Evaluation of Droplet Digital Polymerase Chain Reaction (ddPCR) for the Absolute Quantification of Aspergillus species in the Human Airway

 ,
,
Abstract
1. Introduction
2. Results
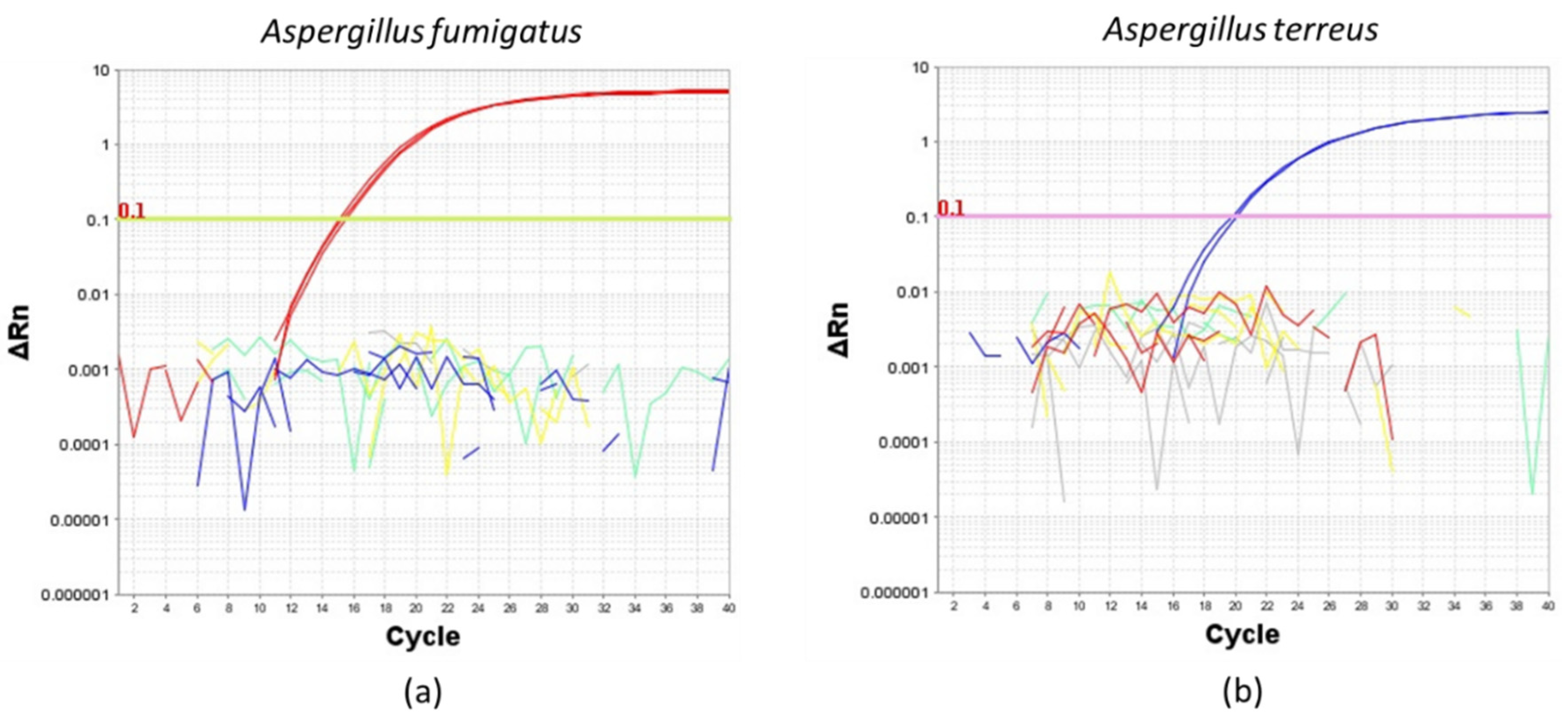
2.1. Assessment for the Specificity of the A. fumigatus and A. terreus Duplex TaqMan Primer and Probe Set Used for ddPCR Evaluation
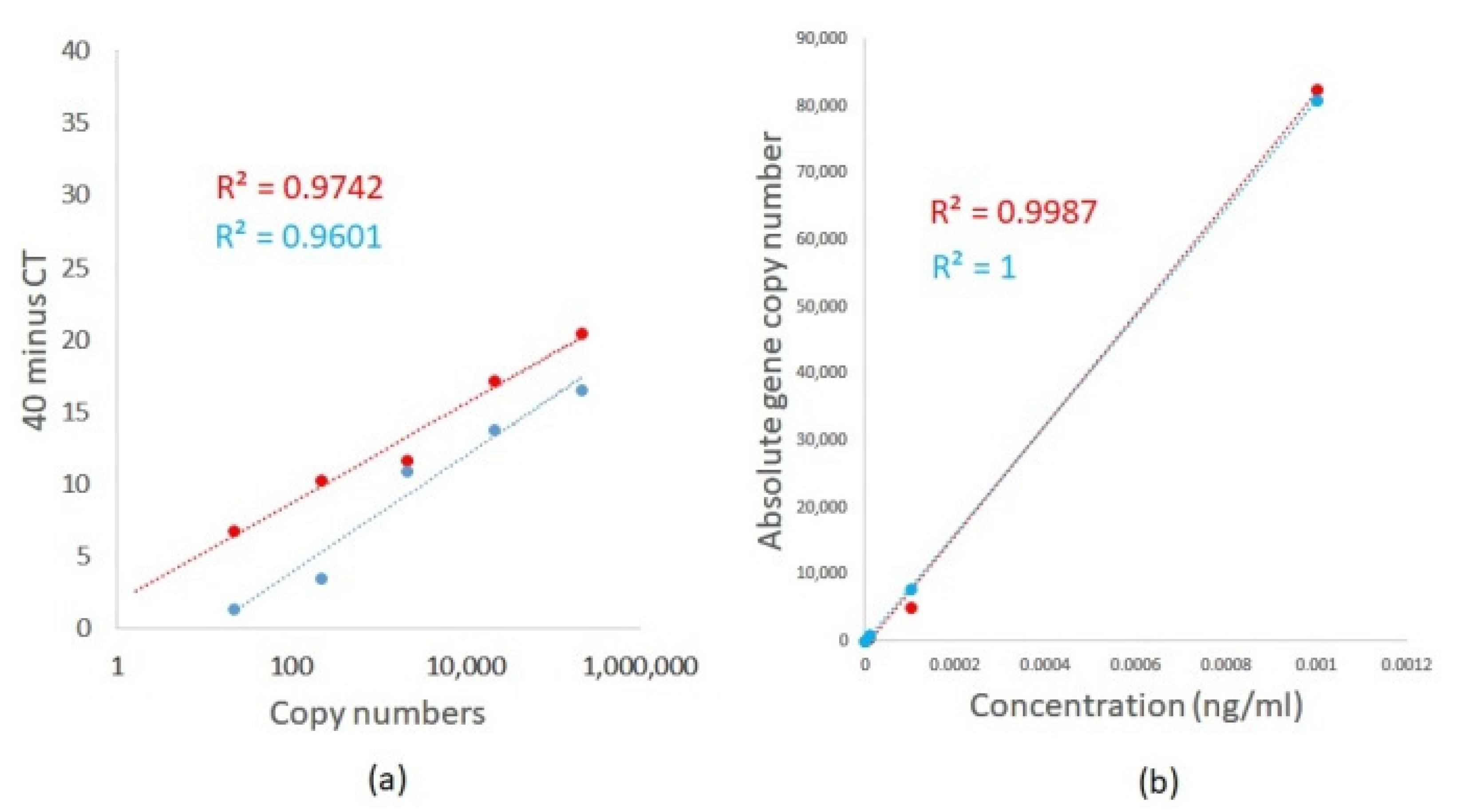
2.2. Evaluation of Limits for the Detection and Quantification of A. fumigatus and A. terreus Using TaqMan Duplex Sets by qPCR and ddPCR
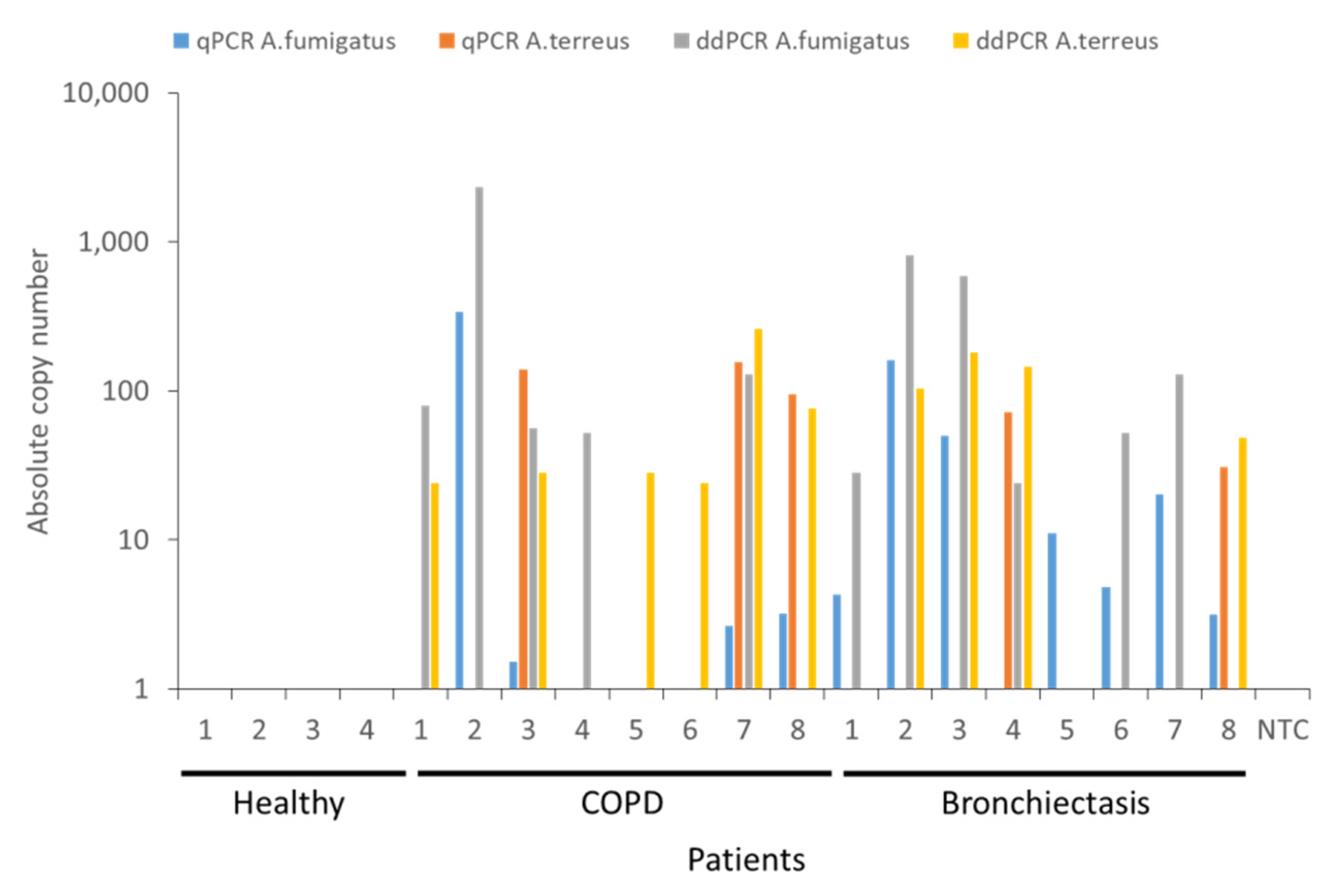
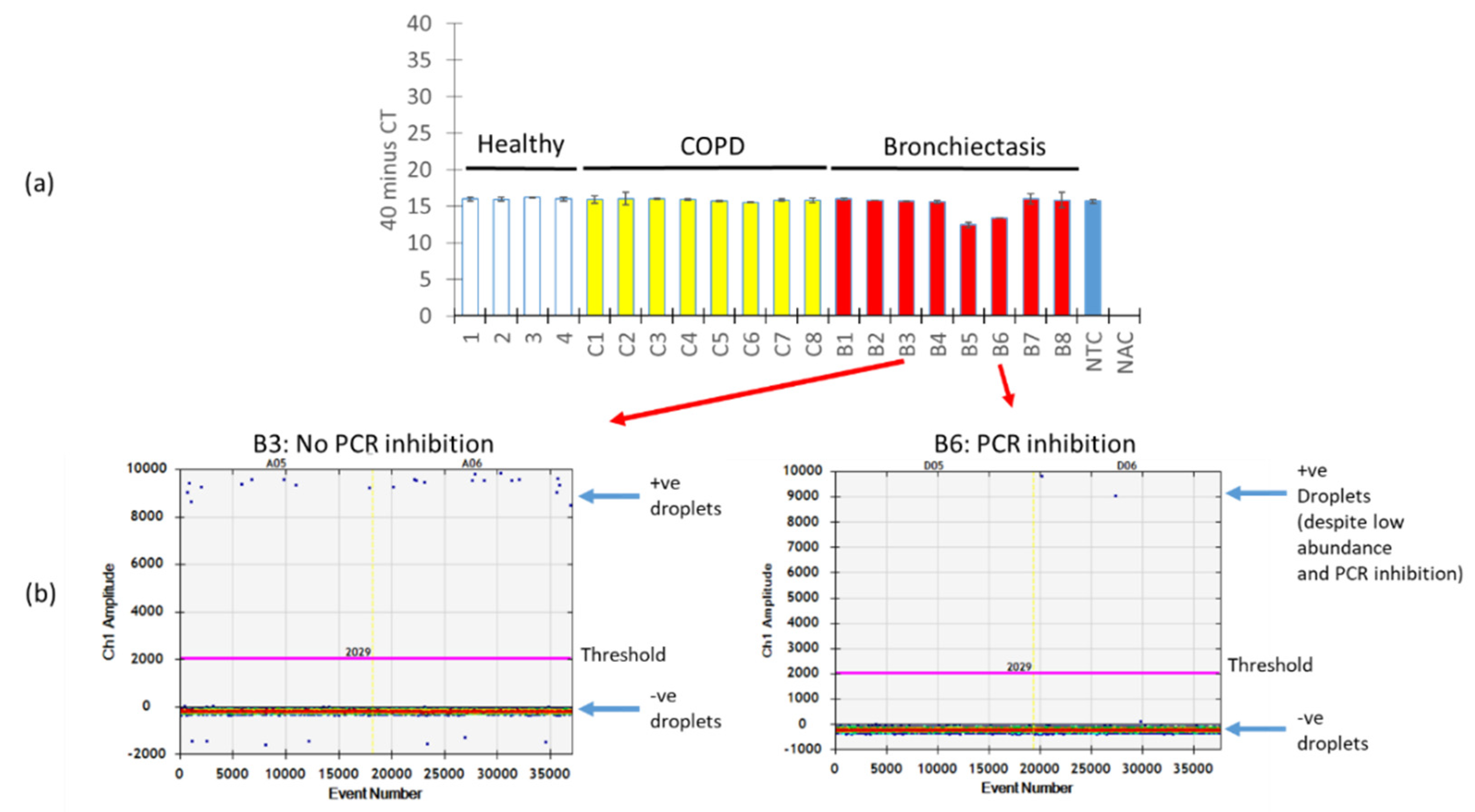
2.3. Quantification of Airway A. fumigatus and A. terreus in Respiratory Specimens Obtained from Non-Diseased (Healthy) Individuals and Patients with Chronic Obstructive Pulmonary Disease (COPD) and Bronchiectasis by qPCR and ddPCR
2.4. Quantification of Airway A. fumigatus by ddPCR is Resistant to PCR Inhibition
3. Discussion
4. Materials and Methods
4.1. Design of Primers and Probes:
Aspergillus
4.2. Growth and Harvesting of Fungal Cultures
4.3. Cloning of A. Fumigatus and A. Terreus ITS
4.4. Study Population
4.5. Ethical Approval
4.6. Specimen (Sputum) Collection and DNA Extraction
4.7. Quantitative-PCR (qPCR) and Digital Droplet PCR (ddPCR) Detection of Aspergillus Species
4.8. Data Analysis (qPCR)
4.9. Data Analysis (ddPCR)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| % | percentage |
| +ve | positive |
| < | less than |
| > | greater than |
| ± | plus/minus |
| ∆Rn | delta normalized reporter value |
| °C | degrees Celsius |
| µL | microliter |
| µM | micro-molar |
| 6-FAM | 6-carboxyfluorescein |
| Asp | Aspergillus |
| BHQ1 | Black Hole Quencher 1 |
| BLAST | Basic Local Alignment Search Tool |
| BMI | body mass index |
| B | bronchiectasis |
| BSI | Bronchiectasis Severity Index |
| CAT | COPD assessment test |
| COPDCF | chronic obstructive pulmonary diseaseCystic Fibrosis |
| Cqs | Quantification cycle |
| ddPCR | droplet digital PCR |
| DNA | deoxyribonucleic acid |
| FEV1 | forced expiratory volume in the first second |
| Fg | femtogram |
| FVC | forced vital capacity |
| g | gravitational force |
| HRCT | high-resolution computed tomography |
| IPC | internal positive control |
| IQR | interquartile range |
| ITS | internal transcribed spacer |
| mins | Minutes |
| Mod | modification |
| N | sample size |
| next-gen | next-generation |
| ng | nanogram |
| nM | nano-molar |
| No. | Number |
| PBS | phosphate-buffered saline |
| pg | picogram |
| Post BD | post bronchodilator |
| Pte Ltd. | Private Limited |
| qPCR | quantitative polymerase chain reaction |
| RNAi | RNA interference |
| rpm | rounds per minute |
| rRNA | ribosomal ribonucleic acid |
| Secs | Seconds |
| siRNA | small interfering ribonucleic acid |
| SNP | single nucleotide polymorphism |
| Taq | Thermus aquaticus |
| -ve | Negative |
Appendix A


References
- Mac Aogain, M.; Chandrasekaran, R.; Lim, A.Y.H.; Low, T.B.; Tan, G.L.; Hassan, T.; Ong, T.H.; Ng, A.H.Q.; Bertrand, D.; Koh, J.Y.; et al. Immunological corollary of the pulmonary mycobiome in bronchiectasis: The CAMEB study. Eur. Respir. J. 2018, 52, 1800766. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.J.; Wissel, M.C.; Grantham, K.J.; Petraitiene, R.; Petraitis, V.; Kasai, M.; Francesconi, A.; Cotton, M.P.; Hughes, J.E.; Greene, L.; et al. Molecular detection and species-specific identification of medically important Aspergillus species by real-time PCR in experimental invasive pulmonary aspergillosis. J. Clin. Microbiol. 2011, 49, 4150–4157. [Google Scholar] [CrossRef] [PubMed]
- Poh, T.Y.; Tiew, P.Y.; Lim, A.Y.H.; Thng, K.X.; Ali, N.A.B.M.; Narayana, J.K.; Aogáin, M.M.; Tien, Z.; Chew, W.M.; Chan, A.K.W.; et al. Increased chitotriosidase is associated with Aspergillus and frequent exacerbations in south-east asians with bronchiectasis. CHEST 2020. [Google Scholar] [CrossRef]
- White, P.L.; Barnes, R.A.; Springer, J.; Klingspor, L.; Cuenca-Estrella, M.; Morton, C.O.; Lagrou, K.; Bretagne, S.; Melchers, W.J.; Mengoli, C.; et al. Clinical Performance of Aspergillus PCR for Testing Serum and Plasma: A Study by the European Aspergillus PCR Initiative. J. Clin. Microbiol. 2015, 53, 2832–2837. [Google Scholar] [CrossRef] [PubMed]
- Buess, M.; Cathomas, G.; Halter, J.; Junker, L.; Grendelmeier, P.; Tamm, M.; Stolz, D. Aspergillus-PCR in bronchoalveolar lavage for detection of invasive pulmonary aspergillosis in immunocompromised patients. BMC Infect. Dis. 2012, 12, 237. [Google Scholar] [CrossRef]
- Mac Aogain, M.; Tiew, P.Y.; Lim, A.Y.H.; Low, T.B.; Tan, G.L.; Hassan, T.; Ong, T.H.; Pang, S.L.; Lee, Z.Y.; Gwee, X.W.; et al. Distinct “Immunoallertypes” of Disease and High Frequencies of Sensitization in Non-Cystic Fibrosis Bronchiectasis. Am. J. Respir. Crit. Care Med. 2019, 199, 842–853. [Google Scholar] [CrossRef]
- Johnson, G.L.; Bibby, D.F.; Wong, S.; Agrawal, S.G.; Bustin, S.A. A MIQE-compliant real-time PCR assay for Aspergillus detection. PLoS ONE 2012, 7, e40022. [Google Scholar] [CrossRef]
- Brankatschk, R.; Bodenhausen, N.; Zeyer, J.; Burgmann, H. Simple absolute quantification method correcting for quantitative PCR efficiency variations for microbial community samples. Appl. Environ. Microbiol. 2012, 78, 4481–4489. [Google Scholar] [CrossRef]
- Bustin, S.A.; Nolan, T. Pitfalls of quantitative real-time reverse-transcription polymerase chain reaction. J. Biomol. Tech. 2004, 15, 155–166. [Google Scholar]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hiddessen, A.L.; Legler, T.C.; et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 2012, 84, 1003–1011. [Google Scholar] [CrossRef]
- Mazaika, E.; Homsy, J. Digital Droplet PCR: CNV Analysis and Other Applications. Curr. Protoc. Hum. Genet. 2014, 82, 7.24.1–7.24.13. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Bhattacharya, S.; Kotsopoulos, S.; Olson, J.; Taly, V.; Griffiths, A.D.; Link, D.R.; Larson, J.W. Multiplex digital PCR: Breaking the one target per color barrier of quantitative PCR. Lab Chip 2011, 11, 2167–2174. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Mac Aogain, M.; Morales, R.F.; Tiew, P.Y.; Chotirmall, S.H. Optimisation and Benchmarking of Targeted Amplicon Sequencing for Mycobiome Analysis of Respiratory Specimens. Int. J. Mol. Sci. 2019, 20, 4991. [Google Scholar] [CrossRef]
- Cui, L.; Morris, A.; Ghedin, E. The human mycobiome in health and disease. Genome Med. 2013, 5, 63. [Google Scholar] [CrossRef]
- El-Jurdi, N.; Ghannoum, M.A. The Mycobiome: Impact on Health and Disease States. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Suhr, M.J.; Hallen-Adams, H.E. The human gut mycobiome: Pitfalls and potentials--a mycologist’s perspective. Mycologia 2015, 107, 1057–1073. [Google Scholar] [CrossRef]
- Tiew, P.Y.; Mac Aogain, M.; Ali, N.; Thng, K.X.; Goh, K.; Lau, K.J.X.; Chotirmall, S.H. The Mycobiome in Health and Disease: Emerging Concepts, Methodologies and Challenges. Mycopathologia 2020. [Google Scholar] [CrossRef]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.R.; Tullis, E.; Castanos, C.; Castellani, C.; et al. The future of cystic fibrosis care: A global perspective. Lancet Respir. Med. 2020, 8, 65–124. [Google Scholar] [CrossRef]
- Chalmers, J.D.; Chotirmall, S.H. Bronchiectasis: New therapies and new perspectives. Lancet Respir. Med. 2018, 6, 715–726. [Google Scholar] [CrossRef]
- Goh, K.J.; Yii, A.C.A.; Lapperre, T.S.; Chan, A.K.; Chew, F.T.; Chotirmall, S.H.; Koh, M.S. Sensitization to Aspergillus species is associated with frequent exacerbations in severe asthma. J. Asthma Allergy 2017, 10, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Chotirmall, S.H.; Gellatly, S.L.; Budden, K.F.; Mac Aogain, M.; Shukla, S.D.; Wood, D.L.; Hugenholtz, P.; Pethe, K.; Hansbro, P.M. Microbiomes in respiratory health and disease: An Asia-Pacific perspective. Respirology 2017, 22, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Auchtung, T.A.; Fofanova, T.Y.; Stewart, C.J.; Nash, A.K.; Wong, M.C.; Gesell, J.R.; Auchtung, J.M.; Ajami, N.J.; Petrosino, J.F. Investigating Colonization of the Healthy Adult Gastrointestinal Tract by Fungi. mSphere 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Tipton, L.; Ghedin, E.; Morris, A. The lung mycobiome in the next-generation sequencing era. Virulence 2017, 8, 334–341. [Google Scholar] [CrossRef]
- Nguyen, L.D.; Viscogliosi, E.; Delhaes, L. The lung mycobiome: An emerging field of the human respiratory microbiome. Front. Microbiol. 2015, 6, 89. [Google Scholar] [CrossRef]
- Cui, L.; Lucht, L.; Tipton, L.; Rogers, M.B.; Fitch, A.; Kessinger, C.; Camp, D.; Kingsley, L.; Leo, N.; Greenblatt, R.M.; et al. Topographic diversity of the respiratory tract mycobiome and alteration in HIV and lung disease. Am. J. Respir. Crit. Care Med. 2015, 191, 932–942. [Google Scholar] [CrossRef]
- Hager, C.L.; Ghannoum, M.A. The mycobiome in HIV. Curr. Opin. HIV AIDS 2018, 13, 69–72. [Google Scholar] [CrossRef]
- McTaggart, L.R.; Copeland, J.K.; Surendra, A.; Wang, P.W.; Husain, S.; Coburn, B.; Guttman, D.S.; Kus, J.V. Mycobiome Sequencing and Analysis Applied to Fungal Community Profiling of the Lower Respiratory Tract During Fungal Pathogenesis. Front. Microbiol. 2019, 10, 512. [Google Scholar] [CrossRef]
- Chotirmall, S.H.; Al-Alawi, M.; Mirkovic, B.; Lavelle, G.; Logan, P.M.; Greene, C.M.; McElvaney, N.G. Aspergillus-associated airway disease, inflammation, and the innate immune response. Biomed. Res. Int. 2013, 2013, 723129. [Google Scholar] [CrossRef]
- Budden, K.F.; Shukla, S.D.; Rehman, S.F.; Bowerman, K.L.; Keely, S.; Hugenholtz, P.; Armstrong-James, D.P.H.; Adcock, I.M.; Chotirmall, S.H.; Chung, K.F.; et al. Functional effects of the microbiota in chronic respiratory disease. Lancet Respir. Med. 2019, 7, 907–920. [Google Scholar] [CrossRef]
- Chotirmall, S.H.; Martin-Gomez, M.T. Aspergillus Species in Bronchiectasis: Challenges in the Cystic Fibrosis and Non-cystic Fibrosis Airways. Mycopathologia 2018, 183, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Chotirmall, S.H.; McElvaney, N.G. Fungi in the cystic fibrosis lung: Bystanders or pathogens? Int. J. Biochem. Cell Biol. 2014, 52, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, C.A.; Chotirmall, S.H.; Renwick, J.; Hassan, T.; Low, T.B.; Bergsson, G.; Eshwika, A.; Bennett, K.; Dunne, K.; Greene, C.M.; et al. The effect of Aspergillus fumigatus infection on vitamin D receptor expression in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 999–1007. [Google Scholar] [CrossRef]
- Leung, J.M.; Tiew, P.Y.; Mac Aogain, M.; Budden, K.F.; Yong, V.F.; Thomas, S.S.; Pethe, K.; Hansbro, P.M.; Chotirmall, S.H. The role of acute and chronic respiratory colonization and infections in the pathogenesis of COPD. Respirology 2017, 22, 634–650. [Google Scholar] [CrossRef]
- Yii, A.C.; Koh, M.S.; Lapperre, T.S.; Tan, G.L.; Chotirmall, S.H. The emergence of Aspergillus species in chronic respiratory disease. Front. Biosci. 2017, 9, 127–138. [Google Scholar] [CrossRef]
- Chotirmall, S.H.; Branagan, P.; Gunaratnam, C.; McElvaney, N.G. Aspergillus/allergic bronchopulmonary aspergillosis in an Irish cystic fibrosis population: A diagnostically challenging entity. Respir. Care 2008, 53, 1035–1041. [Google Scholar]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003–1005. [Google Scholar] [CrossRef]
- Sanders, R.; Huggett, J.F.; Bushell, C.A.; Cowen, S.; Scott, D.J.; Foy, C.A. Evaluation of digital PCR for absolute DNA quantification. Anal. Chem. 2011, 83, 6474–6484. [Google Scholar] [CrossRef]
- Hijano, D.R.; Brazelton de Cardenas, J.; Maron, G.; Garner, C.D.; Ferrolino, J.A.; Dallas, R.H.; Gu, Z.; Hayden, R.T. Clinical correlation of influenza and respiratory syncytial virus load measured by digital PCR. PLoS ONE 2019, 14, e0220908. [Google Scholar] [CrossRef] [PubMed]
- Sze, M.A.; Abbasi, M.; Hogg, J.C.; Sin, D.D. A comparison between droplet digital and quantitative PCR in the analysis of bacterial 16S load in lung tissue samples from control and COPD GOLD 2. PLoS ONE 2014, 9, e110351. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T.; et al. The digital MIQE guidelines: Minimum Information for Publication of Quantitative Digital PCR Experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Nixon, G.; Garson, J.A.; Grant, P.; Nastouli, E.; Foy, C.A.; Huggett, J.F. Comparative study of sensitivity, linearity, and resistance to inhibition of digital and nondigital polymerase chain reaction and loop mediated isothermal amplification assays for quantification of human cytomegalovirus. Anal. Chem. 2014, 86, 4387–4394. [Google Scholar] [CrossRef]
- Imbert, S.; Meyer, I.; Palous, M.; Brossas, J.Y.; Uzunov, M.; Touafek, F.; Gay, F.; Trosini-Desert, V.; Fekkar, A. Aspergillus PCR in Bronchoalveolar Lavage Fluid for the Diagnosis and Prognosis of Aspergillosis in Patients With Hematological and Non-hematological Conditions. Front. Microbiol. 2018, 9, 1877. [Google Scholar] [CrossRef]
- Fonceca, A.M.; Chopra, A.; Levy, A.; Noakes, P.S.; Poh, M.W.; Bear, N.L.; Prescott, S.; Everard, M.L. Infective respiratory syncytial virus is present in human cord blood samples and most prevalent during winter months. PLoS ONE 2017, 12, e0173738. [Google Scholar] [CrossRef]
- Schwartz, S.L.; Lowen, A.C. Droplet digital PCR: A novel method for detection of influenza virus defective interfering particles. J. Virol. Methods 2016, 237, 159–165. [Google Scholar] [CrossRef]
- Vogelmeier, C.F.; Criner, G.J.; Martinez, F.J.; Anzueto, A.; Barnes, P.J.; Bourbeau, J.; Celli, B.R.; Chen, R.; Decramer, M.; Fabbri, L.M.; et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report. GOLD Executive Summary. Am. J. Respir. Crit. Care Med. 2017, 195, 557–582. [Google Scholar] [CrossRef]
- Chalmers, J.D.; Goeminne, P.; Aliberti, S.; McDonnell, M.J.; Lonni, S.; Davidson, J.; Poppelwell, L.; Salih, W.; Pesci, A.; Dupont, L.J.; et al. The bronchiectasis severity index. An international derivation and validation study. Am. J. Respir. Crit. Care Med. 2014, 189, 576–585. [Google Scholar] [CrossRef]
- Pasteur, M.C.; Bilton, D.; Hill, A.T. British Thoracic Society guideline for non-CFbronchiectasis. Thorax 2010, 65, i1–i58. [Google Scholar] [CrossRef]
- Naidich, D.P.; McCauley, D.I.; Khouri, N.F.; Stitik, F.P.; Siegelman, S.S. Computed tomography of bronchiectasis. J. Comput. Assist. Tomogr. 1982, 6, 437–444. [Google Scholar] [CrossRef]
- Chotirmall, S.H.; O’Donoghue, E.; Bennett, K.; Gunaratnam, C.; O’Neill, S.J.; McElvaney, N.G. Sputum Candida albicans presages FEV(1) decline and hospital-treated exacerbations in cystic fibrosis. Chest 2010, 138, 1186–1195. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
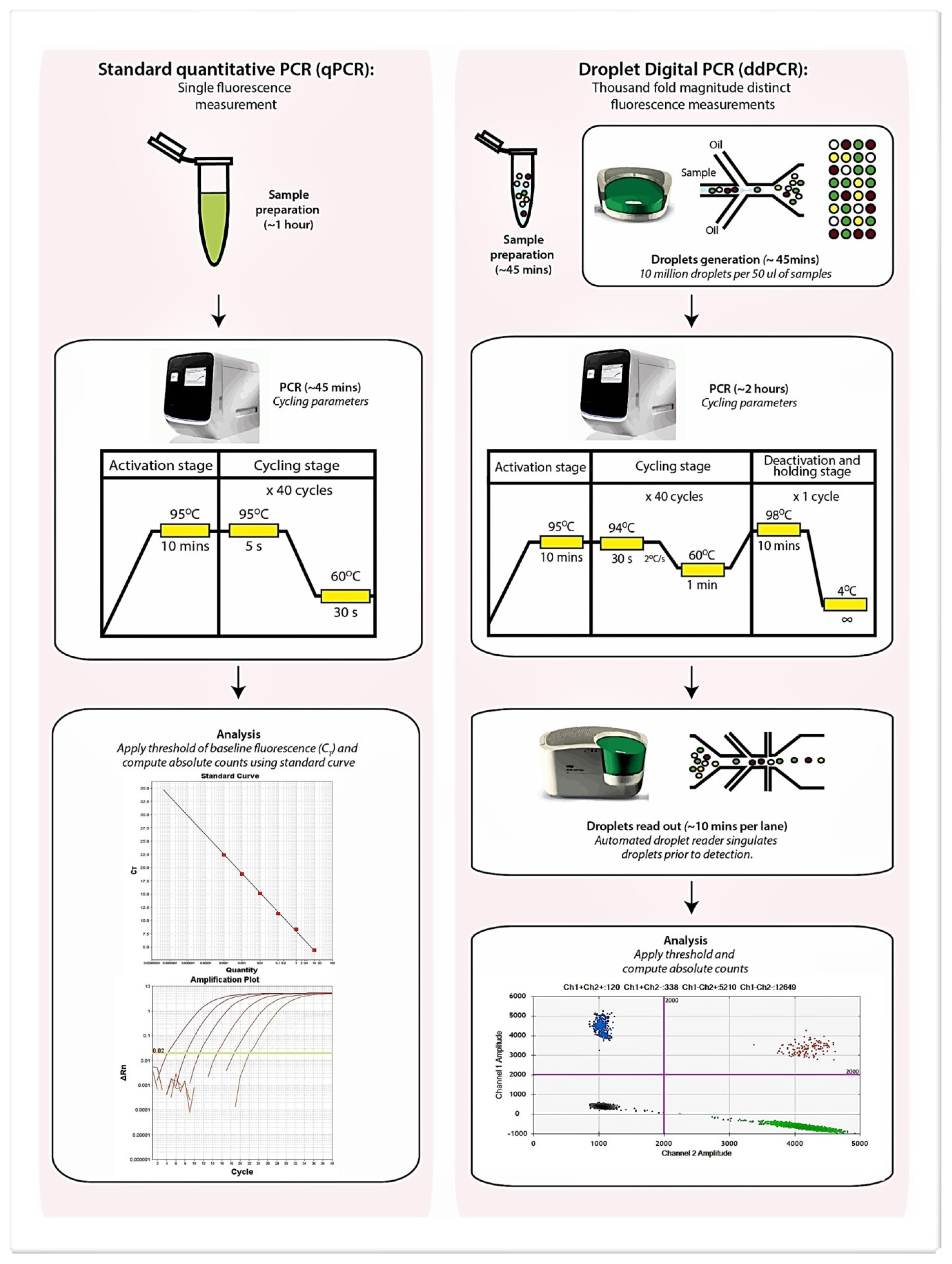
| Real-Time Quantitative PCR | Droplet Digital PCR | |
|---|---|---|
| Overview | Measures PCR amplification as it occurs | Measures the fraction of positive and negative replicates to determine absolute copies |
| Quantitative measurement | Yes | Yes |
| Methods of data collection | Data is collected during the exponential growth (log) phase of the PCR reaction when the quantity of the PCR product is directly proportional to the amount of template nucleic acid | PCR reaction is partitioned into thousands of individual real-time PCR reactions prior to amplification and data only acquired at the end-point |
| Method of calculation | Targets with unknown quantity are compared to a standard curve with known quantities and a value extrapolated | The fraction of positive and negative reactions are used to generate an absolute value for the exact number of target molecules within a sample according to a Poisson distribution statistical algorithm |
| Relative or Absolute quantification | Both, however a standard curve with known absolute quantities of the target is needed for absolute quantification | Absolute, ddPCR provides an absolute count of target DNA copies per input sample without the need for standard curves |
| Reproducibility | Moderate as quantification can be influenced by PCR efficiency bias between runs | High |
| Single-plex or multiplex | Both, up to 5-plex | Both, up to 5-plex [13] |
| Sensitivity | Moderate: with detection limit from 1 to 10 | High: with a detection limit as low as 1 in 2000 |
| Other advantages |
|
|
| Applications |
|
|
| Standards | Aspergillus fumigatus | Aspergillus terreus | ||||
|---|---|---|---|---|---|---|
| Plasmid Concentration, ng/mL (fg/mL) | 40 Minus Cqs | Standard Error | Absolute Copy Number | 40 Minus Cqs | Standard Error | Absolute Copy Number |
| 0.001 (1000 fg/mL) | 20.46 | 1.51 | 216,516.79 | 16.53 | 0.13 | 215,988.54 |
| 0.0001 (100 fg/mL) | 17.18 | 1.63 | 21,651.68 | 13.82 | 0.5 | 21,598.854 |
| 0.00001 (10 fg/mL) | 11.69 | 0.91 | 2165.17 | 10.92 | 0.19 | 2159.8854 |
| 0.000001(1 fg/mL) | 10.22 | 0.06 | 216.52 | 3.45 | 0.85 | 215.98854 |
| 0.0000001 (0.1 fg/mL) | 6.77 | 0.07 | 21.65 | 1.4 | 1.98 | 21.598854 |
| Aspergillus fumigatus | Aspergillus terreus | |
|---|---|---|
| Plasmid Concentration, ng/mL (fg/mL) | Absolute Copy Number | Absolute Copy Number |
| 0.001 (1000 fg/mL) | 82,480 | 80,900 |
| 0.0001 (100 fg/mL) | 5140 | 7940 |
| 0.00001 (10 fg/mL) | 438 | 886 |
| 0.000001 (1 fg/mL) | 68 | 172 |
| 0.0000001 (0.1fg/mL) | 20 | 40 |
| Oligo Name | Sequence (5′ to 3′) | Amplicon | 5′ Mod | 3′ Mod |
|---|---|---|---|---|
| Aspergillus fumigatus forward primer | TTGTCACCTGCTCTGTAGGC | 83 bp | None | None |
| Aspergillus fumigatus reverse primer | TCCCTACCTGATCCGAGGTC | None | None | |
| Aspergillus fumigatus probe | CCGGCGCCAGCCGACACCCA | 6-FAM | BHQ1 | |
| Aspergillus terreus forward primer | CATTACCGAGTGCGGGTCTTTA | 70 bp | None | None |
| Aspergillus terreus reverse primer | CCCGCCGAAGCAACAAG | None | None | |
| Aspergillus terreus probe | CCCAACCTCCCACCCGTGACTATTG | HEX | BHQ1 | |
| Pan Aspergillus ITS forward primer | CGGAAGGATCATTACCGAGT | Unknown | None | None |
| Pan Aspergillus ITS reverse primer | CCTACCTGATCCGAGGTCAA | None | None |
| Characteristics | Non-Diseased (Healthy, n = 4) | COPD (n = 8) | Bronchiectasis (n = 8) |
|---|---|---|---|
| Age (years): Median (IQR) | 64 (63–66) | 71 (68–74) | 67 (58–74) |
| Gender (male): n (%) | 1 (25%) | 8 (100%) | 4 (50%) |
| BMI (kg/m2): Median (IQR) | 23 (21.3–24.6) | 24.3 (22.5–28.6) | 18.3 (16.8–20.5) |
| Smoking history: n (%) | |||
| Never–smoker | 4 (100%) | 0 (0) | 7 (87.5%) |
| Current smoker | 0 (0%) | 3 (37.5%) | 0 (0%) |
| Ex-smoker | 0 (0%) | 5 (62.5%) | 1 (12.5%) |
| COPD assessment test (CAT): Median IQR) | NA | 15 (7.3–24.5) | NA |
| Post BD FEV1 (% predicted): Medium (IQR) | NA | 37.5 (32.8–38) | 57.5 (44–80) |
| Post BD FEV1/FVC (% predicted): Medium (IQR) | NA | 44.5 (39.5–49.8) | 79 (78–81) |
| BSI score | NA | NA | 8 (6.6–16) |
| No. of exacerbations in year preceding recruitment: Median (IQR) | NA | 1 (1–1) | 3 (0–4) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poh, T.Y.; Ali, N.A.B.M.; Chan, L.L.Y.; Tiew, P.Y.; Chotirmall, S.H. Evaluation of Droplet Digital Polymerase Chain Reaction (ddPCR) for the Absolute Quantification of Aspergillus species in the Human Airway. Int. J. Mol. Sci. 2020, 21, 3043. https://doi.org/10.3390/ijms21093043
Poh TY, Ali NABM, Chan LLY, Tiew PY, Chotirmall SH. Evaluation of Droplet Digital Polymerase Chain Reaction (ddPCR) for the Absolute Quantification of Aspergillus species in the Human Airway. International Journal of Molecular Sciences. 2020; 21(9):3043. https://doi.org/10.3390/ijms21093043
Chicago/Turabian StylePoh, Tuang Yeow, Nur A’tikah Binte Mohamed Ali, Louisa L.Y. Chan, Pei Yee Tiew, and Sanjay H. Chotirmall. 2020. "Evaluation of Droplet Digital Polymerase Chain Reaction (ddPCR) for the Absolute Quantification of Aspergillus species in the Human Airway" International Journal of Molecular Sciences 21, no. 9: 3043. https://doi.org/10.3390/ijms21093043
APA StylePoh, T. Y., Ali, N. A. B. M., Chan, L. L. Y., Tiew, P. Y., & Chotirmall, S. H. (2020). Evaluation of Droplet Digital Polymerase Chain Reaction (ddPCR) for the Absolute Quantification of Aspergillus species in the Human Airway. International Journal of Molecular Sciences, 21(9), 3043. https://doi.org/10.3390/ijms21093043