Abstract
Abdominal aortic aneurysm (AAA) refers to the localized dilatation of the infra-renal aorta, in which the diameter exceeds 3.0 cm. Loss of vascular smooth muscle cells, degradation of the extracellular matrix (ECM), vascular inflammation, and oxidative stress are hallmarks of AAA pathogenesis and contribute to the progressive thinning of the media and adventitia of the aortic wall. With increasing AAA diameter, and left untreated, aortic rupture ensues with high mortality. Collective evidence of recent genetic and epigenetic studies has shown that phenotypic modulation of smooth muscle cells (SMCs) towards dedifferentiation and proliferative state, which associate with the ECM remodeling of the vascular wall and accompanied with increased cell senescence and inflammation, is seen in in vitro and in vivo models of the disease. This review critically analyses existing publications on the genetic and epigenetic mechanisms implicated in the complex role of SMCs within the aortic wall in AAA formation and reflects the importance of SMCs plasticity in AAA formation. Although evidence from the wide variety of mouse models is convincing, how this knowledge is applied to human biology needs to be addressed urgently leveraging modern in vitro and in vivo experimental technology.
1. Introduction
Abdominal aortic aneurysm (AAA) is identified as the dilatation of the infra-renal region of the abdominal aorta. It is characterized by >3.0 cm increase in aortic diameter or ≥50% of normal diameter and commonly presented as fusiform-shaped. The prevalence of AAA varies across different population and race with a range of 1.3–5% according to screening programs in the UK and US [1]. The major epidemiological risk factors related to AAA include male gender, age of more than 65 years old, and a history of smoking [2,3]. Other risk factors include atherosclerotic disease, hypertension, ethnicity, and family history of AAA. The AAA shares many risk factors with atherosclerosis but maintains its unique pathological characteristics. Pre-aneurysmal and aneurysmal infra-renal aortic dimension have been reported to correlate with non-AAA related cardiovascular mortality [4]. However, unlike atherosclerosis, AAA susceptibility and progression are negatively associated with diabetes mellitus [5]. However, a correlation between AAA and these conditions are not always positive and linear. Particularly in type II diabetic patients, the risk of developing AAA is reduced by approximately 50% [6].
AAA pathogenesis is a complex process characterized by aortic wall remodeling that leads to its weakening. In summary, it could be divided but not limited to: (1) Alteration of connective tissue protein and their architecture; (2) connective tissue degradation due to the imbalance between matrix metallopeptidases (MMPs) and tissue inhibitor of metallopeptidases (TIMPs); (3) chronic inflammation and cytokine release; (4) vascular smooth muscle cell (VSMC) trans-differentiation and cell death [7]. Moreover, compelling evidence shows that VSMCs are involved in all the above-mentioned pathophysiological changes that take place in the AAA wall. Furthermore, VSMCs in the differentiated state have the remarkable ability to acquire different phenotypes, including synthetic, fibroblast, and macrophage-like phenotypes, which occurs in the aortic wall of AAA patients [8].
Another reason to believe that VSMCs play an important role in the AAA lies in the field of embryology. AAA formation in the specific region of the abdominal aorta correlates with differential embryological development of the descending aorta. One of the main differences during the development of the descending and abdominal aorta is VSMCs origin. VSMCs in coronary arteries are well known to be derived from the epicardium, those in the ascending aorta and arch vessels are derived from the neural crest, and those in the descending aorta are derived from mesodermal somatic precursors layer. In adulthood, though the abdominal aorta presents three distinct layers like other aortic regions, there are histological and genetic differences across the different VSMCs lineages [8,9].
Although AAA is a well-investigated condition, an available therapy that can tackle constant aneurismal growth is currently lacking. From a clinical perspective, a greater understanding of the genetic and epigenetic mechanisms in depth would allow improved medical therapy to stabilize aortic wall, which could lead to the prevention of disease progression. This would, in turn, also help avoid devastating complications such as aortic rupture. There have been a few drug classes that have gone into clinical trials such as doxycycline, angiotensin-converting enzyme (ACE) inhibitors, and a few studies evaluating statins. However, conclusive evidence suggesting effective reduction in size or growth rate has yet to be reported, though some trials are still ongoing [10,11,12]. Moreover, therapeutic advancement would positively affect surgery and stenting application [13]. Thus, we have presented this review with the aim of providing a comprehensive overview of genetic and epigenetic discoveries pertaining to VSMCs underpinning AAA pathobiology.
2. Genetics of AAA
AAA has shown to cluster within certain families and there is evidence showing strong genetic component in AAA risk. Twin studies have reported that heritability could be as high as 70% [14]. The Swedish Twin registry reported that a monozygotic twin has a 24% probability of having an aneurysm as opposed to only 4.8% in dizygotic twins if the other twin has an aneurysm. Another large twin study in Denmark reported that the phenotypic variance determined by genetics is estimated to be 70–80%, whilst non-shared environment effects (such as infections, smoking, or occupational exposure) contributes to 20–30% [14,15]. Positive family history was shown to approximately double the risk of developing AAA [16]. In line with this, population studies have shown that ~19% of AAA patients reported a first-degree relative with the condition as well, considerably higher than 2–10% reported by unaffected controls [17,18,19].
This genetic inheritance appears to be autosomal in nature, though it could present as either dominant or recessive [20]. This highlights the importance of distinguishing familial cases from sporadic AAA when studying genetic risk factors as they could be different genetic entities. There has been evidence showing that familial AAA cases were also more prone to having a ruptured AAA and less likely to have heart disease compared to sporadic cases [21]. A Dutch study reported that although familial AAA cases shared a similar cardiovascular risk profile with sporadic AAA cases, the former group was shown to be more at risk of poorer outcomes after endovascular repair with higher requirement of repeated procedure after primary intervention [22].
The heritability of AAA can be classified as either having a Mendelian, single-gene cause, or having a non-Mendelian form with a more complex cause due to a variation of genes and downstream molecular pathways. Rare genetic diseases such as Ehlers-Danlos syndrome type IV, Marfan syndrome, Loeys-Dietz syndrome, and fibromuscular dysplasia, which can cause abdominal aortic aneurysms, are examples of Mendelian heritability [23]. However, the majority of AAA patients do not have these diseases as epidemiological analyses have revealed that only 10–20% patients with AAA have at least one relative with genetic disease [21]. This could suggest that a more complex mechanism, rather than single-gene variance may underlie AAA heritability. There is more compiling evidence suggesting that non-Mendelian heritability related to epigenetic and downstream translational mechanisms play a major role in vascular disease and VSMC plasticity related to that process [24].
Observations of AAA co-occurring with thoracic aortic aneurysm (TAA) in patients have led to studies focusing on shared genetic risk between dilating arterial conditions such as AAA, TAA, and intracranial aneurysm [25,26]. Several loci such as 9p21 and 15q21 were reported to contribute to potential pan-aneurysmal effects, though substantial genetic overlap between the three conditions was lacking [25].
3. VSMC Development and AAA
In the early stages of vasculogenesis, the recruitment and differentiation of precursor VSMCs is driven by chemoattractants like platelet-derived growth factor-β (PDGFβ) and transforming growth factor-β (TGFβ) and the formation of a network of tracks made of endothelial cells (ECs), leading to vasculature maturation in a highly regulated process. [27]. Vascular maturation involves the activation of many genes and their associated signaling molecules including those encoding sonic hedgehog (Shh), Notch, Ephrin, Tie, and Angiotensin (Ang) in specific locations across the vascular system including the descending and abdominal aorta. VSMC recruitment and differentiation in the descending aorta (dorsal aorta at relevant embryological stage) is associated with the presence of the notch ligand Jagged 1 (Jag1) in ECs and leads to a unique induction of VSMCs through Notch2 and Notch3 activation. The process of VSMC recruitment and media formation in the descending aorta at the latter stages of vasculogenesis leads to the replacement of mesodermal (lateral plate)-derived SM22α+ cells by somite-derived VSMCs [28].
The unique embryological origin of VSMCs in abdominal aorta leads to a specific landscape of the genetic transcript in these cells. The infra-renal abdominal area is different in cellular content, genetic activity, and histological structure, which render it more susceptible to specific clinical conditions [29,30]. Of note, elastin degradation in the infra-renal aorta is one of the most potent mechanisms that promote the dilation of this specific aortic segment. There are also differences in elastin content between the aortic arch and descending aorta due to primitive SMCs of the aortic arch being replaced by VSMCs migrated from the neural crest, which are better withstand the higher pulse pressure and ejection volume by laying down more elastic lamellae during development and vascular remodeling [31,32]. Altogether, there is growing evidence showing that genetic and epigenetic modulation in VSMCs acquired in vasculogenesis during embryological development may play a major role in the pathophysiological mechanisms involved in AAA development. The mesodermal VSMCs precursors of the abdominal aorta respond differently to various cytokines and growth factors compared to the neural crest precursor VSMCs [33]. Human lateral plate derived mesodermal VSMCs uniquely respond to the administration of cytokines like interleukin-1β (Il-1β), leading to MMP9 and TIMP1 upregulation, which causes ECM degradation [34]. Homocysteine, an amino acid containing sulfur is reported to be involved in elastolysis, collagen deposition, and aortic compliance was found to stimulate neural crest vascular SMC proliferation and promote synthetic activity but did not cause any effects on mesoderm-derived SMCs [35]. Transforming growth factor β (TGFβ), a mediator of cell growth and differentiation, which is able to induce DNA synthesis and collagen production in neural crest-derived VSMCs, does not have a similar effect on mesodermal-derived VSMCs [36]. These differences between neural crest-derived VSMC and somite-derived VSMCs have been confirmed in in vivo experiments. Continual infusion of the vasoactive peptide angiotensin II, associated in the previous experiments with vascular remodeling and atherosclerosis, lead to the development of aneurysms in the suprarenal abdominal aorta in apolipoprotein E knock out (ApoE-/-) mice without affecting the thoracic aorta [37]. In the ascending aorta and arch, angiotensin II response by neural crest-derived VSMCs promotes hyperplasia. A distinctly different response by mesoderm-derived VSMC was observed, marked by increased activity of the transcriptional regulator inhibitor of differentiation 3 (ID3), a mediator of mitogenic signaling [38].
4. VSMC-Mediated Extracellular Matrix Production and Degradation in AAA
The ECM, synthesized and processed by fibroblast-like VSMCs, plays a valuable role to maintain the blood containing function of the arterial wall. For instance, the enzyme lysyl oxidase is synthesized and secreted by VSMCs and is crucial for maturation of fibrillar structures such as elastin and collagen, rendering them insoluble. The ECM is comprised of microfibrillar structures such as elastin and collagen, fibronectin, and fibrillin. In healthy vessels, the elastin functions to resist vasodilation, whereas collagen functions to resist rupture [39]. Multiple studies have shown that VSMCs not only produce ECM but also involved in release and maturation of MMPs and TIMPs, which, in turn, control integrity and degradation of extracellular fibrillar structure of aortic wall. Thus, MMP/TIMP production balance plays an important role in the formation of AAA [40,41]. Furthermore, the proteolytic and oxidative environment in the aortic wall can lead to the partial disappearance of VSMCs seen in AAA. Plasmin and elastase, in particular, have been shown to cause cell detachment and death [42,43].
4.1. MMPs in AAA
MMPs play a major role in the progression of both AAA and TAA. In the normal aorta, MMPs are produced in VSMCs, ECs, and adventitial fibroblasts [44]. In AAA, they can also be released by inflammatory cells such as neutrophils and macrophages. VSMCs are also able to produce a wide variety of MMPs elevated in AAA, including MMP-1, -2, -9, -13, and -14, which lead to degradation of the aortic wall (Figure 1). As a result, many genome-wide association studies (GWAS) have studied single nucleotide polymorphisms (SNPs) in specific MMP genes to investigate their association with AAA development.
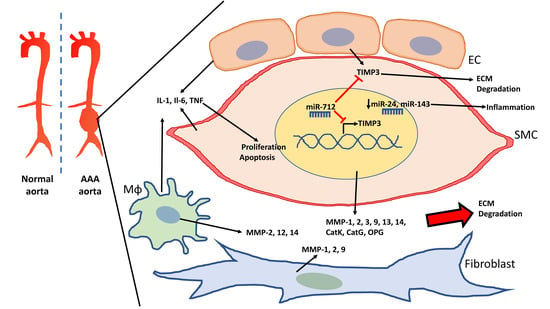
Figure 1.
Schematic of the abdominal aortic aneurysm (AAA) aorta and vascular cells involved in the degradation of the extracellular matrix (ECM). Vascular smooth muscle cells (VSMCs), endothelial cells (EC), and macrophages (Mɸ) release cytokines that elevate inflammation in AAA and promote VSMC proliferation as well as apoptosis. VSMCs, macrophages, and fibroblasts also contribute to the elevation of matrix metallopeptidases (MMPs), which lead to ECM degradation. miR-712 targets tissue inhibitor of metallopeptidase (TIMP)3 to further promote ECM degradation in AAA. Downregulation of miR-24 and miR-143 lead to increased inflammation.
The MMP family of endopeptidases is wide-ranging and consists of members that target specific components of the ECM. Each member has five domain structures: a signal domain, a prodomain, hinge region, a hemopexin domain, and a catalytic site [45]. MMPs are classified into 6 groups based on their substrate specificity: Collagenases, gelatinases, stromelysins, matrilysins, membrane-type MMPs, and other MMPs (Table 1).

Table 1.
Key MMPs that are synthesized by VSMCs and/or mediate ECM degradation in AAA.
4.2. Other Mediators of ECM Degradation: Cathepsins, Osteoprotegerin and Kallikreins
Regulators of protease and MMP activities that cause VSMC apoptosis and tunica media degradation in AAA have also been reported. Cathepsin K (CatK) and CatG are both highly expressed in vascular tissues in AAAs and can activate MMPs, collagenase, and elastase. CatK is expressed by ECs and osteoclasts; CatG is expressed by SMCs, neutrophils, and mast cells. Both CatK and CatG deficiency in AAA mouse models showed reduced VSMC apoptosis, elastin degradation, and lesion formation [61,62]. Osteoprotegerin is another driver of protease and MMP activity that is also positively associated with human AAA growth.
Other proteases produced in vascular tissue include kallikreins. They aid in the releasing of kinins (bradykinin and lysyl-bradykinin), which in turn cause the eicosanoids, nitric oxide (NO), and endotheliu- derived hyperpolarizing factor (EDHF) release. These processes induce vasodilatation, natriuretic and diuretic effects. Lowering of kallikrein levels has led to remodeling of the arterial wall [63]. A SNP (rs5516) in the KLK1 promoter was reported to be enriched in AAA patients compared to controls and in large AAAs compared to small AAAs [64].
4.3. TIMPs
The condition of the ECM and aortic wall is dependent on the balance between metallopeptidases and their inhibitors, TIMPs. Insufficient activity or low expression of TIMPs could promote ECM degradation and VSMC modulation. This is seen in the aneurysmal aortic wall as decreased mRNA levels of TIMPs have been reported [65,66]. TIMP1-deficient mice were shown to develop larger aneurysms after porcine pancreatic elastase (PPE) perfusion compared to wild-type mice [67].
Tilson et al. conducted the first DNA sequencing analysis of TIMP1 in six AAA patients and revealed a silent polymorphism in two of the patients [68]. Another study reported a SNP (C>T) at nucleotide position 434 and showed an association between the C-allele and female AAA patients [69], though the patient cohort was reportedly small (n = 20). A subsequent sequence analysis conducted in 50 patients for the TIMP1 gene failed to detect any mutations associated with AAA [70].
TIMP2 level decreased in AAA patient serum and the ratio of MMP2/TIMP2 mRNA is elevated in AAA tissue compared to control aortic tissue [48,71]. A DNA sequence analysis of the TIMP2 gene revealed a SNP at nt 573G>A, with the G allele being significantly more frequent in male AAA patients (n = 64) compared to male controls (n = 29) [69]. However, a similar analysis of the TIMP2 gene failed to find this association [72]. The role of TIMP2 is rather complex as it also plays a role in MMP2 activation by functioning as a co-activator of proMMP-2 [73,74]. In mice, Timp2 knockout resulted in protection against CaCl2-induced AAA [75].
TIMP3 level is elevated in AAA and may be attributed to its protective role in disease. TIMP3 deficiency was shown to trigger the AAA in AngII-infused mice. The knockout of Timp3 gene led to the adverse remodeling of the abdominal aorta, reduced collagen and elastin proteins, and elevated proteolytic activities, which was not seen in wild-type mice [76]. However, no association between TIMP3 and AAA were identified.
The role of TIMP4 in AAA has been least investigated, though increased expression was shown in hyperhomocysteinemia-associated aortic aneurysm in human and mice [77].
4.4. miRs Regulating ECM Degradation
MiRNAs are short (~20 base pair long), single-stranded non-coding RNA that directly regulate gene expression and exert a wide range of cellular functions [78]. They negatively regulate gene expression by degrading mRNA or mimicking small interfering RNA (siRNA) and inhibiting translation. miRNAs are initially transcribed as pi-miRNAs, longer primary transcripts, that are processed by the ribonuclease enzyme complex Drosha/DGCR8, followed by Dicer, and incorporation into the RNA-induced silencing complex (RISC) [79]. Mature miRNA is able to pair with a highly conserved seed sequence within the 3’ untranslated region (UTR) of potentially multiple target genes [78]. A wide variety of miRNAs have been studied in AAA that target specific genes to regulate VSMC plasticity as well as ECM degradation and inflammation (Table 2).
Table 2.
miRNAs that are key players in the pathophysiology of AAA organized under three categories: ECM degradation inducing, contractile phenotype inducing, and synthetic phenotype inducing.
4.4.1. miR-29b
In both the PPE and AngII infusion models of ApoE-/- mice, AAA development was associated with decreased aortic miR-29b expression [80]. AAA was also associated with increased expression of Col1a1, Col3a1, Col5a1, and Eln genes. Overexpression of miRNA-29b increased AAA expansion and aortic rupture rate, while miRNA-29b inhibition using anti-miR-29b decreased AAA expansion and promoted aortic fibrosis. Both human aortic VSMCs and human fibroblasts express miR-29b, though treatment with transforming growth β (TGFβ) significantly decreased miR-29b expression only in aortic fibroblasts, suggesting that these cells primarily mediate profibrotic effects. Similar to the murine models, human AAA patients displayed downregulated miR29b and upregulated COL1A1, COL3A1, COL5A1, and ELN in aneurysmal tissue [80]. In contrast, miR29b was increased in early aneurysm development in the Marfan syndrome mouse model [81]. This was associated with increased apoptosis presented by elevated cleaved caspase-3 and -9, enhanced caspase-3 activity, and decreased expression of antiapoptotic proteins—myeloid cell leukemia 1 (Mcl-1) and B cell lymphoma 2 (Bcl-2). Interestingly, miR-29 activity was repressed by nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) signaling.
4.4.2. miR-205
miR-712 in mice and its human/murine homolog miR-205 have been shown to suppress TIMP activity in response to AngII-induced enhanced aortic MMP activity and promote AAA formation [82]. Both miR-712 and miR-205 stimulated MMP activity by direct targeting of TIMP3 and reversion inducing cysteine-rich protein with kazal motifs (RECK) in ECs of AngII-infused ApoE-/- mice. The effects of miR-712 were also observed in VSMCs though the effect of miR-205 on VSMCs was not reported. The upregulation of four AngII-sensitive miRNAs, miR-205, -21, -133, and -378, identified in the mouse model, was also observed in aortic samples of AAA patients (Figure 1).
4.5. HDAC Inhibitors Regulating MMPs-2 and -9
Histone deacetylases (HDACs) are potent epigenetic regulators of gene transcription. They comprise of 18 molecules divided into four classes: I, II (comprising IIa and IIb), III, and IV. Class I and IIa HDACs have been shown to modulate the expression of VSMC genes involved in AAA and regulate differentiation, contractility, proliferation, inflammation, and ECM deposition [83]. HDACs and lysine histone acetylase transfers (HATs, also known as KATs), play key roles in post-translational modification as they can attach or remove an acetyl group at the lysine residue. Acetylation neutralizes their positive charge of histone tails and results in the loosening of the histones and associated DNA. This loose chromatin structure allows accessibility for transcription factors and promotes transcriptional activation. In converse, deacetylation leads to a more compact chromatin structure [84].
Galan et al. reported increased expression of HDACs 1 and 2 (from class I) and HDACs 4 and 7 (both from class IIa) in aortic samples of AAA patients compared to the control group [85]. High expression levels of the four HDACs were particularly seen in the tunica media of aneurysmal samples comprised of VSMCs and inflammatory cells. A similar HDAC expression profile was also observed in aortic samples from angII-infused ApoE-/- AAA mice.
4.5.1. MS-275 and MC-1568
The class 1 HDAC inhibitor MS-275 and class IIa inhibitor MC-1568 decreased AAA expansion and improved survival in mice [85]. Both inhibitors were able to reduce systemic inflammatory response such as macrophage and lymphocyte infiltration and elastin and collagen fiber disorganization. Additionally, both MS-275 and MC-1568 inhibited the upregulation of pro-inflammatory markers and MMP-2 and MMP-9 activity observed in the mouse AAA model (Figure 1).
4.5.2. Metacept (MCT)
Similarly, another HDAC inhibitor, metacept-1 (MCT-1), was also shown to reduce AAA incidence in AngII infused ApoE-/- mice [86]. MCT also reduced expression of MMP2 in human VSMCs in vitro and MMP-2 and MMP-9 in AAA mouse tissue samples (Figure 2).
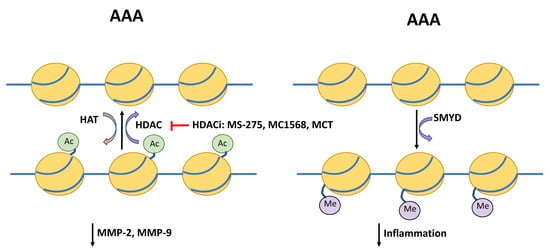
Figure 2.
Schematic depicting regulators of histone acetylation and DNA methylation in AAA. Histone deacetylases (HDAC) and histone acetylase transferases (HAT) provide post-transcriptional modification via removing or attaching an acetyl group at the lysine residue. HDAC inhibitors (HDACi) MS-275, MC1568, and metacept (MCT) inhibit acetylation, which has been shown to reduce levels of matrix metallopeptidases (MMP)-2 and-9, and in turn protect the extracellular matrix from degradation. The SET and MYND domain-containing (SMYD) promoter region is hypomethylated in AAA compared to controls. SMYD itself is able to methylation of promoters of interleukin-6 (IL-6), tumor necrosis factor α (TNFα), and hypoxia inducible factor 1 α (HIF1α) and suppress inflammatory signaling.
4.5.3. HATs in AAA
The activity of myocardin, a cardiac- and muscle-specific coactivator of the transcription factor serum response factor (SRF), was shown to be enhanced by p300, a HAT that associates with the activation domain of myocardin [83]. Class II HDACs have shown to interact with myocardin in separate binding domains and suppress myocardin-induced smooth muscle differentiation.
A study comparing AAA and healthy aortic samples reported a wide variety of HATs that were significantly higher in disease [87]. KAT2B, KAT3A, KAT3B, and KAT6B were among the highest expressed in AAA tissue. KAT6A expression was shown to correlate with the contractile VSMC marker myosin heavy chain 11 (MYH11) as well as inflammatory CD3+ and vascular cell adhesion molecule 1 (VCAM-1)+ ECs. Thus, KAT expression profiles may potentially serve as biomarkers of AAA and risk of rupture but also shed light on the complexity of the balance between HAT and HDAC activity in AAA.
5. Smooth Muscle Cells and Inflammation in AAA
Overall, inflammation is a large component of AAA though the origin of immune cells and the inflammatory cascade has been a much-debated subject [44,88]. There is evidence showing that macrophages have a hemopoietic and tissue-specific origin. More recently, lineage tracing techniques have confirmed that VSMCs are also able to transdifferentiate into macrophage-like cells.
Phenotypic switching of VSMCs to macrophage-like LGALS3+ and CD68+ cells has been shown in human AAA lesion samples. Lineage tracing in the ApoE-/- mouse model showed that phenotypically transdifferentiated VSMCs comprised of ~30% of the cell population within the lesion of the brachiocephalic [34]. Furthermore, phenotype modulation could be assessed through differential expression of key genes such as MYH11, alpha-smooth muscle actin (ACTA2), calponin 1 (CNN1), and transgelin (TAGLN) in VSMCs.
The study using in situ hybridization and proximity ligation assays reported that the dimethylation of histone H3 Lysine K4 (H3K4me2) of the MYH11 locus was specific to the smooth muscle cells in human and mouse sections. Remarkably, this epigenetic mark was retained in the cells in phenotypic modulated cells within atherosclerotic lesions, suggesting the potential robustness of certain epigenetic markers in SMCs [24].
5.1. Upregulated Cytokines in AAA
In general, inflammation is a hallmark AAA formation of aneurysms and its role in AAA is well supported compared to the limited studies shown in TAA. Neutrophils infiltrate to the aortic site very early, though only transiently, and are sources of proteases, MMPs, and reactive oxygen species and can start degradation of the ECM and aortic wall. However, most of the studies on neutrophil involvement include animal experimental work.
The function of smooth muscle is heavily influenced by macrophage activation and infiltration in AAA as these inflammatory cells are responsible for a lot of the production of MMPs, cytokines, chemokines, and ability to remove cellular debris [44]. Macrophages can also release netrin-1, a protein known to promote macrophage retention in tissues, to drive AAA pathogenesis. Netrin-1 deletion resulted in an MMP3-dependent reduction in AAA formation [89].
Upregulation of the cytokines such as interleukin 1α (IL1α), IL1β, IL-6, and tumor necrosis factor α (TNFα) are seen in AAA. Of interest, the very same cytokines are produced by VSMCs [90,91,92]. IL1α, IL1β, and TNFα, which are also produced by macrophages and are potent mitogens of VSMCs, implying that the cytokine in the aortic wall may be produced via autocrine regulation [86,93,94,95,96]. IL1β and TNFα in combination with interferon γ promote VSMC apoptosis [97]. It is also known that IL-1β plasma levels in patients with AAA and coronary artery disease (CAD) are significantly higher than in CAD patients without AAA. IL-1β likely promotes apoptosis via MMP upregulation and activation in patients with CAD and AAA [98,99,100,101,102,103]. Cytokine elevation in AAA patients is well documented, though their precise mechanisms are a matter of great debate. Six SNPs in the IL1A, IL1B, and IL1RN genes have been investigated in a study but no associations with AAA were found [104]. Two small case-control studies and a meta-analysis failed to find an association between a SNP in the IL1B gene (nt3953 C>T) and AAA [104,105,106].
TNFα can induce VSMC apoptosis by causing the increase of caspase-3 activity and inhibiting gap junctions, specifically connexin 43 [107]. The A-allele of a SNP located at nt-308 has been shown to cause a six- to nine-fold increase in TNFα mRNA in B-cells compared to the G-allele, though the association between the A-allele and AAA was not seen [105,108].
IL-6 is predominantly expressed in leukocytes such as macrophages and T-cells and though they are not normally expressed by VSMCs, they can be expressed by aortic VSMCs under conditions of elevated aortic wall tension, as would be the case in AAA [92]. A large case-control study analyzed data identified a single SNP (rs1800796; -572 G>C) in the IL-6 promoter as an independent risk factor for AAA, though it was only present in 1.5% of the cases [109]. Other SNPs were detected in subsequent studies but showed no association [105,110].
TGF-β is the most extensively studied cytokine in TAA, though it may play a role in AAA mechanistically [111,112]. In contrast to how TGF-β signaling drives TAA formation, systemic blockade of TGF-β activity or genetic deletion of its downstream signaling protein SMAD3, a transcriptional modulator and member of TGF-β receptor regulated SMAD family, worsened AngII and CaCl2-induced AAA in mice [113,114]. Other study showed that the peptide antagonized thrombospondin-1 could promote AAA in AngII-infused ApoE deficient mice through inhibition of TGF-β1-mediated Smad2/3 signaling [115]. A study analyzing data from large Australian and New Zealand cohorts examined for 49 SNPs in the TGFBR2 gene and reported one SNP (rs1078985) that showed weak association with AAA, though it did not hold up to multiple testing [116]. Two more SNPs in the TGFRB2 gene (rs1036095 and rs4522809) reported in a large Dutch study also showed association with AAA [117].
5.2. Inflammation-Associated Genes
The SET and MYND domain-containing 2 (SMYD2) is a highly expressed gene in muscle cells that is involved in myofibril organization. It interacts with three functional groups of molecules: (1) chromatin remodeling and histone modification, (2) DNA replication, and (3) repair and molecular chaperones. It methylates heat shock protein 90 (Hsp90) and improves sarcomere stability [118]. The SMYD promoter region of SMCs is significantly hypomethylated in AAAs compared to controls. In line with this, Hsp90 inhibition also reduces AAA formation in murine models [119]. SMYD2 methylation in aortic SMCs correlated with gene expression, with cytoplasmic abundance in smooth muscle in the tunica media being lower in AAA compared to non-aneurysmal controls. Moreover, SMYD2 can methylate the promoters of IL-6 and TNFα gene promoters, leading to the inhibition of NFkB and extracellular signal-regulated kinase (ERK) signaling pathways [120]. Thus, hypomethylation of SMYD may result in decreased vascular structural stability and increased inflammation, promoting the AAA phenotype (Figure 2).
There are several SNPs on the 1p13.3 locus that have been identified by several AAA meta-GWAS studies located near the PSRC1, as well as other genes including CELSR2 and SORT1 [121,122]. Expression quantitative trait loci and chromatin interaction data showed that SORT1 gene may be most likely to be influenced by the SNPs [123]. SORT1 was first seen expressed in the liver regulating plasma LDL-cholesterol through interactions with promoter of pro-protein convertase subtilisin/kexin type 9 (PCSK9) acidic sphingomyelinase and apoB100 in hepatocytes [124]. In 2018, Zhang et al. reported that it is also expressed in VSMCs and could induce VSMC calcification in vitro and in rats [125].
5.3. Reduced Contractile Phenotype/Anti-Inflammatory Inducing miRNA in AAA
5.3.1. miR-24
Downregulation of the miR-23b-24-27b cluster in murine AAA models was reportedly associated with AAA in the PPE-infusion mouse model, in which miR-24 showed the most significant inverse correlation of its predicted targets in array profiling studies [126]. In both the PPE and AngII ApoE AAA mouse models, miR-24 was shown to target chitinase 3-like 1 (Chi3l1), an inflammation marker of AAA disease progression. This limited inflammation by blocking IL-8 and monocyte chemoattractant protein 1 (MCP1/CCL2) production by VSMCs and M1 subtype macrophages and inhibiting new macrophage recruitment and survival. Decreased plasma miR-24 expression was observed in AAA patients as well as in the murine AAA models. Furthermore, IL-6 was shown to decrease miR-24 expression via NFkB-mediated pathway in siRNA-mediated knockdown models of the RelA and NFkb1 genes. CHI3L1 was another gene that showed the ability to promote aortic VSMC migration in vitro via c-Jun N-terminal kinase (JNK) and ERK phosphorylation [127].
5.3.2. miR-143/145
The miR-143/145 cluster highly expressed in VSMCs and has been shown to be most abundantly expressed in the heart and aorta [128]. The miRNAs miR-143 and miR-145 are known to induce a differentiated, contractile phenotype in smooth muscle cells and are also shown to be downregulated in aneurysms compared to control aorta [128]. Their expression in blood is significantly lower in coronary artery disease (CAD) patients compared to healthy individuals [129]. The studies implicate that it may be involved in modulating the pro-inflammatory, MMP-producing VSMC phenotype to stabilize AAA [130].
6. Smooth Muscle Cell Plasticity and Apoptosis in AAA
VSMCs in the aneurysmal wall have impaired contractility and greater susceptibility to apoptosis and senescence [11,131]. In addition, a variety of genetic and epigenetic regulators have been reported to modulate the VSMCs toward either a dedifferentiated synthetic (proliferative) phenotype typically seen in disease or a senescent phenotype. In vitro and animal studies have shown that targeting these regulators could lead to a deeper understanding of AAA pathophysiology and advancements in therapeutics.
6.1. Genetic and Epigenetic Mechanisms Promoting the Synthetic SMC Phenotype
6.1.1. Cyclin-Dependent Kinase 2B Antisense (CDKN2BAS)
There have been a number of alleles discovered through GWAS in the last two decades that directly or indirectly affect smooth muscle proliferation and apoptosis. SNPs at the 9p21.3 locus have been consistently associated with AAA. The non-coding RNA gene CDKN2BAS, also known as antisense non-coding RNA in the INK4 locus (ANRIL), encodes a lncRNA regulating the expression of cyclin-dependent kinase inhibitor 2B associated with AAA development and is located within the p15/CDKN2B-p16/CDKN2A-p14 alternate reading frame (ARF) gene cluster [26,132,133,134]. PPE-infused Cdkn2b-/- mice developed larger aortic aneurysms compared to wild-type mice. The Cdkn2b-/- mice contained fewer VSMCs that exhibited increased apoptosis in the aortic wall [135]. Cdkn2b knockdown in the smooth muscle cells in vitro led to increased growth arrest and apoptosis. Furthermore, immune-histochemical analysis of human tissue samples showed that CDKN2B was mainly expressed in VSMCs and reduced levels of CDKN2B in AAA aortic tissues samples were observed compared to controls. When a 70 kbp region, including the SNP (rs10757278) and portions of the Cdkn2bas gene but not the two closest protein genes Cdkn2a and Cdkn2b, was deleted in mice, cardiac and vascular Cdkn2a and Cdkn2b expressions were reduced. The rate of proliferation of SMCs isolated from aorta of these mice was double of those from same strain controls [136]. Thus, patients with the risk allele at the 9p21 locus have lower CDKN2B expression levels, which in turn enhances apoptosis, causing thinning of the medial layer of the aortic wall and ultimately making it more prone to dilation seen in AAA (Figure 3) [137].
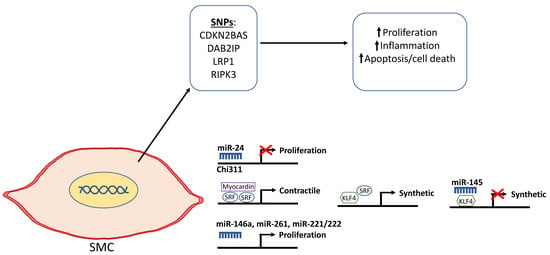
Figure 3.
Schematic depicting the genetic and epigenetic mechanisms that are involved in the phenotypic modulation of smooth muscle cells (SMCs) in AAA. Single nucleotide polymorphisms in selected genes including cyclin dependent kinase 2B antisense (CDKN2BAS/ANRIL), DAB2IP, lipoprotein receptor-related protein 1 (LRP1), and receptor-interacting serine/threonine-protein kinase 3 (RIPK3) have been shown to be associated with the diseased SMC phenotype as depicted by increased proliferation that leads to vascular remodeling, inflammation, and cell death. miR-24 selectively targets Chi311 to inhibit proliferation. Kruppel-like factor 4 (KLF4) normally represses myocardin to downregulate SMC differentiation and promote the synthetic phenotype. miR-145 can repress KLF4 and, in turn, inhibit de-differentiation into the synthetic SMC phenotype. miR-146a, miR-261, and mir-221/222 have been reported to promote SMC proliferation.
6.1.2. Disabled Homologue 2 Interacting Protein (DAB2IP)
Another AAA GWAS studied assessed approximately 300,000 SNPs and located 3 SNPs on chromosome 9p21 associated with AAA at genome-wide significance and 22 other SNPs that were also significantly associated. One SNP (rs7025486) that had a highly significant P-value was within intron 1 of DAB2 interacting protein, also known as apoptosis signal-regulating kinase 1 (ASK1) interacting protein (AIP1) [138]. DAB2IP is a Ras GTPase-activating protein family member that is normally highly expressed in VSMCs and ECs and has been shown to inhibit proliferation and intimal expansion. Knockout or knockdown of DAB2IP in VSMCs was shown to significantly enhance IFN-γ-induced JAK-signal transducer and activator of transcription proteins (STAT) signaling and IFN-γ dependent migration and proliferation of VSMCs [139]. By binding to JAK2, DAB2IP could inhibit IFN-γ signaling through STAT1/STAT3 along with their pro-survival effects (Figure 3).
6.1.3. Lipoprotein Receptor-Related Protein 1 (LRP1)
A large multi-center GWAS in AAA revealed nine loci associated with AAA as well as a single SNP (rs1466535) on chromosome 12q13.3 located within intron 2 of the gene encoding low-density lipoprotein receptor-related protein 1 (LRP1), which showed high association with AAA [133]. This association was also confirmed by a later study [140]. Smooth muscle-specific LRP1 inactivation by crossing SM22Cre transgenic mice with Lrpflox animals has been shown to cause aneurysm, overexpression, and abnormal activation of platelet-derived growth factor (PDGF), disruption of the elastic layer, VSMC proliferation, and cholesterol-induced atherosclerosis [141]. LRP1 plays an important role in maintaining integrity of the vascular wall, which could be via Smad signaling [142]. Thus, in patients where LRP1 signaling is disrupted, its protective effects could be suppressed, leading to AAA development (Figure 3).
6.1.4. Receptor-Interacting Serine/Threonine-Protein Kinase 3 (RIPK3)
Medial VSMC depletion is a hallmark characteristic in both AAA and TAA. Whether VSMC death is an active pathological event or a consequence of growth arrest or apoptosis has not been fully investigated. Cellular apoptosis and necrosis have been categorized as distinctly separate forms of cell death. More recently, studies have pointed towards a third form of death known as necroptosis, where certain types of necrosis are regulated by orchestrated signaling networks [143]. Receptor-interacting serine/threonine-protein kinase 3 (RIPK) has been reported as an important regulator of this programmed necrosis (Figure 3). Ripk deletion reduced VSMC necroptosis and inhibited the development of PPE-induced AAAs in C57BL/6 mice [43].
6.1.5. Notch Homolog 1 Translocation-Associated (NOTCH1)
In humans, the NOTCH1 gene encodes a transmembrane receptor expressed on many cell types including smooth muscle and ECs and is involved in development and differentiation. Notch1 signaling was reported to be activated in the AngII-infused ApoE knockout AAA mouse model. Aneurysm incidence could be reduced via Notch1 haploinsufficiency and pharmacological inhibition of NOTCH1 signaling through the prevention of macrophage accumulation to the aneurysm site [144].
6.1.6. HIF-1α
The transcription factor hypoxia-inducible factor-1α (HIF-1α) regulates multiple biological pathways, including stimulating cellular proliferation, angiogenesis, cell apoptosis, migration, and metabolism to adapt to low oxygen availability [145,146]. Its transcription is low in normoxia and elevated in hypoxic conditions. Its upregulation is prominent in various vascular diseases including pulmonary hypertension, atherosclerosis, and AAA [88]. HIF-1α overexpression is present in the rupture edge of AAA human tissues. VSMCs derived HIF-1α can promote vascular remodeling by upregulating cytokines and chemokines, leading to increased AngII signaling [147,148]. A study by Gabel et al. reported that genes positively associated with progression of larger AAA were also associated with angiogenesis and vascular remodeling and highly expressed in fibroblasts and VSMs of the aneurysmal wall [149]. Additionally, these genes as well as genes exclusively upregulated in ruptured AAA were functionally converged through processes mediated by the HIF-1α pathway.
6.2. Elevated miRNA Promoting Synthetic Phenotype
6.2.1. miR-21
miR-21 has been shown to inhibit VSMC apoptosis and protect against AAA formation in Apoe-/- mice (Figure 3) [150]. Aortic miR-21 expression levels were elevated in both the PPE- and angII-infusion ApoE knockout AAA models compared to controls group. Overexpression of miR-21 through lentiviral transduction reduced phosphatase and tensin homolog (Pten) expression and led to increased phosphorylation and activation of AKT, which regulates a number of kinases, transcription factors, and regulatory molecules, promoting a proliferative and antiapoptotic VSMC phenotype. Furthermore, the miR-21 upregulation was augmented by nicotine, further implicating the role of miR-21 as a driver of vascular remodeling in smoking, a major modifiable risk factor for AAA.
6.2.2. miR-146a
miR-146a was also reported to be elevated in AAA and promote VSMC proliferation and survival and involved in AAA pathogenesis (Figure 3) [151]. Sun and colleagues reported that miR-146a targeted the 3’ untranslated region (UTR) of the Kruppel-like factor 4 (KLF4) transcription factor and caused VSMC proliferation in vitro and vascular neo intimal hyperplasia in balloon-injured rat carotid arteries. KLF4 has an anti-proliferative effect on VSMCs through upregulation of p21 and differentiation genes SM22α and α-smooth muscle actin and downregulation of the dedifferentiation gene SMEMB. Inhibiting miR-146a using anti-miR transfection in VSMCs increased KLF4 expression, while miR-146a overexpression reduced KLF4 levels.
6.2.3. miR-26a
miR-26a promotes VSMC proliferation while inhibiting cellular differentiation and apoptosis and altered TGF-β signaling (Figure 3) [152]. In human aortic SMCs, inhibition of miR-26a via anti-miR transfection accelerated VSMC differentiation, promoted H2O2-induced apoptosis, and inhibited proliferation and migration. Conversely, its overexpression blunted differentiation. miR-26a inhibition led to increased SMAD1 and SMAD4 expression, while overexpression inhibited SMAD-1, suggesting that miR-26a promotes proliferation via altering TGFβ signaling. This would affirm past studies that showed that TGFβ signaling through SMAD-1/5/8 induces VSMC differentiation and inhibits proliferation [153]. Zhang et al. reported that SMAD4 or TGFβ receptor II deficiency in VSMCs led to aortic aneurysm formation in mice and increased cathepsin S and MMP-12 [154]. miR-26a was downregulated in both the PPE and AngII ApoE-/- AAA mouse models [152].
6.2.4. miR-221/222
miR-221 and miR-222, which have been implicated in cancer cell proliferation, have also been shown to regulate VSMC proliferation and neo-intimal hyperplasia (Figure 3) [155]. Their expressions were identified in rat carotid arteries after angioplasty, which were upregulated and localized in VSMCs after injury to the vascular wall. They were also upregulated after PDGF and serum stimulation in VSMCs. Knockdown of miR-221 and miR-222 using antisense oligonucleotide-mediated miRNA depletion led to decreased rat aortic SMC proliferation in vitro via activation of p27 and p57, which are negative regulators of VSMC proliferation. miR-221 was also crucial for PDGF-induced cell proliferation by downregulating p27 and c-Kit expression [156].
6.3. lncRNAs Associated with Pro-Proliferative SMC Upregulated Alongside Apoptosis
6.3.1. H19
H19 was one of the earliest lncRNAs identified in AAA and was upregulated in the AngII and PPE mouse models of AAA compared to controls [157]. H19 knockdown using site-specific antisense oligonucleotides (LNA-GapmeRs) significantly inhibited aneurysm growth in both mouse models. The medial VSMCs and adventitial fibroblasts of normal non-dilated tissue expressed H19. AngII infusion led to H19 upregulation in medial VSMCs of dilated aortic tissues, which was abolished through H19-targeted deletion by GapmeRs. AngII/H19-induced VSMC apoptosis was dependent on the expression of HIF1α, which interacted with proto-oncogene transformed mouse 3T3 cell double minute 2 (Mdm2), and prevented Mdm2-mediated reduction of p53, a factor that triggers aneurysm expansion.
6.3.2. Plasmacytoma Variant Translocation 1 (PVT1)
Plasmacytoma variant translocation 1 (PVT1) is a lncRNA that increased expression in tissues from AAA patients and AngII-infused ApoE-/- mice [158]. PVT1 expression was increased by AngII in VSMCs in vitro. Furthermore, overexpression of PVT1 in the AngII-induced AAA mouse model led to increased VSMC apoptosis, elevated MMP-2, and MMP-9, reduced TIMP-1, and differentiation into the synthetic SMC phenotype. Knockdown in vitro and silencing in vivo using shRNA lentiviral vector against RNA-PVT1 exhibit reversed AAA-induced alterations.
6.4. Downregulation of Protective Genes in AAA
6.4.1. Peroxisome Proliferative-Activated Receptor Gamma (PPARG)
Peroxisome proliferative-activated receptor gamma (PPARγ), a nuclear hormone receptor known for playing a critical role in glucose and lipid metabolism, is expressed in VSMCs. Its activation by thiazolidnediones leads to a contractile VSMC phenotype with reduced proliferation and oxidative stress [159]. PPARγ activation has also been shown to inhibit osteoproteregrin expression in human aortic SMCs [160]. The PPARG c.34G>C polymorphism weakly associated with AAA, while c.1347C>T was strongly associated with increased AAA growth [161]. VSMC-targeted deletion of Pparγ in mice has been shown to cause systemic hypotension possibly via increased β2-adrenergic receptor expression as well as pulmonary hypertension [162,163]. Interfering PPARγ signaling has also shown to induce vascular dysfunction, hypertrophy, and remodeling [164,165]. Thus, a polymorphism affecting PPARγ activation might implicate loss of protective effects on VSMCs, leading to aortic wall degradation.
6.4.2. Serpin Proteinase Inhibitor B9 (SERPINB9)
Serpin proteinase inhibitors are able to protect the aortic wall and their decrease in expression levels has been implicated in AAA. Serpin proteinase inhibitor, clade A, member 1 (SERPINA1), also known as α1-antritrypsin (α1-AT), was shown to inhibit VSMC cell apoptosis and ECM degradation induced by elastase in VSMCs and mammary artery cultured ex vivo [166].
SERPINB9 was reported to inhibit apoptosis of human VSMCs [167]. A genome-wide methylation analysis in peripheral blood mononuclear cell DNA from AAA patients and control subjects identified increased methylated regions in calponin 2 (CNN2) and adenylate cyclase 10 pseudogene 1 (ADCY101). The same study also showed decreased methylation in kelch-like family member 35 (KLHL35) and SERPINB9 in AAA samples compared to controls [168]. Gene expression was higher for SERPINB9 and lower for CNN2 in AAA patients. Immuno-histochemical analysis showed that the aortic media and intima was more prominently stained for CNN in AAA patients and SERPINB9 in control patients. CNN2 is an actin-binding protein, implicated in vascular development and shown to be upregulated in stretched vascular walls [169].
6.5. Reduced miRNAs Promoting Contractile Phenotype
miR-143/145
Both miR-143 and miR-145 induce the contractile VSMCs phenotype and their expression is significantly diminished in aneurysms compared to controls tissue [128]. They are encoded by highly conserved genes that lie in close proximity with each other on chromosome 5 in humans and chromosome 18 in murine [170]. The transcription factors serum response factor (SRF) and cardiac NK-2 transcription factor (Nkx2-5) can independently activate miR-143 and miR-145 expression and synergistically upregulate expression to promote de-differentiation of contractile VSMCs. Myocardin, a transcriptional coactivator of SRF, was shown to augment miR-143/145 expression when in combination with SRF, and cause additive effects with both SRF and Nkx2-5 [171]. The loss of miR-143 in a KO mouse model utilizing a lacZ reporter also led to decreased miR-145 expression, implicating their co-dependence [128].
Phenotypic differentiation caused by miR-143 and miR-145 has been studied in in vitro and in vivo models. Overexpression of miR-145 in VSMCs increased the gene expression of differentiation markers like smooth muscle (SM) α-actin, calponin, and SM-myosin heavy chain. It also helps to maintain a spindle-like shape and inhibits VSMC proliferation [172]. Homozygous miR-143/145 KO mice had a reduced number of contractile and increased number of synthetic VSMCs in the aorta and femoral artery compared to wild-type controls [128]. Smooth muscle layers of aorta and other arteries were remarkably thinner in miR-145 and miR-143/145 KO mice compared to wild-type or miR-143 KO mice due to a greater reduction in actin-based stress fibers, indicating the more prominent role of mir-145. Ultrastructural analyses revealed dilated endoplasmic reticulum that were increased in number in miR-143, -145, and -143/145 knockout mice, indicative of synthetically active VSMCs.
The two miRNAs can work in a myriad of pathways to induce VSMCs differentiation. miR-145 represses KLF4, which is normally able to interact with SRF, repress myocardin and calmodulin kinase II-δ, promote neointimal proliferation, and downregulate VSMC differentiation marker genes (Figure 3) [159,160,161]. Another target of miR-145 is KLF5, which also represses myocardin expression and is activated in AngII-induced VSMC proliferation via the angiotensin I-triggered ERK and p38 mitogen-activated protein kinase (MAPK) pathways [172]. miR-143 was reported to target Ets like gene 1 (Elk-1), which competes with myocardin to bind to SRF and inhibits VSMC differentiation [173]. miR-143 was also shown to attenuate the expression of versican, a chondroitin sulfate proteoglycan of the ECM produced by synthetic VSMCs, upon binding to myocardin [174]. Both miR-143 and miR-145 can target actin-dependent SRF coactivator myocardin-related transcription factor-B (MRTF-B) and Adducin-3 (ADD3), which are membrane skeletal proteins that bridge the membrane and actin cytoskeleton and aid in cell motility [175,176,177,178]. Thus, targeting MRTF-B and ADD3 resulted in inhibition of the migratory phenotype.
7. Smooth Muscle and ROS in AAA
Reactive oxygen species (ROS) plays an integral role in the development of AAA, possibly more so than in TAA, as they result in VSMC apoptosis, MMP activation, and induction of pro-inflammatory genes in AAA [44]. Increased superoxide levels were some of the first observations seen in AAA [183]. Administration of vitamin E as an antioxidant has shown to lead to decreases in AAA size and rupture in animal models [184]. Similarly, catalase overexpression in smooth muscle was shown to prevent mechanical changes in the aortic wall in the AngII model and AAA formation inhibition in the CaCl2 model [185]. Oxidative stress was also shown to be linked with histone acetylation in various models of cardiovascular diseases leading to downstream proinflammatory signaling [83]. Increased ROS production has been associated with molecular changes in vascular cells that lead to increased levels of cytokine release, upregulated adhesion molecules that allow leukocyte infiltration, upregulation of MMPs and other proteases, and induction of apoptotic cell death pathways (ie. Fas and perforin) that culminate towards the structural deterioration of the aortic wall in AAA [186].
7.1. NADPH Oxidase
A major source of ROS in AAA is NADPH oxidase (NOX), which was reported to be upregulated in AAA patient aortic tissue [183]. VSMCs as well as ECs, fibroblasts, and infiltrating leukocytes are able to form O2- and other oxidants such as H2O2 via NADPH oxidase. Mechanical and pulsatile stretch was shown to induce ROS production by NADHPH oxidase in VSMCs, which subsequently led to NFkB activation and MMP-2 expression and activation [187,188]. VSMCs and tissue infiltrating macrophages can release cytokines such as TNFα that can also activate NADPH oxidase [189,190]. Other activators of NADPH oxidase include growth factors such as AngII and platelet-derived growth factor (PDGF), lipid mediators such as leukotrienes, and lysophosphatidic acid, and oxidized low-density lipoproteins LDL [186]. Genetic deletion of the p47 subunit of NADPH oxidase has been reported to attenuate AAA in the ApoE-/- and AngII induced mouse models [191,192]. Nox2 deficiency in the angII-induced AAA mouse model led to increased AAA diameter and formation through increasing vascular inflammation in the LDL receptor and decreased overall ROS production and macrophages polarization towards the anti-inflammatory phenotype [193]. Another study showed that genetic knockdown Nox1, Nox2, Nox4, or p47phox prevented development of AAA in AngII-infused mice. This observation was accompanied by improved NO and tetrabiopterin bioavailability, reduced superoxide production, and recoupled endothelial nitric oxide synthase [194]. In the same study, two novel NOX4 mutations were identified in AAA patients that were associated with elevated H2O2 levels.
7.2. Low Antioxidant Gene Expression in AAA
Antioxidant profile in AAA can also help determine severity of ROS in patients. Reduced levels of catalase expression are observed in animal AAA tissue, while reduced levels of glutathione peroxidase and paroxinase-1 are observed in AAA patients. Both increased and decreased levels of superoxide dismutase (SOD), which catalyze O2- to O2 and hydrogen peroxide, have been reported in patients with AAA [44,195,196]. Specific isoforms of SOD have also been reported with manganese-dependent SOD (MnSOD) expression increased and extracellular SOD decreased in the PPE-induced rat model [197,198].
A family of mitochondrial transporter proteins that are known to provide protection from cells against oxidative damage are uncoupling proteins (UCPs) [199,200]. Ucp mRNA and protein expression are upregulated in AngII-induced AAA of mice [201]. Knockout of the subtype Ucp-2 was reported to increase AAA incidence, promote elastin degradation and destruction of the aorta via upregulation of MMP2 and MMP9, increase oxidative stress, decrease SOD, and induce VSMC apoptosis in ApoE-/- AAA mice compared to Ucp+/+ aneurysm and wild type controls enzyme system.
7.3. ROS and Histone Acetylation
The biological effects of oxidative stress in various cardiovascular diseases including AAA have been shown to be partly mediated by histone acetylation regulated by HAT and HDAC activities, leading to the activation of NFκB signaling [83]. Elevated HAT and decreased HDAC expression levels were associated with increased ROS in vitro studies. ROS was shown to raise HAT activity and, in turn, increase histone acetylation, promoting inflammation in several cell lines [202]. AngII-induced ROS was reported to increase acetylated cyclphilin, a pro-inflammatory agent, in VSMCs. This was associated with increased phosphorylated ERK1/2, MMP-2 activation, elevated ROS production in VSMCs as well as increased intercellular adhesion molecule 1 and VCAM-1, which cause monocyte adhesion, compared to non-acetylated cyclophilin A [203]. H2O2 also was shown to increase gene acetylation by increasing HAT and suppressing HDAC activities [202]. Interestingly, application of the HDAC inhibitor hydroxamic acid inhibited streptozotocin-induced Nox1, Nox 2, and Nox4 expression and ROS production in mouse aorta [204]. Thus, the field of histone acetylation in AAA appears complex but also rapidly expanding.
8. DNA Methylation in AAA
DNA methylation is a powerful epigenetic mechanism that is important in the maintenance of DNA structure, chromosome stability, X chromosome inactivation, and can regulate elements such as transposons and retrotransposons and gene expression [205]. DNA methylation occurs when a methyl group is added to a region where a cytosine base 5’ to a guanine by DNA methyltransferase enzymes forming a CpG dinucleotide [206,207]. A cluster of CpGs is referred to as an CpG island. Repression of genes via DNA methylation can occur by either preventing transcription factor binding to their binding sites through methylation of cytosine (C) or occupying the promoter region by one of the methyl-cytosine binding (MBD) proteins [208]. Though it is classically known to inhibit gene expression, increased methylation is also associated in some instances with increased gene expression, as methylation can be necessary for transcriptional binding [209,210,211].
There has been limited investigation of the role of DNA methylation in AAA, relative to other epigenetic mechanisms. A large study that assessed peripheral blood mononuclear cell DNA from AAA patients controls reported that global DNA methylation was significantly higher in men with large AAA compared to small AAA and controls [212]. VSMCs isolated from AAA patients showed altered DNA methylation levels found in four genes: v-ets avian erythroblastosis virus E26 oncogene homolog (ERG) with 13 hyper-methylated CpGs, IL-6 receptor (IL-6R) with 2 hyper-methylated CpGs, SMYD2 with 4 hypomethylated CpGs, and SERPINB9 with 6 hypo-methylated CpGs. There was also downregulation of SMYD2 and SERPINB9 in AAA, and a direct relationship between the SMYD2 promoter methylation and expression. Downregulated SMYD2 was observed in the AAA wall. The studies suggest that an epigenetic signature of global and CpG-specific hypermethylation of proinflammatory and pro-apoptotic genes and hypomethylation of anti-inflammatory and anti-apoptotic genes is associated with AAA formation.
A recent study showed that methylation of the SM22α gene promoter was significantly higher in AngII and CaCl2-induced AAA mouse tissues compared to control tissues [213]. Genetic knockdown of the SM22α in VSMCS of both models resulted in accelerated AAA formation, elevated ROS production, and NFκB activation in AAA tissues, while overexpression resulted in opposite effects. This would suggest potential association between gene-specific methylation and AAA development.
It should be noted that age, smoking, and inflammation are major risk factors of AAA that can have a substantial impact on DNA methylation patterns. Studies in aging have shown genome-wide hypo-methylation and promoter-specific hyper-methylation [214,215,216]. Cigarette smoking has shown to lower methylation levels than non-smokers. Smoking cessation resulted in partial restoration of DNA methylation status but was never completely reversed to non-smoking levels [217]; conflicting evidence showing both hypo-methylation and hyper-methylation. A genome-wide methylation study reported that inflammation and increased mortality in chronic kidney disease was associated with global DNA hypermethylation. In atherosclerosis, the underlying mechanism was reported to be based on the inactivation of suppressors of cytokine signaling (SOCS) [218]. However, it remains unclear whether DNA methylation changes are causal factors for inflammatory disease or vice versa [219].
9. Conclusions
The past two decades have shown tremendous growth in the understanding of molecular mechanisms underlying AAA pathobiology. Though it is not possible to include every discovery, we have attempted to report a comprehensive overview of both the genetic and epigenetic mechanisms associated with the role of smooth muscle cells in AAA formation. Though there are no curative therapies for AAA, a deeper understanding of the two areas of molecular biology and how they integrate together could help pave the way forward toward better patient care and AAA therapeutics.
Author Contributions
R.G. and V.S. conceptualized the overall manuscript; R.G. and V.S. wrote the abstract, R.F. and A.M.C. contributed to the editing of the abstract; R.G. designed the figures; R.G. and V.S. wrote the first and subsequent drafts; and all authors contributed to the manuscript editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AAA | Abdominal aortic aneurysm |
| ACTA | Actin alpha 2 |
| ADCY101 | Adenylate cyclase 10 pseudogene 1 |
| ADD3 | Adducin-3 |
| Ang | Angiotensin |
| ApoB100 | Apolipoprotein B100 |
| Bcl-2 | B cell lymphoma 2 |
| C>T | Cytosine to Thymine single nucleotide polymorphism |
| CaCl2 | Calcium chloride |
| CAD | Coronary artery disease |
| CatK/G | Cathepsin K/G |
| CDKN | Cyclin-dependent kinase |
| Chi311 | Chitinase 3-like 1 |
| CNN | Calponin |
| COL | Collagen |
| CpG | Cytosine-guanine dinucleotide cluster |
| DNA | Deoxyribonucleic acid |
| EC | Endothelial cell |
| ECM | Extracellular matrix |
| EDHF | Endothelium-derived hyperpolarising factor |
| ELN | Elastin |
| ERG | E26 oncogene homolog |
| ERK | Extracellular signal regulated kinase |
| G>C | Guanine to Cytosine single nucleotide polymorphism |
| H2O2 | Hydrogen peroxide |
| H3K4me2 | Dimethylation of lysin residue 4 on histone 3 |
| HDAC | Histone deacetylases |
| hESC | Human embryonic stem cell |
| HIF1α | Hypoxia-inducible factor 1 α |
| Hsp90 | Heat shock protein 90 |
| ID3 | Inhibitor of differentiation 3 |
| Il-1β | Interleukin-1β |
| Il-6 | Interleukin-6 |
| iNOS | Inducible nitric oxide |
| Jag1 | Jagged1 |
| JNK | c-Jun N-terminal kinase |
| KAT | Lysine histone acetylase transferase |
| KLF | Kruppel-like factor 4 |
| KLHL35 | Kelch-like family member 35 |
| LKL | Kallikrein |
| LncRNA | Long non-coding ribonucleic acid |
| LRP1 | Lipoprotein receptor-related protein 1 |
| MAPK | Mitogen-activated protein kinase |
| MBD | Methyl-cytosine binding |
| Mcl-1 | Myeloid cell leukemia 1 |
| Mdm2 | Mouse 3T3 cell double minute 2 |
| mIR | Micro ribonucleic acid |
| miRNA | Micro ribonucleic acid |
| MMP | Matrix metallopeptidase |
| mRNA | Messenger ribonucleic acid |
| MRTF-B | Myocardin-related transcription factor-B |
| MYH11 | Myosin heavy chain 11 |
| NADPH | Reduced Nicotinamide adenine dinucleotide phosphate |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| Nkx2-5 | NK-2 transcription factor |
| NOX | NADPH oxidase |
| Nt | Nucleotide |
| OPG | Osteoprotegerin |
| PCSK9 | Propretein convertase subtilisin/kexin type 9 |
| PDGF | Platelet-derived growth factor |
| PI3k | Phosphoinositide 3-kinase |
| PPARγ | Peroxisome proliferative-activated receptor gamma |
| PPE | Porcine pancreatic elastase |
| PTEN | Phosphatase and tensin homolog |
| PVT1 | Plasmacytoma variant translocation 1 |
| RECK | Reversion inducing cysteine-rich protein with kazal motifs |
| RIPK3 | Receptor-interacting serine/threonine-protein kinase 3 |
| RNA | Ribonucleic acid |
| ROS | Reactive oxygen species |
| SERPINA1 | Serine peptidase inhibitor, clade A, member 1 |
| Shh | Sonic hedgehog |
| shRNA | Short hairpin RNA |
| SMYD2 | SET and MYND domain-containing 2 |
| SNP | Single nucleotide polymorphism |
| SOD | Superoxide dismutase |
| SRF | Serum response factor |
| STAT | Signal transducer and activator of transcription proteins |
| TAA | Thoracic aortic aneurysm |
| TGFBR2 | Transforming growth factor beta receptor 2 |
| TGF-β | Transforming growth factor β |
| TIMP | Tissue inhibitor of metallopeptidase |
| TNFα | Tumor necrosis factor α |
| UCP | Uncoupling protein |
| UTR | Untranslated region |
| VSMC | Vascular smooth muscle cell |
| α1-AT | α1-antitrypsin |
References
- Wanhainen, A.; Verzini, F.; Van Herzeele, I.; Allaire, E.; Bown, M.; Cohnert, T.; Dick, F.; van Herwaarden, J.; Karkos, C.; Koelemay, M.; et al. Editor’s choice–European society for vascular surgery (ESVS) 2019 clinical practice guidelines on the management of abdominal aorto-iliac artery aneurysms. Eur. J. Vasc. Endovasc. Surg. 2019, 57, 8–93. [Google Scholar] [CrossRef]
- Grøndal, N.; Søgaard, R.; Lindholt, J.S. Baseline prevalence of abdominal aortic aneurysm, peripheral arterial disease and hypertension in men aged 65–74 years from a population screening study (VIVA trial). Br. J. Surg. 2015, 102, 902–906. [Google Scholar] [CrossRef]
- Lee, E.S.; Pickett, E.; Hedayati, N.; Dawson, D.L.; Pevec, W.C. Implementation of an aortic screening program in clinical practice: Implications for the screen for abdominal aortic aneurysms very efficiently (SAAAVE) Act. J. Vasc. Surg. 2009, 49, 1107–1111. [Google Scholar] [CrossRef]
- Forsdahl, S.H.; Solberg, S.; Singh, K.; Jacobsen, B.K. Abdominal aortic aneurysms, or a relatively large diameter of non-aneurysmal aortas, increase total and cardiovascular mortality: The Tromsø study. Int. J. Epidemiol. 2010, 39, 225–232. [Google Scholar] [CrossRef] [PubMed]
- DE Rango, P.; Farchioni, L.; Fiorucci, B.; Lenti, M. Diabetes and abdominal aortic aneurysms. Eur. J. Vasc. Endovasc. Surg. 2013, 47, 243–261. [Google Scholar] [CrossRef] [PubMed]
- Sweeting, M.J.; Thompson, S.G.; Brown, L.C.; Powell, J.T. Meta-analysis of individual patient data to examine factors affecting growth and rupture of small abdominal aortic aneurysms. Br. J. Surg. 2012, 99, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.W. Basic science of abdominal aortic aneurysms: Emerging therapeutic strategies for an unresolved clinical problem. Curr. Opin. Cardiol. 1996, 11, 504–518. [Google Scholar] [CrossRef] [PubMed]
- Petsophonsakul, P.; Furmanik, M.; Forsythe, R.; Dweck, M.; Schurink, G.W.; Natour, E.; Reutelingsperger, C.; Jacobs, M.; Mees, B. Role of vascular smooth muscle cell phenotypic switching and calcification in aortic aneurysm formation involvement of vitamin K-dependent processes. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1351–1368. [Google Scholar] [CrossRef]
- Pfaltzgraff, E.R.; Bader, D.M. Heterogeneity in vascular smooth muscle cell embryonic origin in relation to adult structure, physiology, and disease. Dev. Dyn. 2015, 244, 410–416. [Google Scholar] [CrossRef]
- Bicknell, C.D.; Kiru, G.; Falaschetti, E.; Powell, J.T.; Poulter, N.R.; Ashby, D.; Brown, L.; Dhanjil, S.; Davies, M.; Earnshaw, J.; et al. An evaluation of the effect of an angiotensinconverting enzyme inhibitor on the growth rate of small abdominal aortic aneurysms: A randomized placebo-controlled trial (AARDVARK). Eur. Heart J. 2016, 37, 3213–3221. [Google Scholar] [CrossRef]
- Kokje, V.B.C.; Hamming, J.F.; Lindeman, J.H.N. Editor’s choice-Pharmaceutical management of small abdominal aortic aneurysms: A systematic review of the clinical evidence. Eur. J. Vasc. Endovasc. Surg. 2015, 50, 702–713. [Google Scholar] [CrossRef]
- Baxter, B.T.; Matsumura, J.; Curci, J.A.; McBride, R.; Larson, L.; Blackwelder, W.; Lam, D.; Wijesinha, M.; Terrin, M. Effect of doxycycline on aneurysm growth among patients with small infrarenal abdominal aortic aneurysms: A randomized clinical trial. JAMA 2020, 323, 2029–2038. [Google Scholar] [CrossRef] [PubMed]
- Bogunovic, N.; Meekel, J.P.; Micha, D.; Blankensteijn, J.D. Impaired smooth muscle cell contractility as a novel concept of abdominal aortic aneurysm pathophysiology. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wahlgren, C.M.; Larsson, E.; Magnusson, P.K.E.; Hultgren, R.; Swedenborg, J. Genetic and environmental contributions to abdominal aortic aneurysm development in a twin population. J. Vasc. Surg. 2010, 51, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Svensjö, S.; Björck, M.; Gürtelschmid, M.; Djavani Gidlund, K.; Hellberg, A.; Wanhainen, A. Low prevalence of abdominal aortic aneurysm among 65-year-old swedish men indicates a change in the epidemiology of the disease. Circulation 2011, 124, 1118–1123. [Google Scholar] [CrossRef]
- Larsson, E.; Granath, F.; Hultgren, R.; Swedenborg, J. A population-based case-control study of the familial risk of abdominal aortic aneurysm. J. Vasc. Surg. 2009, 49, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Johansen, K.; Koepsell, T. Familial tendency for abdominal aortic aneurysms. JAMA 1986, 256, 1934–1936. [Google Scholar] [CrossRef]
- Ogata, T.; Mackean, G.L.; Cole, C.W.; Arthur, C.; Andreou, P.; Tromp, G.; Kuivaniemi, H. NIH public access. J. Vasc. Surg. 2005, 42, 891–897. [Google Scholar] [CrossRef]
- Jones, G.T.; Hill, B.G.; Curtis, N.; Kabir, T.D.; Wong, L.E.; Tilyard, M.W.; Williams, M.J.A.; Rij, A.M. Van comparison of three targeted approaches to screening for abdominal aortic aneurysm based on cardiovascular risk. Br. J. Surg. 2016, 103, 1139–1146. [Google Scholar] [CrossRef]
- Sandford, R.M.; Bown, M.J.; London, N.J.; Sayers, R.D. The genetic basis of abdominal aortic aneurysm: A review. Eur. J. Vasc. Endovasc. Surg. 2007, 390, 381–390. [Google Scholar] [CrossRef]
- Sakalihasan, N.; Defraigne, J.O.; Kerstenne, M.A.; Cheramy-Bien, J.P.; Smelser, D.T.; Tromp, G.; Kuivaniemi, H. Family members of patients with abdominal aortic aneurysms are at increased risk for aneurysms: Analysis of 618 probands and their families from the liège AAA family study. Ann. Vasc. Surg. 2014, 28, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Van de Luijtgaarden, K.M.; Bastos Gonçalves, F.; Hoeks, S.E.; Majoor-Krakauer, D.; Rouwet, E.V.; Stolker, R.J.; Verhagen, H.J.M. Familial abdominal aortic aneurysm is associated with more complications after endovascular aneurysm repair. J. Vasc. Surg. 2014, 59, 275–282. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Beckman, J.A. Aortic aneurysm: Pathophysiology, epidmiology and prognosis. In Vascular Medicine: A Companion to Braunwald’s Heart Disease; Creager, M., Dzau, V., Loscalzo, J., Eds.; Saunders Elsevier Inc.: Philadephia, PA, USA, 2006; pp. 457–470. [Google Scholar]
- Gomez, D.; Swiatlowska, P.; Owens, G.K. Epigenetic control of smooth muscle cell identity and lineage memory. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2508–2516. [Google Scholar] [CrossRef] [PubMed]
- Van’t Hof, F.; Ruigrok, Y.; Lee, C.; Ripke, S.; Anderson, G.; de Andrade, M.; Baas, A.; Blankensteijn, J.; Bottinger, E.; Bown, M.J.; et al. shared genetic risk factors of intracranial, abdominal, and thoracic aneurysms. J. Am. Hear. Assoc. 2016, 5, 1–19. [Google Scholar] [CrossRef]
- Helgadottir, A.; Thorleifsson, G.; Magnusson, K.P.; Grétarsdottir, S.; Steinthorsdottir, V.; Manolescu, A.; Jones, G.T.; Rinkel, G.J.E.; Blankensteijn, J.D.; Ronkainen, A.; et al. The same sequence variant on 9p21 associates with myocardial infarction, abdominal aortic aneurysm and intracranial aneurysm. Nat. Genet. 2008, 40, 217–224. [Google Scholar] [CrossRef]
- Yao, Q.; Renault, M.A.; Chapouly, C.; Vandierdonck, S.; Belloc, I.; Jaspard-Vinassa, B.; Daniel-Lamazière, J.M.; Laffargue, M.; Merched, A.; Desgranges, C.; et al. Sonic hedgehog mediates a novel pathway of PDGF-BB-dependent vessel maturation. Blood 2014, 123, 2429–2437. [Google Scholar] [CrossRef]
- High, F.A.; Min, M.L.; Pear, W.S.; Loomes, K.M.; Kaestner, K.H.; Epstein, J.A. Endothelial expression of the Notch ligand Jagged1 is required for vascular smooth muscle development. Proc. Natl. Acad. Sci. USA 2008, 105, 1955–1959. [Google Scholar] [CrossRef]
- Campa, J.S.; Greenhalgh, R.M.; Powell, J.T. Elastin degradation in abdominal aortic aneurysms. Atherosclerosis 1987, 65, 13–21. [Google Scholar] [CrossRef]
- Cheung, C.; Bernardo, A.S.; Trotter, M.W.B.; Pedersen, R.A.; Sinha, S. Generation of human vascular smooth muscle subtypes provides insight into embryological origing-dependent disease susceptibility. Nat. Biotechnol. 2012, 30, 165–173. [Google Scholar] [CrossRef]
- Wiegreffe, C.; Christ, B.; Huang, R.; Scaal, M. Remodeling of aortic smooth muscle during avian embryonic development. Dev. Dyn. 2009, 238, 624–631. [Google Scholar] [CrossRef]
- Tromp, G.; Kuivaniemi, H.; Hinterseher, I.; Carey, D.J. Novel genetic mechanisms for aortic aneurysms. Curr. Atheroscler. Rep. 2010, 12, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Ruddy, J.M.; Jones, J.A.; Ikonomidis, J.S. Pathophysiology of thoracic aortic aneurysm (TAA): Is it not one uniform aorta? Role of embryologic origin. Prog. Cardiovasc. Dis. 2013, 56, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.C.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Dalton, M.L.; Gadson, P.F.; Wrenn, R.W.; Rosenquist, T.H. Homocysteine signal cascade: Production of phospholipids, activation of protein kinase C, and the induction of c-fos and c-myb in smooth muscle cells. FASEB J. 1997, 11, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Gadson, P.F.; Dalton, M.L.; Patterson, E.; Svoboda, D.D.; Hutchinson, L.; Schram, D.; Rosenquist, T.H. Differential response of mesoderm- and neural crest-derived smooth muscle to TGF-β1: Regulation of c-myb and α1 (I) procollagen genes. Exp. Cell Res. 1997, 230, 169–180. [Google Scholar] [CrossRef]
- Rateri, D.L.; Howatt, D.A.; Moorleghen, J.J.; Charnigo, R.; Cassis, L.A.; Daugherty, A. Prolonged infusion of angiotensin II in apoE-/-mice promotes macrophage recruitment with continued expansion of abdominal aortic aneurysm. Am. J. Pathol. 2011, 179, 1542–1548. [Google Scholar] [CrossRef]
- Owens, A.P.; Subramanian, V.; Moorleghen, J.J.; Guo, Z.; McNamara, C.A.; Cassis, L.A.; Daugherty, A. Angiotensin II induces a region-specific hyperplasia of the ascending aorta through regulation of inhibitor of differntiation 3. Circ. Res. 2010, 106, 611–619. [Google Scholar] [CrossRef]
- Sakalihasan, N.; Michel, J.; Katsargyris, A.; Kuivaniemi, H.; Defraigne, J.; Nchimi, A.; Powell, J.T.; Yoshimura, K.; Hultgren, R. Abdominal aortic aneurysms. Nat. Rev. Dis. Prim. 2018, 4, 34. [Google Scholar] [CrossRef]
- Airhart, N.; Brownstein, B.H.; Cobb, J.P.; Schierding, W.; Arif, B.; Ennis, T.L.; Thompson, R.W.; Curci, J.A.; Louis, S. From the society for vascular surgery smooth muscle cells from abdominal aortic aneurysms are unique and can independently and synergistically degrade insoluble elastin. J. Vasc. Surg. 2013, 60, 1033–1042. [Google Scholar] [CrossRef]
- Ailawadi, G.; Moehle, C.W.; Pei, H.; Walton, S.P.; Yang, Z.; Kron, I.L.; Lau, C.L.; Owens, G.K. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2009, 138, 1392–1399. [Google Scholar] [CrossRef]
- Michel, J.B. Anoïkis in the cardiovascular system: Known and unknown extracellular mediators. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 2146–2154. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Z.; Ren, J.; Morgan, S.; Assa, C.; Liu, B. Receptor-interacting protein Kinase 3 contributes to abdominal aortic aneurysms via smooth muscle cell necrosis and inflammation. Circ. Res. 2015, 116, 600–611. [Google Scholar] [CrossRef] [PubMed]
- Quintana, R.A.; Taylor, W.R. Cellular mechanisms of aortic aneurysm formation. Circ. Res. 2019, 124, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Kadoglou, N.P.; Liapis, C.D.; Kadoglou, N.P.; Liapis, C.D. Matrix metalloproteinases: Contribution to pathogenesis, diagnosis, surveillance and treatment of abdominal aortic aneurysms Matrix metalloproteinases: Contribution to pathogenesis, diagnosis, surveillance and treatment of abdominal aortic aneurysms. Curr. Med. Res. Opin. 2004, 20, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Newman, K.M.; Jean-Claude, J.; Li, H.; Scholes, J.V.; Ogata, Y.; Nagase, H.; Tilson, M.D. Cellular localization of matrix metalloproteinases in the abdominal aortic aneurysm wall. J. Vasc. Surg. 1994, 20, 814–820. [Google Scholar] [CrossRef]
- Crowther, M.; Goodall, S.; Jones, J.L.; Bell, P.R.F.; Thompson, M.M. Increased matrix metalloproteinase 2 expression in vascular smooth muscle cells cultured from abdominal aortic aneurysms. J. Vasc. Surg. 2000, 32, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Tamarina, N.A.; McMillan, W.D.; Shively, V.P.; Pearce, W.H. Expression of matrix metalloproteinases and their inhibitors in aneurysms and normal aorta. Surgery 1997, 122, 264–272. [Google Scholar] [CrossRef]
- Goodall, S.; Porter, K.E.; Bell, P.R.; Thompson, M.M. Enhanced invasive properties exhibited by smooth muscle cells are associated with elevated production of MMP-2 in patients with aortic aneurysms. Eur. J. Vasc. Endovasc. Surg. 2002, 24, 72–80. [Google Scholar] [CrossRef][Green Version]
- Davis, V.; Persidskaia, R.; Baca-Regen, L.; Itoh, Y.; Nagase, H.; Persidsky, Y.; Ghorpade, A.; Timothy Baxter, B. Matrix metalloproteinase-2 production and its binding to the matrix are increased in abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1625–1633. [Google Scholar] [CrossRef]
- Saracini, C.; Bolli, P.; Sticchi, E.; Pratesi, G.; Pulli, R.; Sofi, F.; Pratesi, C.; Gensini, G.F.; Abbate, R.; Giusti, B. Polymorphisms of genes involved in extracellular matrix remodeling and abdominal aortic aneurysm. J. Vasc. Surg. 2012, 55, 171–179. [Google Scholar] [CrossRef]
- Jones, G.T.; Phillips, V.L.; Harris, E.L.; Rossaak, J.I.; Van Rij, A.M. Functional matrix metalloproteinase-9 polymorphism (C-1562T) associated with abdominal aortic aneurysm. J. Vasc. Surg. 2003, 38, 1363–1367. [Google Scholar] [CrossRef]
- Thompson, R.W.; Holmes, D.R.; Mertens, R.A.; Liao, S.; Botney, M.D.; Mecham, R.P.; Welgus, H.G.; Parks, W.C. Production and localization of 92-kilodalton gelatinase in abdominal aortic aneurysms. Am. Soc. Clin. Investig. 1995, 96, 318–326. [Google Scholar] [CrossRef]
- Pyo, R.; Lee, J.K.; Shipley, J.M.; Curci, J.A.; Mao, D.; Ziporin, S.J.; Ennis, T.L.; Shapiro, S.D.; Senior, R.M.; Thompson, R.W. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J. Clin. Investig. 2000, 105, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Longo, G.M.; Xiong, W.; Greiner, T.C.; Zhao, Y.; Fiotti, N.; Baxter, B.T. Work in concert to produce aortic aneurysms. J. Clin. Investig. 2002, 110, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Curci, J.A.; Liao, S.; Huffman, M.D.; Shapiro, S.D.; Thompson, R.W. Expression and localization of macrophage elastase (matrix metalloproteinase-12) in abdominal aortic aneurysms. J. Clin. Investig. 1998, 102, 1900–1910. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Xing, L.; Zeng, C.; Wu, T.; Gui, Y.; Li, W.; Lan, T.; Yang, Y.; Gu, Q.; Qi, C.; et al. Inactivation of PI3Kδ induces vascular injury and promotes aneurysm development by upregulating the AP-1/MMP-12 pathway in macrophages. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 368–377. [Google Scholar] [CrossRef]
- Lizarbe, T.R.; Tarín, C.; Gómez, M.; Lavin, B.; Aracil, E.; Orte, L.M.; Zaragoza, C. Nitric oxide induces the progression of abdominal aortic aneurysms through the matrix metalloproteinase inducer EMMPRIN. Am. J. Pathol. 2009, 175, 1421–1430. [Google Scholar] [CrossRef]
- Nollendorfs, A.; Greiner, T.C.; Nagase, H.; Baxter, B.T. The expression and localization of membrane type-1 matrix metalloproteinase in human abdominal aortic aneurysms. J. Vasc. Surg. 2001, 34, 316–322. [Google Scholar] [CrossRef]
- Xiong, W.; Knispel, R.; MacTaggart, J.; Greiner, T.C.; Weiss, S.J.; Baxter, B.T. Membrane-type 1 matrix metalloproteinase regulates macrophage-dependent elastolytic activity and aneurysm formation in vivo. J. Biol. Chem. 2009, 284, 1765–1771. [Google Scholar] [CrossRef]
- Sun, J.; Sukhova, G.K.; Zhang, J.; Chen, H.; Sjoberg, S.; Libby, P.; Xia, M.; Xiong, N.; Gelb, B.D.; Shi, G.-P. Cathepsin K deficiency reduces elastase perfusion-induced abdominal aortic aneurysms in mice. Arter. Thromb. Vasc. Biol. 2012, 32, 15–23. [Google Scholar] [CrossRef]
- Wang, J.; Kukhova, G.K.; Liu, J.; Ozaki, K.; Lesner, A.; Libby, P.; Kovanen, P.T.; Shi, G.-P. Cathepsin G deficiency reduces peri-aortic calcium chloride injury-induced abdominal aortic aneurysms in mice. Physiol. Behav. 2015, 62, 1615–1624. [Google Scholar] [CrossRef]
- Carretero, O.A. Vascular remodelling and the kallikrein-kinin system. J. Clin. Investig. 2005, 115, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Biros, E.; Norman, P.E.; Walker, P.J.; Nataatmadja, M.; West, M.; Golledge, J. A single nucleotide polymorphism in exon 3 of the kallikrein 1 gene is associated with large but not small abdominal aortic aneurysm. Atherosclerosis 2011, 217, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Defawe, O.D.; Colige, A.; Lambert, C.A.; Munaut, C.; Delvenne, P.; Lapière, C.M.; Limet, R.; Nusgens, B.V.; Sakalihasan, N. TIMP-2 and PAI-1 mRNA levels are lower in aneurysmal as compared to athero-occlusive abdominal aortas. Cardiovasc. Res. 2003, 60, 205–213. [Google Scholar] [CrossRef]
- Brophy, C.M.; Marks, W.H.; Reilly, J.M.; David Tilson, M. Decreased tissue inhibitor of metalloproteinases (TIMP) in abdominal aortic aneurysm tissue: A preliminary report. J. Surg. Res. 1991, 50, 653–657. [Google Scholar] [CrossRef]
- Eskandari, M.K.; Vijungco, J.D.; Flores, A.; Borensztajn, J.; Shively, V.; Pearce, W.H. Enhanced abdominal aortic aneurysm in TIMP-1-deficient mice. J. Surg. Res. 2005, 123, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Tilson, M.D.; Reilly, J.M.; Brophy, C.M.; Webster, E.L.; Barnett, T.R. Expression and sequence of the gene for tissue inhibitor of metalloproteinases in patients with abdominal aortic aneurysms. J. Vasc. Surg. 1993, 18, 266–270. [Google Scholar] [CrossRef][Green Version]
- Wang, X.; Tromp, G.; Cole, C.W.; Verloes, A.; Sakalihasan, N.; Yoon, S.; Kuivaniemi, H. Analysis of coding sequences for tissue inhibitor of metalloproteinases 1 (TIMP1)) and 2 (TIMP2) in patients with aneurysms. Matrix Biol. 1999, 18, 121–124. [Google Scholar] [CrossRef]
- Hinterseher, I.; Krex, D.; Kuhlisch, E.; Schmidt, K.G.; Pilarsky, C.; Schneiders, W.; Saeger, H.D.; Bergert, H. Tissue inhibitor of metalloproteinase-1 (TIMP-1) polymorphisms in a Caucasian population with abdominal aortic aneurysm. World J. Surg. 2007, 31, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- Annabi, B.; Shédid, D.; Ghosn, P.; Kenigsberg, R.L.; Desrosiers, R.R.; Bojanowski, M.W.; Beaulieu, E.; Nassif, E.; Moumdjian, R.; Béliveau, R. Differential regulation of matrix metalloproteinase activities in abdominal aortic aneurysms. J. Vasc. Surg. 2002, 35, 539–546. [Google Scholar] [CrossRef]
- Hinterseher, I.; Tromp, G.; Kuivaniemi, H. Genes and abdominal aortic aneurysm. Ann. Vasc. Surg. 2011, 25, 388–412. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Juttermann, R.; Soloway, P.D. TIMP-2 is required for efficient activation of proMMP-2 in vivo. J. Biol. Chem. 2000, 275, 26411–26415. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, M.M.; Fridman, R. TIMP-2 (tissue inhibitor of metalloproteinase-2) regulates MMP-2 (matrix metalloproteinase-2) activity in the extracellular environment after pro-MMP-2 activation by MT1 (membrane type 1)-MMP. Biochem. J. 2003, 374, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Knispel, R.; Mactaggart, J.; Baxter, B.T. Effects of tissue inhibitor of metalloproteinase 2 deficiency on aneurysm formation. J. Vasc. Surg. 2006, 44, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; Fan, D.; Kandalam, V.; Lee, J.; Das, S.K.; Wang, X.; Baldwin, T.A.; Oudit, G.Y.; Kassiri, Z. Loss of Timp3 gene leads to abdominal aortic aneurysm formation in response to angiotensin II *. J. Biol. Chem. 2012, 287, 44083–44096. [Google Scholar] [CrossRef]
- Narayanan, N.; Tyagi, N.; Shah, A.; Pagni, S.; Tyagi, S.C. Hyperhomocysteinemia during aortic aneurysm, a plausible role of epigenetics. Int. J. Physiol. Pathophysiol. Pharmacol. 2013, 5, 32–42. [Google Scholar]
- Mangum, K.D.; Farber, M.A. Genetic and epigenetic regulation of abdominal aortic aneurysms. Clin. Genet. 2020, 97, 1–12. [Google Scholar] [CrossRef]
- Kim, V.N. MicroRNA biogenesis: Coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 2005, 6, 376–385. [Google Scholar] [CrossRef]
- Maegdefessel, L.; Azuma, J.; Toh, R.; Merk, D.R.; Deng, A.; Chin, J.T.; Raaz, U.; Schoelmerich, A.M.; Raiesdana, A.; Leeper, N.J.; et al. Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development. J. Clin. Investig. 2012, 122, 497–506. [Google Scholar] [CrossRef]
- Merk, D.R.; Chin, J.T.; Dake, B.A.; Maegdefessel, L.; Miller, M.O.; Kimura, N.; Tsao, P.S.; Iosef, C.; Berry, G.J.; Mohr, F.W.; et al. miR-29b participates in early aneurysm development in marfan syndrome. Circ. Res. 2012, 110, 312–324. [Google Scholar] [CrossRef]
- Kim, C.W.; Kumar, S.; Son, D.J.; Jang, I.-H.; Griendling, K.K.; Jo, H. Prevention of abdominal aortic aneurysm by anti-miRNA-712 or anti-miR-205 in Angiotensin II infused mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1412–1421. [Google Scholar] [CrossRef]
- Pons, D.; De Vries, F.R.; Van Den Elsen, P.J.; Heijmans, B.T.; Quax, P.H.A.; Jukema, J.W. Epigenetic histone acetylation modifiers in vascular remodelling: New targets for therapy in cardiovascular disease. Eur. Heart J. 2009, 30, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Tekwani, B.L. Histone deacetylases inhibitors in neurodegenerative diseases, neuroprotection and neuronal differentiation. Front. Pharmacol. 2020, 11, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Galán, M.; Varona, S.; Orriols, M.; Rodríguez, J.A.; Aguiló, S.; Dilmé, J.; Camacho, M.; Martínez-González, J.; Rodriguez, C. Induction of histone deacetylases (HDACs) in human abdominal aortic aneurysm: Therapeutic potential of HDAC inhibitors. DMM Dis. Model. Mech. 2016, 9, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Jovinge, S.; Hultgårdh-Nilsson, A.; Regnström, J.; Nilsson, J. Tumor necrosis factor-α activates smooth muscle cell migration in culture and is expressed in the balloon-injured rat aorta. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Tanios, F.; Reeps, C.; Zhang, J.; Schwamborn, K.; Eckstein, H.H.; Zernecke, A.; Pelisek, J. Histone acetylation and histone acetyltransferases show significant alterations in human abdominal aortic aneurysm. Clin. Epigenetics 2016, 8, 1–13. [Google Scholar] [CrossRef]
- Zhang, S.L.; Du, X.; Chen, Y.Q.; Tan, Y.S.; Liu, L. Potential medication treatment according to pathological mechanisms in abdominal aortic aneurysm. J. Cardiovasc. Pharmacol. 2018, 71, 46–57. [Google Scholar] [CrossRef]
- Hadi, T.; Boytard, L.; Silvestro, M.; Alebrahim, D.; Jacob, S.; Feinstein, J.; Barone, K.; Spiro, W.; Hutchison, S.; Simon, R.; et al. Macrophage-derived netrin-1 promotes abdominal aortic aneurysm formation by activating MMP3 in vascular smooth muscle cells. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Boyle, J.J.; Weissberg, P.L.; Bennett, M.R. Tumor necrosis factor-α promotes macrophage-induced vascular smooth muscle cell apoptosis by direct and autocrine mechanisms. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1553–1558. [Google Scholar] [CrossRef]
- Middleton, R.K.; Lloyd, G.M.; Bown, M.J.; Cooper, N.J.; London, N.J.; Sayers, R.D. The pro-inflammatory and chemotactic cytokine microenvironment of the abdominal aortic aneurysm wall: A protein array study. J. Vasc. Surg. 2007, 45, 574–580. [Google Scholar] [CrossRef]
- Akerman, A.W.; Stroud, R.E.; Bans, R.W.; Grespin, R.T.; McDonald, L.T.; LaRue, R.A.C.; Mukerjee, R.; Ikonomidis, J.S.; Jones, J.A.; Ruddy, J.M. Elevated wall tension initiates interleukin-6 expression and abdominal aortic dilation. Ann. Vasc. Surg. 2018, 46, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. The interface of atherosclerosis and thrombosis: Basic mechanisms. Vasc. Med. 1998, 3, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Beasley, D.; McGuiggin, M.E.; Dinarello, C.A. Human vascular smooth muscle cells produce an intracellular form of interleukin-1 receptor antagonist. Am. J. Physiol. Cell Physiol. 1995, 269. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Castresana, M.R.; Newman, W.H. NF-κB is required for TNF-α-directed smooth muscle cell migration. FEBS Lett. 2001, 508, 360–364. [Google Scholar] [CrossRef]
- Clarke, M.C.H.; Talib, S.; Figg, N.L.; Bennett, M.R. Vascular smooth muscle cell apoptosis induces interleukin-1-directed inflammation: Effects of hyperlipidemia-mediated inhibition of phagocytosis. Circ. Res. 2010, 106, 363–372. [Google Scholar] [CrossRef]
- Geng, Y.J.; Wu, Q.; Muszynski, M.; Hansson, G.K.; Libby, P. Apoptosis of vascular smooth muscle cells induced by in vitro stimulation with interferon-γ, tumor necrosis factor-α, and interleukin-1β. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 19–27. [Google Scholar] [CrossRef]
- Juvonen, J.; Surcel, H.M.; Satta, J.; Teppo, A.M.; Bloigu, A.; Syrjälä, H.; Airaksinen, J.; Leinonen, M.; Saikku, P.; Juvonen, T. Elevated circulating levels of inflammatory cytokines in patients with abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2843–2847. [Google Scholar] [CrossRef]
- Esnault, S.; Kelly, E.A.; Johnson, S.H.; Delain, L.P.; Haedt, M.J.; Noll, A.L.; Sandbo, N.; Jarjour, N.N. Matrix Metalloproteinase-9-Dependent Release of IL-1β by Human Eosinophils. Mediators Inflamm. 2019, 2019, 7479107. [Google Scholar] [CrossRef]
- Bauer, J.; Huy, C.; Brenmoehl, J.; Obermeier, F.; Bock, J. Matrix metalloproteinase-1 expression induced by IL-1β requires acid sphingomyelinase. FEBS Lett. 2009, 583, 915–920. [Google Scholar] [CrossRef]
- Xie, S.; Issa, R.; Sukkar, M.B.; Oltmanns, U.; Bhavsar, P.K.; Papi, A.; Caramori, G.; Adcock, I.; Chung, K.F. Induction and regulation of matrix metalloproteinase-12 in human airway smooth muscle cells. Respir. Res. 2005, 6, 1–11. [Google Scholar] [CrossRef]
- Raymond, L.; Eck, S.; Mollmark, J.; Hays, E.; Tomek, I.; Kantor, S.; Elliott, S.; Vincenti, M. Interleukin-1 beta induction of matrix metalloproteinase-1 transcription in chondrocytes requires ERK-dependent activation of CCAAT enhancer-binding protein-beta. J. Cell. Physiol. 2006, 207, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, W.; Huwiler, A.; Beck, K.-F.; Walpen, S.; Pfeilschifter, J. Amplification of IL-1β-induced matrix metalloproteinase-9 expression by superoxide in rat glomerular mesangial cells is mediated by increased activities of NF-κB and activating protein-1 and involves activation of the mitogen-activated protein kinase path. J. Immunol. 2000, 165, 5788–5797. [Google Scholar] [CrossRef] [PubMed]
- Marculescu, R.; Sodeck, G.; Domanovits, H.; Hobusch, G.; Exner, M.; Heinzl, H.; Huber, K.; Mannhalter, C.; Minar, E.; Wagner, O.; et al. Interleukin-1 gene cluster variants and abdmominal aortic aneurysms. Thromb. Haemost. 2005, 94, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Bown, M.J.; Burton, P.R.; Horsburgh, T.; Nicholson, M.L.; Bell, P.R.F.; Sayers, R.D. The role of cytokine gene polymorphisms in the pathogenesis of abdominal aortic aneurysms: A case-control study. J. Vasc. Surg. 2003, 37, 999–1005. [Google Scholar] [CrossRef]
- Thompson, A.R.; Drenos, F.; Hafez, H.; Humphries, S.E. Candidate gene association studies in abdominal aortic aneurysm disease: A review and meta-analysis. Eur. J. Vasc. Endovasc. Surg. 2008, 35, 19–30. [Google Scholar] [CrossRef][Green Version]
- Tang, M.; Fang, J. TNF-α regulates apoptosis of human vascular smooth muscle cells through gap junctions. Mol. Med. Rep. 2017, 15, 1407–1411. [Google Scholar] [CrossRef]
- Wilson, A.G.; Symons, J.A.; Mcdowell, T.L.; Mcdevitt, H.O.; Duff, G.W. Effects of a polymorphism in the human tumor necrosis factor α promoter on transcriptional activation. Proc. Natl. Acad. Sci. USA 1997, 94, 3195–3199. [Google Scholar] [CrossRef]
- Smallwood, L.; Allcock, R.; Van Bockxmeer, F.; Warrington, N.; Palmer, L.J.; Iacopetta, B.; Golledge, J.; Norman, P.E. Polymorphisms of the matrix metalloproteinase 9 gene and abdominal aortic aneurysm. Br. J. Surg. 2008, 95, 1239–1244. [Google Scholar] [CrossRef]
- Jones, G.; Brull, D.; Brown, L.; Sian, M.; Greenhalgh, R.; Humphries, S.; Powell, J. Interleukin-6 (IL-6) and the prognosis of abdominal aortic aneurysms. Circulation 2001, 103, 2260–2265. [Google Scholar] [CrossRef]
- Doyle, A.J.; Redmond, E.M.; Gillespie, D.L.; Knight, P.A.; Cullen, J.P.; Cahill, P.A.; Morrow, D.J. Differential expression of Hedgehog/Notch and transforming growth factor-β in human abdominal aortic aneurysms. Physiol. Behav. 2015, 62, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.M.; Rateri, D.L.; Daugherty, A.; Arbor, A. Abdominal aortic aneurysm: Novel mechanisms and therapies. Curr. Opin. Cardiol. 2015, 30, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tedgui, A.; Mallat, Z.; Wang, Y.; Ait-oufella, H.; Herbin, O.; Bonnin, P.; Ramkhelawon, B. TGF- b activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II –infused mice Find the latest version: TGF-β activity protects against inflammatory aortic aneurysm progression and complications in angiotens. J. Clin. Investig. 2010, 120, 422–432. [Google Scholar] [CrossRef]
- Dai, X.; Shen, J.; Priyanka Annam, N.; Jiang, H.; Levi, E.; Schworer, C.M.; Tromp, G.; Arora, A.; Higgins, M.; Wang, X.F.; et al. SMAD3 deficiency promotes vessel wall remodeling, collagen fiber reorganization and leukocyte infiltration in an inflammatory abdominal aortic aneurysm mouse model. Sci. Rep. 2015, 5, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Krishna, S.M.; Seto, S.W.; Jose, R.J.; Biros, E.; Moran, C.S.; Wang, Y.; Clancy, P.; Golledge, J. A peptide antagonist of thrombospondin-1 promotes abdominal aortic aneurysm progression in the angiotensin II-infused apolipoprotein-E-deficient mouse. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Golledge, J.; Clancy, P.; Jones, G.T.; Cooper, M.; Palmer, L.J.; Van Rij, A.M.; Norman, P.E. Possible association between genetic polymorphisms in transforming growth factor β receptors, serum transforming growth factor β1 concentration and abdominal aortic aneurysm. Br. J. Surg. 2009, 96, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Baas, A.F.; Medic, J.; Van ’T Slot, R.; De Kovel, C.G.; Zhernakova, A.; Geelkerken, R.H.; Kranendonk, S.E.; Van Sterkenburg, S.M.; Grobbee, D.E.; Boll, A.P.; et al. Association of the TGF-Β receptor genes with abdominal aortic aneurysm. Eur. J. Hum. Genet. 2010, 18, 240–244. [Google Scholar] [CrossRef][Green Version]
- Du, S.J.; Tan, X.; Zhang, J. SMYD proteins: Key regulators in skeletal and cardiac muscle development and function. Anat. Rec. 2014, 297, 1650–1662. [Google Scholar] [CrossRef]
- Qi, J.; Yang, P.; Yi, B.; Huo, Y.; Chen, M.; Zhang, J.; Sun, J. Heat shock protein 90 inhibition by 17-DMAG attenuates abdominal aortic aneurysm formation in mice. Am. J. Physiol. Hear. Circ. Physiol. 2015, 308, H841–H852. [Google Scholar] [CrossRef]
- Xu, G.; Liu, G.; Xiong, S.; Liu, H.; Chen, X.; Zheng, B. The histone methyltransferase Smyd2 is a negative regulator of macrophage activation by suppressing interleukin 6 (IL-6) and tumor necrosis factor α (tnf-α). J. Biol. Chem. 2015, 290, 5414–5423. [Google Scholar] [CrossRef]
- Jones, G.T.; Tromp, G.; Kuivaniemi, H.; Gretarsdottir, S.; Baas, A.F.; Giusti, B.; Strauss, E.; Van’T Hof, F.N.G.; Webb, T.R.; Erdman, R.; et al. Meta-analysis of genome-wide association studies for abdominal aortic aneurysm identifies four new disease-specific risk loci. Circ. Res. 2017, 120, 341–353. [Google Scholar] [CrossRef]
- Jones, G.T.; Bown, M.J.; Gretarsdottir, S.; Romaine, S.P.R.; Helgadottir, A.; Yu, G.; Tromp, G.; Norman, P.E.; Jin, C.; Baas, A.F.; et al. A sequence variant associated with sortilin-1 (SORT1) on 1p13.3 is independently associated with abdominal aortic aneurysm. Hum. Mol. Genet. 2013, 22, 2941–2947. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Strong, A.; Frank-Kamenetsky, M.; Lee, N.E.; Ahfeldt, T.; Sachs, K.V.; Li, X.; Li, H.; Kuperwasser, N.; Vera, M.; et al. Cholesterol Locus. Nature 2010, 466, 714–719. [Google Scholar] [CrossRef]
- Kjolby, M.; Andersen, O.M.; Breiderhoff, T.; Fjorback, A.W.; Pedersen, K.M.; Madsen, P.; Jansen, P.; Heeren, J.; Willnow, T.E.; Nykjaer, A. Sort1, encoded by the cardiovascular risk locus 1p13.3, is a regulator of hepatic lipoprotein export. Cell Metab. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Jiang, W.; Yang, H.; Lin, Q.; Qin, X. The miR-182/SORT1 axis regulates vascular smooth muscle cell calcification in vitro and in vivo. Exp. Cell Res. 2018, 362, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Maegdefessel, L.; Dalman, R.L.; Tsao, P.S. Pathogenesis of Abdominal Aortic Aneurysms: MicroRNAs, Proteases, Genetic Associations. Annu. Rev. Med. 2014, 65, 49–62. [Google Scholar] [CrossRef]
- Tang, H.; Sun, Y.; Shi, Z.; Huang, H.; Fang, Z.; Chen, J.; Xiu, Q.; Li, B. YKL-40 Induces IL-8 expression from bronchial epithelium via MAPK (JNK and ERK) and NF-κB pathways, causing bronchial smooth muscle proliferation and migration. J. Immunol. 2013, 190, 438–446. [Google Scholar] [CrossRef]
- Elia, L.; Quintavalle, M.; Zhang, J.; Contu, R.; Cossu, L.; Latronico, M.V.G.; Peterson, K.L.; Indolfi, C.; Catalucci, D.; Chen, J.; et al. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: Correlates with human disease. Cell Death Differ. 2009, 16, 1590–1598. [Google Scholar] [CrossRef]
- Fichtlscherer, S.; De Rosa, S.; Fox, H.; Schwietz, T.; Fischer, A.; Liebetrau, C.; Weber, M.; Hamm, C.W.; Röxe, T.; Müller-Ardogan, M.; et al. Circulating microRNAs in patients with coronary artery disease. Circ. Res. 2010, 107, 677–684. [Google Scholar] [CrossRef]
- Rangrez, A.Y.; Massy, Z.A.; Meuth, V.M.-L.; Metzinger, L. MiR-143 and miR-145 molecular keys to switch the phenotype of vascular smooth muscle cells. Circ. Cardiovasc. Genet. 2011, 4, 197–205. [Google Scholar] [CrossRef]
- Riches, K.; Angelini, T.G.; Mudhar, G.S.; Kaye, J.; Clark, E.; Bailey, M.A.; Sohrabi, S.; Korossis, S.; Walker, P.G.; Scott, D.J.A.; et al. Exploring smooth muscle phenotype and function in a bioreactor model of abdominal aortic aneurysm. J. Transl. Med. 2013, 11, 1. [Google Scholar] [CrossRef]
- Thompson, A.R.; Golledge, J.; Cooper, J.A.; Hafez, H.; Norman, P.E.; Humphries, S.E. Sequence variant on 9p21 is associated with the presence of abdominal aortic aneurysm disease but does not have an impact on aneurysmal expansion. Eur. J. Hum. Genet. 2009, 17, 391–394. [Google Scholar] [CrossRef] [PubMed]
- Bown, M.J.; Braund, P.S.; Thompson, J.; London, N.J.M.; Samani, N.J.; Sayers, R.D. Association between the coronary artery disease risk locus on chromosome 9p21.3 and abdominal aortic aneurysm. Circ. Cardiovasc. Genet. 2008, 1, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Biros, E.; Cooper, M.; Palmer, L.J.; Walker, P.; Norman, P.E.; Golledge, J. Association of an allele on chromosome 9 and abdominal aortic aneurysm aneurysm. Atherosclerosis 2010, 212, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Leeper, N.J.; Raiesdana, A.; Kojima, Y.; Kundu, R.K.; Cheng, H.; Maegdefessel, L.; Toh, R.; Ahn, G.O.; Ali, Z.A.; Anderson, R.D.; et al. Loss of CDKN2B promotes p53-dependent smooth muscle cell apoptosis and aneurysm formation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Zhu, Y.; Afzal, V.; Gong, E.; Attanasio, C.; Blow, M.J.B.; Cohen, J.C.; Rubin, E.M.; Pennacchio, L.A. Targeted deletion of the 9p21 noncoding coronary artery disease risk interval in mice. Nature 2010, 464, 409–412. [Google Scholar] [CrossRef]
- Golledge, J.; Kuivaniemi, H. Genetics of abdominal aortic aneurysm. Curr. Opin. Cardiol. 2013, 28, 290–296. [Google Scholar] [CrossRef]
- Gretarsdottir, S.; Baas, A.F.; Thorleifsson, G.; Holm, H.; Den Heijer, M.; De Vries, J.P.P.M.; Kranendonk, S.E.; Zeebregts, C.J.A.M.; Van Sterkenburg, S.M.; Geelkerken, R.H.; et al. Genome-wide association study identifies a sequence variant within the DAB2IP gene conferring susceptibility to abdominal aortic aneurysm. Nat. Genet. 2010, 42, 692–697. [Google Scholar] [CrossRef]
- Yu, L.; Qin, L.; Zhang, H.; He, Y.; Chen, H.; Pober, J.S.; Tellides, G.; Min, W. AIP1 prevents graft arteriosclerosis by inhibiting interferon-γ- dependent smooth muscle cell proliferation and intimal expansion. Circ. Res. 2011, 109, 418–427. [Google Scholar] [CrossRef]
- Bown, M.J.; Jones, G.T.; Harrison, S.C.; Wright, B.J.; Bumpstead, S.; Baas, A.F.; Gretarsdottir, S.; Badger, S.A.; Bradley, D.T.; Burnand, K.; et al. Abdominal aortic aneurysm is associated with a variant in low-density lipoprotein receptor-related protein 1. Am. J. Hum. Genet. 2011, 89, 619–627. [Google Scholar] [CrossRef]
- Boucher, P.; Gotthardt, M.; Li, W.P.; Anderson, R.G.W.; Herz, J. LRP: Role in vascular wall integrity and protection from atherosclerosis. Science 2003, 300, 329–332. [Google Scholar] [CrossRef]
- Wild, J.B.; Stather, P.W.; Sylvius, N.; Choke, E.; Sayers, R.D.; Bown, M.J. Low Density Lipoprotein Receptor Related Protein 1 and Abdominal Aortic Aneurysms. Eur. J. Vasc. Endovasc. Surg. 2012, 44, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Giusti, C.; Luciani, M.F.; Klein, G.; Aubry, L.; Tresse, E.; Kosta, A.; Golstein, P. Necrotic cell death: From reversible mitochondrial uncoupling to irreversible lysosomal permeabilization. Exp. Cell Res. 2009, 315, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Hans, C.P.; Koenig, S.N.; Huang, N.; Chen, J.; Beceiro, S.; Guggilam, A.; Kuivaniemi, H.; Partida-Sanchez, S.; Garg, V. Inhibition of Notch1 signaling reduces abdominal aortic aneurysm in mice by attenuating macrophage-mediated inflammation. Arter. Thromb. Vasc. Biol. 2012, 32, 3012–3023. [Google Scholar] [CrossRef] [PubMed]
- Hinterseher, I.; Elmore, J.R.; Cindric, M.C.; Bowen, W.D.; Schworer, M.; Chernousov, M.A.; Franklin, P.; Gray, J.L.; Garvin, P.; Gatalica, Z.; et al. Novel pathways in the pathobiology of human abdominal aortic aneurysms. Pathobiology 2013, 80, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Winter, S.C.; Shah, K.A.; Han, C.; Campo, L.; Turley, H.; Leek, R.; Corbridge, R.J.; Cox, G.J.; Harris, A.L. The relation between hypoxia-inducible factor (HIF)=1α and HIF-2α expression with anemia and outcome in surgically treated head and neck cancer. Cancer 2006, 107, 757–766. [Google Scholar] [CrossRef]
- Imanishi, M.; Chiba, Y.; Tomita, N.; Matsunaga, S.; Nakagawa, T. Hypoxia-inducible factor-1 α in smooth muscle cells protects against aortic aneurysms—Brief report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2158–2162. [Google Scholar] [CrossRef]
- Nakayama, T.; Kurobe, H.; Sugasawa, N.; Kinoshita, H.; Higashida, M.; Matsuoka, Y.; Yoshida, Y.; Hirata, Y.; Sakata, M.; Maxfield, M.W.; et al. Role of macrophage-derived hypoxia-inducible factor (HIF)-1α as a mediator of vascular remodelling. Cardiovasc. Res. 2013, 99, 705–715. [Google Scholar] [CrossRef]
- Gäbel, G.; Northoff, B.H.; Weinzierl, I.; Ludwig, S.; Hinterseher, I.; Wilfert, W.; Teupser, D.; Doderer, S.A.; Bergert, H.; Schönleben, F.; et al. Molecular fingerprint for terminal abdominal aortic aneurysm disease. J. Am. Heart Assoc. 2017, 6, 1–18. [Google Scholar] [CrossRef]
- Maegdefessel, L.; Azuma, J.; Toh, R.; Deng, A.; Merk, D.R.; Leeper, N.J.; Raaz, U.; Schoelmerich, A.M.; Michael, V.; Dalman, R.L.; et al. MicroRNA-21 blocks abdominal aortic aneurysm development and nicotine-augmented expansion. Sci. Transl. Med. 2012, 4, 1–27. [Google Scholar] [CrossRef]
- Sun, S.; Zheng, B.; Han, M.; Fang, X.; Li, H.; Miao, S.; Su, M.; Han, Y.; Shi, H.; Wen, J. miR-146a and Kru¨ppel-like factor 4 form a feedback loop to participate in vascular smooth muscle cell proliferation. EMBO Rep. 2011, 12, 56–62. [Google Scholar] [CrossRef]
- Leeper, N.J.; Raiesdana, A.; Kojima, Y.; Chun, H.J.; Azuma, J.; Maegdefessel, L.; Kundu, R.K.; Quertermous, T.; Tsao, P.S.; Spin, J.M. MicroRNA-26a is a novel regulator of vascular smooth muscle cell function. J. Cell. Physiol. 2011, 226, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.B.; Deng, D.Y.; Beppu, H.; Hong, C.C.; Lai, C.; Hoyng, S.A.; Kawai, N.; Bloch, K.D. Bone morphogenetic protein (BMP) type II receptor is required for BMP-mediated growth arrest and differentiation in pulmonary artery smooth muscle cells. J. Biol. Chem. 2008, 283, 3877–3888. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Hou, S.; Chen, J.; Zhang, J.; Lin, F.; Ju, R.; Cheng, X.; Ma, X.; Song, Y.; Zhang, Y.; et al. Smad4 deficiency in smooth muscle cells initiates the formation of aortic aneurysm. Circ. Res. 2016, 118, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cheng, Y.; Zhang, S.; Lin, Y.; Yang, J.; Zhang, C. A Necessary Role of miR-221 and miR-222 in Vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ. Res. 2009, 104, 476–487. [Google Scholar] [CrossRef]
- Davis, B.N.; Hilyard, A.C.; Nguyen, P.H.; Lagna, G.; Hata, A. Induction of MicroRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J. Biol. Chem. 2009, 284, 3728–3738. [Google Scholar] [CrossRef]
- Li, D.Y.; Busch, A.; Jin, H.; Chernogubova, E.; Pelisek, J.; Karlsson, J.; Sennblad, B.; Lao, S.; Hofmann, P.; Suzanne, M.; et al. H19 induces abdominal aortic aneurysm development and progression. Circulation 2018, 138, 1551–1568. [Google Scholar] [CrossRef]
- Zhang, Z.; Zou, G.; Chen, X.; Lu, W.; Liu, J.; Zhai, S. Molecules and cells knockdown of lncRNA PVT1 inhibits vascular smooth muscle cell apoptosis and extracellular matrix disruption in a murine abdominal aortic aneurysm model. Mol. Cells 2019, 42, 218–227. [Google Scholar]
- Kleinhenz, J.M.; Murphy, T.C.; Pokutta-Paskaleva, A.P.; Gleason, R.L.; Lyle, A.N.; Taylor, W.R.; Blount, M.A.; Cheng, J.; Yang, Q.; Sutliff, R.L.; et al. Smooth muscle-targeted overexpression of peroxisome proliferator activated receptor-γ disrupts vascular wall structure and function. PLoS ONE 2015, 10, 1–25. [Google Scholar] [CrossRef][Green Version]
- Fu, M.; Zhang, J.; Lin, Y.G.; Zhu, X.; Willson, T.M.; Chen, Y.E. Activation of peroxisome proliferator-activated receptor γ inhibits osteoprotegerin gene expression in human aortic smooth muscle cells. Biochem. Biophys. Res. Commun. 2002, 294, 597–601. [Google Scholar] [CrossRef]
- Moran, C.S.; Clancy, P.; Biros, E.; Blanco-Martin, B.; McCaskie, P.; Palmer, L.J.; Coomans, D.; Norman, P.E.; Golledge, J. Association of PPARγ allelic variation, osteoprotegerin and abdominal aortic aneurysm. Clin. Endocrinol. Oxf. 2010, 72, 128–132. [Google Scholar] [CrossRef]
- Hansmann, G.; Perez, V.A.D.J.; Alastalo, T.; Alvira, C.M.; Guignabert, C.; Bekker, J.M.; Schellong, S.; Urashima, T.; Wang, L.; Morrell, N.W.; et al. An antiproliferative BMP-2 / PPARγ / apoE axis in human and murine SMCs and its role in pulmonary hypertension. J. Clin. Investig. 2008, 118, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Villacorta, L.; Zhang, J.; Garcia-Barrio, M.T.; Yang, K.; Hamblin, M.; Whitesall, S.E.; D’Alecy, L.G.; Chen, Y.E. Vascular smooth muscle cell-selective PPARγ deletion leads to hypotension. Circulation 2009, 119, 2161–2169. [Google Scholar] [CrossRef] [PubMed]
- Beyer, A.M.; Baumbach, G.L.; Halabi, C.M.; Modrick, M.L.; Cynthia, M.; Gerhold, T.D.; Ghoneim, S.M.; De Lange, W.J.; Keen, H.L.; Tsai, Y.; et al. Interference with PPARγ signaling causes cerebral vascular dysfunction, hypertrophy, and remodeling. Hypertension 2008, 51, 867–871. [Google Scholar] [CrossRef] [PubMed]
- Halabi, C.M.; Beyer, A.M.; de Lange, W.J.; Keen, H.L.; Baumbach, G.L.; Faraci, F.M.; Sigmund, C.D. Interference with PPARγ function in smooth muscle causes vascular dysfunction and hypertension. Cell Metab. 2008, 7, 215–226. [Google Scholar] [CrossRef]
- Ortiz-Muñoz, G.; Houard, X.; Martín-Ventura, J.-L.; Ishida, B.Y.; Loyau, S.; Rossignol, P.; Moreno, J.-A.; Kane, J.P.; Chalkley, R.J.; Burlingame, A.L.; et al. HDL antielastase activity prevents smooth muscle cell anoikis, a potential new antiatherogenic property. FASEB J. 2009, 23, 3129–3139. [Google Scholar] [CrossRef]
- Young, J.L.; Sukhova, G.K.; Foster, D.; Kisiel, W.; Libby, P.; Schönbeck, U. The serpin proteinase inhibitor 9 is an endogenous inhibitor of interleukin 1β-converting enzyme (caspase-1) activity in human vascular smooth muscle cells. J. Exp. Med. 2000, 191, 1535–1544. [Google Scholar] [CrossRef]
- Ryer, E.J.; Ronning, K.E.; Erdman, R.; Schworer, C.M.; Elmore, J.R.; Peeler, T.C.; Nevius, C.D.; Lillvis, J.H.; Garvin, R.P.; Franklin, D.P.; et al. The potential role of DNA methylation in abdominal aortic aneurysms. Int. J. Mol. Sci. 2015, 16, 11259–11275. [Google Scholar] [CrossRef] [PubMed]
- Albinsson, S.; Nordström, I.; Hellstrand, P. Stretch of the vascular wall induces smooth muscle differentiation by promoting actin polymerization. J. Biol. Chem. 2004, 279, 34849–34855. [Google Scholar] [CrossRef]
- Xin, M.; Small, E.M.; Sutherland, L.B.; Qi, X.; McAnally, J.; Plato, C.F.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009, 23, 2166–2178. [Google Scholar] [CrossRef] [PubMed]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.-H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Liu, X.; Yang, J.; Lin, Y.; Xu, D.Z.; Lu, Q.; Deitch, E.A.; Huo, Y.; Delphin, E.S.; Zhang, C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ. Res. 2009, 105, 158–166. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, D.Z.; Hockemeyer, D.; McAnally, J.; Nordheim, A.; Olson, E.N. Myocardin and ternary complex factors compete for SRF to control smooth muscle gene expression. Nature 2004, 428, 185–189. [Google Scholar] [CrossRef]
- Wang, X.; Hu, G.; Zhou, J. Repression of versican expression by microRNA-143. J. Biol. Chem. 2010, 285, 23241–23250. [Google Scholar] [CrossRef] [PubMed]
- Kuwahara, K.; Barrientos, T.; Pipes, G.C.T.; Li, S.; Olson, E.N. Muscle-Specific Signaling Mechanism That Links Actin Dynamics to Serum Response Factor. Mol. Cell. Biol. 2005, 25, 3173–3181. [Google Scholar] [CrossRef] [PubMed]
- Miralles, F.; Posern, G.; Zaromytidou, A.I.; Treisman, R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef]
- Barkalow, K.L.; Italiano, J.E.; Chou, D.E.; Matsuoka, Y.; Bennett, V.; Hartwig, J.H. α-adducin dissociates from F-actin and spectrin during platelet activation. J. Cell Biol. 2003, 161, 557–570. [Google Scholar] [CrossRef]
- Gardner, K.; Bennett, V. Modulation of spectrin-actin assembly by erythrocyte adducin. Nature 1987, 328, 359–362. [Google Scholar] [CrossRef]
- Maegdefessel, L.; Spin, J.M.; Raaz, U.; Eken, S.M.; Toh, R.; Azuma, J.; Adam, M.; Nagakami, F.; Heymann, H.M.; Chernugobova, E.; et al. MiR-24 limits aortic vascular inflammation and murine abdominal aneurysm development. Nat. Commun. 2014, 5, 1–14. [Google Scholar] [CrossRef]
- Venkatesh, P.; Phillippi, J.; Chukkapalli, S.; Rivera-Kweh, M.; Velsko, I.; Gleason, T.; VanRyzin, P.; Aalaei-Andabili, S.H.; Ghanta, R.K.; Beaver, T.; et al. Aneurysm-specific miR-221 and miR-146a participates in human thoracic and abdominal aortic aneurysms. Int. J. Mol. Sci. 2017, 18, 875. [Google Scholar] [CrossRef]
- Kin, K.; Miyagawa, S.; Fukushima, S.; Shirakawa, Y.; Torikai, K.; Shimamura, K.; Daimon, T.; Kawahara, Y.; Kuratani, T.; Sawa, Y. Tissue-and plasma-specific microRNA signatures for atherosclerotic abdominal aortic aneurysm. Am. Hear. Assoc. J. 2012, 1, 1–10. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, Y.; Yang, J.; Xu, L.; Zhang, C. Cell-specific effects of miR-221/222 in vessels: Molecular mechanism and therapeutic application. J. Mol. Cell. Cardiol. 2012, 52, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.J.; Sharp, W.J.; Fang, X.; Oberley, L.W.; Oberley, T.D.; Weintraub, N.L. Oxidative stress in human abdominal aortic aneurysms: A potential mediator of aneurysmal remodeling. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 560–565. [Google Scholar] [CrossRef] [PubMed]
- Gavrila, D.; Li, W.G.; Mccormick, M.L.; Thomas, M.; Daugherty, A.; Cassis, L.A.; Miller, F.J., Jr.; Oberley, L.W.; Dellsperger, K.C.; Weintraub, N.L. Vitamin E inhibits abdominal aortic aneurysm formation in angiotensin II–Infused apolipoprotein E–Deficient mice. Arterioscler. Thromb. 2005, 25, 1671–1677. [Google Scholar] [CrossRef] [PubMed]
- Maiellaro-Rafferty, K.; Weiss, D.; Joseph, G.; Wan, W.; Gleason, R.L.; Taylor, W.R. Catalase overexpression in aortic smooth muscle prevents pathological mechanical changes underlying abdominal aortic aneurysm formation. Am. J. Physiol. Hear. Circ. Physiol. 2011, 301, 1–14. [Google Scholar] [CrossRef] [PubMed]
- McCormick, M.L.; Gavrila, D.; Weintraub, N.L. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 461–469. [Google Scholar] [CrossRef]
- Grote, K.; Flach, I.; Luchtefeld, M.; Akin, E.; Holland, S.M.; Drexler, H.; Schieffer, B. Mechanical stretch enhances mRNA expression and proenzyme release of matrix metalloproteinase-2 (MMP-2) via NAD(P)H oxidase-derived reactive oxygen species. Circ. Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Hishikawa, K.; Oemar, B.S.; Yang, Z.; Lüscher, T.F. Pulsatile stretch stimulates superoxide production and activates nuclear factor-κb in human coronary smooth muscle. Circ. Res. 1997, 81, 797–803. [Google Scholar] [CrossRef]
- Brandes, R.P.; Kreuzer, J. Vascular NADPH oxidases: Molecular mechanisms of activation. Cardiovasc. Res. 2005, 65, 16–27. [Google Scholar] [CrossRef]
- De Keulenaer, G.W.; Alexander, R.W.; Ushio-Fukai, M.; Ishizaka, N.; Griendling, K.K. Tumour necrosis factor α activates a p22phox -based NADH oxidase in vascular smooth muscle. Biochem. J. 1998, 329, 653–657. [Google Scholar] [CrossRef]
- Thomas, M.; Gavrila, D.; McCormick, M.L.; Miller, F.J.; Daugherty, A.; Cassis, L.A.; Dellsperger, K.C.; Weintraub, N.L. Deletion of p47phox attenuates angiotensin II–Induced abdominal aortic aneurysm formation in apolipoprotein E–deficient mice. Circulation 2006, 114, 404–413. [Google Scholar] [CrossRef]
- Siu, K.L.; Miao, X.N.; Cai, H. Recoupling of eNOS with folic acid prevents abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E null mice. PLoS ONE 2014, 9, e0088899. [Google Scholar] [CrossRef] [PubMed]
- Kigawa, Y.; Miyazaki, T.; Lei, X.F.; Nakamachi, T.; Oguchi, T.; Kim-Kaneyama, J.R.; Taniyama, M.; Tsunawaki, S.; Shioda, S.; Miyazaki, A. NADPH oxidase deficiency exacerbates angiotensin II-induced abdominal aortic aneurysms in mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2413–2420. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.L.; Li, Q.; Zhang, Y.; Guo, J.; Youn, J.Y.; Du, J.; Cai, H. NOX isoforms in the development of abdominal aortic aneurysm. Redox Biol. 2017, 11, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.L.; Carraro, C.C.; Belló-Klein, A.; Kalil, A.N.; Aerts, N.R.; Carvalho, F.B.; Fernandes, M.C.; Zettler, C.G. Oxidative stress in aortas of patients with advanced occlusive and aneurysmal diseases. Ann. Vasc. Surg. 2018, 52, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Dubick, M.A.; Keen, C.L.; DiSilvestro, R.A.; Eskelson, C.D.; Ireton, J.; Hunter, G.C. Antioxidant enzyme activity in human abdominal aortic aneurysmal and occlusive disease. Proc. Soc. Exp. Biol. Med. 1999, 220, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Sinha, I.; Pearce, C.G.; Cho, B.S.; Hannawa, K.K.; Roelofs, K.J.; Stanley, J.C.; Henke, P.K.; Upchurch, G.R. Differential regulation of the superoxide dismutase family in experimental aortic aneurysms and rat aortic explants. J. Surg. Res. 2007, 138, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Yajima, N.; Masuda, M.; Miyazaki, M.; Nakajima, N.; Chien, S.; Shyy, J.Y.J. Oxidative stress is involved in the development of experimental abdominal aortic aneurysm: A study of the transcription profile with complementary DNA microarray. J. Vasc. Surg. 2002, 36, 379–385. [Google Scholar] [CrossRef]
- Pecqueur, C.; Alves-Guerra, C.; Ricquier, D.; Bouillaud, F. UCP2, a metabolic sensor coupling glucose oxidation to mitochondrial metabolism? IUBMB Life 2009, 61, 762–767. [Google Scholar] [CrossRef]
- Bouillaud, F.; Alves-Guerra, M.C.; Ricquier, D. UCPs, at the interface between bioenergetics and metabolism. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2443–2456. [Google Scholar] [CrossRef]
- Yan, P.; Chen, K.; Wang, Q.; Yang, D.; Li, D.; Yang, Y. UCP-2 is involved in angiotensin-II-induced abdominal aortic aneurysm in apolipoprotein E-knockout mice. PLoS ONE 2017, 12, e0179743. [Google Scholar] [CrossRef]
- Kim, H.W.; Stansfield, B.K. Genetic and Epigenetic Regulation of Aortic Aneurysms. BioMed Res. Int. 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Soe, N.N.; Sowden, M.; Baskaran, P.; Kim, Y.; Nigro, P.; Smolock, E.M.; Berk, B.C. Acetylation of cyclophilin A is required for its secretion and vascular cell activation. Cardiovasc. Res. 2014, 101, 444–453. [Google Scholar] [CrossRef]
- Manea, S.A.; Antonescu, M.L.; Fenyo, I.M.; Raicu, M.; Simionescu, M.; Manea, A. Epigenetic regulation of vascular NADPH oxidase expression and reactive oxygen species production by histone deacetylase-dependent mechanisms in experimental diabetes. Redox Biol. 2018, 16, 332–343. [Google Scholar] [CrossRef]
- Krishna, S.M.; Dear, A.E.; Norman, P.E.; Golledge, J. Genetic and epigenetic mechanisms and their possible role in abdominal aortic aneurysm. Atherosclerosis 2010, 212, 16–29. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Webster, A.L.H.; Yan, M.S.C.; Marsden, P.A. Epigenetics and cardiovascular disease. Can. J. Cardiol. 2013, 29, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Matouk, C.C.; Marsden, P.A. Epigenetic regulation of vascular endothelial gene expression. Circ. Res. 2008, 102, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Bestor, T.H. The DNA methyltransferases of mammals. Hum. Mol. Genet. 2000, 9, 2395–2402. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A Landscape Takes Shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef]
- Yang, X.; Han, H.; de Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef]
- Toghill, B.J.; Saratzis, A.; Freeman, P.J.; Sylvius, N.; Bown, M.J. SMYD2 promoter DNA methylation is associated with abdominal aortic aneurysm (AAA) and SMYD2 expression in vascular smooth muscle cells. Clin. Epigenetics 2018, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; He, X.; Si, X.; Wang, H.; Li, B.; Hu, Y.; Li, M.; Chen, X.; Liao, W.; Liao, Y.; et al. SM22 (smooth muscle 22) prevents aortic aneurysm formation by inhibiting smooth muscle cell phenotypic switching through suppressing reactive oxygen species/NF-κB (nuclear factor-κB). Arterioscler. Thromb. Vasc. Biol. 2019, 39, e10–e25. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Zykovich, A.; Hubbard, A.; Flynn, J.M.; Tarnopolsky, M.; Fraga, M.F.; Kerksick, C.; Ogborn, D.; MacNeil, L.; Mooney, S.D.; Melov, S. Genome-wide DNA methylation changes with age in disease-free human skeletal muscle. Aging Cell 2014, 13, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Toghill, B.J.; Saratzis, A.; Harrison, S.C.; Verissimo, A.R.; Mallon, E.B.; Bown, M.J. The potential role of DNA methylation in the pathogenesis of abdominal aortic aneurysm. Atherosclerosis 2015, 241, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Tsaprouni, L.G.; Yang, T.P.; Bell, J.; Dick, K.J.; Kanoni, S.; Nisbet, J.; Viñuela, A.; Grundberg, E.; Nelson, C.P.; Meduri, E.; et al. Cigarette smoking reduces DNA methylation levels at multiple genomic loci but the effect is partially reversible upon cessation. Epigenetics 2014, 9, 1382–1396. [Google Scholar] [CrossRef] [PubMed]
- Schleithoff, C.; Voelter-Mahlknecht, S.; Dahmke, I.; Mahlknecht, U. On the epigenetics of vascular regulation and disease. Clin. Epigenetics 2012, 4, 7. [Google Scholar] [CrossRef]
- Stenvinkel, P.; Karimi, M.; Johansson, S.; Axelsson, J.; Suliman, M.; Lindholm, B.; Heimbürger, O.; Barany, P.; Alvestrand, A.; Nordfors, L.; et al. Impact of inflammation on epigenetic DNA methylation-A novel risk factor for cardiovascular disease? J. Intern. Med. 2007, 261, 488–499. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).