DNA Methyltransferase 1 (DNMT1) Acts on Neurodegeneration by Modulating Proteostasis-Relevant Intracellular Processes

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. DNMT1 Modulated Retrograde Trafficking
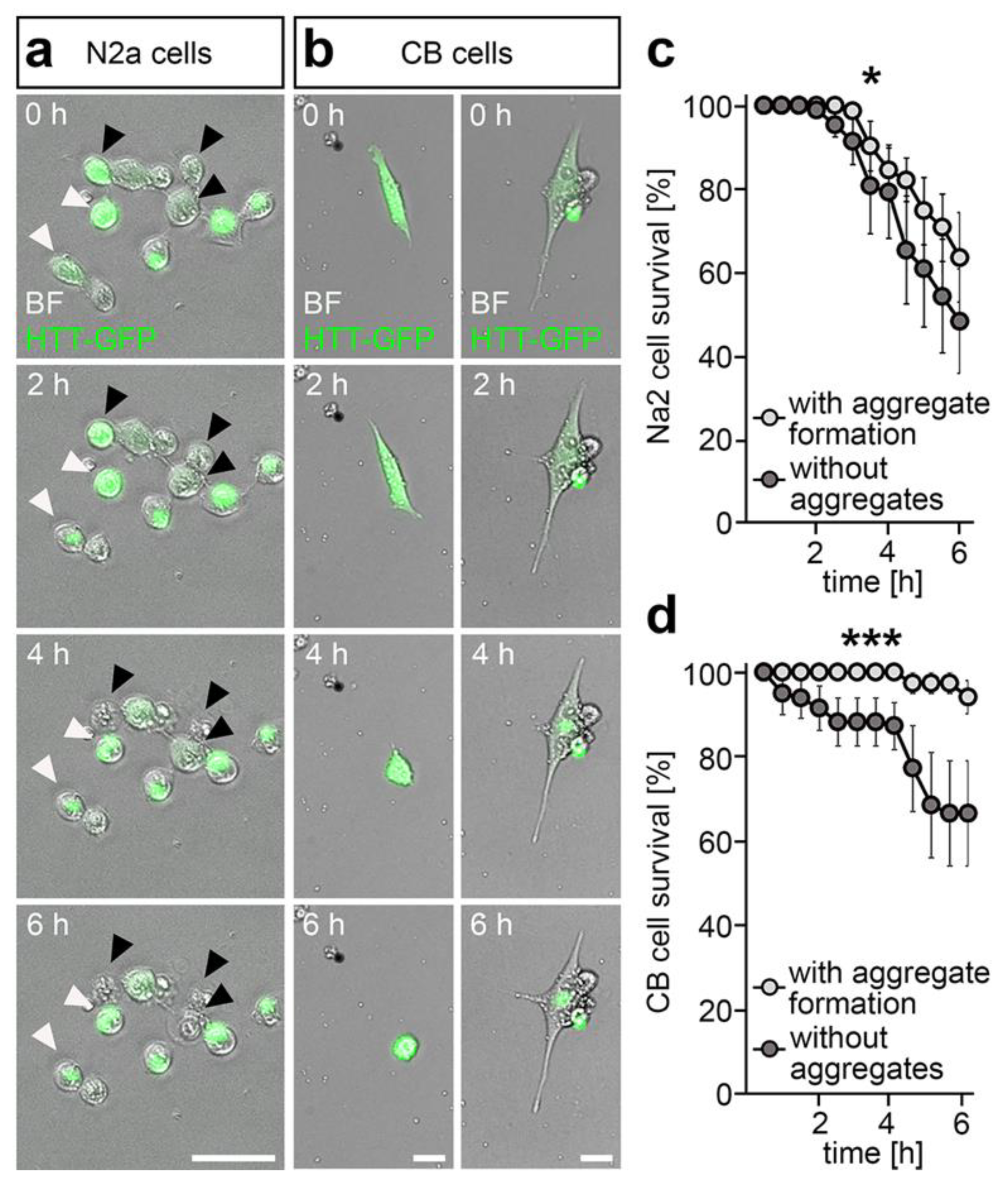
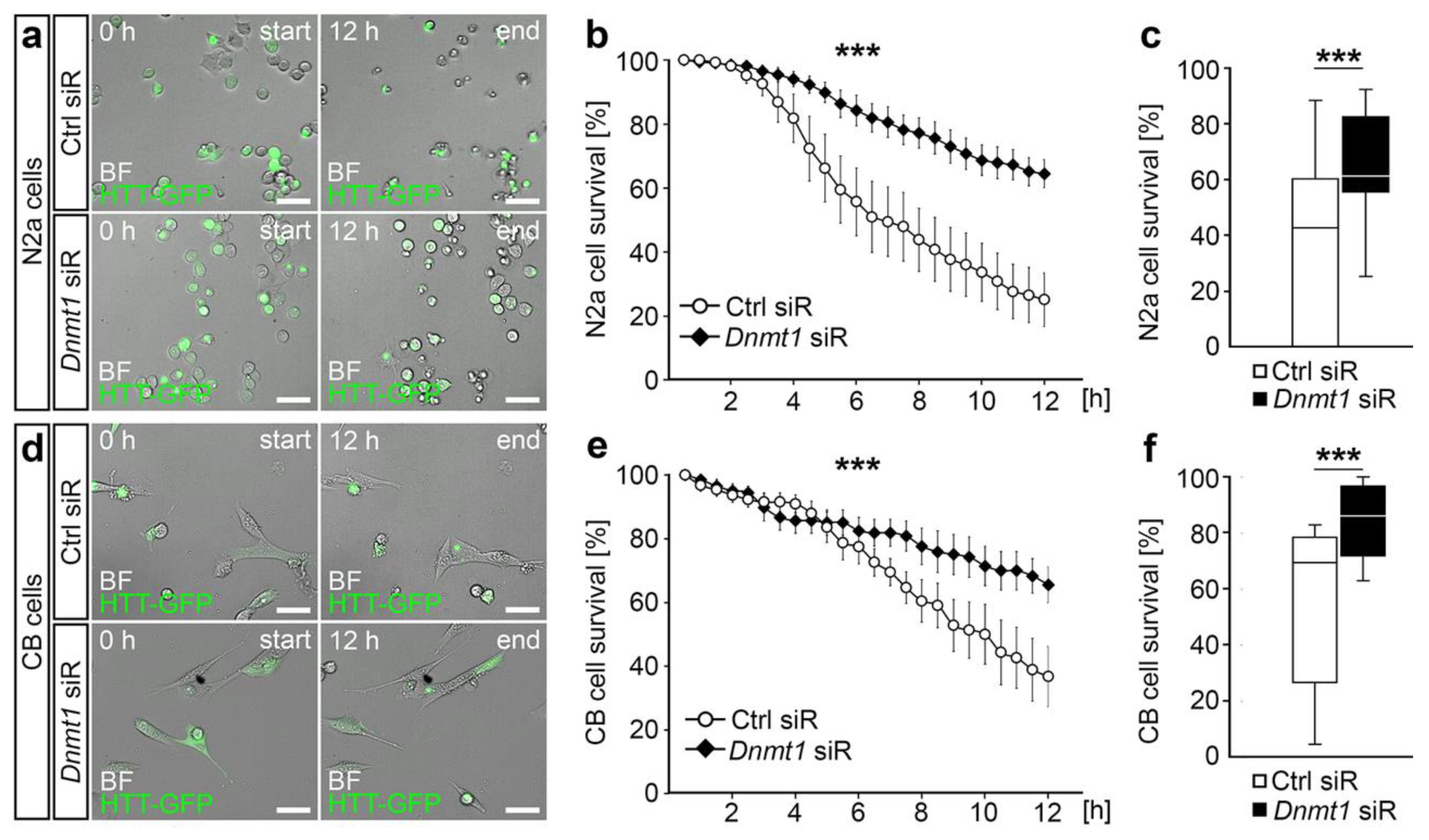
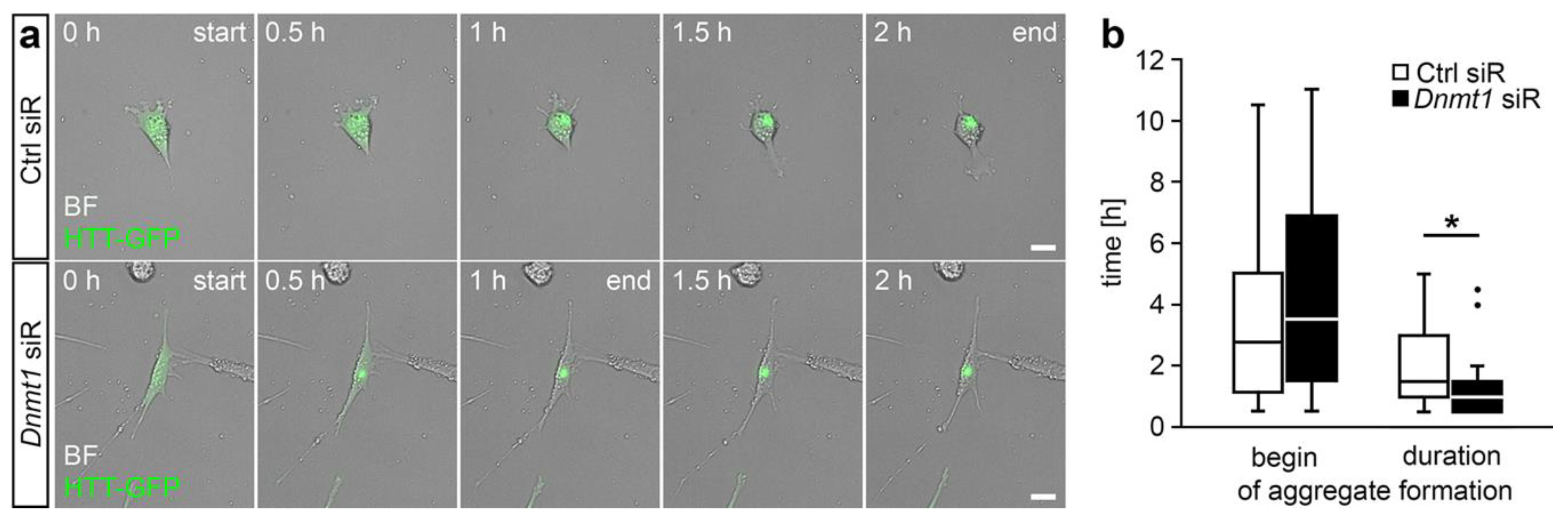
2.2. DNMT1-Mediated Retrograde Trafficking to Perinuclear Regions Influenced Mutant HTT-Induced Cytotoxicity
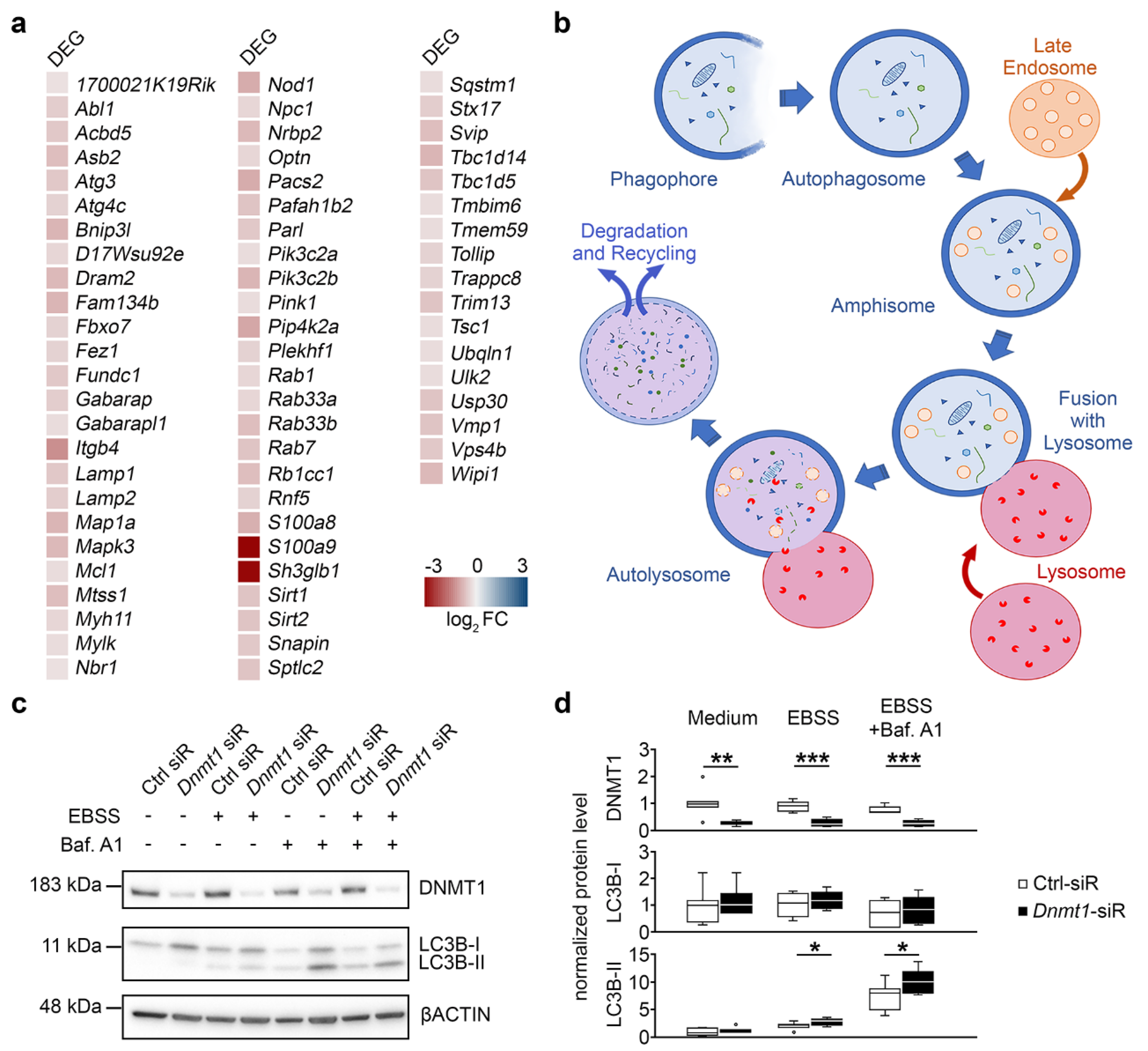
2.3. DNMT1 Is Relevant for Proper Autophagy
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Transfection with siRNA and Co-Transfection with siRNA and Plasmid DNA
4.3. Monitoring of Endo-Lysosomal Vesicle Trafficking
4.4. Huntingtin Cytotoxicity Assay
4.5. Autophagy Assay
4.6. Protein Preparation, Western Blotting, and Detection
4.7. Plasmid Information
4.8. Sequencing Data Analysis
4.9. Microscopy and Data Analysis
4.10. Data and Materials Availability:
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Götz, M.; Nakafuku, M.; Petrik, D. Neurogenesis in the developing and adult brain—Similarities and key differences. Cold Spring Harb. Perspect. Biol. 2016, 8, a018853. [Google Scholar] [CrossRef]
- Symmank, J.; Zimmer, G. Regulation of neuronal survival by DNA methyltransferases. Neural Regen. Res. 2017, 12, 1768. [Google Scholar]
- Zimmer-Bensch, G. Functional Implications of Dynamic DNA Methylation for the Developing, Aging and Diseased Brain. In The DNA, RNA, and Histone Methylomes; Springer: Cham, Switzerland, 2019; pp. 141–163. [Google Scholar]
- Chestnut, B.A.; Chang, Q.; Price, A.; Lesuisse, C.; Wong, M.; Martin, L.J. Epigenetic regulation of motor neuron cell death through DNA methylation. J. Neurosci. 2011, 31, 16619–16636. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Martinowich, K.; Chin, M.H.; He, F.; Fouse, S.D.; Hutnick, L.; Hattori, D.; Ge, W.; Shen, Y.; Wu, H. DNA methylation controls the timing of astrogliogenesis through regulation of JAK-STAT signaling. Development 2005, 132, 3345–3356. [Google Scholar] [CrossRef]
- Kadriu, B.; Guidotti, A.; Chen, Y.; Grayson, D.R. DNA methyltransferases1 (DNMT1) and 3a (DNMT3a) colocalize with GAD67-positive neurons in the GAD67-GFP mouse brain. J. Comp. Neurol. 2012, 520, 1951–1964. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 6146. [Google Scholar] [CrossRef] [PubMed]
- Hutnick, L.K.; Golshani, P.; Namihira, M.; Xue, Z.; Matynia, A.; Yang, X.W.; Silva, A.J.; Schweizer, F.E.; Fan, G. DNA hypomethylation restricted to the murine forebrain induces cortical degeneration and impairs postnatal neuronal maturation. Hum. Mol. Genet. 2009, 18, 2875–2888. [Google Scholar] [CrossRef]
- Rhee, K.; Yu, J.; Zhao, C.; Fan, G.; Yang, X. Dnmt1-dependent DNA methylation is essential for photoreceptor terminal differentiation and retinal neuron survival. Cell Death Dis. 2012, 3, e427. [Google Scholar] [CrossRef]
- Pensold, D.; Symmank, J.; Hahn, A.; Lingner, T.; Salinas-Riester, G.; Downie, B.R.; Ludewig, F.; Rotzsch, A.; Haag, N.; Andreas, N.; et al. The DNA methyltransferase 1 (DNMT1) controls the shape and dynamics of migrating POA-derived interneurons fated for the murine cerebral cortex. Cereb. Cortex 2017, 27, 5696–5714. [Google Scholar] [CrossRef]
- Hahn, A.; Bayer, C.; Pensold, D.; Tittelmeier, J.; Marx-Bluemel, L.; González-Bermúdez, L.; Linde, J.; Gross, J.; Salinas-Riester, G.; von Maltzahn, J.; et al. DNA methyltransferase 1 (DNMT1) function is implicated in the age-related loss of cortical interneurons. Front. Cell Dev. Biol. 2020, 8, 639. [Google Scholar] [CrossRef]
- Pensold, D.; Reichard, J.; Van Loo, K.M.; Ciganok, N.; Hahn, A.; Bayer, C.; Liebmann, L.; Groß, J.; Tittelmeier, J.; Lingner, T.; et al. DNA methylation-mediated modulation of endocytosis as potential mechanism for synaptic function regulation in murine inhibitory cortical interneurons. Cereb. Cortex 2020, 30, 3921–3937. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P.M.; Dillin, A. Protein homeostasis and aging in neurodegeneration. J. Cell Biol. 2010, 190, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Winckler, B.; Faundez, V.; Maday, S.; Cai, Q.; Almeida, C.G.; Zhang, H. The endolysosomal system and proteostasis: From development to degeneration. J. Neurosci. 2018, 38, 9364–9374. [Google Scholar] [CrossRef] [PubMed]
- Nixon, R.A.; Cataldo, A.M. The endosomal-lysosomal system of neurons: New roles. Trends Neurosci. 1995, 18, 489–496. [Google Scholar] [CrossRef]
- Nixon, R.A.; Cataldo, A.M.; Mathews, P.M. The endosomal-lysosomal system of neurons in Alzheimer’s disease pathogenesis: A review. Neurochem. Res. 2000, 25, 1161–1172. [Google Scholar] [CrossRef]
- Zhang, L.; Sheng, R.; Qin, Z. The lysosome and neurodegenerative diseases. Acta Biochim. Et Biophys. Sin. 2009, 41, 437–445. [Google Scholar] [CrossRef]
- Carmona-Gutierrez, D.; Hughes, A.L.; Madeo, F.; Ruckenstuhl, C. The crucial impact of lysosomes in aging and longevity. Ageing Res. Rev. 2016, 32, 2–12. [Google Scholar] [CrossRef]
- Nixon, R.A. The role of autophagy in neurodegenerative disease. Nat. Med. 2013, 19, 983–997. [Google Scholar] [CrossRef]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in neurodegenerative diseases: Pathogenesis and therapy. Brain Pathol. 2018, 28, 3–13. [Google Scholar] [CrossRef]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.; Aronin, N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef]
- Gutekunst, C.-A.; Li, S.-H.; Yi, H.; Mulroy, J.S.; Kuemmerle, S.; Jones, R.; Rye, D.; Ferrante, R.J.; Hersch, S.M.; Li, X.-J. Nuclear and neuropil aggregates in Huntington’s disease: Relationship to neuropathology. J. Neurosci. 1999, 19, 2522–2534. [Google Scholar] [CrossRef] [PubMed]
- Lunkes, A.; Lindenberg, K.S.; Ben-Haïem, L.; Weber, C.; Devys, D.; Landwehrmeyer, G.B.; Mandel, J.-L.; Trottier, Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol. Cell 2002, 10, 259–269. [Google Scholar] [CrossRef]
- Schilling, G.; Klevytska, A.; Tebbenkamp, A.T.; Juenemann, K.; Cooper, J.; Gonzales, V.; Slunt, H.; Poirer, M.; Ross, C.A.; Borchelt, D.R. Characterization of huntingtin pathologic fragments in human Huntington disease, transgenic mice, and cell models. J. Neuropathol. Exp. Neurol. 2007, 66, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Juenemann, K.; Weisse, C.; Reichmann, D.; Kaether, C.; Calkhoven, C.F.; Schilling, G. Modulation of mutant huntingtin N-terminal cleavage and its effect on aggregation and cell death. Neurotox. Res. 2011, 20, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.; Li, L.; Chin, L. Aggresome formation and neurodegenerative diseases: Therapeutic implications. Curr. Med. Chem. 2008, 15, 47–60. [Google Scholar] [PubMed]
- Ravikumar, B.; Duden, R.; Rubinsztein, D.C. Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 2002, 11, 1107–1117. [Google Scholar] [CrossRef]
- Iwata, A.; Riley, B.E.; Johnston, J.A.; Kopito, R.R. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J. Biol. Chem. 2005, 280, 40282–40292. [Google Scholar] [CrossRef]
- Iwata, A.; Christianson, J.C.; Bucci, M.; Ellerby, L.M.; Nukina, N.; Forno, L.S.; Kopito, R.R. Increased susceptibility of cytoplasmic over nuclear polyglutamine aggregates to autophagic degradation. Proc. Natl. Acad. Sci. USA 2005, 102, 13135–13140. [Google Scholar] [CrossRef]
- Taylor, J.P.; Tanaka, F.; Robitschek, J.; Sandoval, C.M.; Taye, A.; Markovic-Plese, S.; Fischbeck, K.H. Aggresomes protect cells by enhancing the degradation of toxic polyglutamine-containing protein. Hum. Mol. Genet. 2003, 12, 749–757. [Google Scholar] [CrossRef]
- Fortun, J.; Dunn, W.A.; Joy, S.; Li, J.; Notterpek, L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J. Neurosci. 2003, 23, 10672–10680. [Google Scholar] [CrossRef]
- Johnston, J.A.; Ward, C.L.; Kopito, R.R. Aggresomes: A cellular response to misfolded proteins. J. Cell Biol. 1998, 143, 1883–1898. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mata, R.; Gao, Y.S.; Sztul, E. Hassles with taking out the garbage: Aggravating aggresomes. Traffic 2002, 3, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Kopito, R.R. Aggresomes, inclusion bodies and protein aggregation. Trends Cell Biol. 2000, 10, 524–530. [Google Scholar] [CrossRef]
- Muchowski, P.J.; Ning, K.; D’Souza-Schorey, C.; Fields, S. Requirement of an intact microtubule cytoskeleton for aggregation and inclusion body formation by a mutant huntingtin fragment. Proc. Natl. Acad. Sci. USA 2002, 99, 727–732. [Google Scholar] [CrossRef]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef]
- Pols, M.S.; Klumperman, J. Trafficking and function of the tetraspanin CD63. Exp. Cell Res. 2009, 315, 1584–1592. [Google Scholar] [CrossRef]
- Tooze, S.A.; Abada, A.; Elazar, Z. Endocytosis and autophagy: Exploitation or cooperation? Cold Spring Harb. Perspect. Biol. 2014, 6, a018358. [Google Scholar] [CrossRef]
- Johannes, L.; Popoff, V. Tracing the retrograde route in protein trafficking. Cell 2008, 135, 1175–1187. [Google Scholar] [CrossRef]
- Tammineni, P.; Cai, Q. Defective retrograde transport impairs autophagic clearance in Alzheimer disease neurons. Autophagy 2017, 13, 982–984. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. Autophagosome dynamics in neurodegeneration at a glance. J. Cell Sci. 2015, 128, 1259–1267. [Google Scholar] [CrossRef]
- Klement, I.A.; Skinner, P.J.; Kaytor, M.D.; Yi, H.; Hersch, S.M.; Clark, H.B.; Zoghbi, H.Y.; Orr, H.T. Ataxin-1 nuclear localization and aggregation: Role in polyglutamine-induced disease in SCA1 transgenic mice. Cell 1998, 95, 41–53. [Google Scholar] [CrossRef]
- Pan, Y.; Daito, T.; Sasaki, Y.; Chung, Y.H.; Xing, X.; Pondugula, S.; Swamidass, S.J.; Wang, T.; Kim, A.H.; Yano, H. Inhibition of DNA methyltransferases blocks mutant huntingtin-induced neurotoxicity. Sci. Rep. 2016, 6, 1–18. [Google Scholar]
- Benito-Cuesta, I.; Diez, H.; Ordoñez, L.; Wandosell, F. Assessment of autophagy in neurons and brain tissue. Cells 2017, 6, 25. [Google Scholar] [CrossRef]
- Lovrečić, L.; Maver, A.; Zadel, M.; Peterlin, B. The role of epigenetics in neurodegenerative diseases. Neurodegener. Dis. 2013, 345. [Google Scholar] [CrossRef]
- Xylaki, M.; Atzler, B.; Outeiro, T.F. Epigenetics of the Synapse in Neurodegeneration. Curr. Neurol. Neurosci. Rep. 2019, 19, 72. [Google Scholar] [CrossRef] [PubMed]
- Zimmer-Bensch, G. Epigenomic remodeling in Huntington’s disease-master or servant? bioRxiv 2020. [Google Scholar] [CrossRef]
- Lardenoije, R.; Iatrou, A.; Kenis, G.; Kompotis, K.; Steinbusch, H.W.; Mastroeni, D.; Coleman, P.; Lemere, C.A.; Hof, P.R.; van den Hove, D.L. The epigenetics of aging and neurodegeneration. Prog. Neurobiol. 2015, 131, 21–64. [Google Scholar] [CrossRef]
- Lee, J.; Hwang, Y.J.; Kim, K.Y.; Kowall, N.W.; Ryu, H. Epigenetic mechanisms of neurodegeneration in Huntington’s disease. Neurotherapeutics 2013, 10, 664–676. [Google Scholar] [CrossRef]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef]
- Sugars, K.L.; Rubinsztein, D.C. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003, 19, 233–238. [Google Scholar] [CrossRef]
- Cha, J.-H.J. Transcriptional signatures in Huntington’s disease. Prog. Neurobiol. 2007, 83, 228–248. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2011, 12, 517. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.-i.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Wolfe, D.M.; Lee, J.h.; Kumar, A.; Lee, S.; Orenstein, S.J.; Nixon, R.A. Autophagy failure in A lzheimer’s disease and the role of defective lysosomal acidification. Eur. J. Neurosci. 2013, 37, 1949–1961. [Google Scholar] [CrossRef]
- Menezes, R.; Tenreiro, S.; Macedo, D.; Santos, C.N.; Outeiro, T.F. From the baker to the bedside: Yeast models of Parkinson’s disease. Microb. Cell 2015, 2, 262. [Google Scholar] [CrossRef]
- McBrayer, M.; Nixon, R.A. Lysosome and Calcium Dysregulation in Alzheimer’s Disease: Partners in Crime; Portland Press Ltd.: London, UK, 2013. [Google Scholar]
- Büttner, S.; Faes, L.; Reichelt, W.; Broeskamp, F.; Habernig, L.; Benke, S.; Kourtis, N.; Ruli, D.; Carmona-Gutierrez, D.; Eisenberg, T. The Ca2+/Mn2+ ion-pump PMR1 links elevation of cytosolic Ca 2+ levels to α-synuclein toxicity in Parkinson’s disease models. Cell Death Differ. 2013, 20, 465–477. [Google Scholar] [CrossRef]
- Jiang, C.H.; Tsien, J.Z.; Schultz, P.G.; Hu, Y. The effects of aging on gene expression in the hypothalamus and cortex of mice. Proc. Natl. Acad. Sci. USA 2001, 98, 1930–1934. [Google Scholar] [CrossRef]
- Arrasate, M.; Finkbeiner, S. Automated microscope system for determining factors that predict neuronal fate. Proc. Natl. Acad. Sci. USA 2005, 102, 3840–3845. [Google Scholar] [CrossRef]
- Cheng, X.-T.; Zhou, B.; Lin, M.-Y.; Cai, Q.; Sheng, Z.-H. Axonal autophagosomes recruit dynein for retrograde transport through fusion with late endosomes. J. Cell Biol. 2015, 209, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Johansen, T. p62/SQSTM1: A missing link between protein aggregates and the autophagy machinery. Autophagy 2006, 2, 138–139. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed]
- van Mierlo, G.; Veenstra, G.J.C.; Vermeulen, M.; Marks, H. The complexity of PRC2 subcomplexes. Trends Cell Biol. 2019, 29, 660–671. [Google Scholar] [CrossRef]
- Seong, I.S.; Woda, J.M.; Song, J.-J.; Lloret, A.; Abeyrathne, P.D.; Woo, C.J.; Gregory, G.; Lee, J.-M.; Wheeler, V.C.; Walz, T. Huntingtin facilitates polycomb repressive complex 2. Hum. Mol. Genet. 2010, 19, 573–583. [Google Scholar] [CrossRef]
- Biagioli, M.; Ferrari, F.; Mendenhall, E.M.; Zhang, Y.; Erdin, S.; Vijayvargia, R.; Vallabh, S.M.; Solomos, N.; Manavalan, P.; Ragavendran, A. Htt CAG repeat expansion confers pleiotropic gains of mutant huntingtin function in chromatin regulation. Hum. Mol. Genet. 2015, 24, 2442–2457. [Google Scholar] [CrossRef]
- von Schimmelmann, M.; Feinberg, P.A.; Sullivan, J.M.; Ku, S.M.; Badimon, A.; Duff, M.K.; Wang, Z.; Lachmann, A.; Dewell, S.; Ma’ayan, A. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci. 2016, 19, 1321–1330. [Google Scholar] [CrossRef]
- Symmank, J.; Bayer, C.; Reichard, J.; Pensold, D.; Zimmer-Bensch, G. Neuronal Lhx1 expression is regulated by DNMT1-dependent modulation of histone marks. Epigenetics 2020, 1–16. [Google Scholar] [CrossRef]
- Symmank, J.; Bayer, C.; Schmidt, C.; Hahn, A.; Pensold, D.; Zimmer-Bensch, G. DNMT1 modulates interneuron morphology by regulating Pak6 expression through crosstalk with histone modifications. Epigenetics 2018, 13, 536–556. [Google Scholar] [CrossRef]
- Reddington, J.P.; Perricone, S.M.; Nestor, C.E.; Reichmann, J.; Youngson, N.A.; Suzuki, M.; Reinhardt, D.; Dunican, D.S.; Prendergast, J.G.; Mjoseng, H. Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb target genes. Genome Biol. 2013, 14, R25. [Google Scholar] [CrossRef]
- Wu, X.; Gong, Y.; Yue, J.; Qiang, B.; Yuan, J.; Peng, X. Cooperation between EZH2, NSPc1-mediated histone H2A ubiquitination and Dnmt1 in HOX gene silencing. Nucleic Acids Res. 2008, 36, 3590–3599. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Zimmer-Bensch, G. Emerging Roles of Long Non-Coding RNAs as Drivers of Brain Evolution. Cells 2019, 8, 1399. [Google Scholar] [CrossRef] [PubMed]
- Buckley, N.J.; Johnson, R.; Zuccato, C.; Bithell, A.; Cattaneo, E. The role of REST in transcriptional and epigenetic dysregulation in Huntington’s disease. Neurobiol. Dis. 2010, 39, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Di Ruscio, A.; Ebralidze, A.K.; Benoukraf, T.; Amabile, G.; Goff, L.A.; Terragni, J.; Figueroa, M.E.; Pontes, L.L.D.F.; Alberich-Jorda, M.; Zhang, P. DNMT1-interacting RNAs block gene-specific DNA methylation. Nature 2013, 503, 371–376. [Google Scholar] [CrossRef]
- Chalei, V.; Sansom, S.N.; Kong, L.; Lee, S.; Montiel, J.F.; Vance, K.W.; Ponting, C.P. The long non-coding RNA Dali is an epigenetic regulator of neural differentiation. Elife 2014, 3, e04530. [Google Scholar] [CrossRef]
- Tobin, A.J.; Signer, E.R. Huntington’s disease: The challenge for cell biologists. Trends Cell Biol. 2000, 10, 531–536. [Google Scholar] [CrossRef]
- Fossale, E.; Wolf, P.; Espinola, J.A.; Lubicz-Nawrocka, T.; Teed, A.M.; Gao, H.; Rigamonti, D.; Cattaneo, E.; MacDonald, M.E.; Cotman, S.L. Membrane trafficking and mitochondrial abnormalities precede subunit c deposition in a cerebellar cell model of juvenile neuronal ceroid lipofuscinosis. BMC Neurosci. 2004, 5, 57. [Google Scholar] [CrossRef]
- Zimmer, G.; Rudolph, J.; Landmann, J.; Gerstmann, K.; Steinecke, A.; Gampe, C.; Bolz, J. Bidirectional ephrinB3/EphA4 signaling mediates the segregation of medial ganglionic eminence-and preoptic area-derived interneurons in the deep and superficial migratory stream. J. Neurosci. 2011, 31, 18364–18380. [Google Scholar] [CrossRef]
- Van Engelenburg, S.B.; Palmer, A.E. Imaging type-III secretion reveals dynamics and spatial segregation of Salmonella effectors. Nat. Methods 2010, 7, 325–330. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayer, C.; Pitschelatow, G.; Hannemann, N.; Linde, J.; Reichard, J.; Pensold, D.; Zimmer-Bensch, G. DNA Methyltransferase 1 (DNMT1) Acts on Neurodegeneration by Modulating Proteostasis-Relevant Intracellular Processes. Int. J. Mol. Sci. 2020, 21, 5420. https://doi.org/10.3390/ijms21155420
Bayer C, Pitschelatow G, Hannemann N, Linde J, Reichard J, Pensold D, Zimmer-Bensch G. DNA Methyltransferase 1 (DNMT1) Acts on Neurodegeneration by Modulating Proteostasis-Relevant Intracellular Processes. International Journal of Molecular Sciences. 2020; 21(15):5420. https://doi.org/10.3390/ijms21155420
Chicago/Turabian StyleBayer, Cathrin, Georg Pitschelatow, Nina Hannemann, Jenice Linde, Julia Reichard, Daniel Pensold, and Geraldine Zimmer-Bensch. 2020. "DNA Methyltransferase 1 (DNMT1) Acts on Neurodegeneration by Modulating Proteostasis-Relevant Intracellular Processes" International Journal of Molecular Sciences 21, no. 15: 5420. https://doi.org/10.3390/ijms21155420
APA StyleBayer, C., Pitschelatow, G., Hannemann, N., Linde, J., Reichard, J., Pensold, D., & Zimmer-Bensch, G. (2020). DNA Methyltransferase 1 (DNMT1) Acts on Neurodegeneration by Modulating Proteostasis-Relevant Intracellular Processes. International Journal of Molecular Sciences, 21(15), 5420. https://doi.org/10.3390/ijms21155420