N-Palmitoylethanolamide-Oxazoline Protects against Middle Cerebral Artery Occlusion Injury in Diabetic Rats by Regulating the SIRT1 Pathway



 ,
,  ,
,  ,
,  ,
, 
 and
and
Abstract
1. Introduction
2. Results
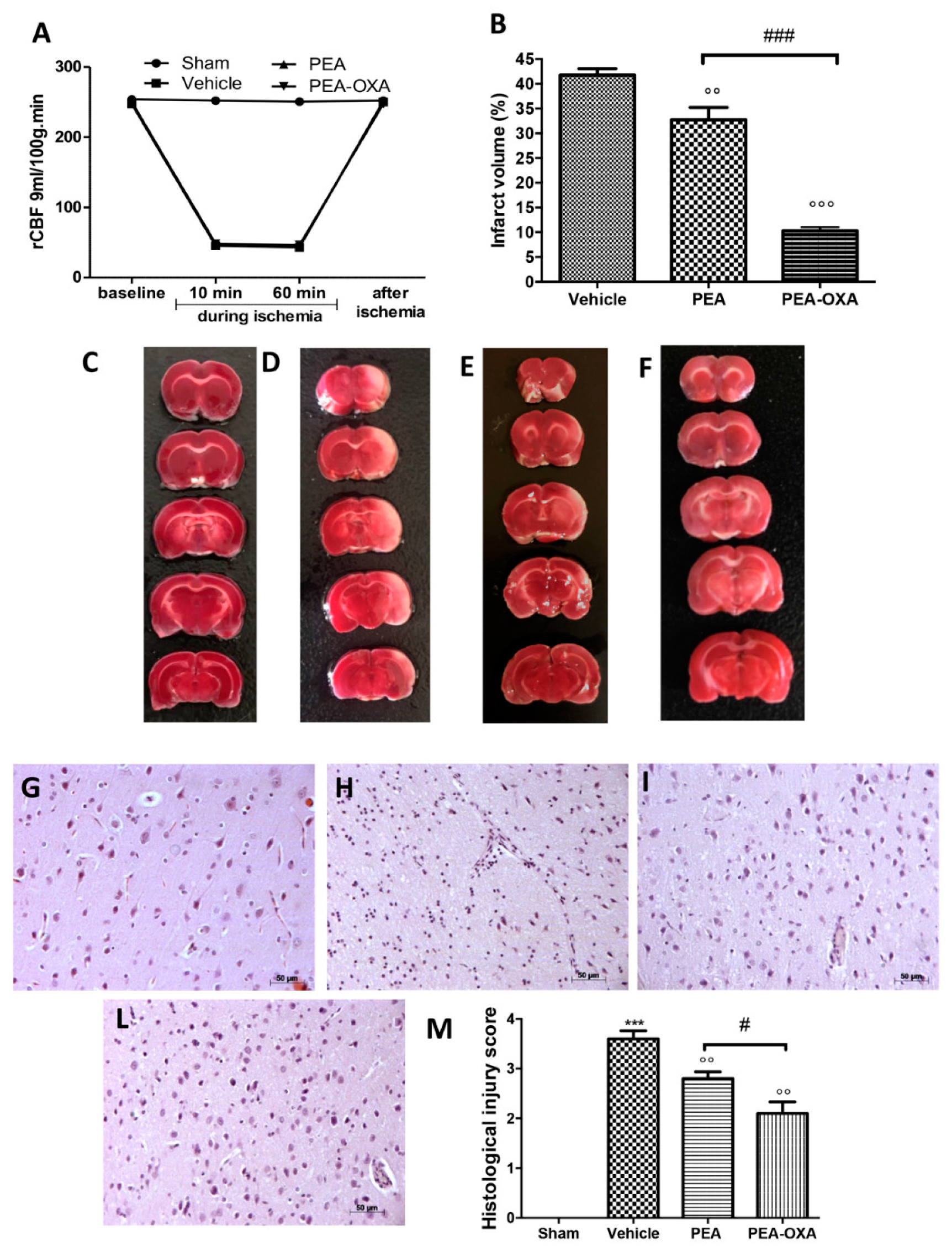
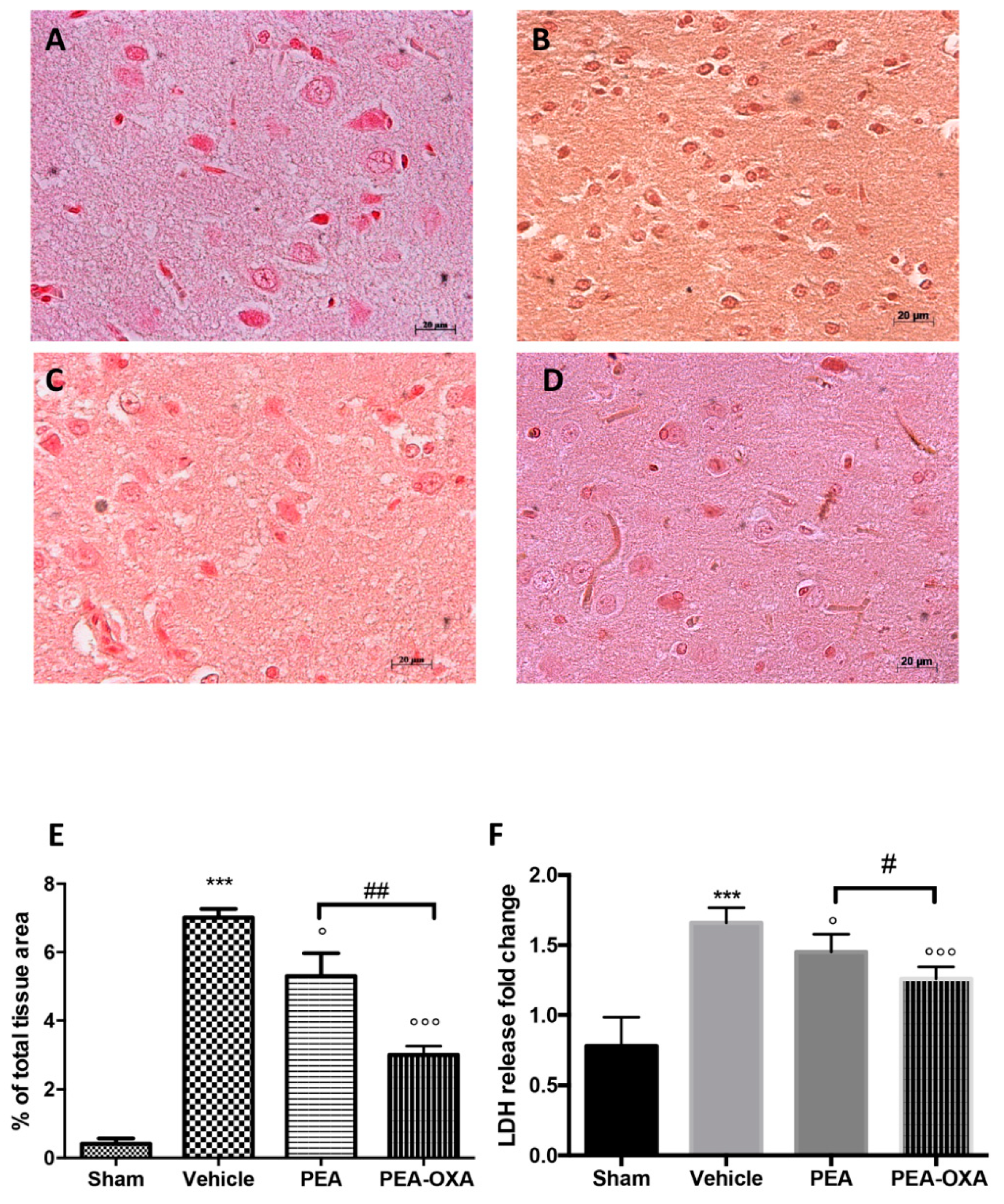
2.1. Effect of PEA-OXA Treatment on Regional Cerebral Blood Flow (rCBF) and Ischemic Brain Damage
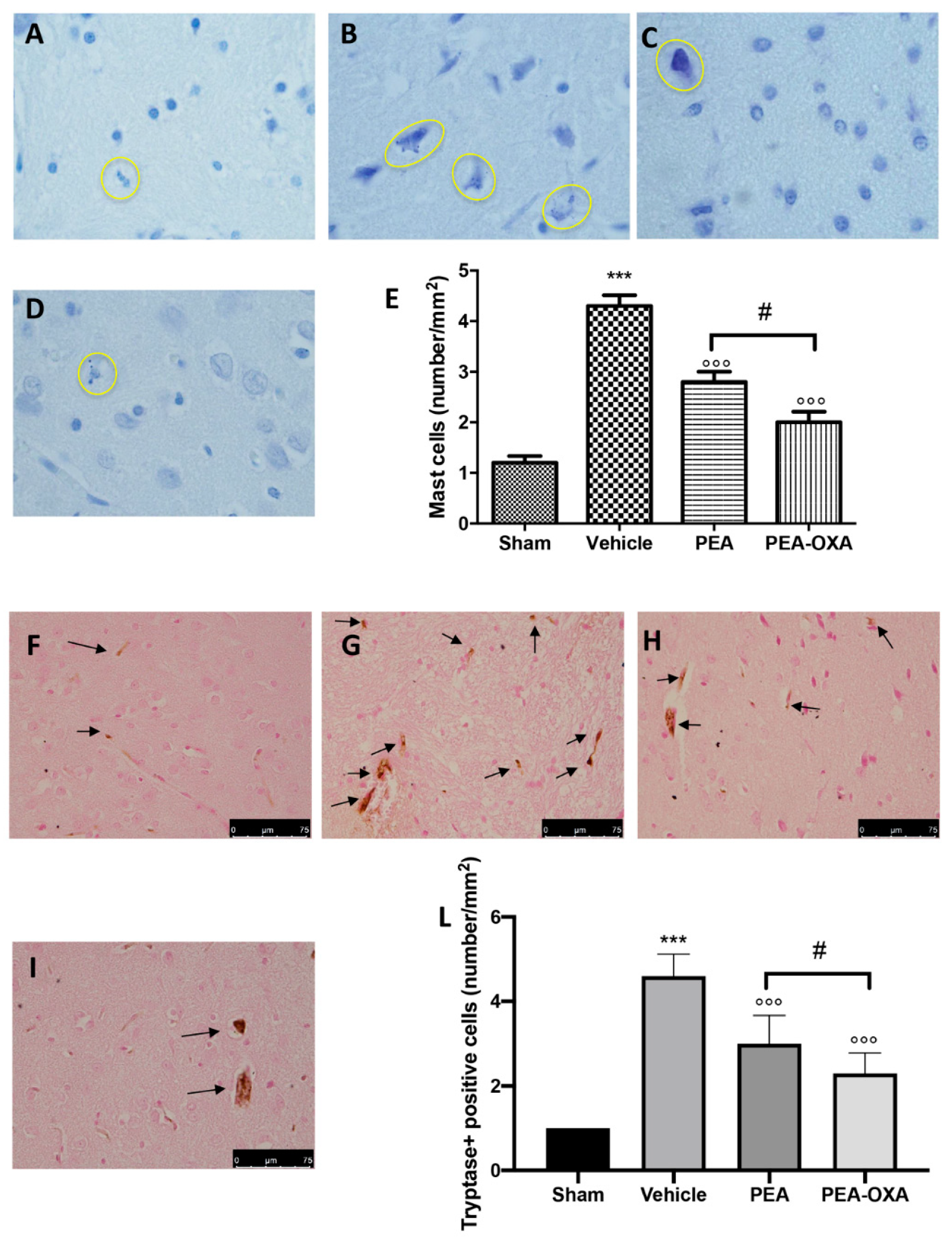
2.2. Effect of PEA-OXA Treatment on MCAo-Induced Mast Cell Degranulation
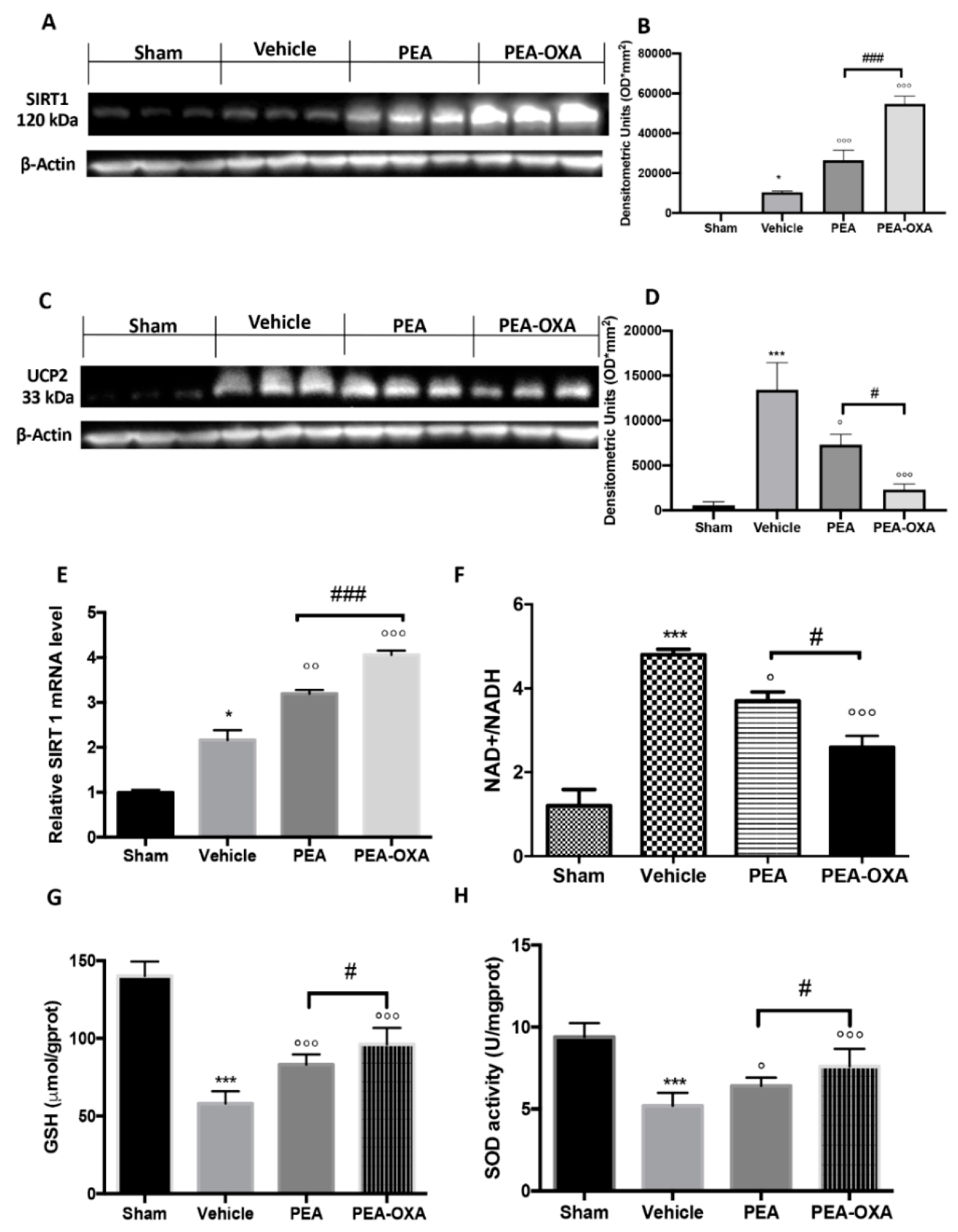
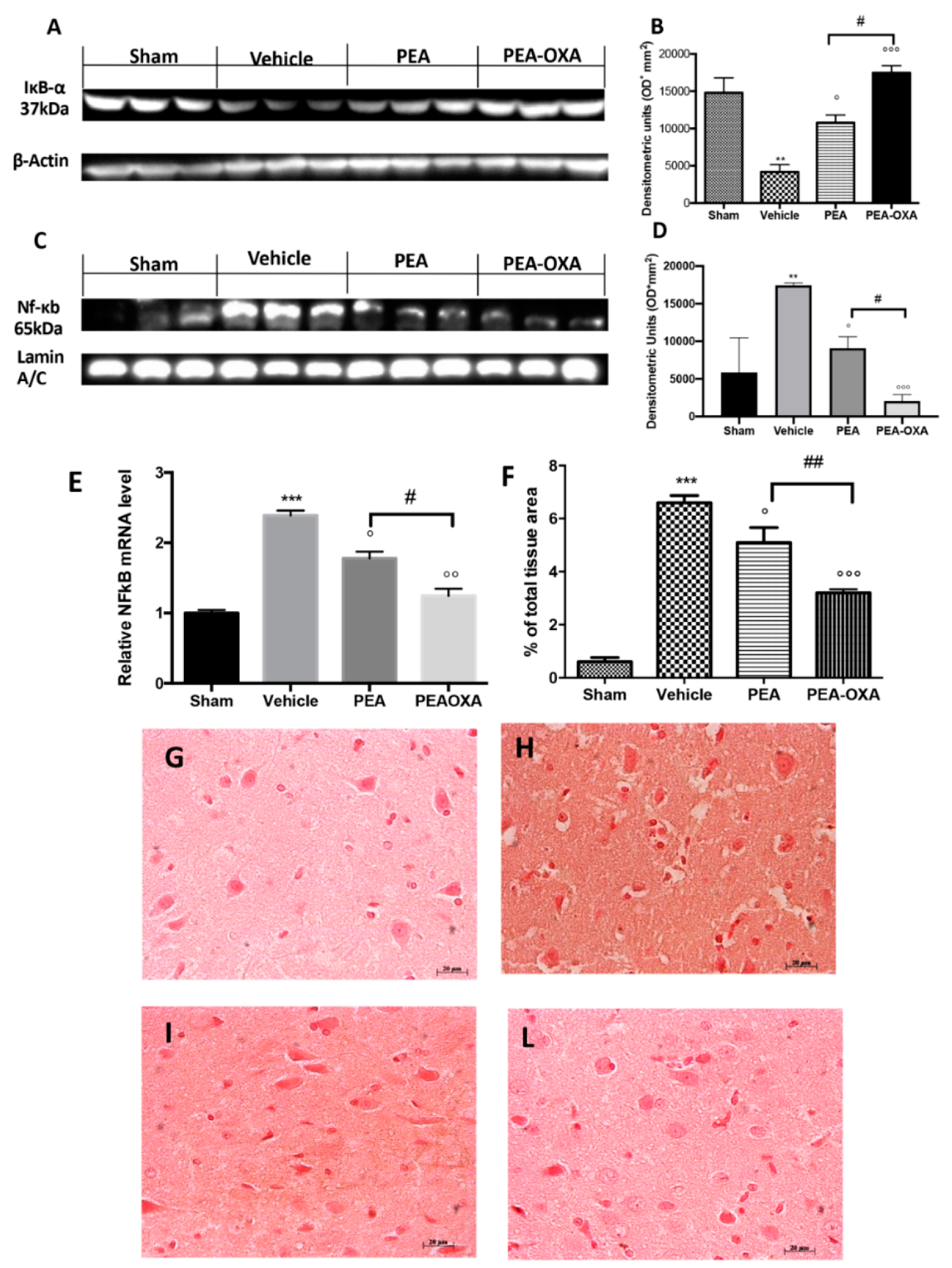
2.3. Effect of PEA-OXA Treatment on MCAo-Induced SIRT1 and UCP2 Expression and Redox Status
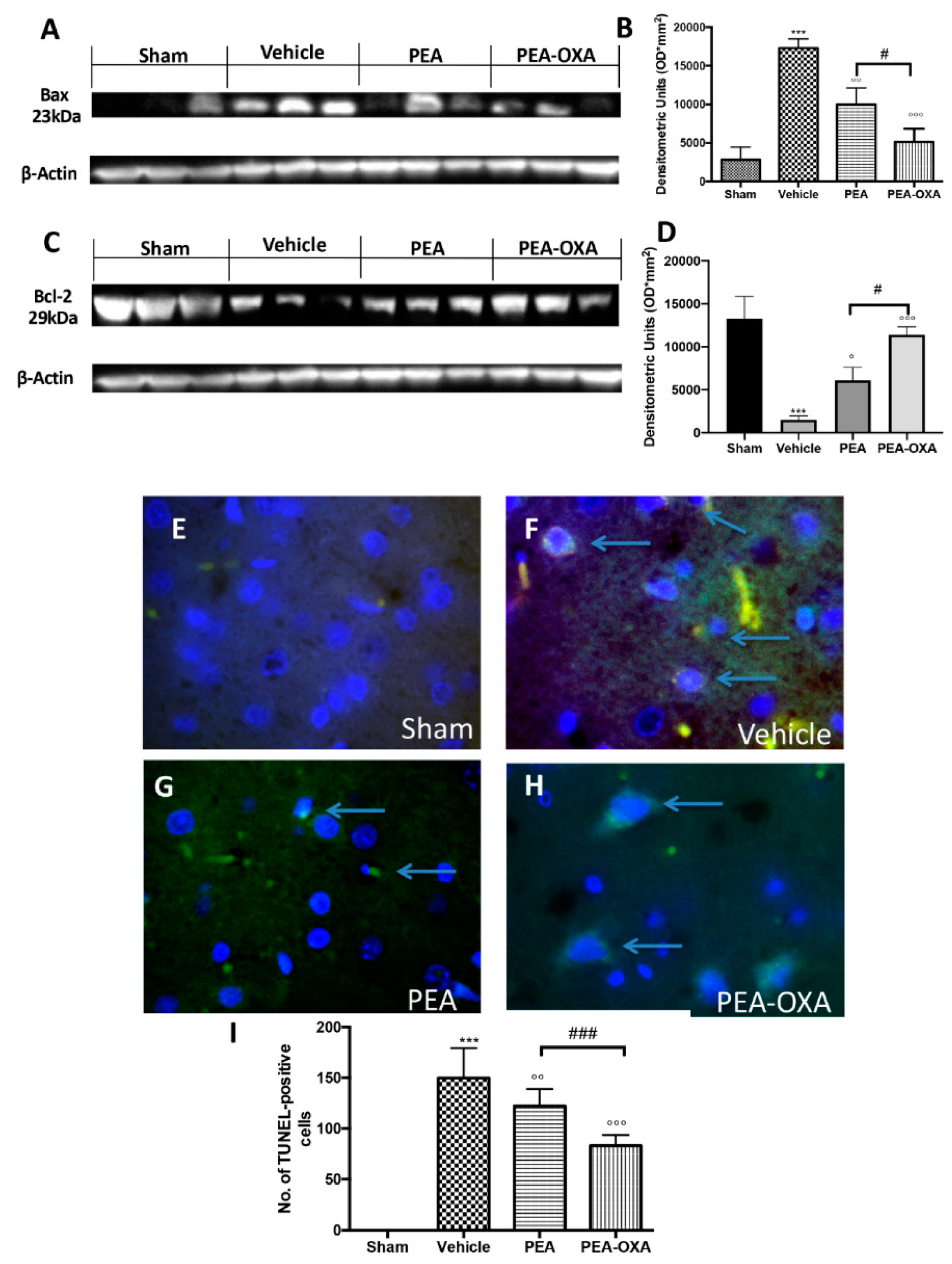
2.4. Effects of PEA-OXA Treatment on MCAo-Induced Apoptosis
2.5. Effect of PEA-OXA Treatment on MCAo-Induced IκB-α Degradation, NF-κB Translocation, and TGF-β Expression
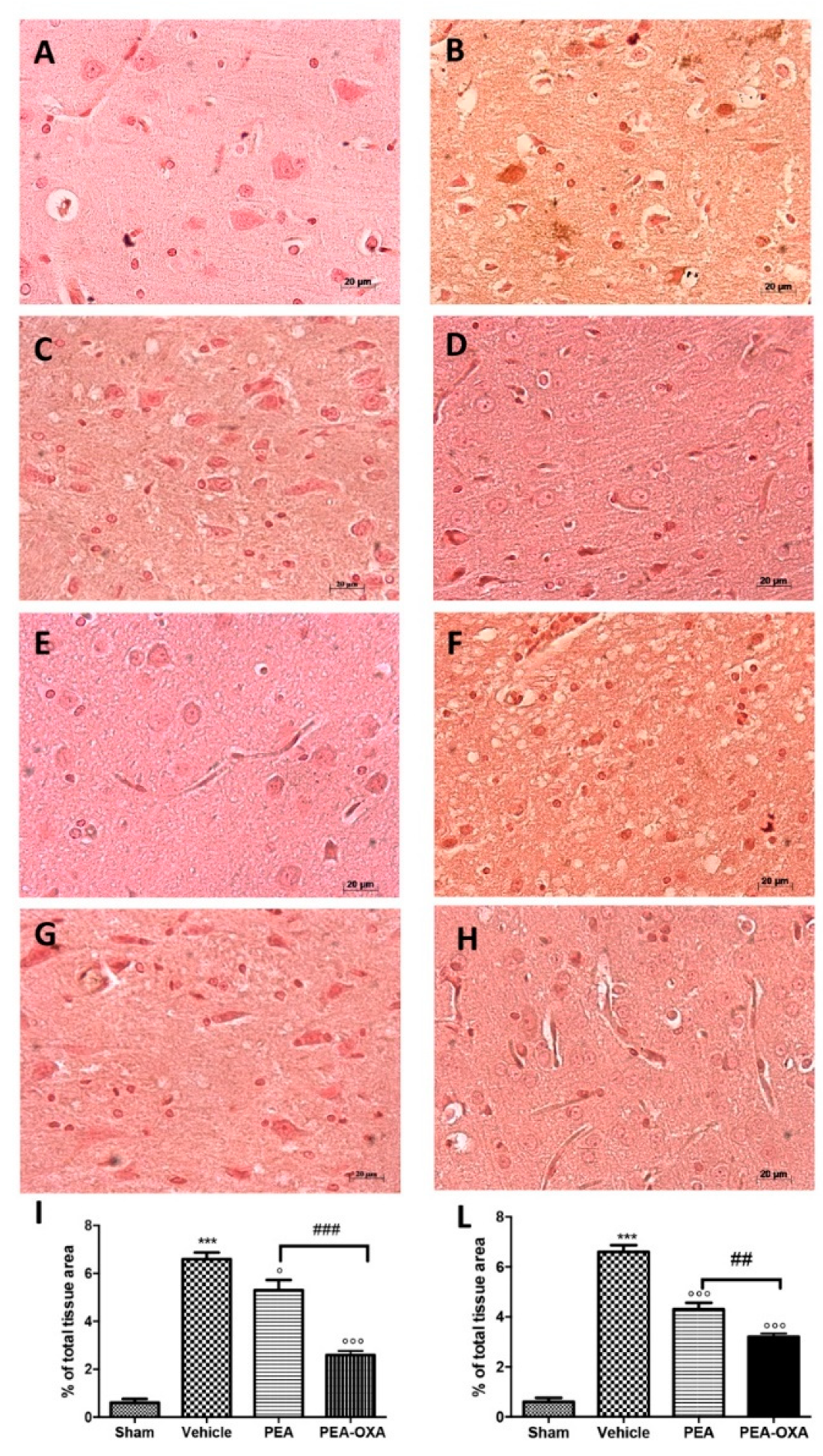
2.6. Effect of PEA-OXA Treatment on MCAo-Induced Cytokine Production
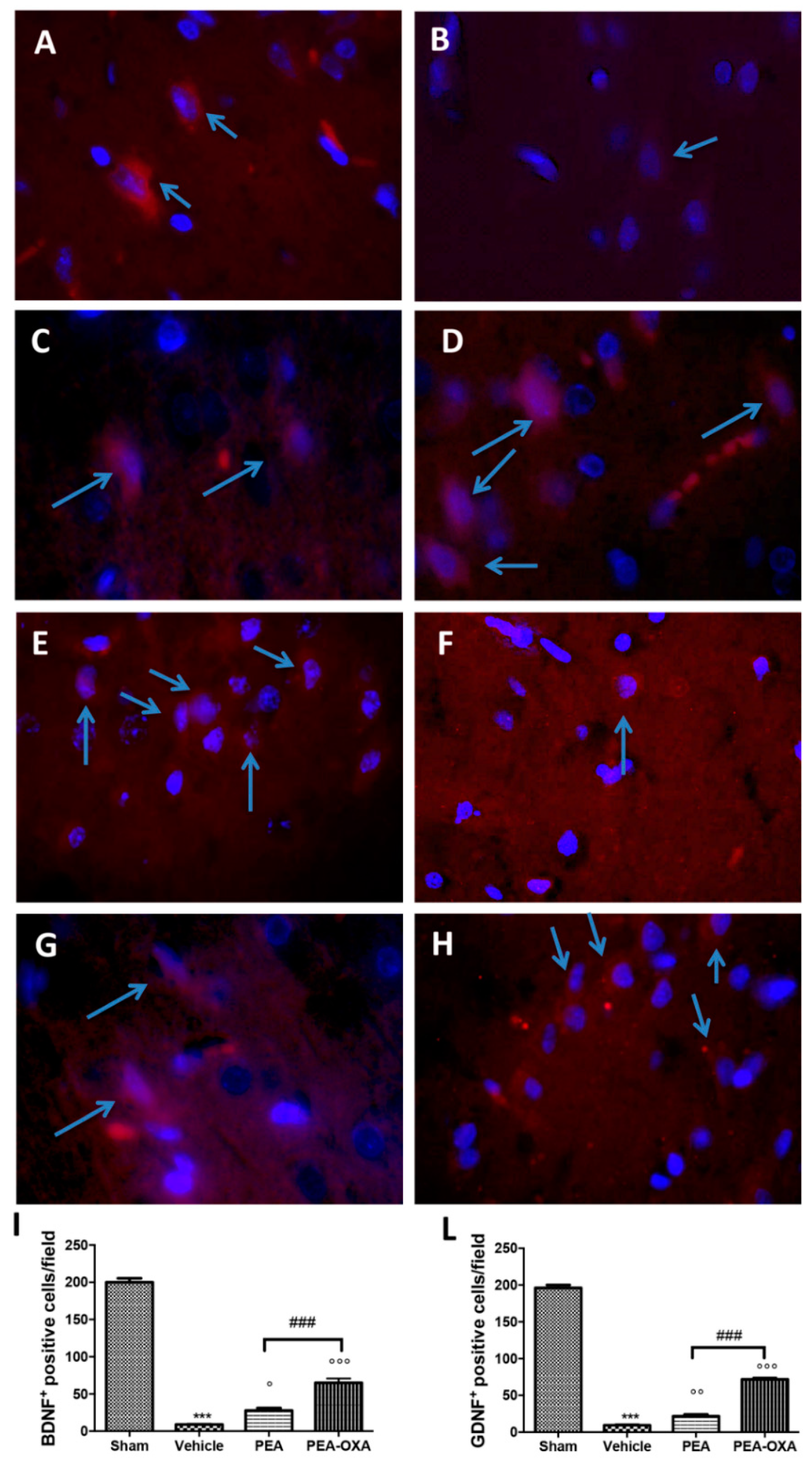
2.7. Effect of PEA-OXA Treatment on MCAo-Induced Reduced BDNF and GDNF Expression
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Middle Cerebral Artery Occlusion
4.3. Synthesis of PEA and PEA-OXA
4.4. Induction of Diabetes
4.5. Experimental Groups
4.6. Quantification of Infarct Volume
4.7. Histological Evaluation
4.8. Toluidine Blue Staining
4.9. Immunohistochemical Localization of Tryptase, Caspase-3, Interleukin-1 Beta (IL-1β), Tumor Necrosis Factor-Alpha (TNF-α), and Transforming Growth Factor Beta (TGF-β)
4.10. Immunofluorescence of Brain-Derived Neurotrophic Factor (BDNF) and Glial Cell-derived Neurotrophic Factor (GDNF)
4.11. Western Blot Analysis for SIRT1, UCP2, IkB-α, NF-kB, Bax, and Bcl-2
4.12. TUNEL Staining
4.13. NAD+/NADH Assay
4.14. GSH Levels Assay
4.15. SOD Activity Assay
4.16. LDH Release Detection
4.17. RT-PCR
4.18. Materials
4.19. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HE | hematoxylin and eosin |
| GFAP | antiglial fibrillary acidic protein |
| Iba-1 | ionized calcium-binding adaptor molecule 1 |
| BDNF | brain-derived neurotrophic factor |
| GDNF | glial cell-derived neurotrophic factor |
| IL-1β | interleukin 1 beta |
| IκB-α | nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha |
| NAAA | N-acylethanolamine-hydrolyzing acid amidase |
| NFκB | nuclear factor-κB |
| NGF | nerve growth factor |
| PEA | N-palmitoylethanolamide |
| PEA-OXA | 2-pentadecyl-2-oxazoline |
| rCBF | regional cerebral blood flow |
| ROS | reactive oxygen species |
| SIRT1 | silent information regulator 1 |
| STZ | streptozotocin |
| TGF- β | transforming growth factor beta |
| TNF-α | tumor necrosis factor-alpha |
| UCP2 | uncoupling protein 2 |
| VEGF | vascular endothelial growth factor |
References
- Emerging Risk Factors, C.; Sarwar, N.; Gao, P.; Seshasai, S.R.; Gobin, R.; Kaptoge, S.; di Angelantonio, E.; Ingelsson, E.; Lawlor, D.A.; Selvin, E.; et al. Diabetes mellitus, fasting blood glucose concentration, and risk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet 2010, 375, 2215–2222. [Google Scholar] [CrossRef]
- Bourne, R.R.; Stevens, G.A.; White, R.A.; Smith, J.L.; Flaxman, S.R.; Price, H.; Jonas, J.B.; Keeffe, J.; Leasher, J.; Naidoo, K.; et al. Vision Loss Expert, G. Causes of vision loss worldwide, 1990-2010: A systematic analysis. Lancet Glob. Health 2013, 1, 339–439. [Google Scholar] [CrossRef]
- Biller, J.; Love, B.B. Diabetes and stroke. Med. Clin. North. Am. 1993, 77, 95–110. [Google Scholar] [CrossRef]
- Vinik, A.; Flemmer, M. Diabetes and macrovascular disease. J. Diabetes Complicat. 2002, 16, 235–245. [Google Scholar] [CrossRef]
- Iwata, N.; Takayama, H.; Xuan, M.; Kamiuchi, S.; Matsuzaki, H.; Okazaki, M.; Hibino, Y. Effects of Etanercept against Transient Cerebral Ischemia in Diabetic Rats. Biomed. Res. Int. 2015, 2015, 189292. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Silverman, A.J.; Vannucci, S.J. Mast cells are early responders after hypoxia-ischemia in immature rat brain. Stroke; A J. Cereb. Circ. 2009, 40, 3107–3112. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.; Curley, J.P. Mast cells on the mind: New insights and opportunities. Trends Neurosci. 2013, 36, 513–521. [Google Scholar] [CrossRef]
- Wang, Q.; Tang, X.N.; Yenari, M.A. The inflammatory response in stroke. J. Neuroimmunol. 2007, 184, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Kawabori, M.; Yenari, M.A. Inflammatory responses in brain ischemia. Curr. Med. Chem. 2015, 22, 1258–1277. [Google Scholar] [CrossRef]
- Bramlett, H.M.; Dietrich, W.D. Pathophysiology of cerebral ischemia and brain trauma: Similarities and differences. J. Cereb. Blood Flow Metab. 2004, 24, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.M.; Kitchen, P.; Woodroofe, M.N.; Bill, R.M.; Conner, A.C.; Heath, P.R.; Conner, M.T. Transcriptome analysis of gene expression provides new insights into the effect of mild therapeutic hypothermia on primary human cortical astrocytes cultured under hypoxia. Front. Cell Neurosci. 2017, 11, 386. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, M.D. Adventures in the pathophysiology of brain ischemia: Penumbra, gene expression, neuroprotection: The 2002 Thomas Willis Lecture. Stroke 2003, 34, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J.; Karin, M. Nuclear factor-kappaB: A pivotal transcription factor in chronic inflammatory diseases. New Engl. J. Med. 1997, 336, 1066–1071. [Google Scholar] [CrossRef] [PubMed]
- Bowie, A.; O’Neill, L.A. Oxidative stress and nuclear factor-kappaB activation: A reassessment of the evidence in the light of recent discoveries. Biochem. Pharmacol. 2000, 59, 13–23. [Google Scholar] [CrossRef]
- Calabresi, P.; Cupini, L.M.; Centonze, D.; Pisani, F.; Bernardi, G. Antiepileptic drugs as a possible neuroprotective strategy in brain ischemia. Ann. Neurol. 2003, 53, 693–702. [Google Scholar] [CrossRef]
- ArunaDevi, R.; Lata, S.; Bhadoria, B.K.; Ramteke, V.D.; Kumar, S.; Sankar, P.; Kumar, D.; Tandan, S.K. Neuroprotective effect of 5,7,3’,4’,5’-pentahydroxy dihydroflavanol-3-O-(2’’-O-galloyl)-beta-D-glucopyranoside, a polyphenolic compound in focal cerebral ischemia in rat. Eur. J. Pharm. 2010, 626, 205–212. [Google Scholar] [CrossRef]
- ArunaDevi, R.; Ramteke, V.D.; Kumar, S.; Shukla, M.K.; Jaganathan, S.; Kumar, D.; Sharma, A.K.; Tandan, S.K. Neuroprotective effect of s-methylisothiourea in transient focal cerebral ischemia in rat. Nitric Oxide Biol. Chem. 2010, 22, 1–10. [Google Scholar] [CrossRef]
- Ye, Q.; Li, Q.; Zhou, Y.; Xu, L.; Mao, W.; Gao, Y.; Li, C.; Xu, Y.; Xu, Y.; Liao, H.; et al. Synthesis and Evaluation of 3-(furo[2 –b]pyridin-3-yl)-4-(1H-indol-3-yl)-maleimides as Novel GSK-3beta Inhibitors and Anti-Ischemic Agents. Chem. Biol. Drug Des. 2015, 86, 746–752. [Google Scholar] [CrossRef]
- Jin, Z.; Liang, J.; Wang, J.; Kolattukudy, P.E. MCP-induced protein 1 mediates the minocycline-induced neuroprotection against cerebral ischemia/reperfusion injury in vitro and in vivo. J. Neuroinflammation 2015, 12, 39. [Google Scholar] [CrossRef]
- Corbett, D.; Jeffers, M.; Nguemeni, C.; Gomez-Smith, M.; Livingston-Thomas, J. Lost in translation: Rethinking approaches to stroke recovery. Prog. Brain Res. 2015, 218, 413–434. [Google Scholar] [PubMed]
- Sughrue, M.E.; Grobelny, B.T.; Ducruet, A.F.; Komotar, R.J.; Mocco, J.; Sciacca, R.R.; Sander Connolly, E. Data presentation in rodent stroke studies and the predictive value of confidence intervals. J. Clin. Neurosci.: Off. J. Neurosurg. Soc. Australas. 2010, 17, 11–15. [Google Scholar] [CrossRef]
- Hussain, M.S.; Shuaib, A. Research into neuroprotection must continue... But with a different approach. Stroke; A J. Cereb. Circ. 2008, 39, 521–522. [Google Scholar] [CrossRef]
- Prestigiacomo, C.J.; Kim, S.C.; Connolly, E.S., Jr.; Liao, H.; Yan, S.F.; Pinsky, D.J. CD18-mediated neutrophil recruitment contributes to the pathogenesis of reperfused but not nonreperfused stroke. Stroke A J. Cereb. Circ. 1999, 30, 1110–1117. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Z.G.; Zhang, R.L.; Lu, M.; Krams, M.; Chopp, M. Effects of a selective CD11b/CD18 antagonist and recombinant human tissue plasminogen activator treatment alone and in combination in a rat embolic model of stroke. Stroke; A J. Cereb. Circ. 2003, 34, 1790–1795. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.J. Anti-leukocyte antibodies: LeukArrest (Hu23F2G) and Enlimomab (R6.5) in acute stroke. Curr. Med. Res. Opin. 2002, 18, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Investigators, E.A.S.T. Use of anti-ICAM-1 therapy in ischemic stroke results of the Enlimomab Acute Stroke Trial. Neurology 2001, 57, 1428–1434. [Google Scholar]
- Pacher, P.; Batkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharm. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef]
- Lambert, D.M.; Vandevoorde, S.; Jonsson, K.O.; Fowler, C.J. The palmitoylethanolamide family: A new class of anti-inflammatory agents? Curr. Med. Chem. 2002, 9, 663–674. [Google Scholar] [CrossRef]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N.; Stockinger, B.; Tak, P.P. The resolution of inflammation. Nat. Rev. Immunol. 2013, 13, 59–66. [Google Scholar] [CrossRef]
- Skaper, S.D.; Facci, L. Mast cell-glia axis in neuroinflammation and therapeutic potential of the anandamide congener palmitoylethanolamide. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 2012, 367, 3312–3325. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Giusti, P. Mast cells, glia and neuroinflammation: Partners in crime? Immunology 2014, 141, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Impellizzeri, D.; Esposito, E.; Attley, J.; Cuzzocrea, S. Targeting inflammation: New therapeutic approaches in chronic kidney disease (CKD). Pharmacol. Res. 2014, 81, 91–102. [Google Scholar] [CrossRef]
- Alhouayek, M.; Muccioli, G.G. Harnessing the anti-inflammatory potential of palmitoylethanolamide. Drug Discov. Today 2014, 19, 1632–1639. [Google Scholar] [CrossRef]
- Esposito, E.; Cordaro, M.; Cuzzocrea, S. Roles of fatty acid ethanolamides (FAE) in traumatic and ischemic brain injury. Pharmacol. Res. 2014, 86, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Fidaleo, M.; Fanelli, F.; Ceru, M.P.; Moreno, S. Neuroprotective properties of peroxisome proliferator-activated receptor alpha (PPARalpha) and its lipid ligands. Curr. Med. Chem. 2014, 21, 2803–2821. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Tsuboi, K.; Uyama, T. N-acylethanolamine metabolism with special reference to N-acylethanolamine-hydrolyzing acid amidase (NAAA). Prog. Lipid Res. 2010, 49, 299–315. [Google Scholar] [CrossRef] [PubMed]
- Solorzano, C.; Zhu, C.; Battista, N.; Astarita, G.; Lodola, A.; Rivara, S.; Mor, M.; Russo, R.; Maccarrone, M.; Antonietti, F.; et al. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc. Natl. Acad. Sci. USA 2009, 106, 20966–20971. [Google Scholar] [CrossRef]
- Yamano, Y.; Tsuboi, K.; Hozaki, Y.; Takahashi, K.; Jin, X.H.; Ueda, N.; Wada, A. Lipophilic amines as potent inhibitors of N-acylethanolamine-hydrolyzing acid amidase. Bioorganic Med. Chem. 2012, 20, 3658–3665. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, L.; Chen, L.; Li, Y.; Chen, H.; Li, Y.; Ji, G.; Lin, D.; Liu, Z.; Qiu, Y. Potential analgesic effects of a novel N-acylethanolamine acid amidase inhibitor F96 through PPAR-alpha. Sci. Rep. 2015, 5, 13565. [Google Scholar] [CrossRef]
- Ribeiro, A.; Pontis, S.; Mengatto, L.; Armirotti, A.; Chiurchiu, V.; Capurro, V.; Fiasella, A.; Nuzzi, A.; Romeo, E.; Moreno-Sanz, G.; et al. A Potent Systemically Active N-Acylethanolamine Acid Amidase Inhibitor that Suppresses Inflammation and Human Macrophage Activation. Acs Chem. Biol. 2015, 10, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Barbierato, M.; Zusso, M.; Bruschetta, G.; Impellizzeri, D.; Cuzzocrea, S.; Giusti, P. N-Palmitoylethanolamine and Neuroinflammation: A Novel Therapeutic Strategy of Resolution. Mol. Neurobiol. 2015, 52, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Impellizzeri, D.; Cordaro, M.; Bruschetta, G.; Crupi, R.; Pascali, J.; Alfonsi, D.; Marcolongo, G.; Cuzzocrea, S. 2-pentadecyl-2-oxazoline: Identification in coffee, synthesis and activity in a rat model of carrageenan-induced hindpaw inflammation. Pharm. Res. 2016, 108, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Impellizzeri, D.; Cordaro, M.; Bruschetta, G.; Siracusa, R.; Crupi, R.; Esposito, E.; Cuzzocrea, S. N-Palmitoylethanolamine-Oxazoline as a New Therapeutic Strategy to Control Neuroinflammation: Neuroprotective Effects in Experimental Models of Spinal Cord and Brain Injury. J. Neurotrauma 2017, 34, 2609–2623. [Google Scholar] [CrossRef] [PubMed]
- Cordaro, M.; Siracusa, R.; Crupi, R.; Impellizzeri, D.; Peritore, A.F.; D’Amico, R.; Gugliandolo, E.; di Paola, R.; Cuzzocrea, S. 2-Pentadecyl-2-Oxazoline Reduces Neuroinflammatory Environment in the MPTP Model of Parkinson Disease. Mol. Neurobiol. 2018, 55, 9251–9266. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Wang, Y.; Zhang, X.; Zhang, X.; Qian, Y.; Ding, H.; Zhang, S. Stabilization of Brain Mast Cells Alleviates LPS-Induced Neuroinflammation by Inhibiting Microglia Activation. Front. Cell Neurosci 2019, 13, 191. [Google Scholar] [CrossRef]
- Yao, X.; Miao, W.; Li, M.; Wang, M.; Ma, J.; Wang, Y.; Miao, L.; Feng, H. Protective effect of albumin on VEGF and brain edema in acute ischemia in rats. Neurosci Lett 2010, 472, 179–183. [Google Scholar] [CrossRef]
- Ali, C.; Docagne, F.; Nicole, O.; Lesne, S.; Toutain, J.; Young, A.; Chazalviel, L.; Divoux, D.; Caly, M.; Cabal, P.; et al. Increased expression of transforming growth factor-beta after cerebral ischemia in the baboon: An endogenous marker of neuronal stress? J. Cereb Blood Flow Metab 2001, 21, 820–827. [Google Scholar] [CrossRef]
- Chu, H.X.; Kim, H.A.; Lee, S.; Broughton, B.R.; Drummond, G.R.; Sobey, C.G. Evidence of CCR2-independent transmigration of Ly6C(hi) monocytes into the brain after permanent cerebral ischemia in mice. Brain Res. 2016, 1637, 118–127. [Google Scholar] [CrossRef]
- Shi, S.S.; Yang, W.Z.; Chen, Y.; Chen, J.P.; Tu, X.K. Propofol reduces inflammatory reaction and ischemic brain damage in cerebral ischemia in rats. Neurochem. Res. 2014, 39, 793–799. [Google Scholar] [CrossRef]
- Shukla, V.; Shakya, A.K.; Perez-Pinzon, M.A.; Dave, K.R. Cerebral ischemic damage in diabetes: An inflammatory perspective. J. Neuroinflammation 2017, 14, 21. [Google Scholar] [CrossRef]
- Famakin, B.M. The immune response to acute focal cerebral ischemia and associated post-stroke immunodepression: A focused review. Aging Dis. 2014, 5, 307. [Google Scholar] [PubMed]
- Chen, H.; Yoshioka, H.; Kim, G.S.; Jung, J.E.; Okami, N.; Sakata, H.; Maier, C.M.; Narasimhan, P.; Goeders, C.E.; Chan, P.H. Oxidative stress in ischemic brain damage: Mechanisms of cell death and potential molecular targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Paschen, W. Glutamate excitotoxicity in transient global cerebral ischemia. Acta. Neurobiol. Exp. 1996, 56, 313–322. [Google Scholar]
- Abdullahi, W.; Tripathi, D.; Ronaldson, P.T. Blood-brain barrier dysfunction in ischemic stroke: Targeting tight junctions and transporters for vascular protection. Am. J. Physiol. -Cell Physiol. 2018, 315, C343–C356. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, A.F.; Lucke-Wold, B.P.; Turner, R.C.; Huber, J.D.; Rosen, C.L.; Simpkins, J.W. Role of microvascular disruption in brain damage from traumatic brain injury. Compr. Physiol. 2011, 5, 1147–1160. [Google Scholar]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Trendelenburg, G. Molecular regulation of cell fate in cerebral ischemia: Role of the inflammasome and connected pathways. J. Cereb. Blood Flow Metab: Off. J. Int. Soc. Cereb. Blood Flow Metab. 2014, 34, 1857–1867. [Google Scholar] [CrossRef]
- Moretti, R.; Pansiot, J.; Bettati, D.; Strazielle, N.; Ghersi-Egea, J.F.; Damante, G.; Fleiss, B.; Titomanlio, L.; Gressens, P. Blood-brain barrier dysfunction in disorders of the developing brain. Front. Neurosci. 2015, 9, 40. [Google Scholar] [CrossRef]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef]
- Costa, B.; Conti, S.; Giagnoni, G.; Colleoni, M. Therapeutic effect of the endogenous fatty acid amide, palmitoylethanolamide, in rat acute inflammation: Inhibition of nitric oxide and cyclo-oxygenase systems. Br. J. Pharmacol. 2002, 137, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Lo Verme, J.; Fu, J.; Astarita, G.; La Rana, G.; Russo, R.; Calignano, A.; Piomelli, D. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol. Pharmacol. 2005, 67, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Ueda, N.; Yamanaka, K.; Yamamoto, S. Purification and characterization of an acid amidase selective for N-palmitoylethanolamine, a putative endogenous anti-inflammatory substance. J. Biol. Chem. 2001, 276, 35552–35557. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, K.; Takezaki, N.; Ueda, N. The N-acylethanolamine-hydrolyzing acid amidase (NAAA). Chem. Biodivers. 2007, 4, 1914–1925. [Google Scholar] [CrossRef] [PubMed]
- McKinney, M.K.; Cravatt, B.F. Structure and function of fatty acid amide hydrolase. Annu. Rev. Biochem. 2005, 74, 411–432. [Google Scholar] [CrossRef] [PubMed]
- Tai, T.; Tsuboi, K.; Uyama, T.; Masuda, K.; Cravatt, B.F.; Houchi, H.; Ueda, N. Endogenous molecules stimulating N-acylethanolamine-hydrolyzing acid amidase (NAAA). Acs Chem. Neurosci. 2012, 3, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Alhouayek, M.; Bottemanne, P.; Subramanian, K.V.; Lambert, D.M.; Makriyannis, A.; Cani, P.D.; Muccioli, G.G. N-Acylethanolamine-hydrolyzing acid amidase inhibition increases colon N-palmitoylethanolamine levels and counteracts murine colitis. Faseb J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2015, 29, 650–661. [Google Scholar]
- Petrosino, S.; Ahmad, A.; Marcolongo, G.; Esposito, E.; Allarà, M.; Verde, R.; Cuzzocrea, S.; Di Marzo, V. Diacerein is a potent and selective inhibitor of palmitoylethanolamide inactivation with analgesic activity in a rat model of acute inflammatory pain. Pharmacol. Res. 2015, 91, 9–14. [Google Scholar] [CrossRef]
- Della Valle, F.; Della Valle, M.F.; Marcolongo, G.; Di Marzo, V.; Cuzzocrea, S. Compositions and Methods for the Modulation of Specific Amidases for n-Acylethanolamines for Use in the Therapy of Inflammatory Diseases. Google Patents. 2016. Available online: https://patents.google.com/patent/US9512091B2/en URL (accessed on 27 September 2019).
- Bandiera, T.; Ponzano, S.; Piomelli, D. Advances in the discovery of N-acylethanolamine acid amidase inhibitors. Pharmacol. Res. 2014, 86, 11–17. [Google Scholar] [CrossRef]
- Migliore, M.D.; Pontis, S.D.; Fuentes de Arriba, A.L.; Realini, N.; Torrente, E.; Armirotti, A.; Romeo, E.; Di Martino, S.; Russo, D.; Pizzirani, D.; et al. Second-Generation Non-Covalent NAAA Inhibitors are Protective in a Model of Multiple Sclerosis. Angew. Chem. 2016, 55, 11193–11197. [Google Scholar] [CrossRef]
- Xu, J.; Jackson, C.W.; Khoury, N.; Escobar, I.; Perez-Pinzon, M.A. Brain SIRT1 Mediates Metabolic Homeostasis and Neuroprotection. Front. Endocrinol 2018, 9, 702. [Google Scholar] [CrossRef] [PubMed]
- Smart, D.; Jonsson, K.O.; Vandevoorde, S.; Lambert, D.M.; Fowler, C.J. ‘Entourage’ effects of N-acyl ethanolamines at human vanilloid receptors. Comparison of effects upon anandamide-induced vanilloid receptor activation and upon anandamide metabolism. Brit. J. Pharm. 2002, 136, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Mazzari, S.; Canella, R.; Petrelli, L.; Marcolongo, G.; Leon, A. N-(2-Hydroxyethyl)hexadecanamide is orally active in reducing edema formation and inflammatory hyperalgesia by down-modulating mast cell activation. Eur J. Pharm. 1996, 300, 227–236. [Google Scholar] [CrossRef]
- Farquhar-Smith, W.P.; Rice, A.S.C. Administration of endocannabinoids prevents a referred hyperalgesia associated with inflammation of the urinary bladder. Anesthesiology 2001, 94, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Picard, F.; Kurtev, M.; Chung, N.J.; Topark-Ngarm, A.; Senawong, T.; de Oliveira, R.M.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef] [PubMed]
- De Bilbao, F.; Arsenijevic, D.; Vallet, P.; Hjelle, O.P.; Ottersen, O.P.; Bouras, C.; Raffin, Y.; Abou, K.; Langhans, W.; Collins, S. Resistance to cerebral ischemic injury in UCP2 knockout mice: Evidence for a role of UCP2 as a regulator of mitochondrial glutathione levels. J. Neurochem. 2004, 89, 1283–1292. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.S.; Sun, L.K.; Jiang, X.R.; Zhang, Y.; Kang, J.S.; Meng, H.; Li, H.Y.; Su, J. Genipin protects against cerebral ischemia-reperfusion injury by regulating the UCP2-SIRT3 signaling pathway. Eur J. Pharm. 2019, 845, 56–64. [Google Scholar] [CrossRef]
- Della-Morte, D.; Dave, K.R.; DeFazio, R.A.; Bao, Y.C.; Raval, A.P.; Perez-Pinzon, M.A. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1–uncoupling protein 2 pathway. Neuroscience 2009, 159, 993–1002. [Google Scholar] [CrossRef]
- Brandi, J.; Cecconi, D.; Cordani, M.; Torrens-Mas, M.; Pacchiana, R.; Dalla Pozza, E.; Butera, G.; Manfredi, M.; Marengo, E.; Oliver, J.; et al. The antioxidant uncoupling protein 2 stimulates hnRNPA2/B1, GLUT1 and PKM2 expression and sensitizes pancreas cancer cells to glycolysis inhibition. Free Radic. Biol. Med. 2016, 101, 305–316. [Google Scholar] [CrossRef]
- Cardoso, S.; Seica, R.M.; Moreira, P.I. Uncoupling Protein 2 Inhibition Exacerbates Glucose Fluctuation-Mediated Neuronal Effects. Neurotox Res. 2018, 33, 388–401. [Google Scholar] [CrossRef]
- Caltagirone, C.; Cisari, C.; Schievano, C.; Di Paola, R.; Cordaro, M.; Bruschetta, G.; Esposito, E.; Cuzzocrea, S.; Grp, S.S. Co-ultramicronized Palmitoylethanolamide/Luteolin in the Treatment of Cerebral Ischemia: From Rodent to Man. Transl Stroke Res. 2016, 7, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappa B-dependent transcription and cell survival by the SIRT1 deacetylase. Embo J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Toledo-Pereyra, L.H.; Toledo, A.H.; Walsh, J.; Lopez-Neblina, F. Molecular signaling pathways in ischemia/reperfusion. Exp. Clin. Transplant.: Off. J. Middle East. Soc. Organ. Transplant. 2004, 2, 174–177. [Google Scholar]
- Liu, T.; Clark, R.K.; Mcdonnell, P.C.; Young, P.R.; White, R.F.; Barone, F.C.; Feuerstein, G.Z. Tumor-Necrosis-Factor-Alpha Expression in Ischemic Neurons. Stroke 1994, 25, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Pal, G.; Vincze, C.; Renner, E.; Wappler, E.A.; Nagy, Z.; Lovas, G.; Dobolyi, A. Time Course, Distribution and Cell Types of Induction of Transforming Growth Factor Betas following Middle Cerebral Artery Occlusion in the Rat Brain. PLoS ONE 2012, 7, e46731. [Google Scholar] [CrossRef]
- Galvin, K.A.; Oorschot, D.E. Continuous low-dose treatment with brain-derived neurotrophic factor or neurotrophin-3 protects striatal medium spiny neurons from mild neonatal hypoxia/ischemia: A stereological study. Neuroscience 2003, 118, 1023–1032. [Google Scholar] [CrossRef]
- Ahmad, A.; Genovese, T.; Impellizzeri, D.; Crupi, R.; Velardi, E.; Marino, A.; Esposito, E.; Cuzzocrea, S. Reduction of ischemic brain injury by administration of palmitoylethanolamide after transient middle cerebral artery occlusion in rats. Brain Res. 2012, 1477, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke; A J. Cereb. Circ. 1989, 20, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Melani, A.; Pantoni, L.; Corsi, C.; Bianchi, L.; Monopoli, A.; Bertorelli, R.; Pepeu, G.; Pedata, F. Striatal outflow of adenosine, excitatory amino acids, gamma-aminobutyric acid, and taurine in awake freely moving rats after middle cerebral artery occlusion: Correlations with neurological deficit and histopathological damage. Stroke; A J. Cereb. Circ. 1999, 30, 2448–2454; discussion 2455. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Yin, Y.; Duan, J.; Zhu, Y.; Yan, J.; Wei, G.; Guan, Y.; Wu, X.; Wang, Y.; Xi, M.; et al. Neuroprotective effect and underlying mechanism of sodium danshensu [3-(3,4-dihydroxyphenyl) lactic acid from Radix and Rhizoma Salviae miltiorrhizae = Danshen] against cerebral ischemia and reperfusion injury in rats. Phytomedicine: Int. J. Phytother. Phytopharm. 2015, 22, 283–289. [Google Scholar] [CrossRef]
- Di Paola, R.; Fusco, R.; Gugliandolo, E.; D’Amico, R.; Campolo, M.; Latteri, S.; Carughi, A.; Mandalari, G.; Cuzzocrea, S. The Antioxidant Activity of Pistachios Reduces Cardiac Tissue Injury of Acute Ischemia/Reperfusion (I/R) in Diabetic Streptozotocin (STZ)-Induced Hyperglycaemic Rats. Front. Pharm. 2018, 9, 51. [Google Scholar] [CrossRef]
- Maleki, S.N.; Aboutaleb, N.; Souri, F. Berberine confers neuroprotection in coping with focal cerebral ischemia by targeting inflammatory cytokines. J. Chem. Neuroanat. 2018, 87, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Ivers, N.M.; Halperin, I.J.; Barnsley, J.; Grimshaw, J.M.; Shah, B.R.; Tu, K.; Upshur, R.; Zwarenstein, M. Allocation techniques for balance at baseline in cluster randomized trials: A methodological review. Trials 2012, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, S.; Campolo, M.; Impellizzeri, D.; Paterniti, I.; Allarà, M.; Gugliandolo, E.; D’Amico, R.; Siracusa, R.; Cordaro, M.; Esposito, E. 2-pentadecyl-2-oxazoline, the oxazoline of pea, modulates carrageenan-induced acute inflammation. Front. Pharmacol. 2017, 8, 308. [Google Scholar] [CrossRef] [PubMed]
- Schomacher, M.; Muller, H.D.; Sommer, C.; Schwab, S.; Schabitz, W.R. Endocannabinoids mediate neuroprotection after transient focal cerebral ischemia. Brain Res. 2008, 1240, 213–220. [Google Scholar] [CrossRef]
- Hara, H.; Friedlander, R.M.; Gagliardini, V.; Ayata, C.; Fink, K.; Huang, Z.; Shimizu-Sasamata, M.; Yuan, J.; Moskowitz, M.A. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc. Natl. Acad. Sci. USA 1997, 94, 2007–2012. [Google Scholar] [CrossRef] [PubMed]
- Schabitz, W.R.; Li, F.; Irie, K.; Sandage, B.W., Jr.; Locke, K.W.; Fisher, M. Synergistic effects of a combination of low-dose basic fibroblast growth factor and citicoline after temporary experimental focal ischemia. Stroke A J. Cereb. Circ. 1999, 30, 427–431. [Google Scholar] [CrossRef]
- Barber, P.A.; Hoyte, L.; Colbourne, F.; Buchan, A.M. Temperature-regulated model of focal ischemia in the mouse: A study with histopathological and behavioral outcomes. Stroke; A J. Cereb. Circ. 2004, 35, 1720–1725. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Sanabria, F.; Rull, A.; Beltran-Debon, R.; Aragones, G.; Camps, J.; Mackness, B.; Mackness, M.; Joven, J. Tissue distribution and expression of paraoxonases and chemokines in mouse: The ubiquitous and joint localisation suggest a systemic and coordinated role. J. Mol. Histol 2010, 41, 379–386. [Google Scholar] [CrossRef]
- Hernandez-Aguilera, A.; Sepulveda, J.; Rodriguez-Gallego, E.; Guirro, M.; Garcia-Heredia, A.; Cabre, N.; Luciano-Mateo, F.; Fort-Gallifa, I.; Martin-Paredero, V.; Joven, J.; et al. Immunohistochemical analysis of paraoxonases and chemokines in arteries of patients with peripheral artery disease. Int. J. Mol. Sci. 2015, 16, 11323–11338. [Google Scholar] [CrossRef]
- Fusco, R.; D’Amico, R.; Cordaro, M.; Gugliandolo, E.; Siracusa, R.; Peritore, A.F.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; di Paola, R. Absence of formyl peptide receptor 1 causes endometriotic lesion regression in a mouse model of surgically-induced endometriosis. Oncotarget 2018, 9, 31355–31366. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experimental Group | Blood Glucose | ||
|---|---|---|---|
| Day 0 | Day 15 | Day 60 | |
| Sham | 120 ± 3.1 | 136 ± 2.5 | 126 ± 3.0 |
| I/R | 130 ± 2.7 | 459 ± 2.9 | 455 ± 2.4 |
| PEA | 125 ± 2.5 | 463 ± 2.2 | 459 ± 2.5 |
| PEA-OXA | 131 ± 1.9 | 466 ± 2.7 | 462 ± 2.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fusco, R.; Scuto, M.; Cordaro, M.; D’Amico, R.; Gugliandolo, E.; Siracusa, R.; Peritore, A.F.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; et al. N-Palmitoylethanolamide-Oxazoline Protects against Middle Cerebral Artery Occlusion Injury in Diabetic Rats by Regulating the SIRT1 Pathway. Int. J. Mol. Sci. 2019, 20, 4845. https://doi.org/10.3390/ijms20194845
Fusco R, Scuto M, Cordaro M, D’Amico R, Gugliandolo E, Siracusa R, Peritore AF, Crupi R, Impellizzeri D, Cuzzocrea S, et al. N-Palmitoylethanolamide-Oxazoline Protects against Middle Cerebral Artery Occlusion Injury in Diabetic Rats by Regulating the SIRT1 Pathway. International Journal of Molecular Sciences. 2019; 20(19):4845. https://doi.org/10.3390/ijms20194845
Chicago/Turabian StyleFusco, Roberta, Maria Scuto, Marika Cordaro, Ramona D’Amico, Enrico Gugliandolo, Rosalba Siracusa, Alessio Filippo Peritore, Rosalia Crupi, Daniela Impellizzeri, Salvatore Cuzzocrea, and et al. 2019. "N-Palmitoylethanolamide-Oxazoline Protects against Middle Cerebral Artery Occlusion Injury in Diabetic Rats by Regulating the SIRT1 Pathway" International Journal of Molecular Sciences 20, no. 19: 4845. https://doi.org/10.3390/ijms20194845
APA StyleFusco, R., Scuto, M., Cordaro, M., D’Amico, R., Gugliandolo, E., Siracusa, R., Peritore, A. F., Crupi, R., Impellizzeri, D., Cuzzocrea, S., & Di Paola, R. (2019). N-Palmitoylethanolamide-Oxazoline Protects against Middle Cerebral Artery Occlusion Injury in Diabetic Rats by Regulating the SIRT1 Pathway. International Journal of Molecular Sciences, 20(19), 4845. https://doi.org/10.3390/ijms20194845