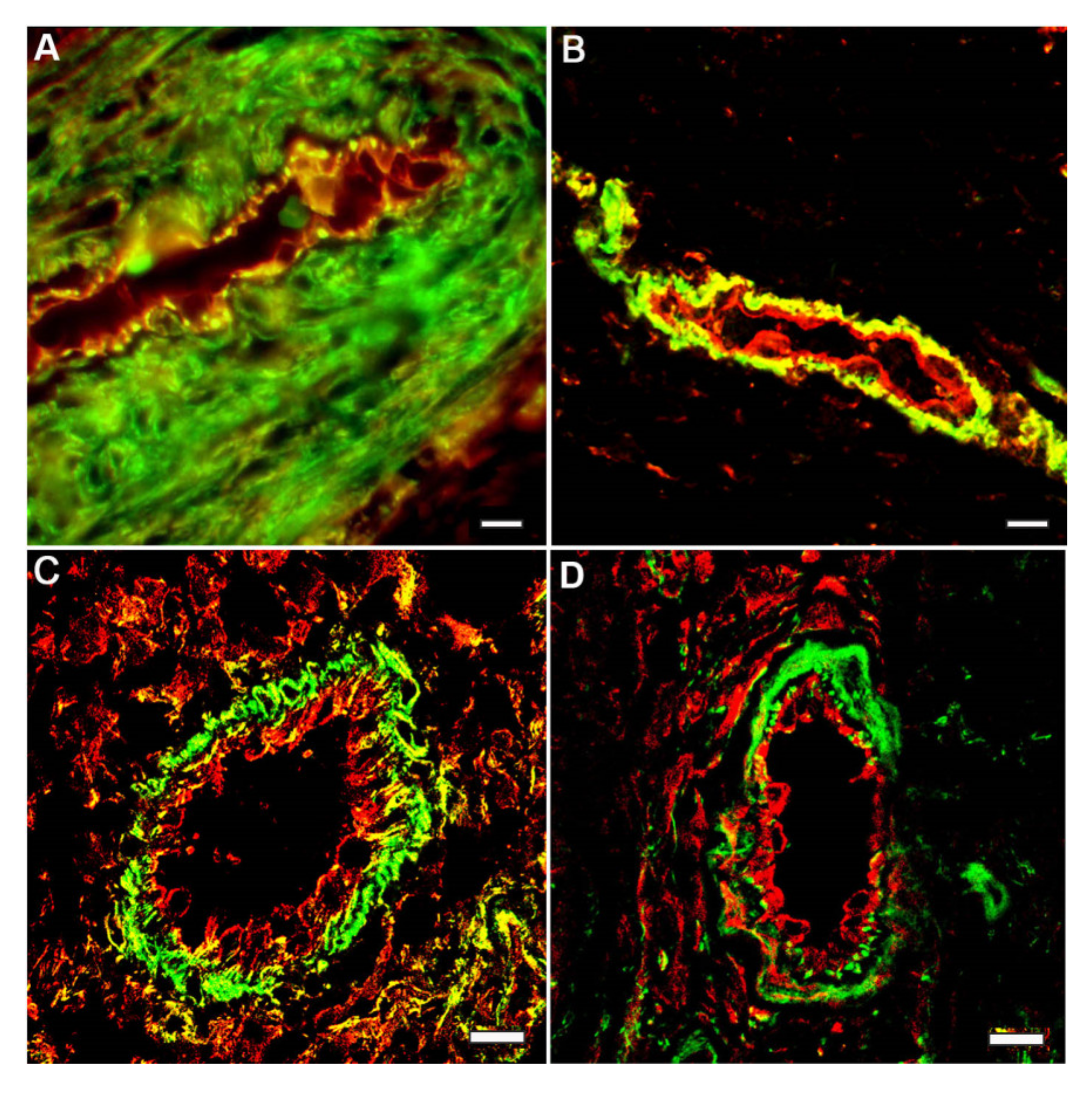
5. Endothelial-to-Mesenchymal Transition in Cardiovascular Diseases: More Questions Than Answers
6. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Perrin, B.J.; Ervasti, J.M. The actin gene family: Function follows isoform. Cytoskeleton 2010, 67, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Ampe, C.; Van Troys, M. Mammalian Actins: Isoform-Specific Functions and Diseases. In Handbook of Experimental Pharmacology; Springer: Cham, Switzerland, 2016. [Google Scholar] [CrossRef]
- Vandekerckhove, J.; Weber, K. At least six different actins are expressed in a higher mammal: An analysis based on the amino acid sequence of the amino-terminal tryptic peptide. J. Mol. Biol. 1978, 126, 783–802. [Google Scholar] [CrossRef]
- Vandekerckhove, J.; Leavitt, J.; Kakunaga, T.; Weber, K. Coexpression of a mutant beta-actin and the two normal beta- and gamma-cytoplasmic actins in a stably transformed human cell line. Cell 1980, 22, 893–899. [Google Scholar] [CrossRef]
- Otey, C.A.; Kalnoski, M.H.; Bulinski, J.C. Identification and quantification of actin isoforms in vertebrate cells and tissues. J. Cell. Biochem. 1987, 34, 113–124. [Google Scholar] [CrossRef]
- Sheterline, P.; Clayton, J.; Sparrow, J. Actin. Protein Profile 1995, 2, 1–103. [Google Scholar]
- Bunnell, T.M.; Burbach, B.J.; Shimizu, Y.; Ervasti, J.M. β-Actin specifically controls cell growth, migration, and the G-actin pool. Mol. Biol. Cell 2011, 22, 4047–4058. [Google Scholar] [CrossRef]
- Bunnell, T.M.; Ervasti, J.M. Delayed embryonic development and impaired cell growth and survival in Actg1 null mice. Cytoskeleton 2010, 67, 564–572. [Google Scholar] [CrossRef] [Green Version]
- Dugina, V.; Zwaenepoel, I.; Gabbiani, G.; Clement, S.; Chaponnier, C.; Clément, S.; Chaponnier, C. Beta and gamma-cytoplasmic actins display distinct distribution and functional diversity. J. Cell Sci. 2009, 122, 2980–2988. [Google Scholar] [CrossRef] [Green Version]
- Lechuga, S.; Baranwal, S.; Li, C.; Naydenov, N.G.; Kuemmerle, J.F.; Dugina, V.; Chaponnier, C.; Ivanov, A.I. Loss of γ-cytoplasmic actin triggers myofibroblast transition of human epithelial cells. Mol. Biol. Cell 2014, 25, 3133–3146. [Google Scholar] [CrossRef]
- Shum, M.S.Y.; Pasquier, E.; Po’uha, S.T.; O’Neill, G.M.; Chaponnier, C.; Gunning, P.W.; Kavallaris, M. γ-Actin regulates cell migration and modulates the ROCK signaling pathway. FASEB J. 2011, 25, 4423–4433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malek, N.; Mrówczyńska, E.; Michrowska, A.; Mazurkiewicz, E.; Pavlyk, I.; Mazur, A.J. Knockout of ACTB and ACTG1 with CRISPR/Cas9(D10A) technique shows that non-muscle β and γ actin are not equal in relation to human melanoma cells’ motility and focal adhesion formation. Int. J. Mol. Sci. 2020, 21, 2746. [Google Scholar] [CrossRef] [Green Version]
- Baranwal, S.; Naydenov, N.G.; Harris, G.; Dugina, V.; Morgan, K.G.; Chaponnier, C.; Ivanov, A.I. Nonredundant roles of cytoplasmic β- and γ-actin isoforms in regulation of epithelial apical junctions. Mol. Biol. Cell 2012, 23, 3542–3553. [Google Scholar] [CrossRef]
- Latham, S.L.; Chaponnier, C.; Dugina, V.; Couraud, P.; Grau, G.E.R.; Combes, V. Cooperation between β- and γ-cytoplasmic actins in the mechanical regulation of endothelial microparticle formation. FASEB J. 2013, 27, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Shakhov, A.S.; Verin, A.D.; Alieva, I.B. Reorganization of endothelial cells cytoskeleton during formation of functional monolayer in vitro. Cell Tissue Biol. 2014, 8, 138–151. [Google Scholar] [CrossRef]
- Shakhov, A.S.; Dugina, V.B.; Alieva, I.B. Reorganization of actin and microtubule systems in human vein endothelial cells during intercellular contact formation. Cell Tissue Biol. 2015, 9, 299–309. [Google Scholar] [CrossRef]
- Shakhov, A.S.; Dugina, V.B.; Alieva, I.B. Structural Features of Actin Cytoskeleton Required for Endotheliocyte Barrier Function. Biochemistry 2019, 84, 358–369. [Google Scholar] [CrossRef]
- Piccin, A.; Murphy, W.G.; Smith, O.P. Circulating microparticles: Pathophysiology and clinical implications. Blood Rev. 2007, 21, 157–171. [Google Scholar] [CrossRef]
- George, F.D. Microparticles in vascular diseases. Thromb. Res. 2008, 122, S55–S59. [Google Scholar] [CrossRef]
- Pasquier, E.; Tuset, M.-P.; Sinnappan, S.; Carnell, M.; Macmillan, A.; Kavallaris, M. γ-Actin plays a key role in endothelial cell motility and neovessel maintenance. Vasc. Cell 2015, 7, 2. [Google Scholar] [CrossRef] [Green Version]
- Alieva, I.B.; Zemskov, E.A.; Smurova, K.M.; Kaverina, I.N.; Verin, A.D. The leading role of microtubules in endothelial barrier dysfunction: Disassembly of peripheral microtubules leaves behind the cytoskeletal reorganization. J. Cell. Biochem. 2013, 114, 2258–2272. [Google Scholar] [CrossRef] [Green Version]
- Small, J.V.; Kaverina, I. Microtubules meet substrate adhesions to arrange cell polarity. Curr. Opin. Cell Biol. 2003, 15, 40–47. [Google Scholar] [CrossRef]
- Preciado López, M.; Huber, F.; Grigoriev, I.; Steinmetz, M.O.; Akhmanova, A.; Dogterom, M.; Koenderink, G.H. In vitro reconstitution of dynamic microtubules interacting with actin filament networks. In Methods in Enzymology; Academic Press Inc.: Cambridge, MA, USA, 2014; Volume 540, pp. 301–320. ISBN 9780123979247. [Google Scholar]
- Po‘uha, S.T.; Kavallaris, M. Gamma-actin is involved in regulating centrosome function and mitotic progression in cancer cells. Cell Cycle 2015, 14, 3908–3919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugina, V.; Alieva, I.; Khromova, N.; Kireev, I.; Gunning, P.W.; Kopnin, P. Interaction of microtubules with the actin cytoskeleton via cross-talk of EB1-containing +TIPs and γ-actin in epithelial cells. Oncotarget 2016, 7, 72699–72715. [Google Scholar] [CrossRef] [Green Version]
- Shagieva, G.S.; Alieva, I.B.; Chaponnier, C.; Dugina, V.B. Divergent Impact of Actin Isoforms on Division of Epithelial Cells. Biochemistry 2020, 85, 1072–1081. [Google Scholar] [CrossRef]
- Chen, A.; Ulloa Severino, L.; Panagiotou, T.C.; Moraes, T.F.; Yuen, D.A.; Lavoie, B.D.; Wilde, A. Inhibition of polar actin assembly by astral microtubules is required for cytokinesis. Nat. Commun. 2021, 12, 2409. [Google Scholar] [CrossRef] [PubMed]
- Dugina, V.; Khromova, N.; Rybko, V.; Blizniukov, O.; Shagieva, G.; Chaponnier, C.; Kopnin, B.; Kopnin, P. Tumor promotion by γ and suppression by β non-muscle actin isoforms. Oncotarget 2015, 6, 14556–14571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugina, V.; Shagieva, G.; Khromova, N.; Kopnin, P. Divergent impact of actin isoforms on cell cycle regulation. Cell Cycle 2018, 17, 2610–2621. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.J.; Tao, B.B.; Wang, M.J.; Jin, H.M.; Zhu, Y.C. PI3K p110α Isoform-Dependent Rho GTPase Rac1 Activation Mediates H2S-Promoted Endothelial Cell Migration via Actin Cytoskeleton Reorganization. PLoS ONE 2012, 7, e44590. [Google Scholar] [CrossRef]
- Markwald, R.; Eisenberg, C.; Eisenberg, L.; Trusk, T.; Sugi, Y. Epithelial-mesenchymal transformations in early avian heart development. Cells Tissues Organs 1996, 156, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Ranchoux, B.; Antigny, F.; Rucker-Martin, C.; Hautefort, A.; Péchoux, C.; Bogaard, H.J.; Dorfmüller, P.; Remy, S.; Lecerf, F.; Planté, S.; et al. Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 2015, 131, 1006–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Good, R.B.; Gilbane, A.J.; Trinder, S.L.; Denton, C.P.; Coghlan, G.; Abraham, D.J.; Holmes, A.M. Endothelial to Mesenchymal Transition Contributes to Endothelial Dysfunction in Pulmonary Arterial Hypertension. Am. J. Pathol. 2015, 185, 1850–1858. [Google Scholar] [CrossRef]
- Tang, H.; Babicheva, A.; McDermott, K.M.; Gu, Y.; Ayon, R.J.; Song, S.; Wang, Z.; Gupta, A.; Zhou, T.; Sun, X.; et al. Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L256–L275. [Google Scholar] [CrossRef]
- Wesseling, M.; Sakkers, T.R.; de Jager, S.C.A.; Pasterkamp, G.; Goumans, M.J. The morphological and molecular mechanisms of epithelial/endothelial-to-mesenchymal transition and its involvement in atherosclerosis. Vascul. Pharmacol. 2018, 106, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Dudley, A.C. Fine-tuning vascular fate during endothelial–mesenchymal transition. J. Pathol. 2017, 241, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Dejana, E.; Hirschi, K.K.; Simons, M. The molecular basis of endothelial cell plasticity. Nat. Commun. 2017, 8, 14361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Wang, F.; Zha, S.; Cao, Q.; Sheng, J.; Chen, S. SIRT1 inhibits TGF-β-induced endothelial-mesenchymal transition in human endothelial cells with Smad4 deacetylation. J. Cell. Physiol. 2018, 233, 9007–9014. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A. Endothelial to mesenchymal transition: Role in physiology and in the pathogenesis of human diseases. Physiol. Rev. 2019, 99, 1281–1324. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Duffhues, G.; García de Vinuesa, A.; ten Dijke, P. Endothelial-to-mesenchymal transition in cardiovascular diseases: Developmental signaling pathways gone awry. Dev. Dyn. 2018, 247, 492–508. [Google Scholar] [CrossRef] [Green Version]
- Reis, M.; Czupalla, C.J.; Ziegler, N.; Devraj, K.; Zinke, J.; Seidel, S.; Heck, R.; Thom, S.; Macas, J.; Bockamp, E.; et al. Endothelial Wnt/β-catenin signaling inhibits glioma angiogenesis and normalizes tumor blood vessels by inducing PDGF-B expression. J. Exp. Med. 2012, 209, 1611–1627. [Google Scholar] [CrossRef] [PubMed]
- Noseda, M.; McLean, G.; Niessen, K.; Chang, L.; Pollet, I.; Montpetit, R.; Shahidi, R.; Dorovini-Zis, K.; Li, L.; Beckstead, B.; et al. Notch Activation Results in Phenotypic and Functional Changes Consistent with Endothelial-to-Mesenchymal Transformation. Circ. Res. 2004, 94, 910–917. [Google Scholar] [CrossRef] [Green Version]
- Garside, V.C.; Chang, A.C.; Karsan, A.; Hoodless, P.A. Co-ordinating Notch, BMP, and TGF-β signaling during heart valve development. Cell. Mol. Life Sci. 2013, 70, 2899–2917. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Gong, J.; Dennery, P.A.; Yao, H. Endothelial-to-mesenchymal transition: Pathogenesis and therapeutic targets for chronic pulmonary and vascular diseases. Biochem. Pharmacol. 2019, 168, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lu, W.; Luo, X.; Liu, S.; Li, Y.; Zheng, Q.; Liu, W.; Wu, X.; Chen, Y.; Jiang, Q.; et al. Mitomycin C induces pulmonary vascular endothelial-to-mesenchymal transition and pulmonary veno-occlusive disease via Smad3-dependent pathway in rats. Br. J. Pharmacol. 2021, 178, 217–235. [Google Scholar] [CrossRef] [PubMed]
- Pardali, E.; Sanchez-Duffhues, G.; Gomez-Puerto, M.C.; ten Dijke, P. TGF-β-induced endothelial-mesenchymal transition in fibrotic diseases. Int. J. Mol. Sci. 2017, 18, 2157. [Google Scholar] [CrossRef] [Green Version]
- Souilhol, C.; Harmsen, M.C.; Evans, P.C.; Krenning, G. Endothelial-mesenchymal transition in atherosclerosis. Cardiovasc. Res. 2018, 114, 565–577. [Google Scholar] [CrossRef]
- Moonen, J.R.A.J.; Lee, E.S.; Schmidt, M.; Maleszewska, M.; Koerts, J.A.; Brouwer, L.A.; Van Kooten, T.G.; Van Luyn, M.J.A.; Zeebregts, C.J.; Krenning, G.; et al. Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc. Res. 2015, 108, 377–386. [Google Scholar] [CrossRef] [Green Version]
- Mahmoud, M.M.; Serbanovic-Canic, J.; Feng, S.; Souilhol, C.; Xing, R.; Hsiao, S.; Mammoto, A.; Chen, J.; Ariaans, M.; Francis, S.E.; et al. Shear stress induces endothelial-To-mesenchymal transition via the transcription factor Snail. Sci. Rep. 2017, 7, 3375. [Google Scholar] [CrossRef]
- Ten Dijke, P.; Egorova, A.D.; Goumans, M.J.T.H.; Poelmann, R.E.; Hierck, B.P. TGF-β signaling in endothelial-to-mesenchymal transition: The role of shear stress and primary cilia. Sci. Signal. 2012, 5. [Google Scholar] [CrossRef]
- Zhong, A.; Mirzaei, Z.; Simmons, C.A. The Roles of Matrix Stiffness and ß-Catenin Signaling in Endothelial-to-Mesenchymal Transition of Aortic Valve Endothelial Cells. Cardiovasc. Eng. Technol. 2018, 9, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Ji, Y.; Zhang, G.; Qu, Y.; Zhang, L.; Jiang, W. Ponatinib attenuates experimental pulmonary arterial hypertension by modulating Wnt signaling and vasohibin-2/vasohibin-1. Life Sci. 2016, 148, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kryczka, J.; Przygodzka, P.; Bogusz, H.; Boncela, J. HMEC-1 adopt the mixed amoeboid-mesenchymal migration type during EndMT. Eur. J. Cell Biol. 2017, 96, 289–300. [Google Scholar] [CrossRef]
- Jiang, X.; Li, T.; Li, B.; Wei, W.; Li, F.; Chen, S.; Xu, R.; Sun, K. SOX7 suppresses endothelial-to-mesenchymal transitions by enhancing VE-cadherin expression during outflow tract development. Clin. Sci. 2021, 135, 829–846. [Google Scholar] [CrossRef]
- Schevzov, G.; Lloyd, C.; Gunning, P. Impact of Altered Actin Gene Expression on Vinculin, Talin, Cell Spreading, and Motility. DNA Cell Biol. 1995, 14, 689–700. [Google Scholar] [CrossRef]
- Peckham, M.; Miller, G.; Wells, C.; Zicha, D.; Dunn, G.A. Specific changes to the mechanism of cell locomotion induced by overexpression of (β)-actin. J. Cell Sci. 2001, 114, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Shmerling, D.; Danzer, C.P.; Mao, X.; Boisclair, J.; Haffner, M.; Lemaistre, M.; Schuler, V.; Kaeslin, E.; Korn, R.; Bürki, K.; et al. Strong and ubiquitous expression of transgenes targeted into the β-actin locus by Cre/lox cassette replacement. Genesis 2005, 42, 229–235. [Google Scholar] [CrossRef]
- Belyantseva, I.A.; Perrin, B.J.; Sonnemann, K.J.; Zhu, M.; Stepanyan, R.; McGee, J.; Frolenkov, G.I.; Walsh, E.J.; Friderici, K.H.; Friedman, T.B.; et al. Gamma-actin is required for cytoskeletal maintenance but not development. Proc. Natl. Acad. Sci. USA 2009, 106, 9703–9708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tondeleir, D.; Lambrechts, A.; Müller, M.; Jonckheere, V.; Doll, T.; Vandamme, D.; Bakkali, K.; Waterschoot, D.; Lemaistre, M.; Debeir, O.; et al. Cells lacking β-actin are genetically reprogrammed and maintain conditional migratory capacity. Mol. Cell. Proteomics 2012, 11, 255–271. [Google Scholar] [CrossRef] [Green Version]
- Bergeron, S.E.; Zhu, M.; Thiem, S.M.; Friderici, K.H.; Rubenstein, P.A. Ion-dependent polymerization differences between mammalian beta- and gamma-nonmuscle actin isoforms. J. Biol. Chem. 2010, 285, 16087–16095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollard, T.D. Actin and actin-binding proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, a018226. [Google Scholar] [CrossRef] [Green Version]
- Boiero Sanders, M.; Antkowiak, A.; Michelot, A. Diversity from similarity: Cellular strategies for assigning particular identities to actin filaments and networks: Actin filaments and networks identities. Open Biol. 2020, 10, 200157. [Google Scholar] [CrossRef] [PubMed]
- Verrills, N.M.; Po’uha, S.T.; Liu, M.L.M.; Liaw, T.Y.E.; Larsen, M.R.; Ivery, M.T.; Marshall, G.M.; Gunning, P.W.; Kavallaris, M. Alterations in γ-actin and tubulin-targeted drug resistance in childhood leukemia. J. Natl. Cancer Inst. 2006, 98, 1363–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dugina, V.B.; Shagieva, G.S.; Shakhov, A.S.; Alieva, I.B. The Cytoplasmic Actins in the Regulation of Endothelial Cell Function. Int. J. Mol. Sci. 2021, 22, 7836. https://doi.org/10.3390/ijms22157836
Dugina VB, Shagieva GS, Shakhov AS, Alieva IB. The Cytoplasmic Actins in the Regulation of Endothelial Cell Function. International Journal of Molecular Sciences. 2021; 22(15):7836. https://doi.org/10.3390/ijms22157836
Chicago/Turabian StyleDugina, Vera B., Galina S. Shagieva, Anton S. Shakhov, and Irina B. Alieva. 2021. "The Cytoplasmic Actins in the Regulation of Endothelial Cell Function" International Journal of Molecular Sciences 22, no. 15: 7836. https://doi.org/10.3390/ijms22157836
APA StyleDugina, V. B., Shagieva, G. S., Shakhov, A. S., & Alieva, I. B. (2021). The Cytoplasmic Actins in the Regulation of Endothelial Cell Function. International Journal of Molecular Sciences, 22(15), 7836. https://doi.org/10.3390/ijms22157836