Opportunities for Overcoming Mycobacterium tuberculosis Drug Resistance: Emerging Mycobacterial Targets and Host-Directed Therapy



Abstract
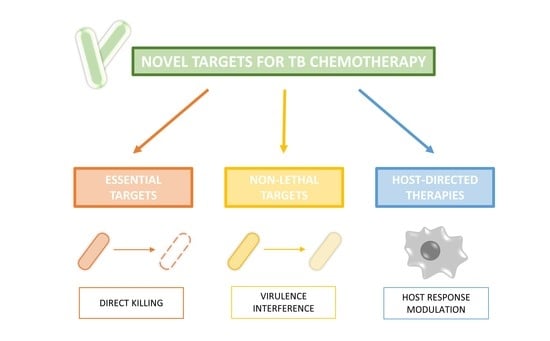
1. Introduction
2. Antimycobacterial Drug Resistance
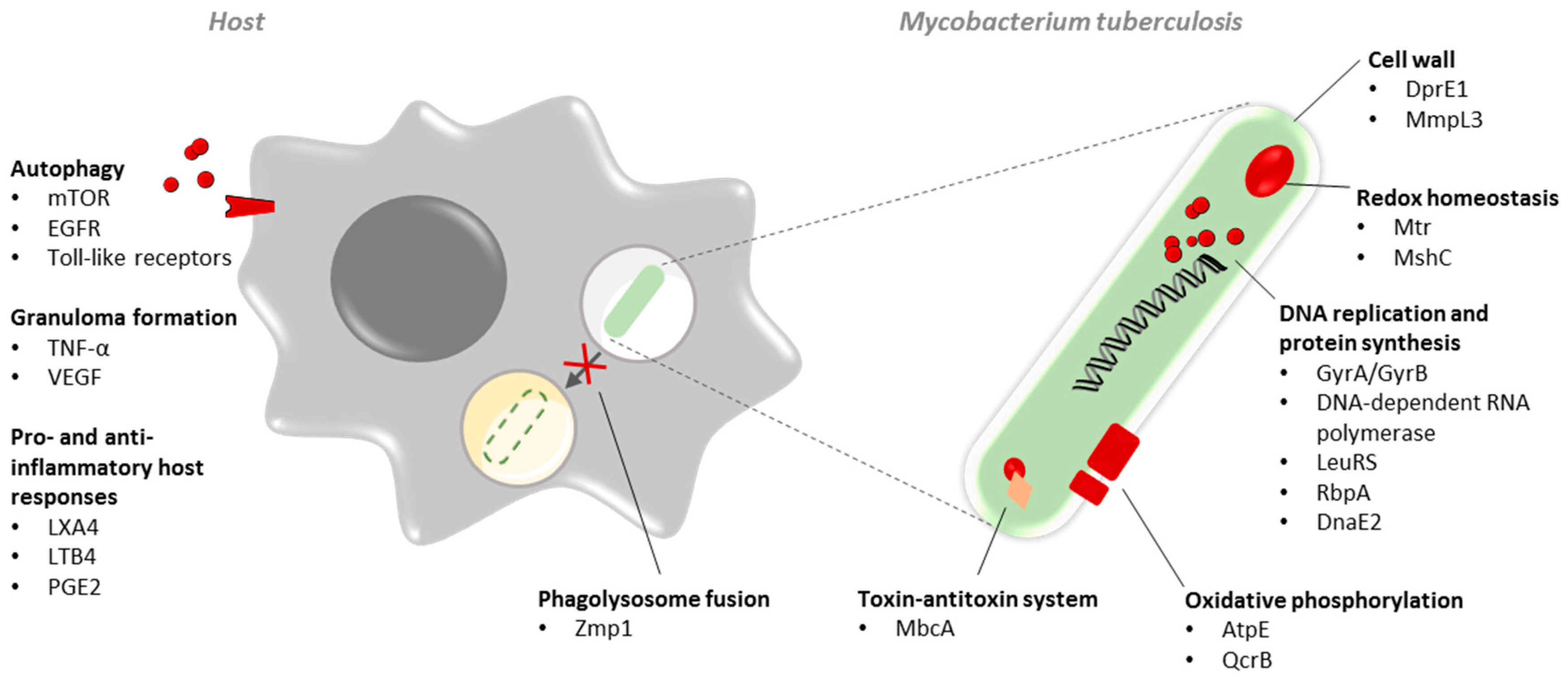
3. Novel Mycobacterial Drug Targets
3.1. Cell Wall Biosynthesis
3.2. Oxidative Phosphorylation
3.3. Protein Synthesis
3.4. Bacillary Redox Homeostasis
3.5. Toxin–Antitoxin System
4. Non-Essential Targets
5. Host-Directed Therapy
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAP | Nα-aroyl-N-aryl-phenylalaninamide |
| AARS | aminoacyl-tRNA synthase |
| AcCys-GlcN-Ins | acetylcysteine-glucosamine-inositol |
| BDQ | bedaquiline |
| BTZ | 1,3-benzothiazin-4-one |
| COX | cyclooxygenase |
| DARQ | diarylquinoline |
| Dnd | deazaflavin (or cofactor F420)-dependent nitroreductase |
| DPA | decaprenylphosphoryl-β-d-arabinofuranose |
| DPR | decaprenylphosphoryl-β-d-ribofuranose |
| DprE1 | decaprenylphosphoryl-β-d-ribose-2′-oxidase |
| DprE2 | decaprenylphosphoryl-2-keto-β-d-erythro-pentose reductase |
| DPX | decaprenylphosphoryl-d-2′-keto-erythro-pentofuranose |
| EGFR | epidermal growth factor receptor |
| ETC | electron transport chain |
| FGD1 | coenzyme F420-dependent glucose-6-phosphate dehydrogenase |
| GlcN-Ins | glucosamine-inositol |
| GyrA | DNA gyrase domain A |
| GyrB | DNA gyrase domain B |
| hERG | human ether-a-go-go related gene |
| HDT | host-directed therapy |
| IFN-γ | interferon-γ |
| IL | interleukine |
| LeuRS | leucyl-tRNA synthase |
| LTB4 | leukotriene B4 |
| LXA4 | lipoxin A4 |
| MbcA | mycobactericidal antitoxin |
| MbcT | mycobactericidal toxin |
| MDR | multidrug resistant |
| MmpL | mycobacterial membrane protein large |
| MmpS | mycobacterial membrane protein small |
| MSH | mycothiol |
| MSSM | mycothione or mycothiol disulfide |
| mTOR | mammalian target of rapamycin |
| Mtr | mycothione reductase |
| Mtb | Mycobacterium tuberculosis |
| NSAID | non-steroidal anti-inflammatory drug |
| PBTZ | piperazine-containing BTZ |
| PGE2 | prostaglandin E2 |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| TA | toxin–antitoxin |
| TB | tuberculosis |
| TDR | total drug resistant |
| TDM | trehalose dimycolate |
| TMM | trehalose monomycolate |
| TNF | tumor necrosis factor |
| VEGF | vascular endothelial growth factor |
| WHO | World Health Organization |
| XDR | Extensively drug resistant |
| Zmp1 | Zn2+ metalloprotease |
References
- World Health Organization (WHO). Global Tuberculosis Report 2018; World Health Organization (WHO): Geneva, Switzerland, 2018. [Google Scholar]
- Gygli, S.M.; Borrell, S.; Trauner, A.; Gagneux, S. Antimicrobial resistance in Mycobacterium tuberculosis: Mechanistic and evolutionary perspectives. FEMS Microbiol. Rev. 2017, 41, 354–373. [Google Scholar] [CrossRef] [PubMed]
- Lönnroth, K.; Mor, Z.; Erkens, C.; Bruchfeld, J.; Nathavitharana, R.R.; van der Werf, M.J.; Lange, C. Tuberculosis in migrants in low-incidence countries: Epidemiology and intervention entry points. Int. J. Tuberc. Lung Dis. 2017, 21, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, S.; Lönnroth, K.; Nellums, L.B.; Olaru, I.D.; Nathavitharana, R.R.; Norredam, M.; Friedland, J.S. Multidrug-resistant tuberculosis and migration to Europe. Clin. Microbiol. Infect. 2017, 23, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Draper, P. The outer parts of the mycobacterial envelope as permeability barriers. Front. Biosci. 1998, 3, D1253–D1261. [Google Scholar] [CrossRef] [PubMed]
- Vilchèze, C.; Jacobs, W.R. The mechanism of isoniazid killing: Clarity through the scope of genetics. Annu. Rev. Microbiol. 2007, 61, 35–50. [Google Scholar] [CrossRef]
- Takayama, K.; Kilburn, J.O. Inhibition of synthesis of arabinogalactan by ethambutol in Mycobacterium smegmatis. Antimicrob. Agents Chemother. 1989, 33, 1493–1499. [Google Scholar] [CrossRef]
- Prosser, G.A.; de Carvalho, L.P.S. Metabolomics Reveal d-Alanine:d-Alanine Ligase As the Target of d-Cycloserine in Mycobacterium tuberculosis. ACS Med. Chem. Lett. 2013, 4, 1233–1237. [Google Scholar] [CrossRef]
- Makarov, V.; Manina, G.; Mikusova, K.; Mollmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones Kill Mycobacterium tuberculosis by Blocking Arabinan Synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef]
- Trefzer, C.; Rengifo-Gonzalez, M.; Hinner, M.J.; Schneider, P.; Makarov, V.; Cole, S.T.; Johnsson, K. Benzothiazinones: Prodrugs That Covalently Modify the Decaprenylphosphoryl-β-d-ribose 2′-epimerase DprE1 of Mycobacterium tuberculosis. J. Am. Chem. Soc. 2010, 132, 13663–13665. [Google Scholar] [CrossRef]
- Mikusova, K.; Huang, H.; Yagi, T.; Holsters, M.; Vereecke, D.; D’Haeze, W.; Scherman, M.S.; Brennan, P.J.; McNeil, M.R.; Crick, D.C. Decaprenylphosphoryl Arabinofuranose, the Donor of the D-Arabinofuranosyl Residues of Mycobacterial Arabinan, Is Formed via a Two-Step Epimerization of Decaprenylphosphoryl Ribose. J. Bacteriol. 2005, 187, 8020–8025. [Google Scholar] [CrossRef]
- Trefzer, C.; Škovierová, H.; Buroni, S.; Bobovská, A.; Nenci, S.; Molteni, E.; Pojer, F.; Pasca, M.R.; Makarov, V.; Cole, S.T.; et al. Benzothiazinones Are Suicide Inhibitors of Mycobacterial Decaprenylphosphoryl-β-d-ribofuranose 2′-Oxidase DprE1. J. Am. Chem. Soc. 2012, 134, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Comprehensive identification of conditionally essential genes in mycobacteria. Proc. Natl. Acad. Sci. USA 2001, 98, 12712–12717. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; van der Sar, A.M.; Raadsen, S.A.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef] [PubMed]
- The Working Group for New TB Drugs OPC-167832. Available online: https://www.newtbdrugs.org/pipeline/compound/opc-167832 (accessed on 6 June 2019).
- Shirude, P.S.; Shandil, R.; Sadler, C.; Naik, M.; Hosagrahara, V.; Hameed, S.; Shinde, V.; Bathula, C.; Humnabadkar, V.; Kumar, N.; et al. Azaindoles: Noncovalent DprE1 Inhibitors from Scaffold Morphing Efforts, Kill Mycobacterium tuberculosis and Are Efficacious in Vivo. J. Med. Chem. 2013, 56, 9701–9708. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Shandil, R.K.; Manjunatha, M.R.; Sadler, C.; Panda, M.; Panduga, V.; Reddy, J.; Saralaya, R.; Nanduri, R.; Ambady, A.; et al. Lead Optimization of 1,4-Azaindoles as Antimycobacterial Agents. J. Med. Chem. 2014, 57, 5728–5737. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, M.; Shandil, R.; Manjunatha, M.R.; Solapure, S.; Ramachandran, V.; Kumar, N.; Saralaya, R.; Panduga, V.; Reddy, J.; KR, P.; et al. 1,4-Azaindole, a Potential Drug Candidate for Treatment of Tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 5325–5331. [Google Scholar] [CrossRef]
- Paulsen, I.T.; Brown, M.H.; Skurray, R.A. Proton-dependent multidrug efflux systems. Microbiol. Rev. 1996, 60, 575–608. [Google Scholar]
- Putman, M.; van Veen, H.W.; Konings, W.N. Molecular properties of bacterial multidrug transporters. Microbiol. Mol. Biol. Rev. 2000, 64, 672–693. [Google Scholar] [CrossRef]
- Nikaido, H.; Takatsuka, Y. Mechanisms of RND multidrug efflux pumps. Biochim. Biophys. Acta 2009, 1794, 769–781. [Google Scholar] [CrossRef]
- Tullius, M.V.; Harmston, C.A.; Owens, C.P.; Chim, N.; Morse, R.P.; McMath, L.M.; Iniguez, A.; Kimmey, J.M.; Sawaya, M.R.; Whitelegge, J.P.; et al. Discovery and characterization of a unique mycobacterial heme acquisition system. Proc. Natl. Acad. Sci. USA 2011, 108, 5051–5056. [Google Scholar] [CrossRef] [PubMed]
- Tahlan, K.; Wilson, R.; Kastrinsky, D.B.; Arora, K.; Nair, V.; Fischer, E.; Barnes, S.W.; Walker, J.R.; Alland, D.; Barry, C.E.; et al. SQ109 Targets MmpL3, a Membrane Transporter of Trehalose Monomycolate Involved in Mycolic Acid Donation to the Cell Wall Core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Protopopova, M.; Hanrahan, C.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Einck, L.; Nacy, C.A. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J. Antimicrob. Chemother. 2005, 56, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.E.; Protopopova, M.; Crooks, E.; Slayden, R.A.; Terrot, M.; Barry, C.E. Combinatorial lead optimization of [1,2]-diamines based on ethambutol as potential antituberculosis preclinical candidates. J. Comb. Chem. 2003, 5, 172–187. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Tomaszewski, J.E.; Hanrahan, C.; Coward, L.; Noker, P.; Gorman, G.; Nikonenko, B.; Protopopova, M. Pharmacodynamics and pharmacokinetics of SQ109, a new diamine-based antitubercular drug. Br. J. Pharmacol. 2005, 144, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Noker, P.E.; Coward, L.; Gorman, G.S.; Protopopova, M.; Tomaszewski, J.E. Interspecies pharmacokinetics and in vitro metabolism of SQ109. Br. J. Pharmacol. 2006, 147, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Nikonenko, B.V.; Protopopova, M.; Samala, R.; Einck, L.; Nacy, C.A. Drug therapy of experimental tuberculosis (TB): Improved outcome by combining SQ109, a new diamine antibiotic, with existing TB drugs. Antimicrob. Agents Chemother. 2007, 51, 1563–1565. [Google Scholar] [CrossRef]
- Barry, C.E.; Lee, R.E.; Mdluli, K.; Sampson, A.E.; Schroeder, B.G.; Slayden, R.A.; Yuan, Y. Mycolic acids: Structure, biosynthesis and physiological functions. Prog. Lipid Res. 1998, 37, 143–179. [Google Scholar] [CrossRef]
- Minnikin, D.E.; Kremer, L.; Dover, L.G.; Besra, G.S. The methyl-branched fortifications of Mycobacterium tuberculosis. Chem. Biol. 2002, 9, 545–553. [Google Scholar] [CrossRef]
- Belisle, J.T.; Vissa, V.D.; Sievert, T.; Takayama, K.; Brennan, P.J.; Besra, G.S. Role of the major antigen of Mycobacterium tuberculosis in cell wall biogenesis. Science 1997, 276, 1420–1422. [Google Scholar] [CrossRef]
- Jackson, M.; Raynaud, C.; Lanéelle, M.A.; Guilhot, C.; Laurent-Winter, C.; Ensergueix, D.; Gicquel, B.; Daffé, M. Inactivation of the antigen 85C gene profoundly affects the mycolate content and alters the permeability of the Mycobacterium tuberculosis cell envelope. Mol. Microbiol. 1999, 31, 1573–1587. [Google Scholar] [CrossRef]
- Li, W.; Upadhyay, A.; Fontes, F.L.; North, E.J.; Wang, Y.; Crans, D.C.; Grzegorzewicz, A.E.; Jones, V.; Franzblau, S.G.; Lee, R.E.; et al. Novel Insights into the Mechanism of Inhibition of MmpL3, a Target of Multiple Pharmacophores in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 6413–6423. [Google Scholar] [CrossRef]
- Li, W.; Obregón-Henao, A.; Wallach, J.B.; North, E.J.; Lee, R.E.; Gonzalez-Juarrero, M.; Schnappinger, D.; Jackson, M. Therapeutic Potential of the Mycobacterium tuberculosis Mycolic Acid Transporter, MmpL3. Antimicrob. Agents Chemother. 2016, 60, 5198–5207. [Google Scholar] [CrossRef]
- Li, K.; Schurig-Briccio, L.A.; Feng, X.; Upadhyay, A.; Pujari, V.; Lechartier, B.; Fontes, F.L.; Yang, H.; Rao, G.; Zhu, W.; et al. Multitarget drug discovery for tuberculosis and other infectious diseases. J. Med. Chem. 2014, 57, 3126–3129. [Google Scholar] [CrossRef]
- Domenech, P.; Reed, M.B.; Barry, C.E. Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance. Infect. Immun. 2005, 73, 3492–3501. [Google Scholar] [CrossRef]
- Stover, C.K.; Warrener, P.; VanDevanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 2006, 3, 2131–2144. [Google Scholar] [CrossRef]
- Manjunatha, U.H.; Boshoff, H.; Dowd, C.S.; Zhang, L.; Albert, T.J.; Norton, J.E.; Daniels, L.; Dick, T.; Pang, S.S.; Barry III, C.E. Identification of a Nitroimidazo-Oxazine-Specific Protein Involved in PA-824 Resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2006, 103, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Haver, H.L.; Chua, A.; Ghode, P.; Lakshminarayana, S.B.; Singhal, A.; Mathema, B.; Wintjens, R.; Bifani, P. Mutations in genes for the F420 biosynthetic pathway and a nitroreductase enzyme are the primary resistance determinants in spontaneous in vitro-selected PA-824-resistant mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 5316–5323. [Google Scholar] [CrossRef] [PubMed]
- Global Alliance for TB Drug Development. Handbook of Anti-Tuberculosis Agents; Elsevier: Amsterdam, The Netherlands, 2008; PA-824; pp. 134–136. [Google Scholar]
- Tyagi, S.; Nuermberger, E.; Yoshimatsu, T.; Williams, K.; Rosenthal, I.; Lounis, N.; Bishai, W.; Grosset, J. Bactericidal activity of the nitroimidazopyran PA-824 in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2005, 49, 2289–2293. [Google Scholar] [CrossRef]
- Liu, Y.; Fujiwara, M.; Hariguchi, N.; Matsumoto, M.; Kawasaki, M. Mechanisms of resistance to delamanid, a drug for Mycobacterium tuberculosis. Tuberculosis 2017, 108, 186–194. [Google Scholar]
- Global Alliance for TB Drug Development. Handbook of Anti-Tuberculosis Agents; Elsevier: Amsterdam, The Netherlands, 2008; OPC-67683; pp. 112–116. [Google Scholar]
- Machado, D.; Girardini, M.; Viveiros, M.; Pieroni, M. Challenging the drug-likeness dogma for new drug discovery in Tuberculosis. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Wessels, H.J.C.T.; de Almeida, N.M.; Kartal, B.; Keltjens, J.T. Bacterial Electron Transfer Chains Primed by Proteomics. Adv. Microb. Physiol. 2016, 68, 219–352. [Google Scholar] [PubMed]
- Iqbal, I.; Bajeli, S.; Akela, A.; Kumar, A. Bioenergetics of Mycobacterium: An Emerging Landscape for Drug Discovery. Pathogens 2018, 7, 24. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Lill, H.; Bald, D. ATP synthase in mycobacteria: Special features and implications for a function as drug target. Biochim. Biophys. Acta 2014, 1837, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E.; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Diez, M.; Zimmermann, B.; Börsch, M.; König, M.; Schweinberger, E.; Steigmiller, S.; Reuter, R.; Felekyan, S.; Kudryavtsev, V.; Seidel, C.A.M.; et al. Proton-powered subunit rotation in single membrane-bound F0F1-ATP synthase. Nat. Struct. Mol. Biol. 2004, 11, 135–141. [Google Scholar] [CrossRef]
- Lu, P.; Villellas, C.; Koul, A.; Andries, K.; Lill, H.; Bald, D. The ATP synthase inhibitor bedaquiline interferes with small-molecule efflux in Mycobacterium smegmatis. J. Antibiot. 2014, 67, 835–837. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.H.; Neefs, J.M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Pethe, K.; Roquet-Banères, F.; Kremer, L.; Herrmann, J.-L.; Dupont, C.; Viljoen, A. Bedaquiline Inhibits the ATP Synthase in Mycobacterium abscessus and Is Effective in Infected Zebrafish. Antimicrob. Agents Chemother. 2017, 61, 1–15. [Google Scholar]
- Hards, K.; Robson, J.R.; Berney, M.; Shaw, L.; Bald, D.; Koul, A.; Andries, K.; Cook, G.M. Bactericidal mode of action of bedaquiline. J. Antimicrob. Chemother. 2015, 70, 2028–2037. [Google Scholar] [CrossRef] [PubMed]
- Kundu, S.; Biukovic, G.; Grüber, G.; Dick, T. Bedaquiline Targets the ε Subunit of Mycobacterial F-ATP Synthase. Antimicrob. Agents Chemother. 2016, 60, 6977–6979. [Google Scholar] [CrossRef] [PubMed]
- Basak, S.; Rao, S.P.S.; Rishikesan, S.; Manimekalai, M.S.S.; Dick, T.; Grüber, G.; Roessle, M.; Hunke, C.; Biuković, G. Variations of Subunit ε of the Mycobacterium tuberculosis F1Fo ATP Synthase and a Novel Model for Mechanism of Action of the Tuberculosis Drug TMC207. Antimicrob. Agents Chemother. 2012, 57, 168–176. [Google Scholar]
- Koul, A.; Dendouga, N.; Vergauwen, K.; Molenberghs, B.; Vranckx, L.; Willebrords, R.; Ristic, Z.; Lill, H.; Dorange, I.; Guillemont, J.; et al. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 2007. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.V.A.; Anthony, R.M.; Bañuls, A.-L.; Nguyen, T.V.A.; Vu, D.H.; Alffenaar, J.-W.C. Bedaquiline Resistance: Its Emergence, Mechanism, and Prevention. Clin. Infect. Dis. 2018, 66, 1625–1630. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, H.S.; Tong, A.S.T.; Choi, P.J.; Blaser, A.; Conole, D.; Franzblau, S.G.; Lotlikar, M.U.; Cooper, C.B.; Upton, A.M.; Denny, W.A.; et al. 3,5-Dialkoxypyridine analogues of bedaquiline are potent antituberculosis agents with minimal inhibition of the hERG channel. Bioorg. Med. Chem. 2019, 27, 1292–1307. [Google Scholar] [CrossRef] [PubMed]
- Tantry, S.J.; Markad, S.D.; Shinde, V.; Bhat, J.; Balakrishnan, G.; Gupta, A.K.; Ambady, A.; Raichurkar, A.; Kedari, C.; Sharma, S.; et al. Discovery of Imidazo[1,2-a]pyridine Ethers and Squaramides as Selective and Potent Inhibitors of Mycobacterial Adenosine Triphosphate (ATP) Synthesis. J. Med. Chem. 2017, 60, 1379–1399. [Google Scholar] [CrossRef] [PubMed]
- Alonso, S.; Cho, S.-N.; Oh, S.; Chua, A.; Jung, J.; Jang, J.; Bifani, P.; Kempf, M.; Kim, Y.M.; Lenaerts, A.J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157–1160. [Google Scholar]
- Kang, S.; Kim, R.Y.; Seo, M.J.; Lee, S.; Kim, Y.M.; Seo, M.; Seo, J.J.; Ko, Y.; Choi, I.; Jang, J.; et al. Lead optimization of a novel series of imidazo[1,2-a]pyridine amides leading to a clinical candidate (Q203) as a multi- and extensively-drug- resistant anti-tuberculosis agent. J. Med. Chem. 2014, 57, 5293–5305. [Google Scholar] [CrossRef]
- Bown, L.; Srivastava, S.K.; Piercey, B.M.; McIsaac, C.K.; Tahlan, K. Mycobacterial Membrane Proteins QcrB and AtpE: Roles in Energetics, Antibiotic Targets, and Associated Mechanisms of Resistance. J. Membr. Biol. 2018, 251, 105–117. [Google Scholar] [CrossRef]
- Crofts, A.R. The Cytochrome bc1 Complex: Function in the Context of Structure. Ann. Rev. Physiol. 2004, 66, 689–733. [Google Scholar] [CrossRef] [PubMed]
- Foo, C.S.; Lupien, A.; Kienle, M.; Vocat, A.; Benjak, A.; Sommer, R.; Lamprecht, D.A.; Steyn, A.J.C.; Pethe, K.; Piton, J.; et al. Arylvinylpiperazine amides, a new class of potent inhibitors targeting QcrB of mycobacterium tuberculosis. MBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, E.B.; Grabowska, A.D.; Cortes, T. Survey and summary: Translational regulation in mycobacteria and its implications for pathogenicity. Nucleic Acids Res. 2018, 46, 6950–6961. [Google Scholar] [CrossRef] [PubMed]
- Drlica, K. Mechanism of fluoroquinolone action. Curr. Opin. Microbiol. 1999, 2, 504–508. [Google Scholar] [CrossRef]
- Floss, H.G.; Yu, T.-W. Rifamycin: Mode of Action, Resistance, and Biosynthesis. Chem. Rev. 2005, 105, 621–632. [Google Scholar] [CrossRef]
- Moazed, D.; Noller, H.F. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature 1987, 327, 389–394. [Google Scholar] [CrossRef]
- Chan, P.F.; Germe, T.; Bax, B.D.; Huang, J.; Thalji, R.K.; Bacqué, E.; Checchia, A.; Chen, D.; Cui, H.; Ding, X.; et al. Thiophene antibacterials that allosterically stabilize DNA-cleavage complexes with DNA gyrase. Proc. Natl. Acad. Sci. USA 2017, 114, E4492–E4500. [Google Scholar] [CrossRef]
- Locher, C.P.; Jones, S.M.; Hanzelka, B.L.; Perola, E.; Shoen, C.M.; Cynamon, M.H.; Ngwane, A.H.; Wiid, I.J.; van Helden, P.D.; Betoudji, F.; et al. A novel inhibitor of gyrase B is a potent drug candidate for treatment of tuberculosis and nontuberculosis mycobacterial infections. Antimicrob. Agents Chemother. 2015, 59, 1455–1465. [Google Scholar] [CrossRef]
- Kashyap, A.; Singh, P.K.; Silakari, O. Chemical classes targeting energy supplying GyrB domain of Mycobacterium tuberculosis. Tuberculosis 2018, 113, 43–54. [Google Scholar] [CrossRef]
- Ma, C.; Yang, X.; Lewis, P.J. Bacterial Transcription as a Target for Antibacterial Drug Development. Microbiol. Mol. Biol. Rev. 2016, 80, 139–160. [Google Scholar] [CrossRef]
- Lin, W.; Mandal, S.; Degen, D.; Liu, Y.; Ebright, Y.W.; Li, S.; Feng, Y.; Zhang, Y.; Mandal, S.; Jiang, Y.; et al. Structural Basis of Mycobacterium tuberculosis Transcription and Transcription Inhibition. Mol. Cell 2017, 66, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hernandez, V.; Rock, F.L.; Choi, W.; Mak, Y.S.L.; Mohan, M.; Mao, W.; Zhou, Y.; Easom, E.E.; Plattner, J.J.; et al. Discovery of a Potent and Specific M. tuberculosis Leucyl-tRNA Synthetase Inhibitor: (S)-3-(Aminomethyl)-4-chloro-7-(2-hydroxyethoxy)benzo[c][1,2]oxaborol-1(3H)-ol (GSK656). J. Med. Chem. 2017, 60, 8011–8026. [Google Scholar] [CrossRef] [PubMed]
- Palencia, A.; Li, X.; Bu, W.; Choi, W.; Ding, C.Z.; Easom, E.E.; Feng, L.; Hernandez, V.; Houston, P.; Liu, L.; et al. Discovery of Novel Oral Protein Synthesis Inhibitors of Mycobacterium tuberculosis That Target Leucyl-tRNA Synthetase. Antimicrob. Agents Chemother. 2016, 60, 6271–6280. [Google Scholar] [CrossRef] [PubMed]
- Soto, R.; Perez-Herran, E.; Rodriguez, B.; Duma, B.M.; Cacho-Izquierdo, M.; Mendoza-Losana, A.; Lelievre, J.; Aguirre, D.B.; Ballell, L.; Cox, L.R.; et al. Identification and characterization of aspartyl-tRNA synthetase inhibitors against Mycobacterium tuberculosis by an integrated whole-cell target-based approach. Sci. Rep. 2018, 8, 12664. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Lin, Y.; Li, D.; Gao, N.; Liu, C.; You, X.; Jiang, J.; Jiang, W.; Si, S. Identification of an anti-TB compound targeting the tyrosyl-tRNA synthetase. J. Antimicrob. Chemother. 2015, 70, 2287–2294. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Guidry, L.; Saini, V.; Hondalus, M.; Steyn, A.J.C.; Farhana, A. Redox homeostasis in mycobacteria: The key to tuberculosis control? Expert Rev. Mol. Med. 2011, 13, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Si, M.; Chen, K.; Zhang, B.; Xiao, H.; Zhao, C.; Shen, X.; Wei, D. Overexpression of Mycothiol Disulfide Reductase Enhances Corynebacterium glutamicum Robustness by Modulating Cellular Redox Homeostasis and Antioxidant Proteins under Oxidative Stress. Sci. Rep. 2016, 6, 1–14. [Google Scholar]
- Haywardi, D.; Wiid, I.; Van Helden, P. Differential expression of mycothiol pathway genes: Are they affected by antituberculosis drugs? IUBMB Life 2004, 56, 131–138. [Google Scholar] [CrossRef]
- Reductase, M.; Patel, M.P.; Blanchard, J.S. Expression, Purification, and Characterization of Mycobacterium tuberculosis Mycothione Reductase. Biochemistry 1999, 38, 11827–11833. [Google Scholar]
- Newton, G.L.; Buchmeier, N.; Fahey, R.C. Biosynthesis and Functions of Mycothiol, the Unique Protective Thiol of Actinobacteria. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. [Google Scholar] [CrossRef]
- Fan, F.; Blanchard, J.S. Toward the catalytic mechanism of a cysteine ligase (MshC) from Mycobacterium smegmatis: An enzyme involved in the biosynthetic pathway of mycothiol. Biochemistry 2009, 48, 7150–7159. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.P.; Blanchard, J.S. Mycobacterium tuberculosis Mycothione Reductase: pH Dependence of the Kinetic Parameters and Kinetic Isotope Effects. Biochemistry 2007, 40, 5119–5126. [Google Scholar] [CrossRef]
- Nkambule, C.M. Great South African molecules: The case for mycothiol. South African J. Chem. 2017, 70, 67–81. [Google Scholar] [CrossRef][Green Version]
- Holsclaw, C.M.; Muse, W.B.; Carroll, K.S.; Leary, J.A. Mass spectrometric analysis of mycothiol levels in wild-type and mycothiol disulfide reductase mutant Mycobacterium smegmatis. Int. J. Mass Spectrom. 2011. [Google Scholar] [CrossRef] [PubMed]
- Rawat, M.; Johnson, C.; Cadiz, V.; Av-Gay, Y. Comparative analysis of mutants in the mycothiol biosynthesis pathway in Mycobacterium smegmatis. Biochem. Biophys. Res. Commun. 2007, 363, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Patel, K.; Song, F.; Andreana, P.R. Synthesis of substrate analogues as potential inhibitors for Mycobacterium tuberculosis enzyme MshC. Carbohydr. Res. 2017, 453–454, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, A.; Mativandlela, S.P.N.; Binneman, B.; Fourie, P.B.; Hamilton, C.J.; Meyer, J.J.M.; van der Kooy, F.; Houghton, P.; Lall, N. Activity of 7-methyljuglone derivatives against Mycobacterium tuberculosis and as subversive substrates for mycothiol disulfide reductase. Bioorganic Med. Chem. 2007, 15, 7638–7646. [Google Scholar] [CrossRef] [PubMed]
- Cappoen, D.; Jacobs, J.; Nguyen Van, T.; Claessens, S.; Diels, G.; Anthonissen, R.; Einarsdottir, T.; Fauville, M.; Verschaeve, L.; Huygen, K.; et al. Straightforward palladium-mediated synthesis and biological evaluation of benzo[j]phenanthridine-7,12-diones as anti-tuberculosis agents. Eur. J. Med. Chem. 2012, 48, 57–68. [Google Scholar] [CrossRef]
- Smets, R.J.; Torfs, E.; Lemière, F.; Cos, P.; Cappoen, D.; Abbaspour Tehrani, K. Synthesis and antitubercular activity of 1- and 3-substituted benzo[g]isoquinoline-5,10-diones. Org. Biomol. Chem. 2019, 17, 2923–2939. [Google Scholar] [CrossRef]
- Cappoen, D.; Claes, P.; Jacobs, J.; Anthonissen, R.; Mathys, V.; Verschaeve, L.; Huygen, K.; Kimpe, N. De 1,2,3,4,8,9,10,11-Octahydrobenzo[j]phenanthridine-7,12-diones as new leads against Mycobacterium tuberculosis. J. Med. Chem. 2014, 57, 2895–2907. [Google Scholar] [CrossRef]
- Slayden, R.A.; Dawson, C.C.; Cummings, J.E. Toxin-antitoxin systems and regulatory mechanisms in Mycobacterium tuberculosis. Pathog. Dis. 2018, 76, fty039. [Google Scholar] [CrossRef] [PubMed]
- Freire, D.M.; Gutierrez, C.; Garza-Garcia, A.; Grabowska, A.D.; Sala, A.J.; Ariyachaokun, K.; Panikova, T.; Beckham, K.S.H.; Colom, A.; Pogenberg, V.; et al. An NAD+ Phosphorylase Toxin Triggers Mycobacterium tuberculosis Cell Death. Mol. Cell 2019, 73, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Cumming, B.M.; Mao, C.; Zhu, Y.; Lu, P.; Steyn, A.J.C.; Chen, S.; Hu, Y. RbpA and σ B association regulates polyphosphate levels to modulate mycobacterial isoniazid-tolerance. Mol. Microbiol. 2018, 108, 627–640. [Google Scholar] [CrossRef]
- Davis, E.O.; Dullaghan, E.M.; Rand, L. Definition of the mycobacterial SOS box and use to identify LexA-regulated genes in Mycobacterium tuberculosis. J. Bacteriol. 2002, 184, 3287–3295. [Google Scholar] [CrossRef] [PubMed]
- Boshoff, H.I.M.; Reed, M.B.; Barry, C.E.; Mizrahi, V. DnaE2 polymerase contributes to in vivo survival and the emergence of drug resistance in Mycobacterium tuberculosis. Cell 2003, 113, 183–193. [Google Scholar] [CrossRef]
- Warner, D.F.; Venclovas, C.; Ndwandwe, D.E.; Abrahams, G.L.; Machowski, E.E.; Kana, B.D.; Mizrahi, V. Essential roles for imuA’- and imuB-encoded accessory factors in DnaE2-dependent mutagenesis in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 13093–13098. [Google Scholar] [CrossRef] [PubMed]
- Jadaun, A.; Sudhakar, D.R.; Subbarao, N.; Dixit, A. In silico screening for novel inhibitors of DNA polymerase III alpha subunit of Mycobacterium tuberculosis (MtbDnaE2, H37Rv). PLoS ONE 2015, 10, 1–15. [Google Scholar]
- Master, S.S.; Rampini, S.K.; Davis, A.S.; Keller, C.; Ehlers, S.; Springer, B.; Timmins, G.S.; Sander, P.; Deretic, V. Mycobacterium tuberculosis Prevents Inflammasome Activation. Cell Host Microbe 2008. [Google Scholar] [CrossRef]
- Ferraris, D.M.; Sbardella, D.; Petrera, A.; Marini, S.; Amstutz, B.; Coletta, M.; Sander, P.; Rizzi, M. Crystal structure of mycobacterium tuberculosis zinc-dependent metalloprotease-1 (Zmp1), a metalloprotease involved in pathogenicity. J. Biol. Chem. 2011, 286, 32475–32482. [Google Scholar] [CrossRef]
- Muttucumaru, D.G.N.; Smith, D.A.; McMinn, E.J.; Reese, V.; Coler, R.N.; Parish, T. Mycobacterium tuberculosis Rv0198c, a putative matrix metalloprotease is involved in pathogenicity. Tuberculosis 2011, 91, 111–116. [Google Scholar] [CrossRef]
- Johansen, P.; Fettelschoss, A.; Amstutz, B.; Selchow, P.; Waeckerle-Men, Y.; Keller, P.; Deretic, V.; Held, L.; Kündig, T.M.; Böttger, E.C.; et al. Relief from Zmp1-Mediated Arrest of Phagosome Maturation Is Associated with Facilitated Presentation and Enhanced Immunogenicity of Mycobacterial Antigens. Clin. Vaccine Immunol. 2011, 18, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Moraca, F.; Deodato, D.; Ferraris, D.M.; Selchow, P.; Sander, P.; Rizzi, M.; Botta, M. Discovery of the first potent and selective Mycobacterium tuberculosis Zmp1 inhibitor. Bioorganic Med. Chem. Lett. 2014, 24, 2508–2511. [Google Scholar] [CrossRef] [PubMed]
- Subhedar, D.D.; Shaikh, M.H.; Nawale, L.; Yeware, A.; Sarkar, D.; Khan, F.A.K.; Sangshetti, J.N.; Shingate, B.B. Novel tetrazoloquinoline-rhodanine conjugates: Highly efficient synthesis and biological evaluation. Bioorganic Med. Chem. Lett. 2016, 26, 2278–2283. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Brindisi, M.; Vallone, A.; Butini, S.; Campiani, G.; Nannicini, C.; Giuliani, G.; Anzini, M.; Lamponi, S.; Giorgi, G.; et al. Development of Potent Inhibitors of the Mycobacterium tuberculosis Virulence Factor Zmp1 and Evaluation of Their Effect on Mycobacterial Survival inside Macrophages. ChemMedChem 2018, 13, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.S.; Hafner, R. Advancing host-directed therapy for tuberculosis. Nat. Rev. Immunol. 2015, 15, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Palucci, I.; Delogu, G. Host Directed Therapies for Tuberculosis: Futures Strategies for an Ancient Disease. Chemotherapy 2018, 63, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Van Crevel, R.; Ottenhoff, T.H.M.; Van der Meer, J.W.M. Innate immunity to Mycobacterium tuberculosis. Clin. Microbiol. Rev. 2002, 15, 294–309. [Google Scholar]
- Paik, S.; Kim, J.K.; Chung, C.; Jo, E.K. Autophagy: A new strategy for host-directed therapy of tuberculosis. Virulence 2018. [Google Scholar] [CrossRef]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 2018, 17, 35. [Google Scholar] [CrossRef]
- Kolloli, A.; Subbian, S. Host-Directed Therapeutic Strategies for Tuberculosis. Front. Med. 2017, 4. [Google Scholar] [CrossRef]
- Chakravarty, S.D.; Zhu, G.; Tsai, M.C.; Mohan, V.P.; Marino, S.; Kirschner, D.E.; Huang, L.; Flynn, J.; Chan, J. Tumor necrosis factor blockade in chronic murine tuberculosis enhances granulomatous inflammation and disorganizes granulomas in the lungs. Infect. Immun. 2008, 76, 916–926. [Google Scholar] [CrossRef] [PubMed]
- Skerry, C.; Harper, J.; Klunk, M.; Bishai, W.R.; Jain, S.K. Adjunctive TNF inhibition with standard treatment enhances bacterial clearance in a murine model of necrotic TB granulomas. PLoS ONE 2012, 7, e39680. [Google Scholar] [CrossRef] [PubMed]
- Bourigault, M.-L.; Vacher, R.; Rose, S.; Olleros, M.L.; Janssens, J.-P.; Quesniaux, V.F.; Garcia, I. Tumor necrosis factor neutralization combined with chemotherapy enhances Mycobacterium tuberculosis clearance and reduces lung pathology. Am. J. Clin. Exp. Immunol. 2013, 2, 124–134. [Google Scholar] [PubMed]
- Wallis, R.S.; Kyambadde, P.; Johnson, J.L.; Horter, L.; Kittle, R.; Pohle, M.; Ducar, C.; Millard, M.; Mayanja-Kizza, H.; Whalen, C.; et al. A study of the safety, immunology, virology, and microbiology of adjunctive etanercept in HIV-1-associated tuberculosis. AIDS 2004, 18, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Oehlers, S.H.; Cronan, M.R.; Scott, N.R.; Thomas, M.I.; Okuda, K.S.; Walton, E.M.; Beerman, R.W.; Crosier, P.S.; Tobin, D.M. Interception of host angiogenic signalling limits mycobacterial growth. Nature 2015, 517, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Datta, M.; Via, L.E.; Kamoun, W.S.; Liu, C.; Chen, W.; Seano, G.; Weiner, D.M.; Schimel, D.; England, K.; Martin, J.D.; et al. Anti-vascular endothelial growth factor treatment normalizes tuberculosis granuloma vasculature and improves small molecule delivery. Proc. Natl. Acad. Sci. USA 2015, 112, 1827–1832. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Bento, C.F.; Empadinhas, N.; Mendes, V. Autophagy in the Fight Against Tuberculosis. DNA Cell Biol. 2015, 34, 228–242. [Google Scholar] [CrossRef]
- Lam, A.; Prabhu, R.; Gross, C.M.; Riesenberg, L.A.; Singh, V.; Aggarwal, S. Role of apoptosis and autophagy in tuberculosis. Am. J. Physiol. Cell. Mol. Physiol. 2017, 313, L218–L229. [Google Scholar] [CrossRef]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef]
- Schiebler, M.; Brown, K.; Hegyi, K.; Newton, S.M.; Renna, M.; Hepburn, L.; Klapholz, C.; Coulter, S.; Obregón-Henao, A.; Henao Tamayo, M.; et al. Functional drug screening reveals anticonvulsants as enhancers of mTOR-independent autophagic killing of Mycobacterium tuberculosis through inositol depletion. EMBO Mol. Med. 2015, 7, 127–139. [Google Scholar] [CrossRef]
- Stanley, S.A.; Barczak, A.K.; Silvis, M.R.; Luo, S.S.; Sogi, K.; Vokes, M.; Bray, M.-A.; Carpenter, A.E.; Moore, C.B.; Siddiqi, N.; et al. Identification of host-targeted small molecules that restrict intracellular Mycobacterium tuberculosis growth. PLoS Pathog. 2014, 10, e1003946. [Google Scholar] [CrossRef]
- Liu, P.T.; Stenger, S.; Li, H.; Wenzel, L.; Tan, B.H.; Krutzik, S.R.; Ochoa, M.T.; Schauber, J.; Wu, K.; Meinken, C.; et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science 2006, 311, 1770–1773. [Google Scholar] [CrossRef] [PubMed]
- Yuk, J.-M.; Shin, D.-M.; Lee, H.-M.; Yang, C.-S.; Jin, H.S.; Kim, K.-K.; Lee, Z.-W.; Lee, S.-H.; Kim, J.-M.; Jo, E.-K. Vitamin D3 induces autophagy in human monocytes/macrophages via cathelicidin. Cell Host Microbe 2009, 6, 231–243. [Google Scholar] [CrossRef]
- Chun, R.F.; Adams, J.S.; Hewison, M. Immunomodulation by vitamin D: Implications for TB. Expert Rev. Clin. Pharmacol. 2011, 4, 583–591. [Google Scholar] [CrossRef]
- Harishankar, M.; Afsal, K.; Banurekha, V.V.; Meenakshi, N.; Selvaraj, P. 1,25-Dihydroxy vitamin D3 downregulates pro-inflammatory cytokine response in pulmonary tuberculosis. Int. Immunopharmacol. 2014, 23, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, P.; Harishankar, M.; Singh, B.; Banurekha, V.V.; Jawahar, M.S. Effect of vitamin D3 on chemokine expression in pulmonary tuberculosis. Cytokine 2012, 60, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Chandra, G.; Selvaraj, P.; Jawahar, M.S.; Banurekha, V.V.; Narayanan, P.R. Effect of vitamin D3 on phagocytic potential of macrophages with live Mycobacterium tuberculosis and lymphoproliferative response in pulmonary tuberculosis. J. Clin. Immunol. 2004, 24, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Prabhu Anand, S.; Selvaraj, P.; Narayanan, P.R. Effect of 1,25 dihydroxyvitamin D3 on intracellular IFN-gamma and TNF-alpha positive T cell subsets in pulmonary tuberculosis. Cytokine 2009, 45, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Tobin, D.M.; Roca, F.J.; Oh, S.F.; McFarland, R.; Vickery, T.W.; Ray, J.P.; Ko, D.C.; Zou, Y.; Bang, N.D.; Chau, T.T.H.; et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell 2012, 148, 434–446. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Divangahi, M.; Gan, H.; Shin, D.S.J.; Hong, S.; Lee, D.M.; Serhan, C.N.; Behar, S.M.; Remold, H.G. Lipid mediators in innate immunity against tuberculosis: Opposing roles of PGE2 and LXA4 in the induction of macrophage death. J. Exp. Med. 2008, 205, 2791–2801. [Google Scholar] [CrossRef] [PubMed]
- Morris, T.; Stables, M.; Hobbs, A.; de Souza, P.; Colville-Nash, P.; Warner, T.; Newson, J.; Bellingan, G.; Gilroy, D.W. Effects of low-dose aspirin on acute inflammatory responses in humans. J. Immunol. 2009, 183, 2089–2096. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.T.; Denkin, S.M.; Zhang, Y. Aspirin and ibuprofen enhance pyrazinamide treatment of murine tuberculosis. J. Antimicrob. Chemother. 2007, 59, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Dutta, N.K.; Mazumdar, K.; Dastidar, S.G.; Park, J.-H. Activity of diclofenac used alone and in combination with streptomycin against Mycobacterium tuberculosis in mice. Int. J. Antimicrob. Agents 2007, 30, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Vilaplana, C.; Marzo, E.; Tapia, G.; Diaz, J.; Garcia, V.; Cardona, P.-J. Ibuprofen therapy resulted in significantly decreased tissue bacillary loads and increased survival in a new murine experimental model of active tuberculosis. J. Infect. Dis. 2013, 208, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, B.; Fu, L.; Zhu, H.; Guo, S.; Huang, H.; Yin, D.; Zhang, Y.; Lu, Y. In vitro and in vivo activities of the riminophenazine TBI-166 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2019. [Google Scholar] [CrossRef] [PubMed]
- The Working Group for New TB Drugs TBI-223. Available online: https://www.newtbdrugs.org/pipeline/compound/tbi-223 (accessed on 6 June 2019).
- Gordeev, M.F.; Yuan, Z.Y. New potent antibacterial oxazolidinone (MRX-I) with an improved class safety profile. J. Med. Chem. 2014, 57, 4487–4497. [Google Scholar] [CrossRef] [PubMed]
- Eckburg, P.B.; Ge, Y.; Hafkin, B. Single- and Multiple-Dose Study To Determine the Safety, Tolerability, Pharmacokinetics, and Food Effect of Oral MRX-I versus Linezolid in Healthy Adult Subjects. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef]
- Shoen, C.; DeStefano, M.; Hafkin, B.; Cynamon, M. In Vitro and In Vivo Activities of Contezolid (MRX-I) against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2018, 62. [Google Scholar] [CrossRef]
- Andrews, J. To be or not to be exclusive: The sutezolid story. Lancet. Glob. Heal. 2016, 4, e89–e90. [Google Scholar] [CrossRef]
- Wallis, R.S.; Jakubiec, W.; Kumar, V.; Bedarida, G.; Silvia, A.; Paige, D.; Zhu, T.; Mitton-Fry, M.; Ladutko, L.; Campbell, S.; et al. Biomarker-Assisted Dose Selection for Safety and Efficacy in Early Development of PNU-100480 for Tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Zong, Z.; Jing, W.; Shi, J.; Wen, S.; Zhang, T.; Huo, F.; Shang, Y.; Liang, Q.; Huang, H.; Pang, Y. Comparison of In Vitro Activity and MIC Distributions between the Novel Oxazolidinone Delpazolid and Linezolid against Multidrug-Resistant and Extensively Drug-Resistant Mycobacterium tuberculosis in China. Antimicrob. Agents Chemother. 2018, 62, e00165-18. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Choe, J.H.; Kim, Y.J.; Yang, C.-S.; Kwon, H.-J.; Jeong, J.; Kim, G.; Park, D.E.; Jo, E.-K.; Cho, Y.-L.; et al. Activity of LCB01-0371, a Novel Oxazolidinone, against Mycobacterium abscessus. Antimicrob. Agents Chemother. 2017, 61, e02752-16. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Targeted Pathway | Target | Relevant Compound (Class) |
|---|---|---|
| Cell wall biosynthesis | DprE1 | BTZ043 (BTZ) |
| DprE1 | PBTZ169 (PBTZ) | |
| DprE1 | TBA7371 (1,4-azaindole) | |
| DprE1 | OPC167832 (1,3-dihydrocarbostyril) | |
| MmpL3 | SQ109 (1,2-ethylenediamine) | |
| Cell wall biosynthesis and Protein synthesis | - | Pretomanid/PA-824 (nitroimidazole) |
| - | Delamanid (nitroimidazole) | |
| Oxidative phosphorylation | AtpE | BDQ (DARQ) |
| ATP synthase | Squaramides | |
| QcrB | Q203 (imidazopyridine) | |
| QcrB | Pyrrolo[3,4-c]pyridine-1,3(2H)-diones | |
| QcrB | 2-(Quinolin-4-yloxy)acetamides | |
| QcrB | AX-35 (arylvinylpiperazine amide) | |
| Protein synthesis | GyrA | Thiophenes |
| GyrB | SPR720 (aminobenzimidazole) | |
| DNA-dependent RNA polymerase | AAPs | |
| LeuRS | GSK656 (oxaborole) | |
| Bacillary redox homeostasis | MshC | Dequalinum |
| Mtr | Benzo[g]isoquinoline-5,10-diones | |
| Mtr | Benzo[j]phenanthridine-7,12-diones |
| Targeted Pathway | Target | Compound (Class) |
|---|---|---|
| Replication | DNAE2 | 6-anilino-1H-pyrimidine-2,4-diones |
| Phagosome maturation | Zmp1 | ZTB23(R) |
| Zmp1 | Arylidene-rhodanines | |
| Zmp1 | N-(benzyloxy)-8-hydroxyquinoline-2-carboxamide |
| Targeted Pathway | Target | Compound (Class) |
|---|---|---|
| Granuloma formation | TNF-α | Etanercept |
| VEGF | Bevacizumab | |
| Autophagy | mTOR | Rapamycin |
| - | Carbamazepine | |
| - | Valproic acid | |
| EGFR | Gefitinib | |
| Cathelicidin (and others) biosynthesis | Vitamin D a | |
| Pro- and anti-inflammatory host responses | LXA4 production | Acetylsalicylic acid |
| COX-1; COX-2 | NSAIDs |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torfs, E.; Piller, T.; Cos, P.; Cappoen, D. Opportunities for Overcoming Mycobacterium tuberculosis Drug Resistance: Emerging Mycobacterial Targets and Host-Directed Therapy. Int. J. Mol. Sci. 2019, 20, 2868. https://doi.org/10.3390/ijms20122868
Torfs E, Piller T, Cos P, Cappoen D. Opportunities for Overcoming Mycobacterium tuberculosis Drug Resistance: Emerging Mycobacterial Targets and Host-Directed Therapy. International Journal of Molecular Sciences. 2019; 20(12):2868. https://doi.org/10.3390/ijms20122868
Chicago/Turabian StyleTorfs, Eveline, Tatiana Piller, Paul Cos, and Davie Cappoen. 2019. "Opportunities for Overcoming Mycobacterium tuberculosis Drug Resistance: Emerging Mycobacterial Targets and Host-Directed Therapy" International Journal of Molecular Sciences 20, no. 12: 2868. https://doi.org/10.3390/ijms20122868
APA StyleTorfs, E., Piller, T., Cos, P., & Cappoen, D. (2019). Opportunities for Overcoming Mycobacterium tuberculosis Drug Resistance: Emerging Mycobacterial Targets and Host-Directed Therapy. International Journal of Molecular Sciences, 20(12), 2868. https://doi.org/10.3390/ijms20122868