
Heterocyclic Iminoquinones and Quinones from the National Cancer Institute (NCI, USA) COMPARE Analysis


 , and
, and 
Abstract
1. Introduction
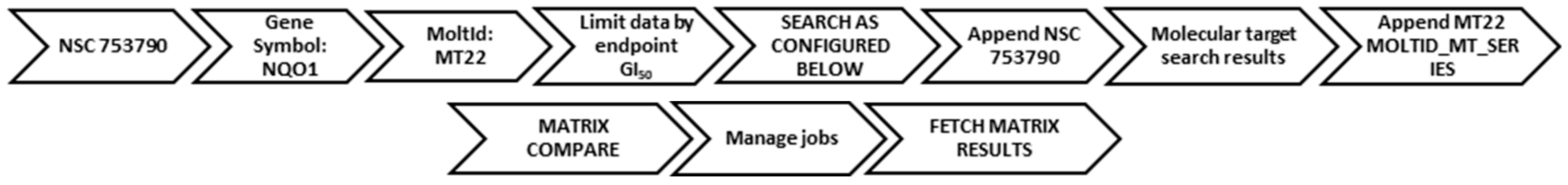
2. Compound Search Methods
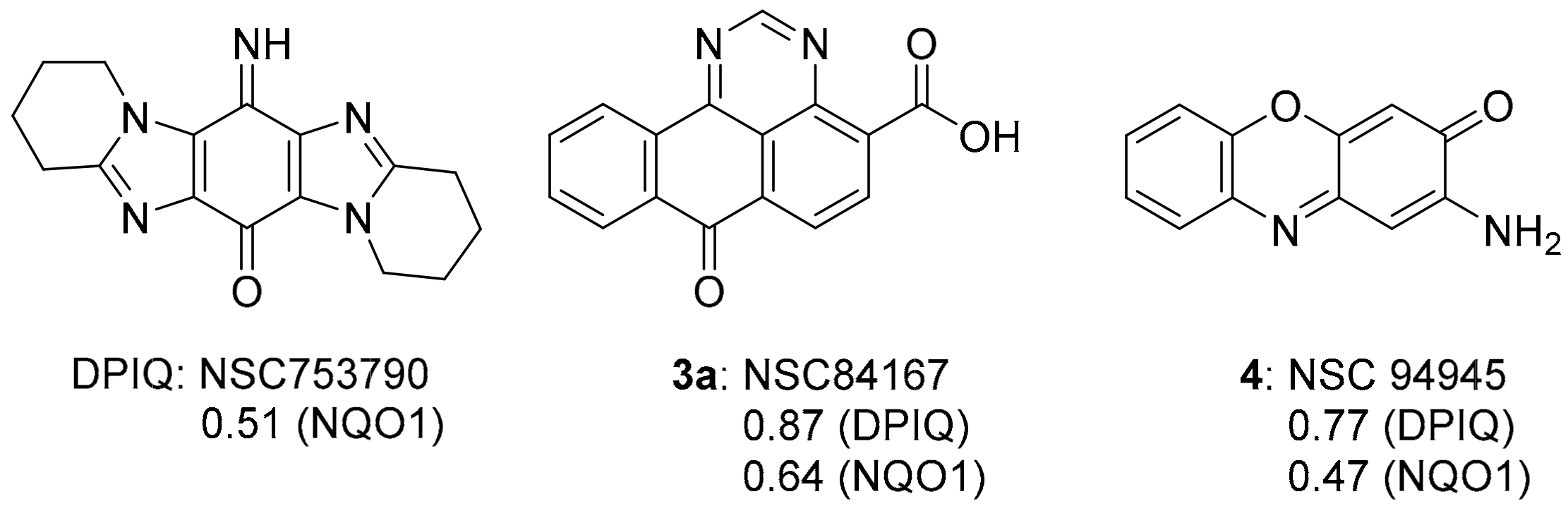
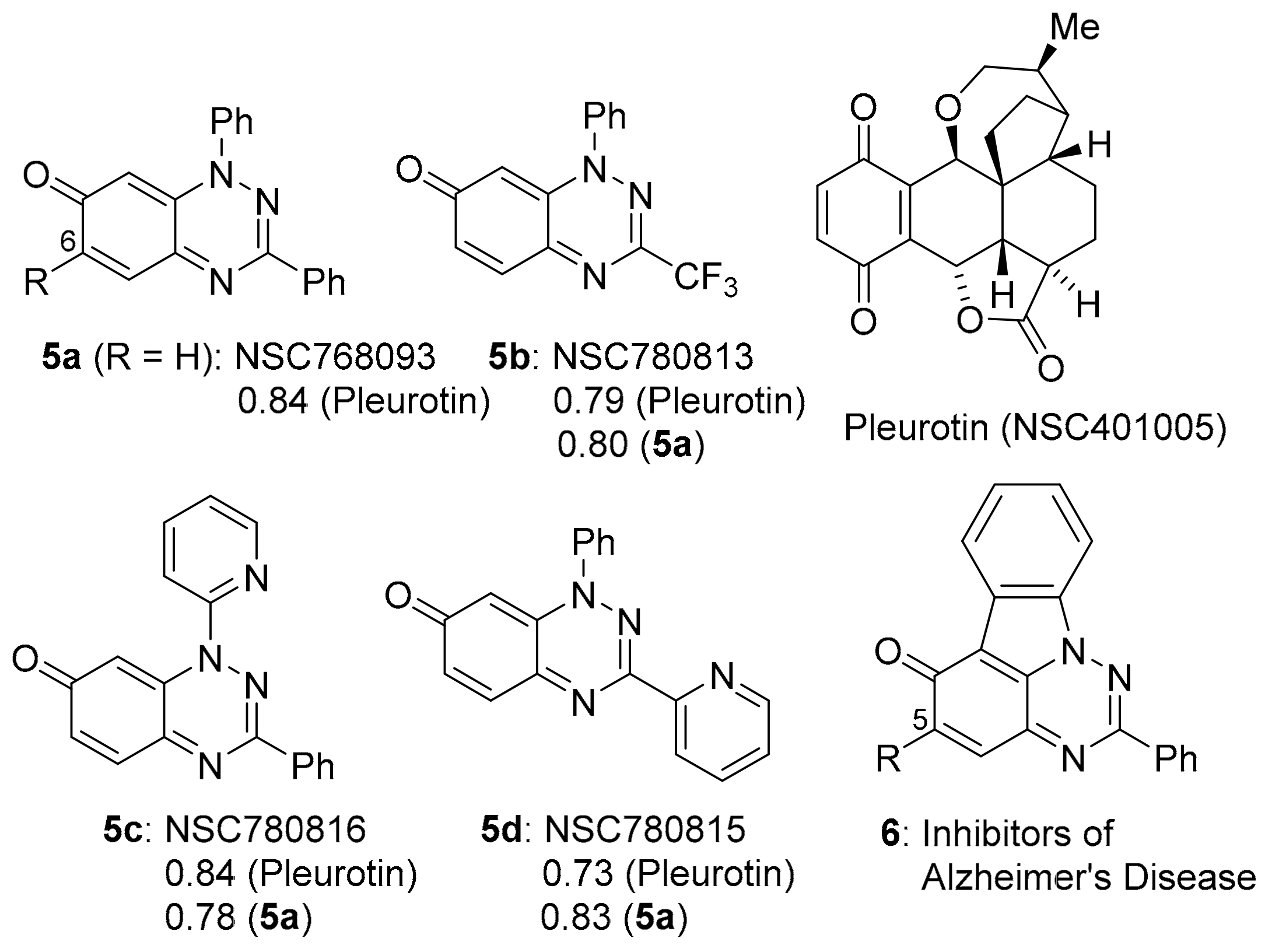
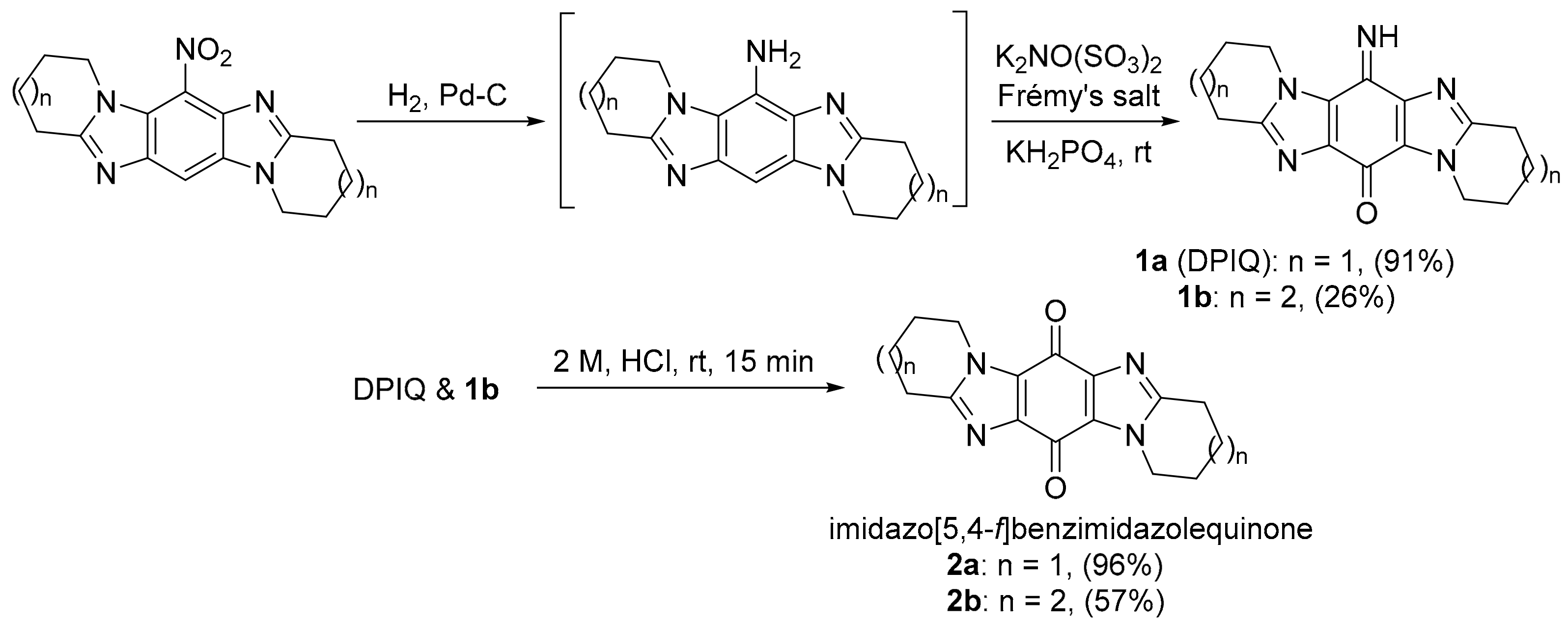
2.1. COMPARE Using DPIQ and 5a as the Seed
2.2. COMPARE Using Molecular Target Expression
3. Discussion
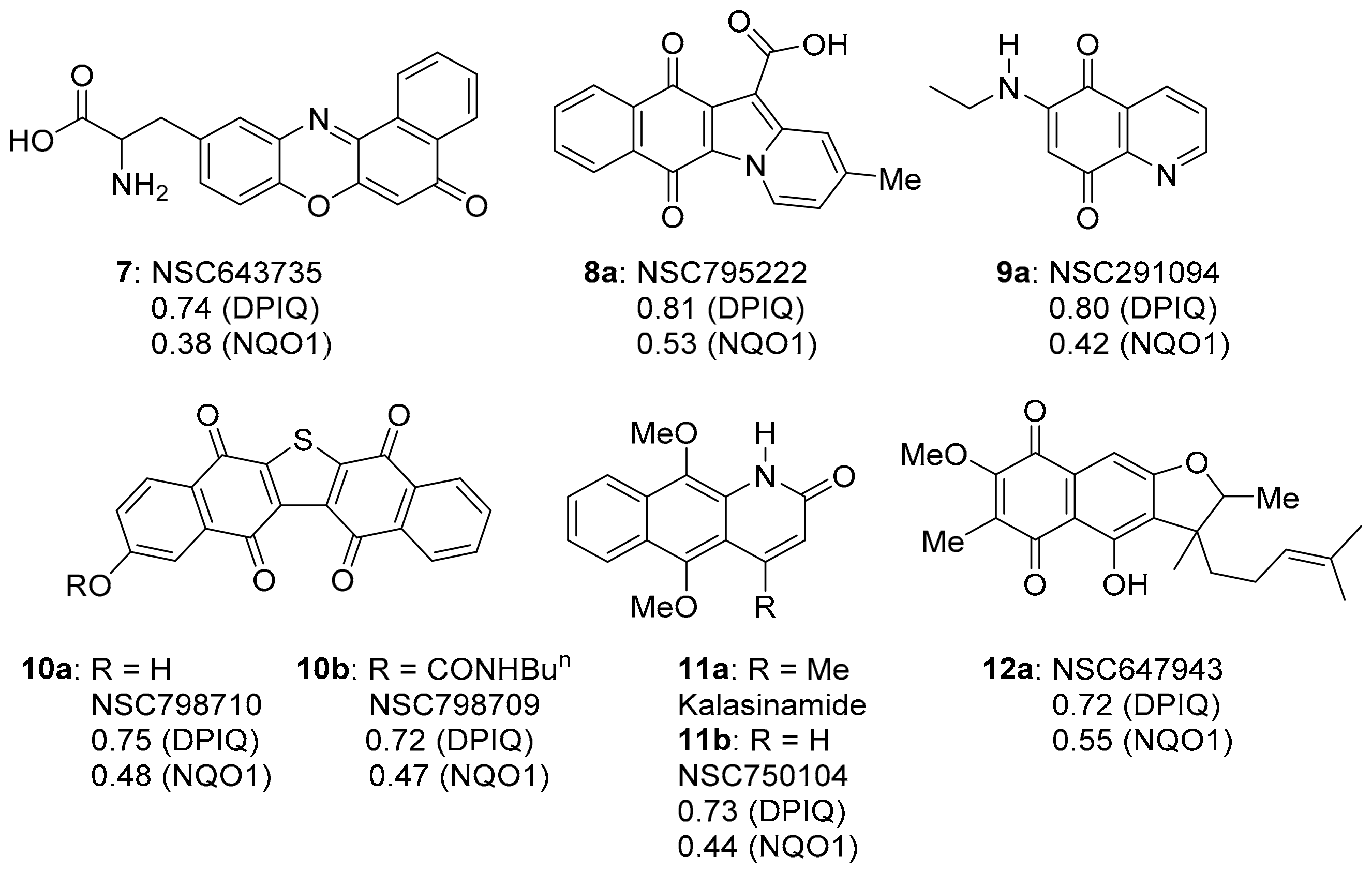
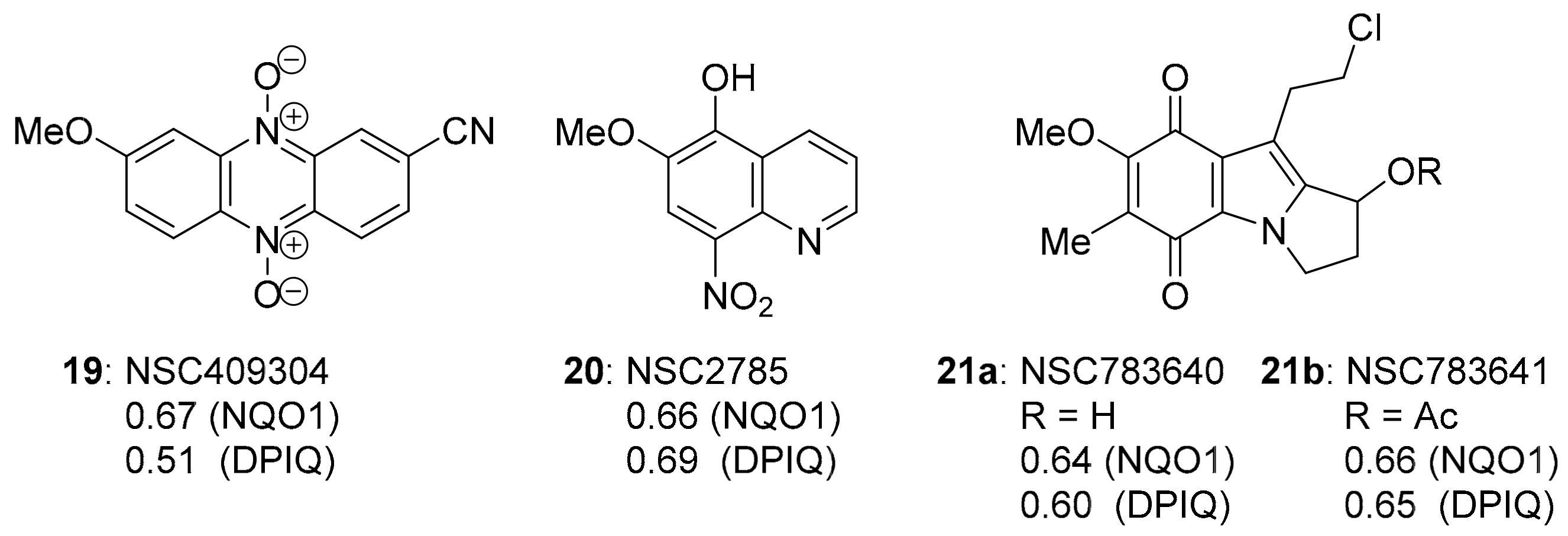
3.1. COMPARE Analysis: Strong Correlations to DPIQ as the Seed
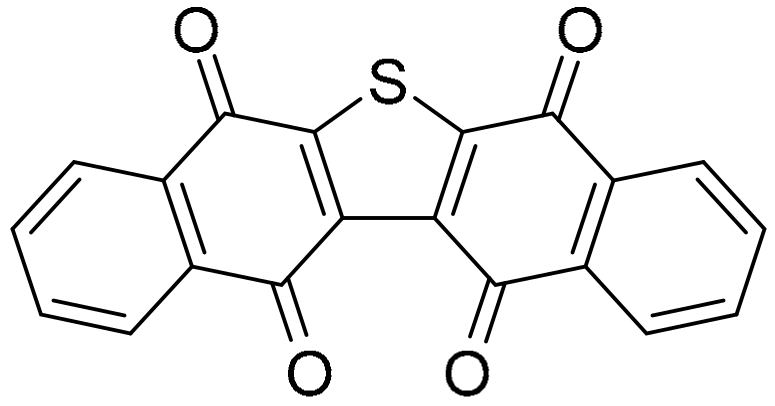
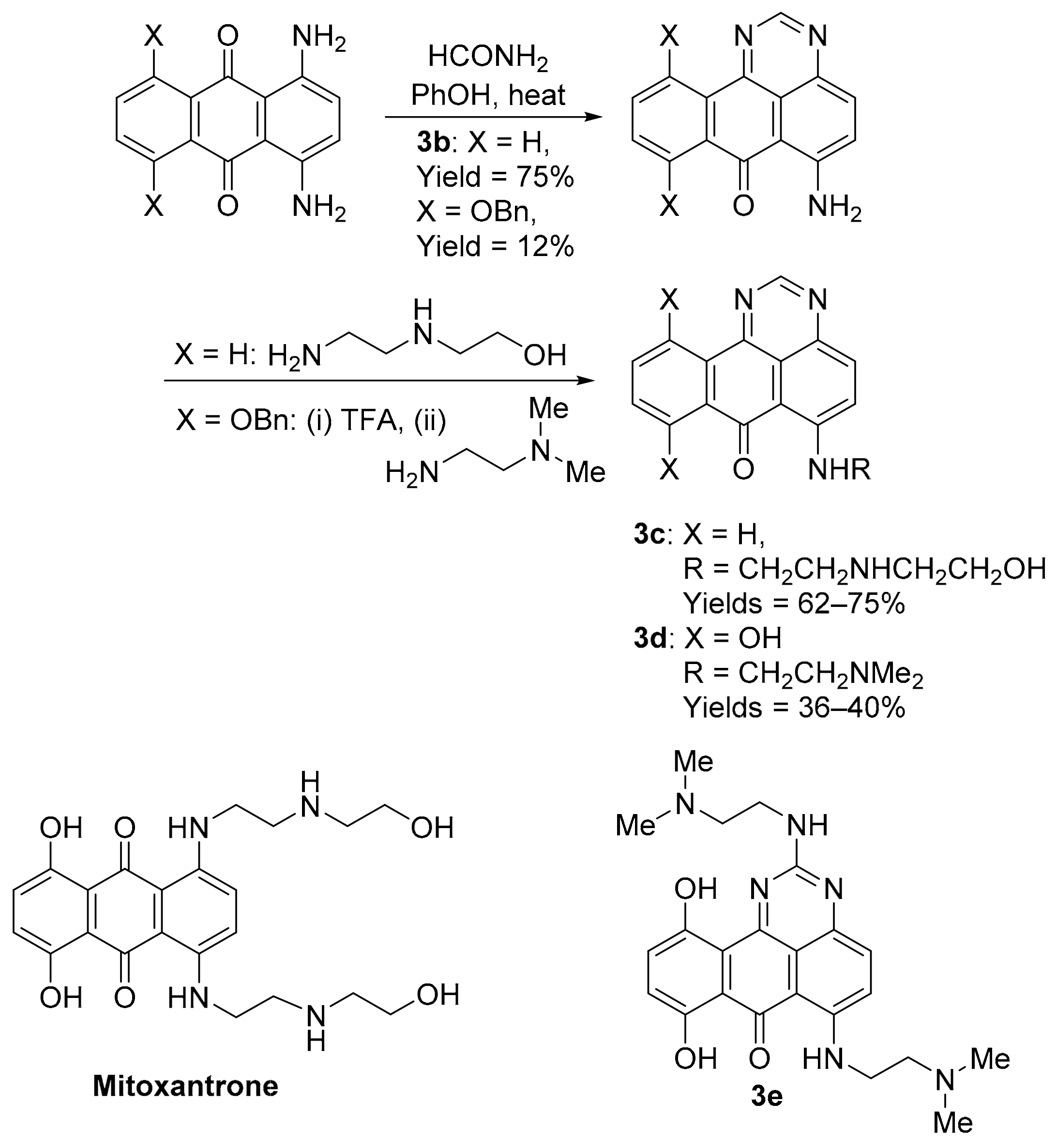
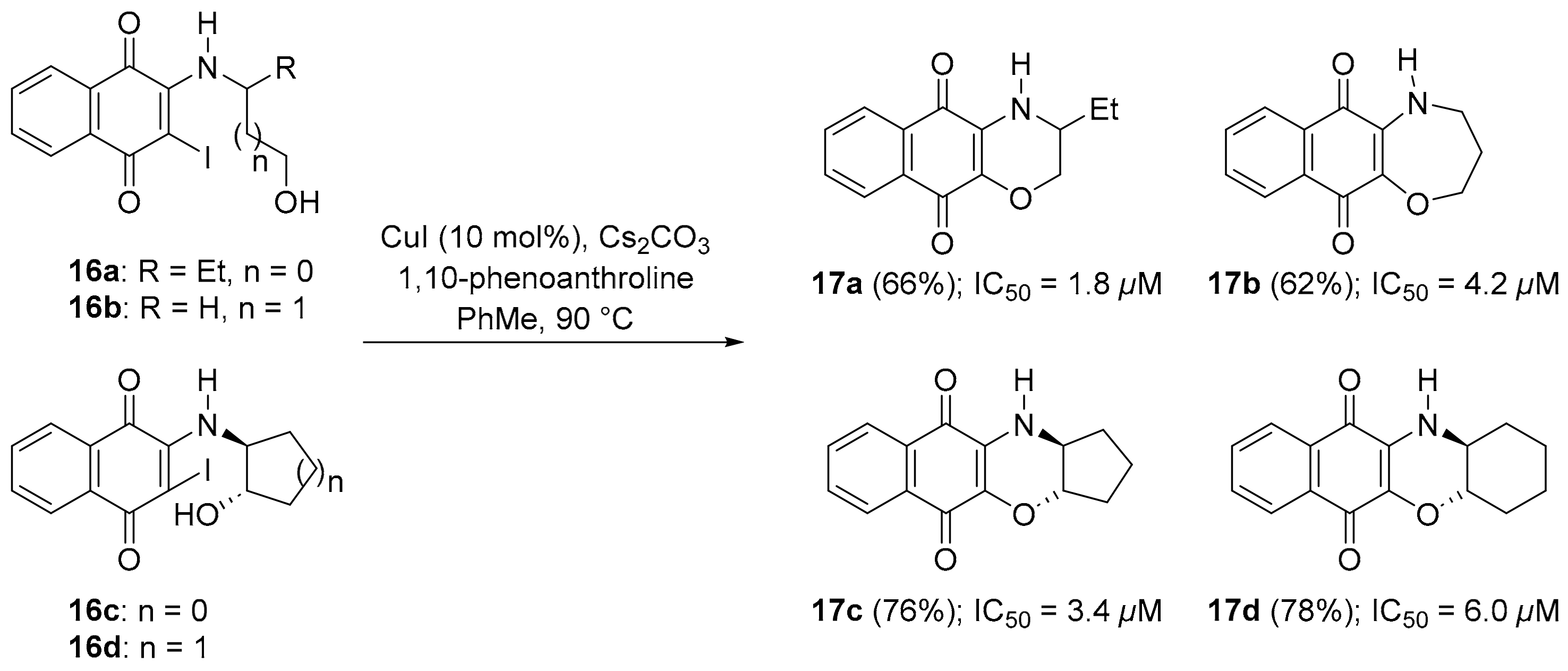
3.1.1. Benzo[e]perimidines
3.1.2. Phenoxazinones
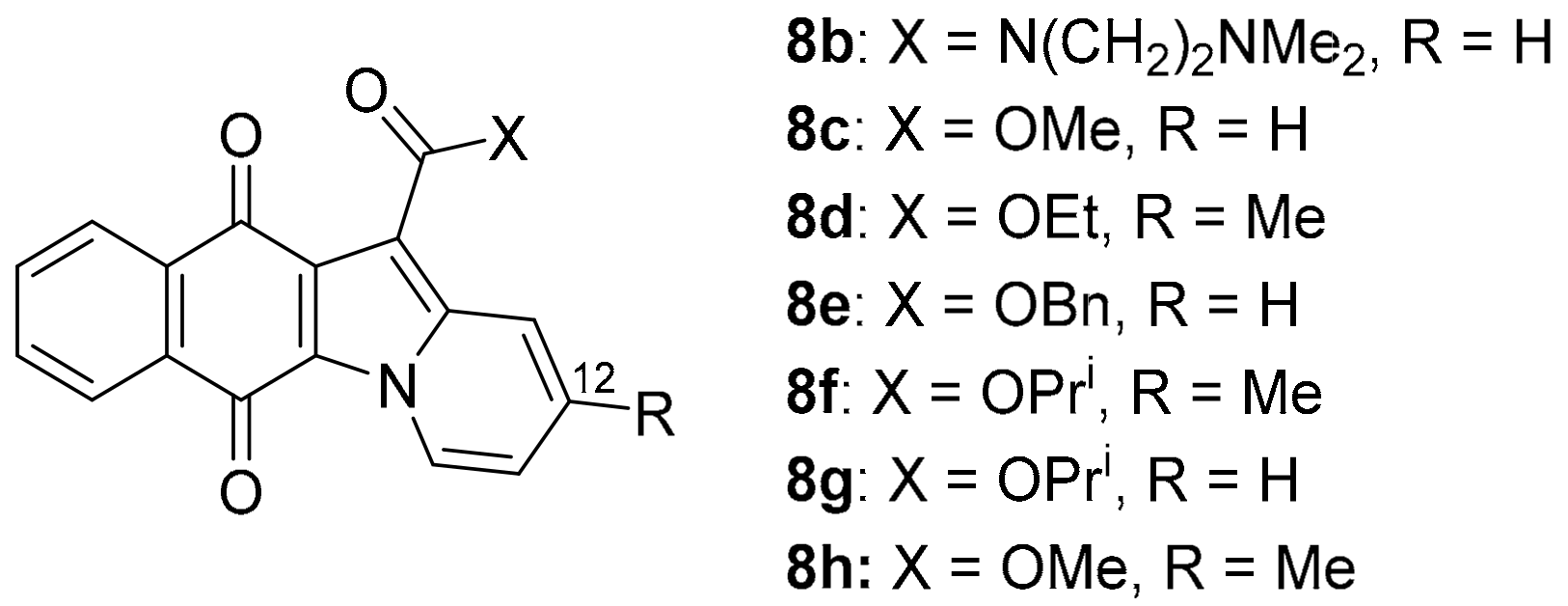
3.1.3. Benz[f]pyrido[1,2-a]indole-6,11-quinone
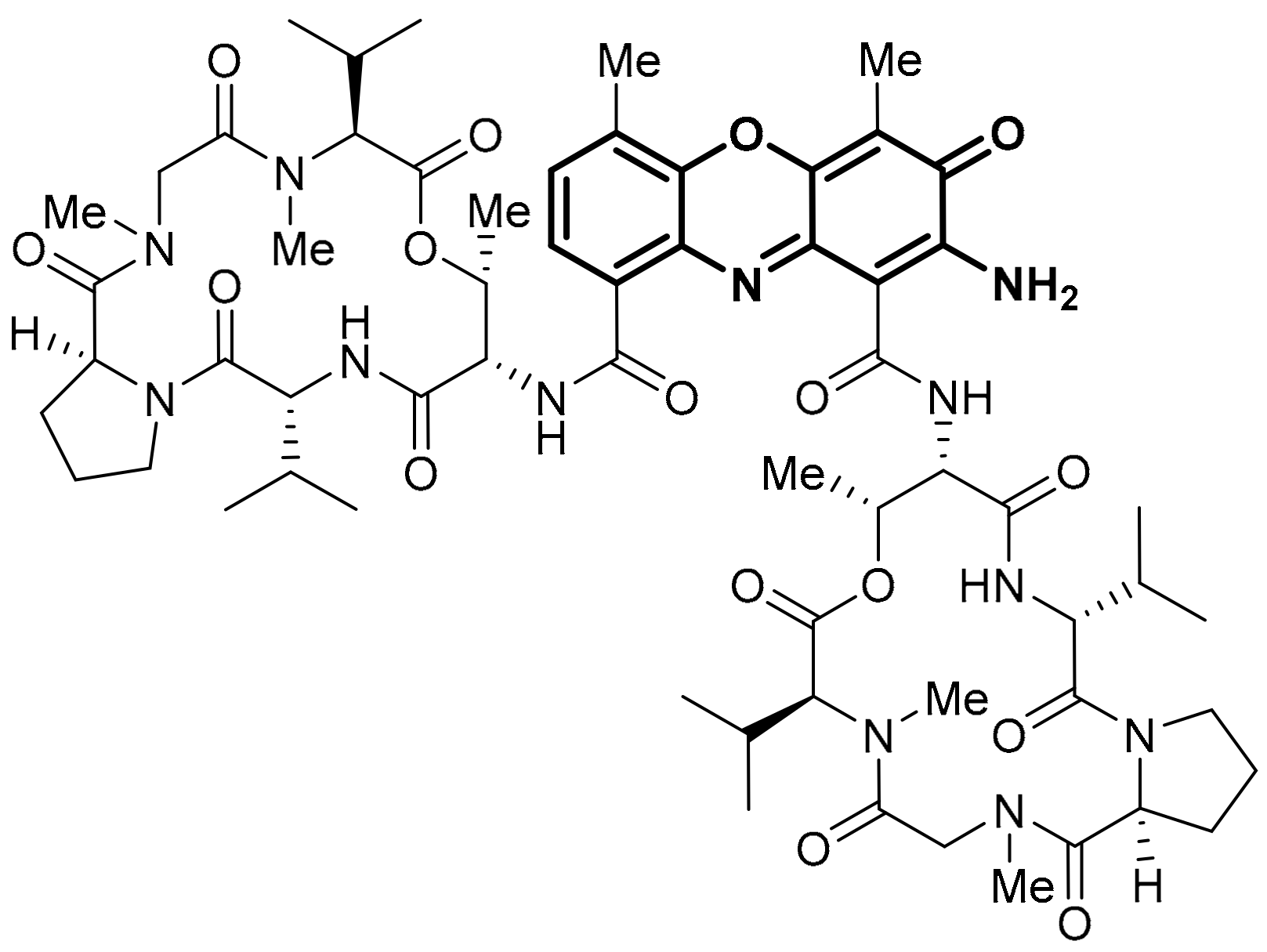
3.1.4. Analogues of Seriniquinone
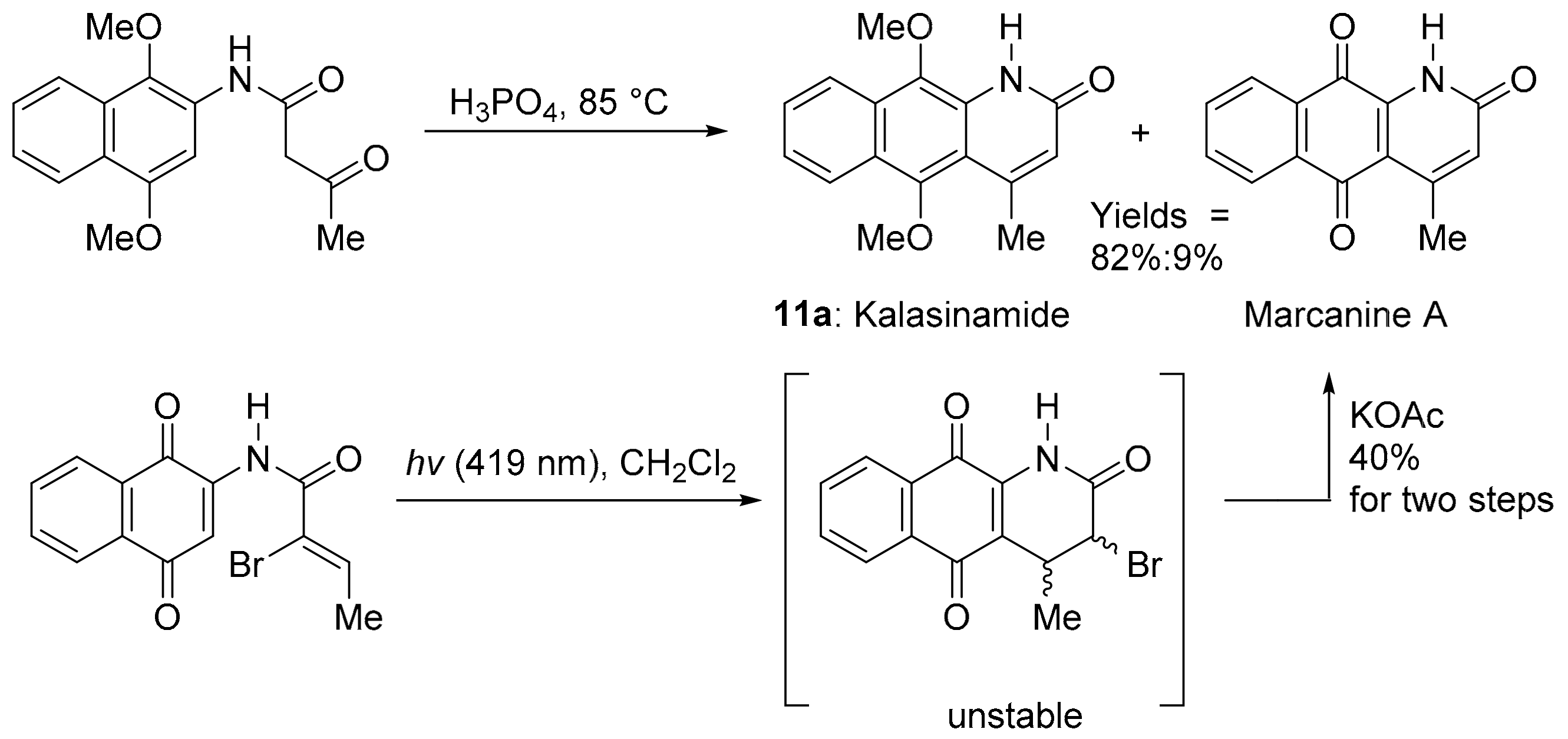
3.1.5. Azaanthracenone
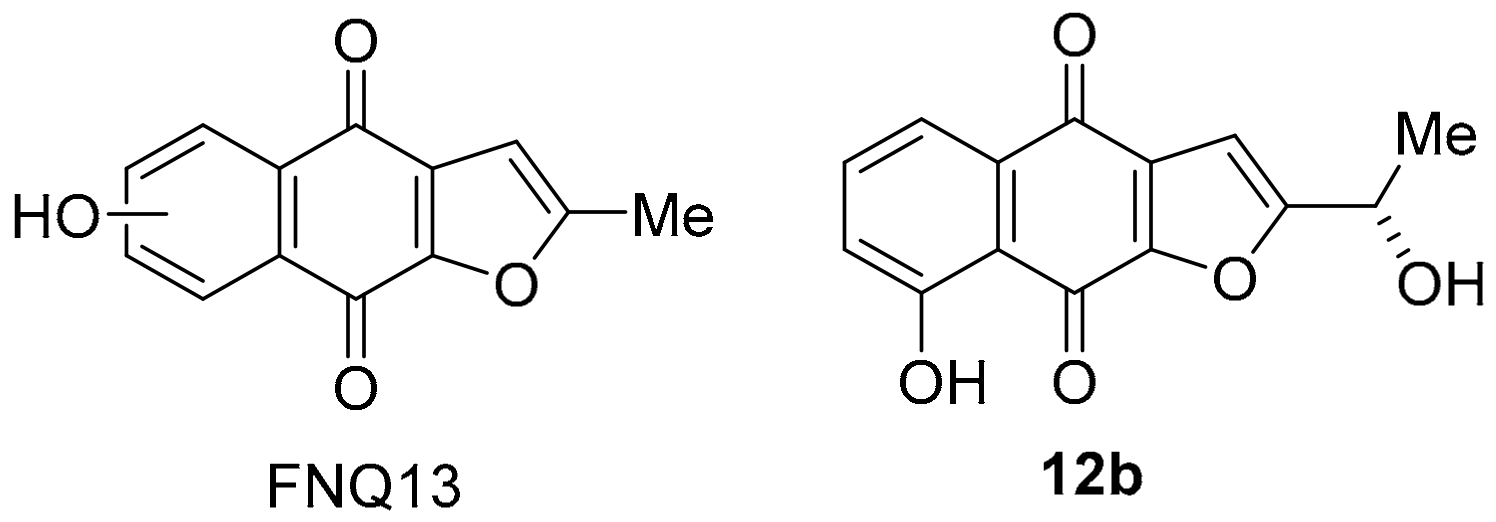
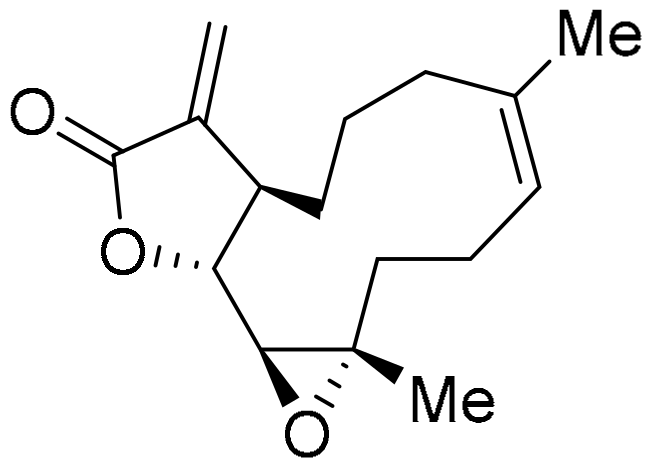
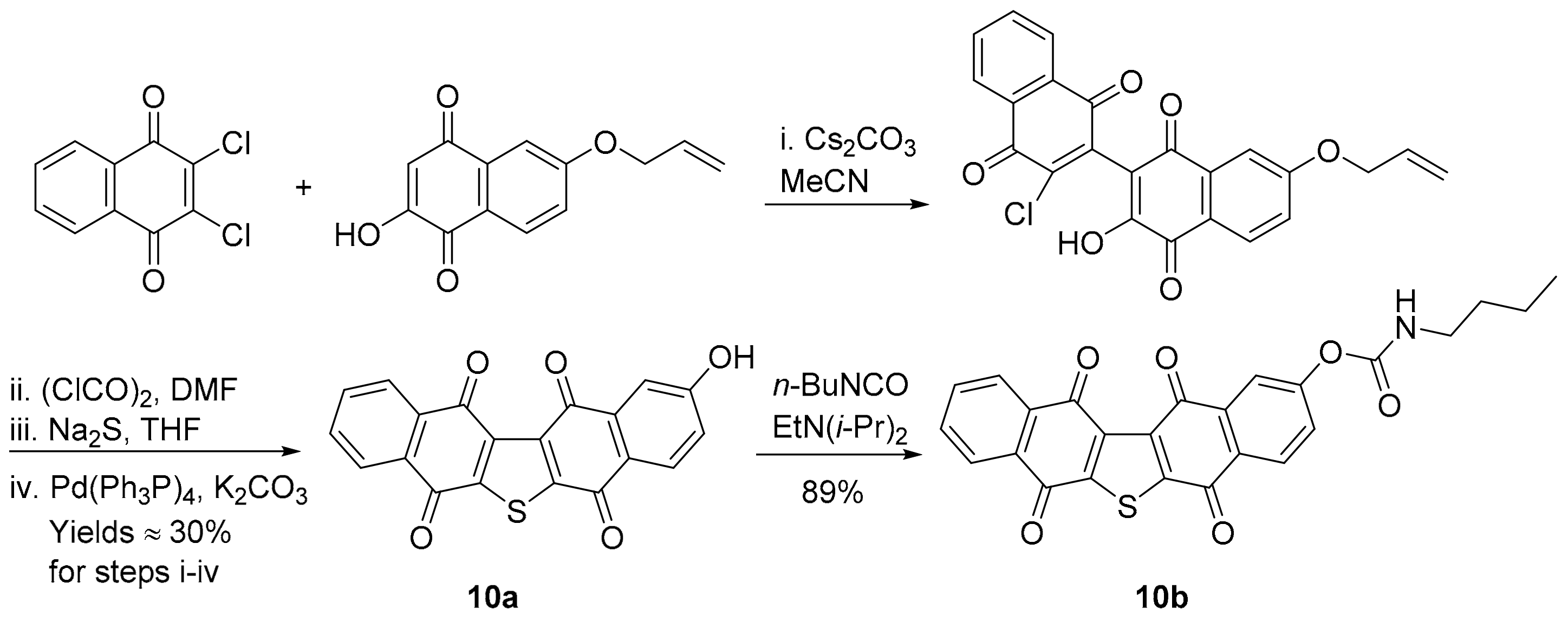
3.1.6. Furano[2,3-b]naphthoquinone (FNQ)
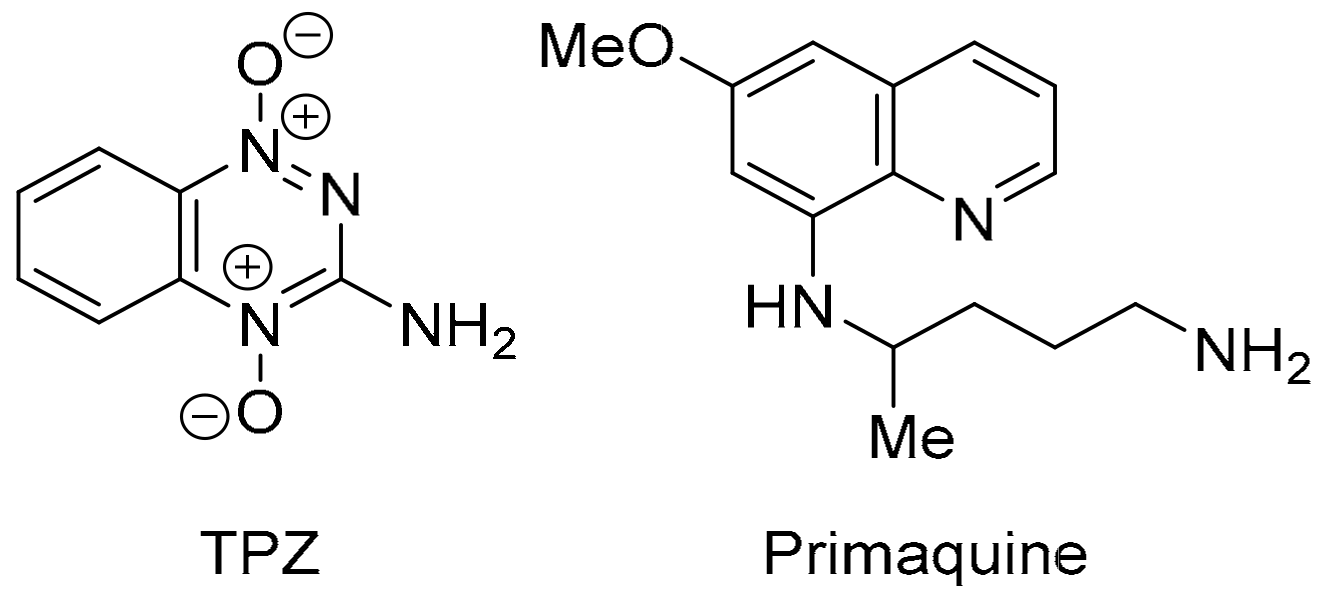
3.2. COMPARE Analysis: Strong Correlations to Benzo[1,2,4]triazin-7-one 5a as the Seed
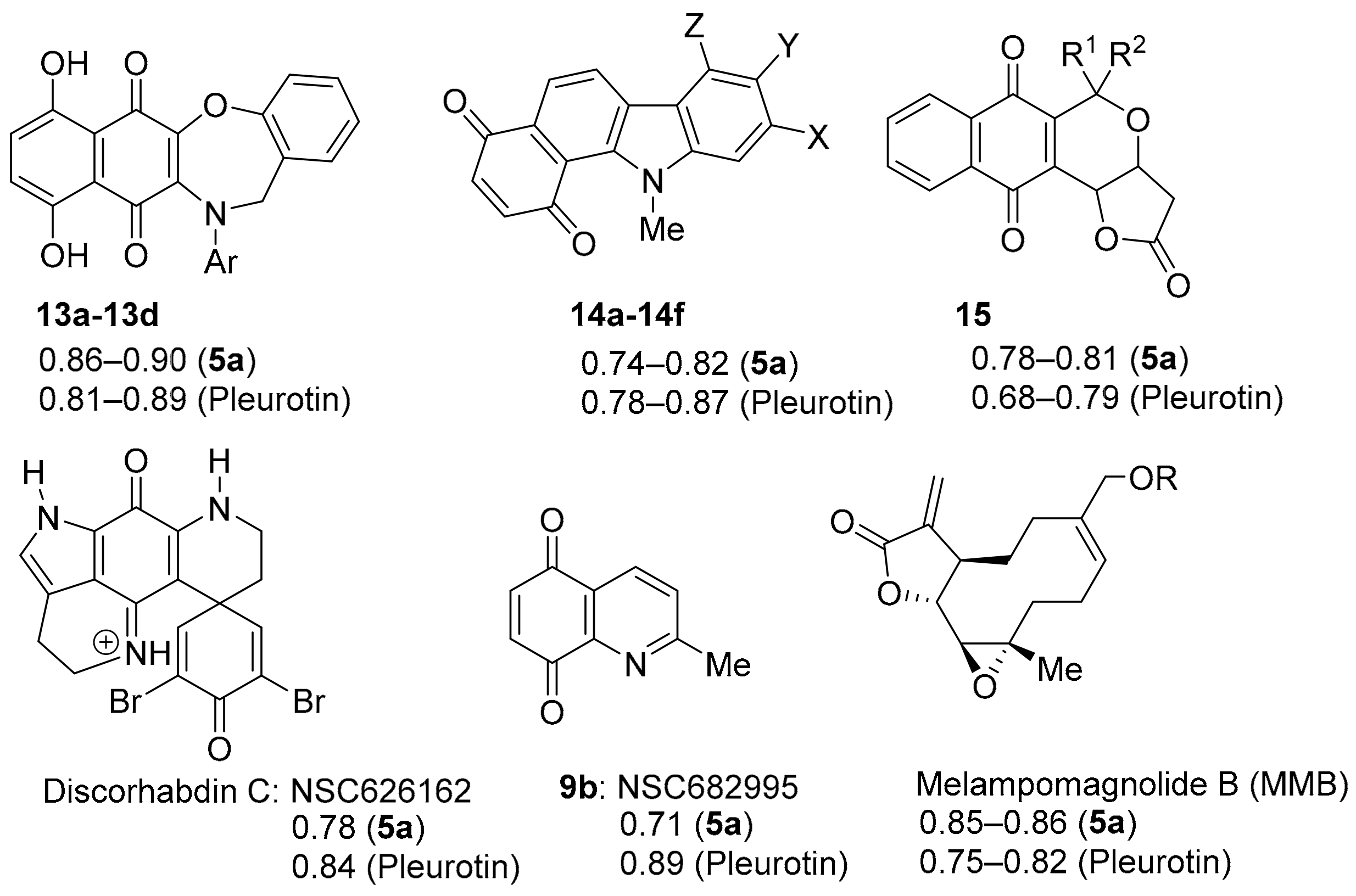
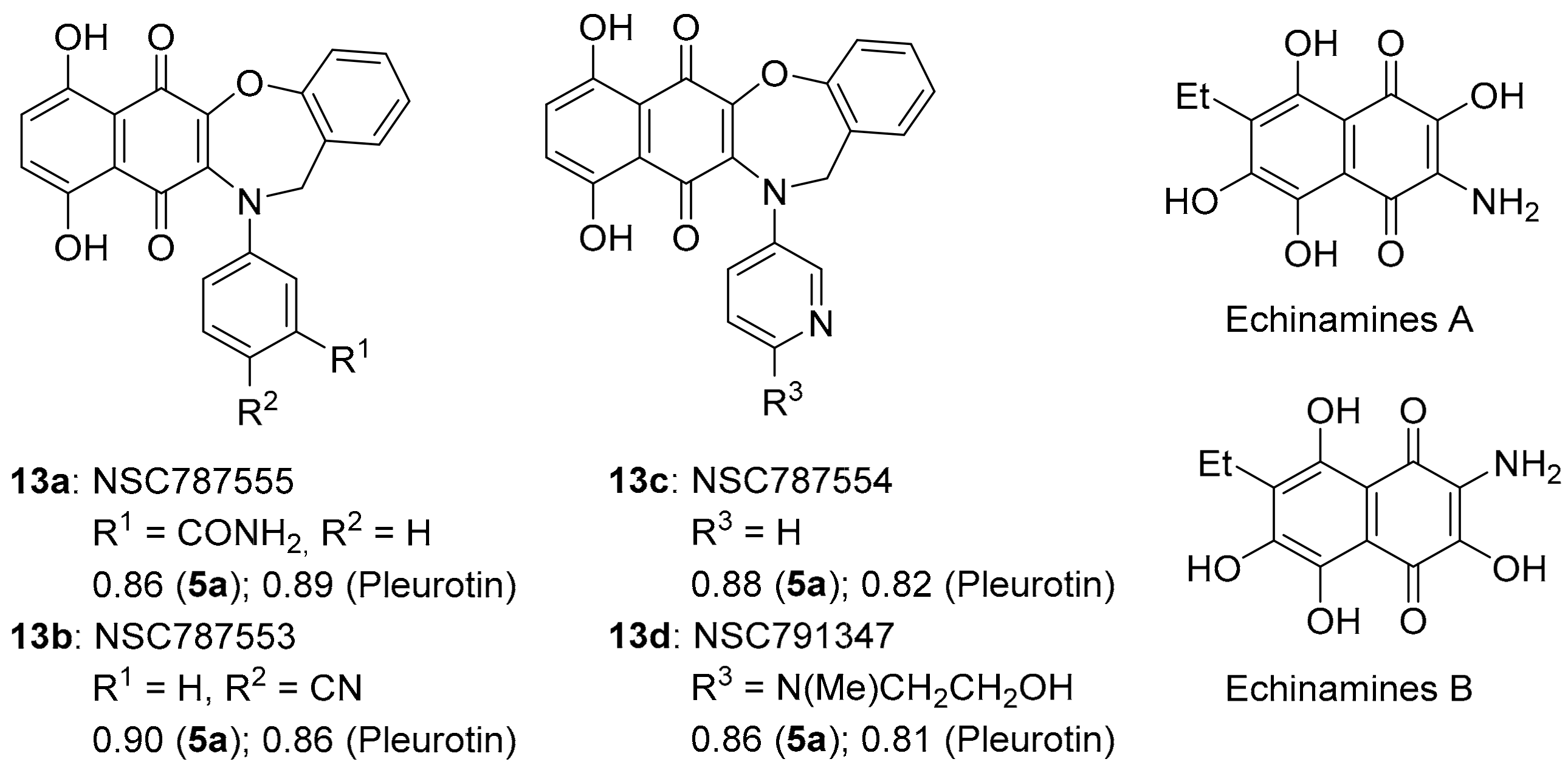
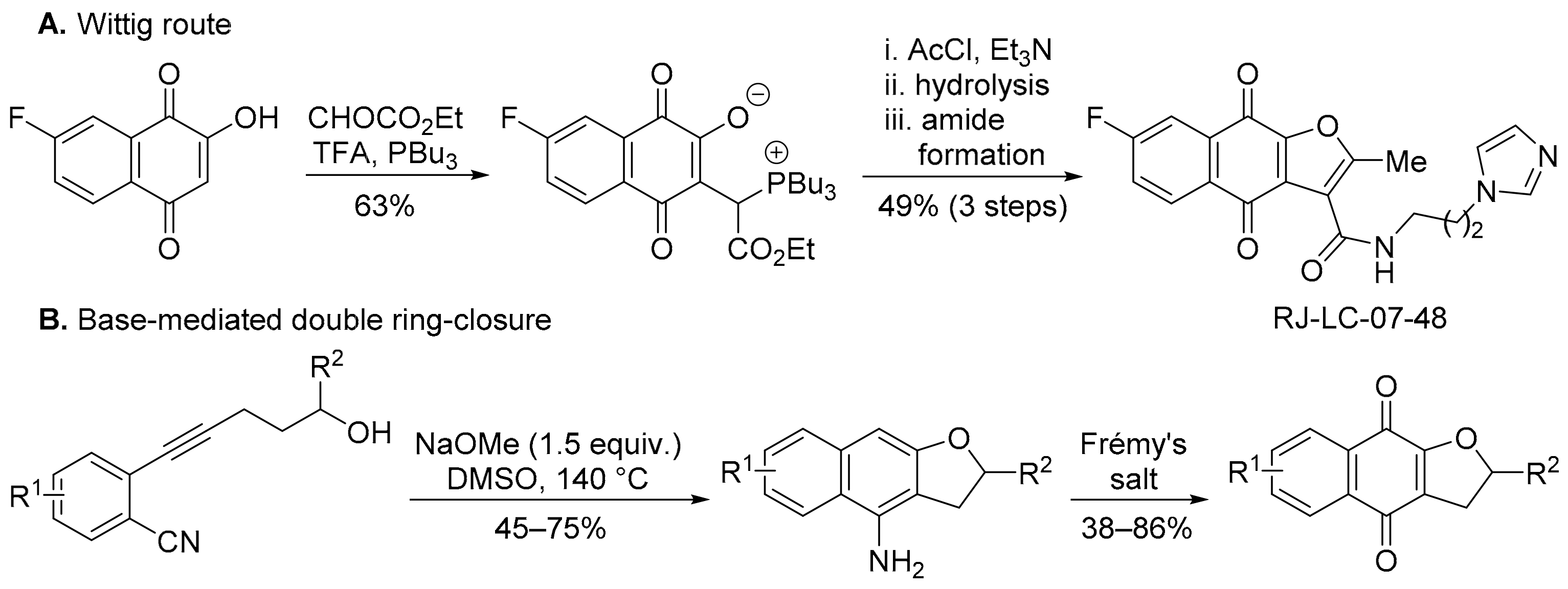
3.2.1. Naphtho[2,3-b][1,4]oxazepine-6,11-dione
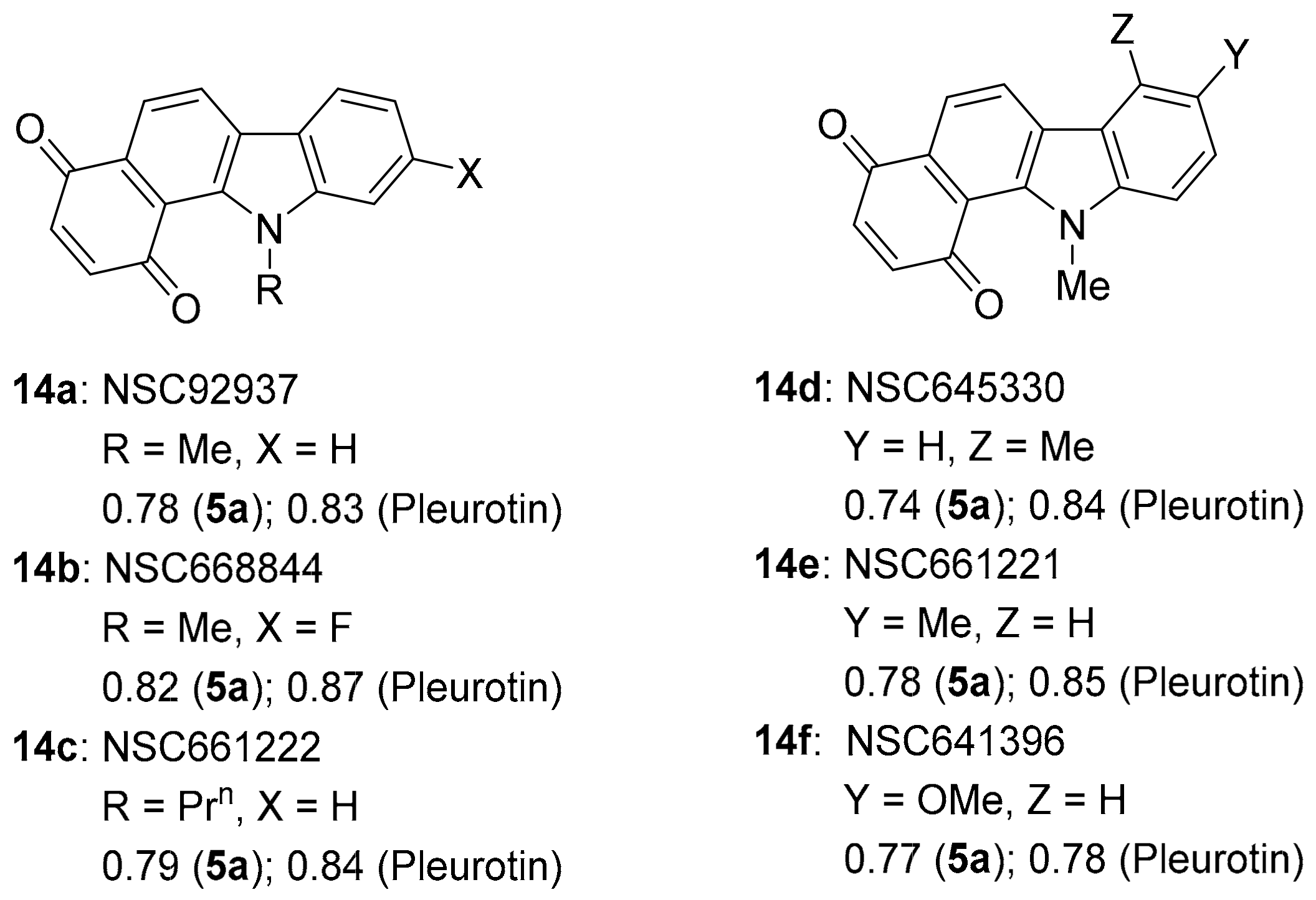
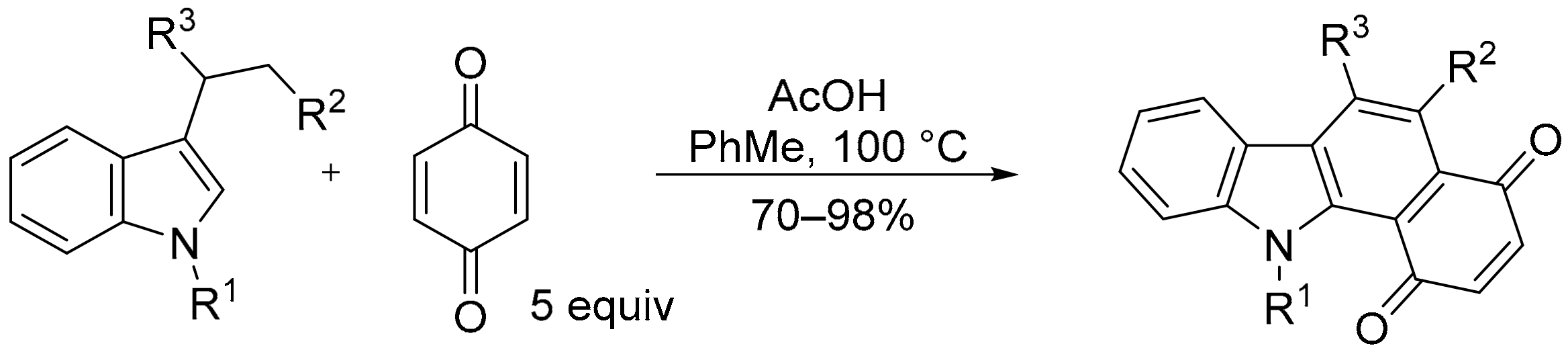
3.2.2. Benzo[a]carbazole-1,4-dione
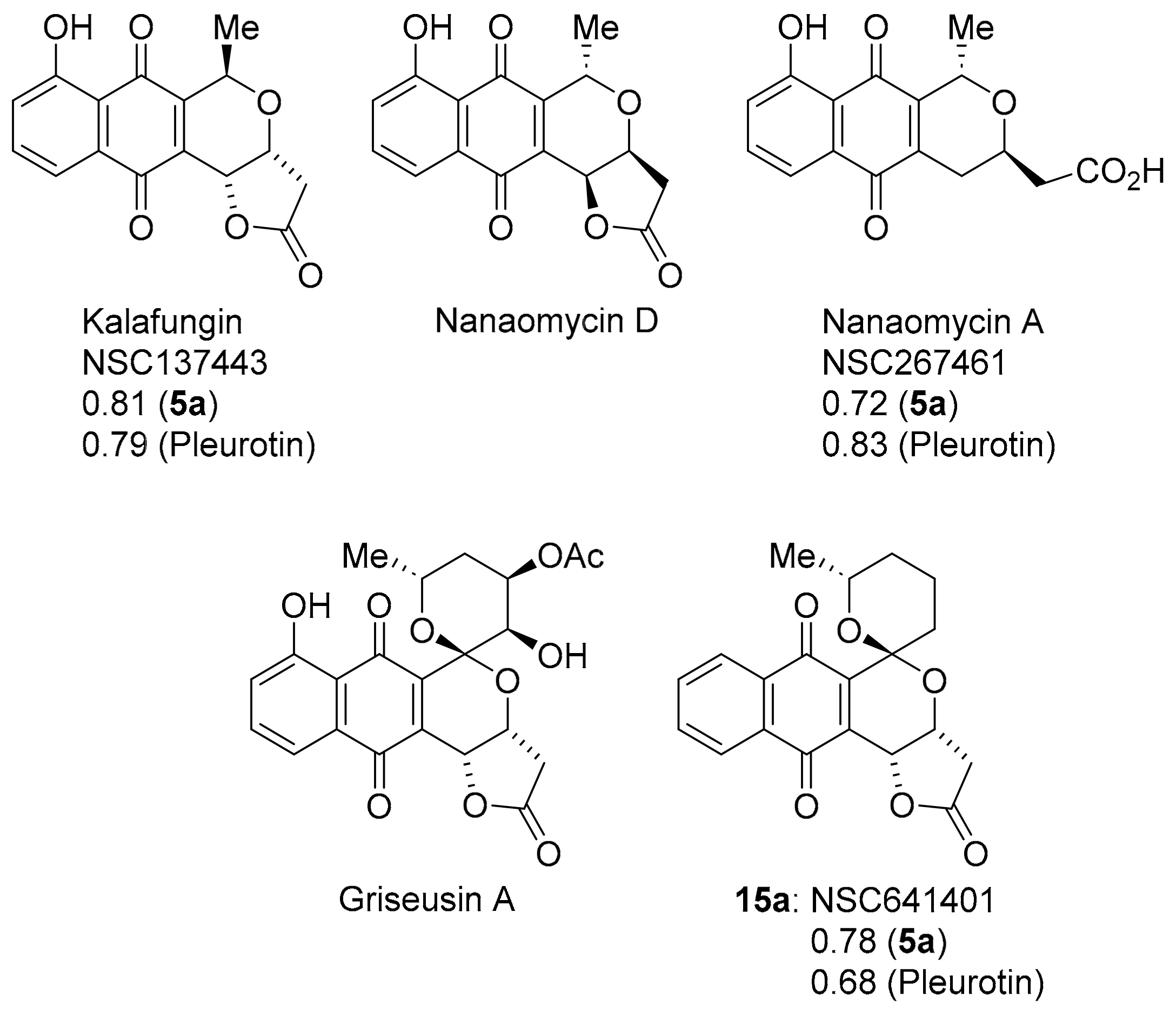
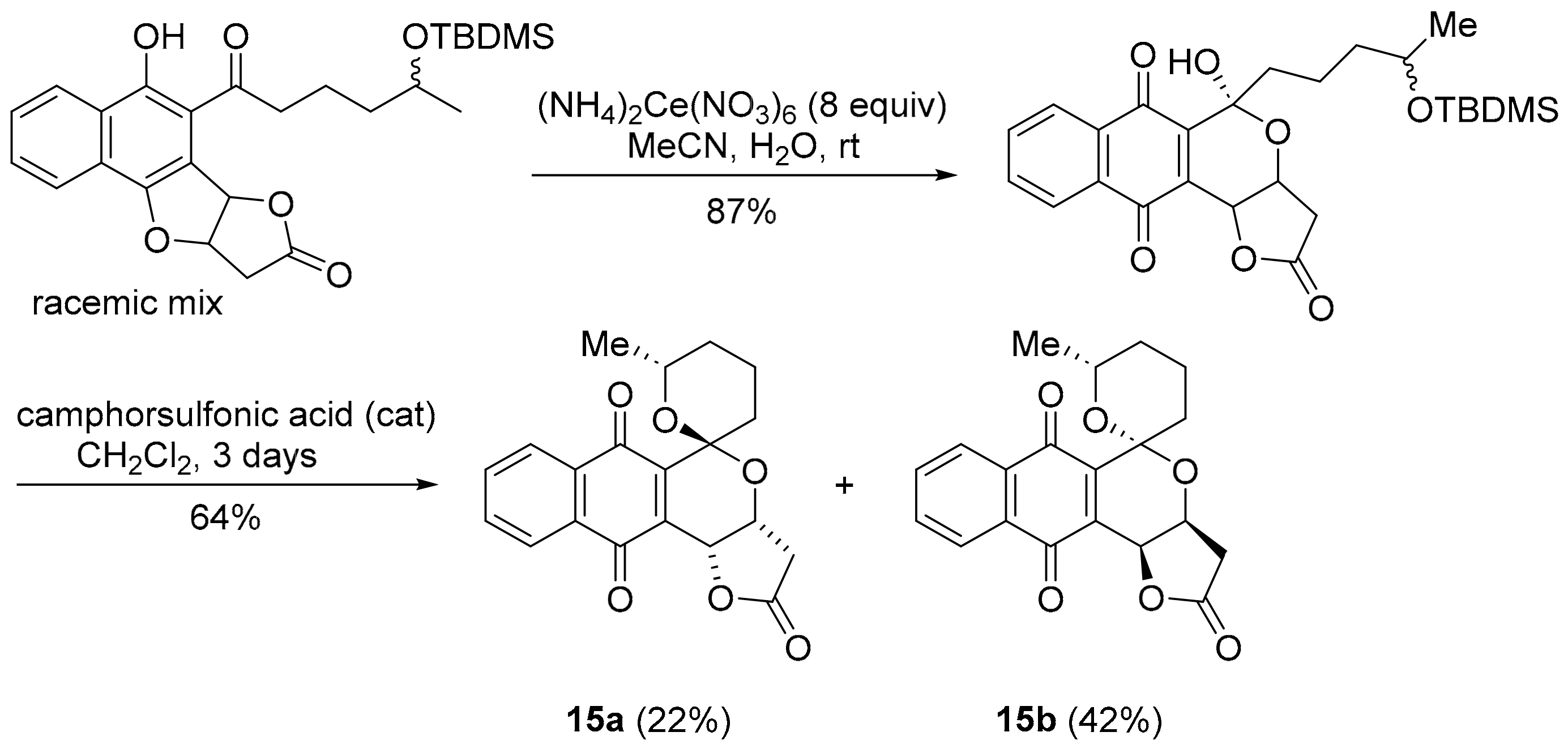
3.2.3. Pyranonaphthoquinones
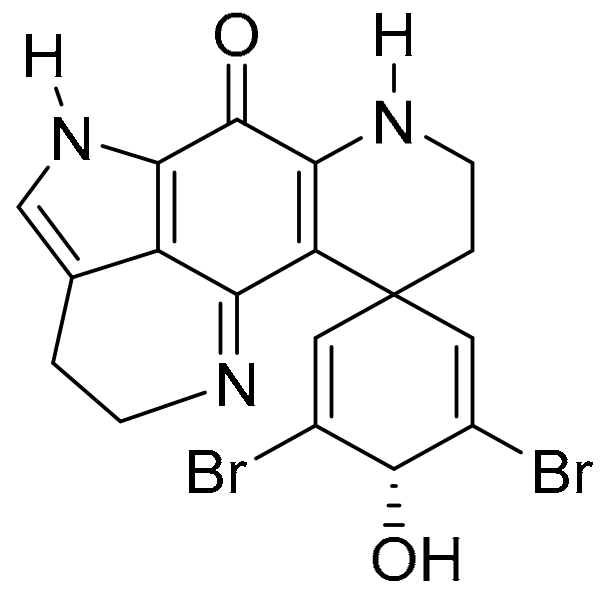
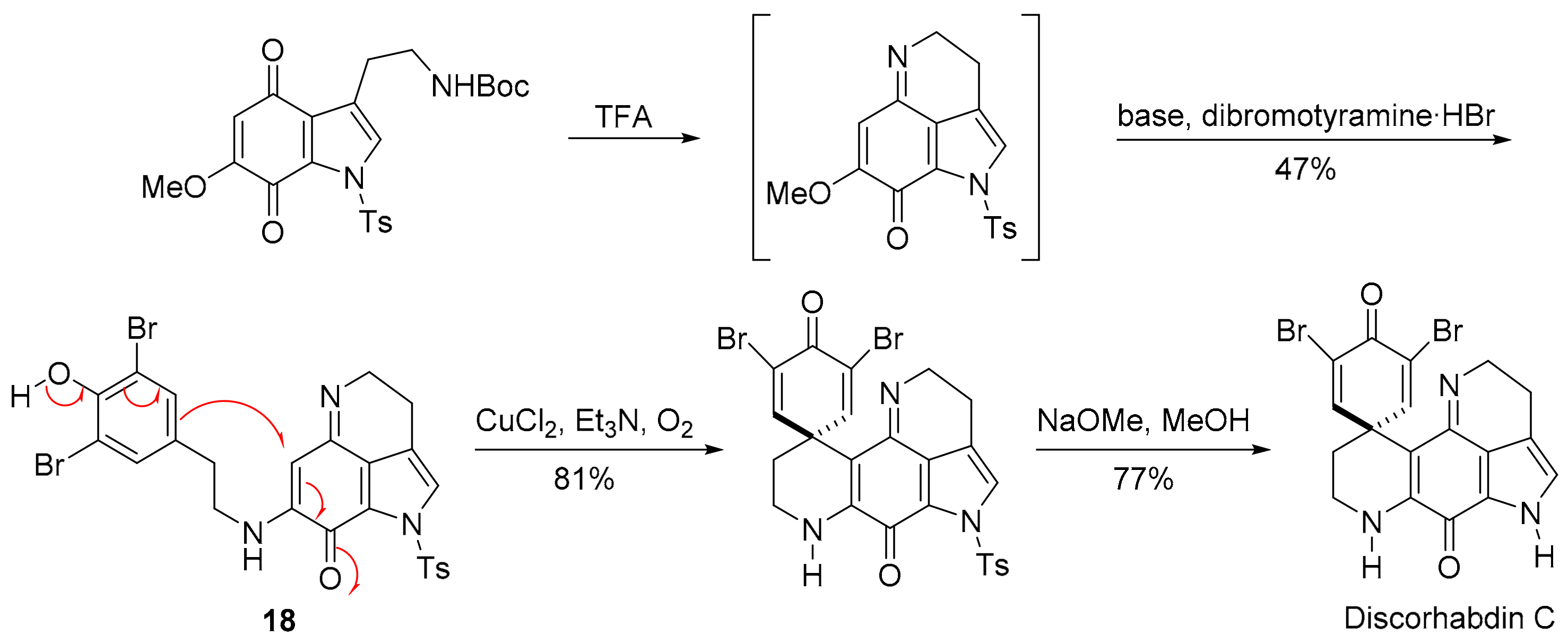
3.2.4. Discorhabdin C
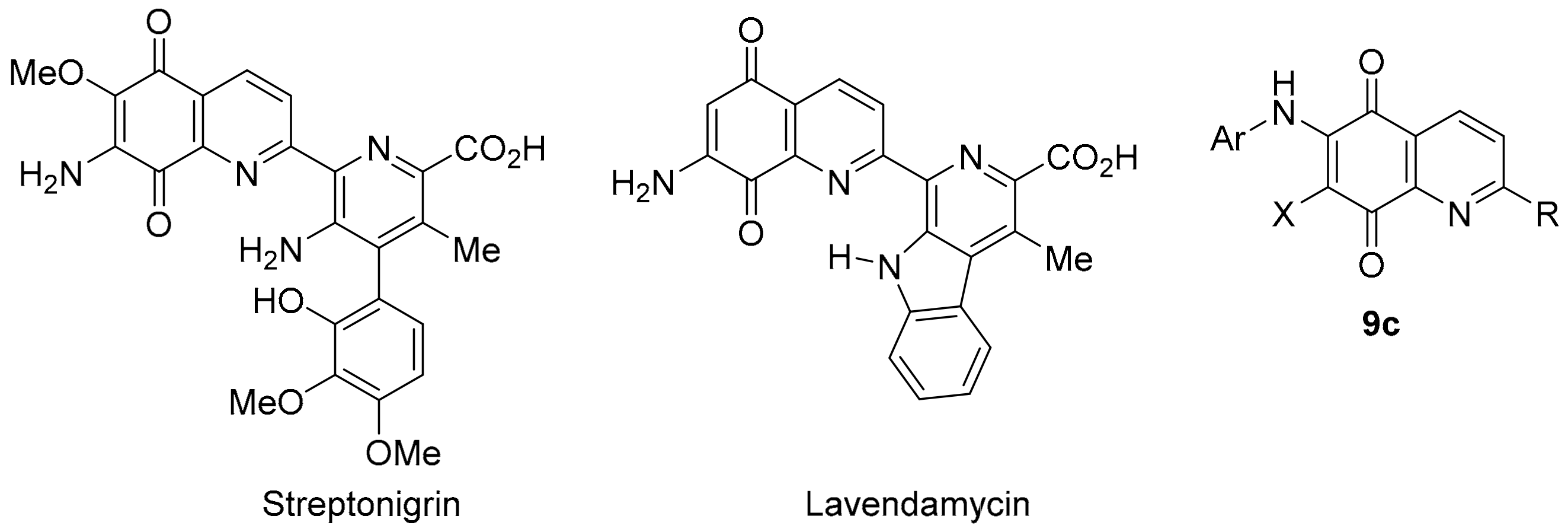
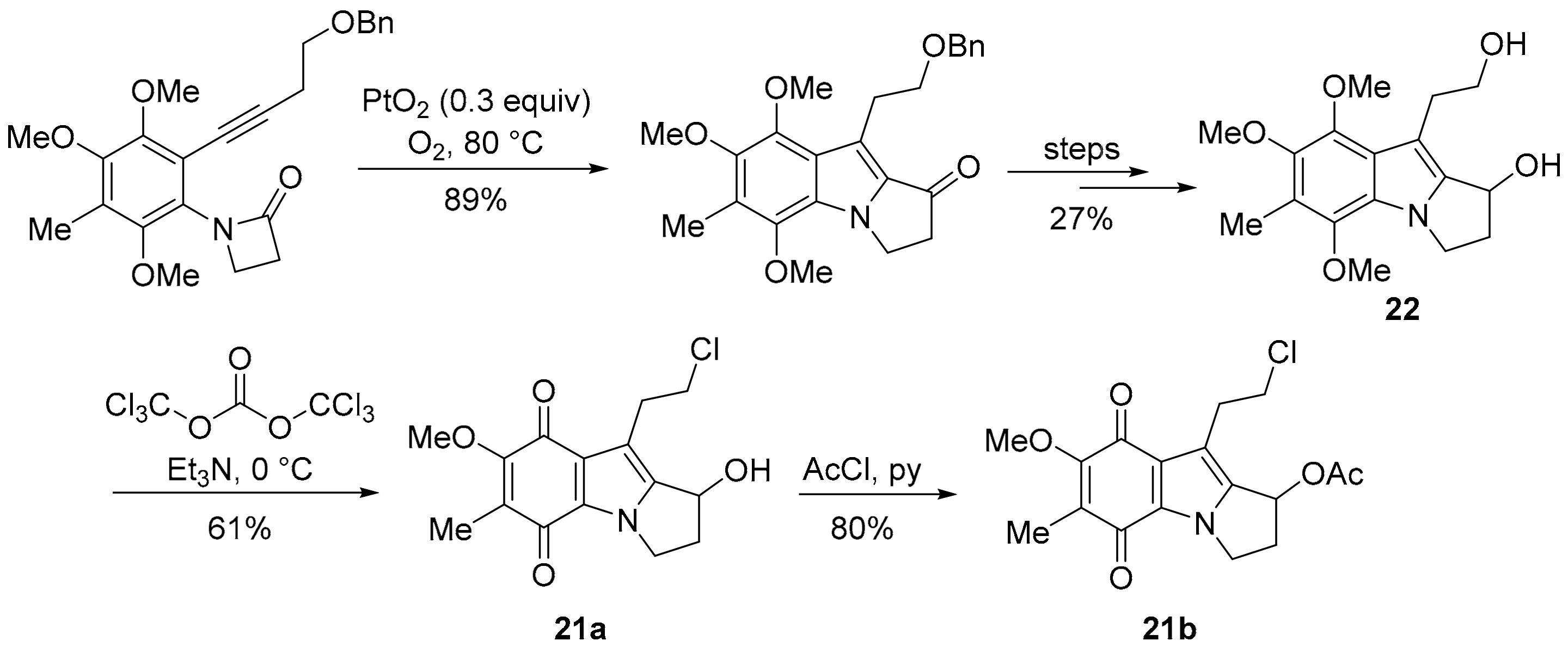
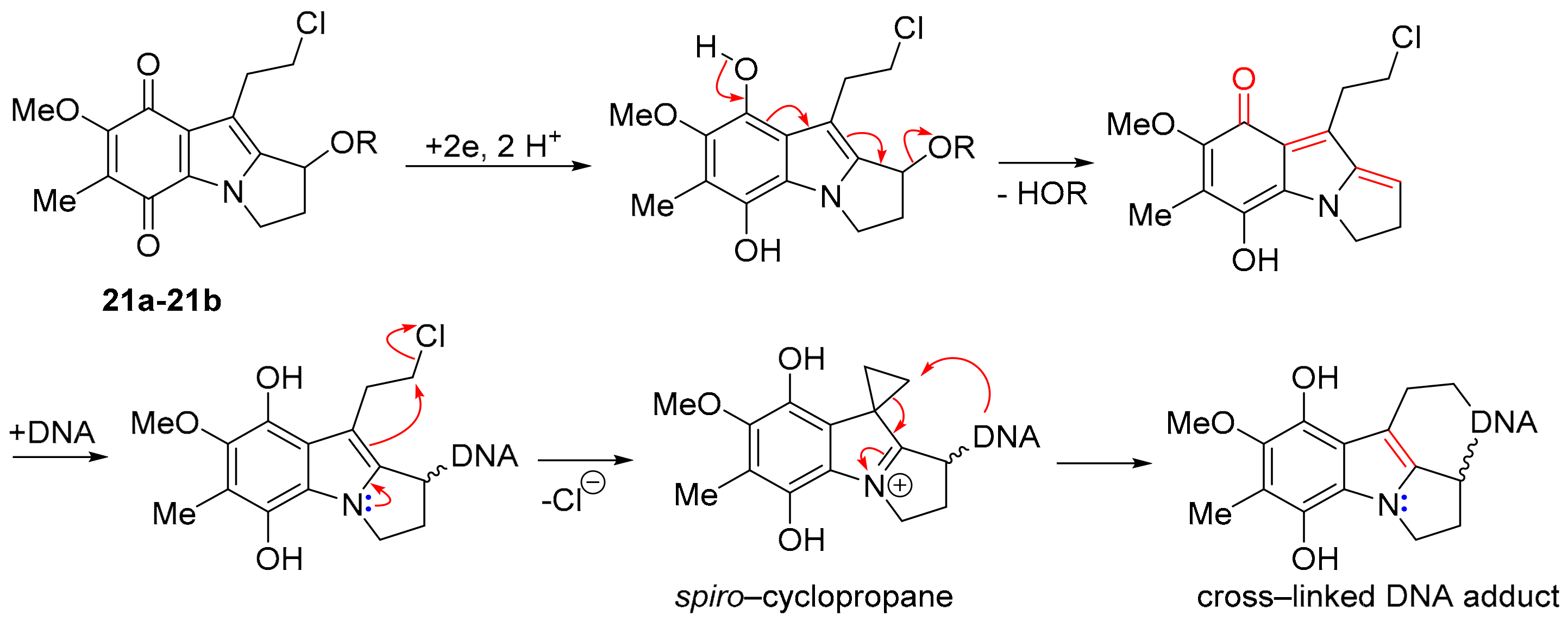
3.3. COMPARE Analysis: Strong Correlations to DPIQ and Benzo[1,2,4]triazin-7-one 5a as the Seed: Quinoline-5,8-diones
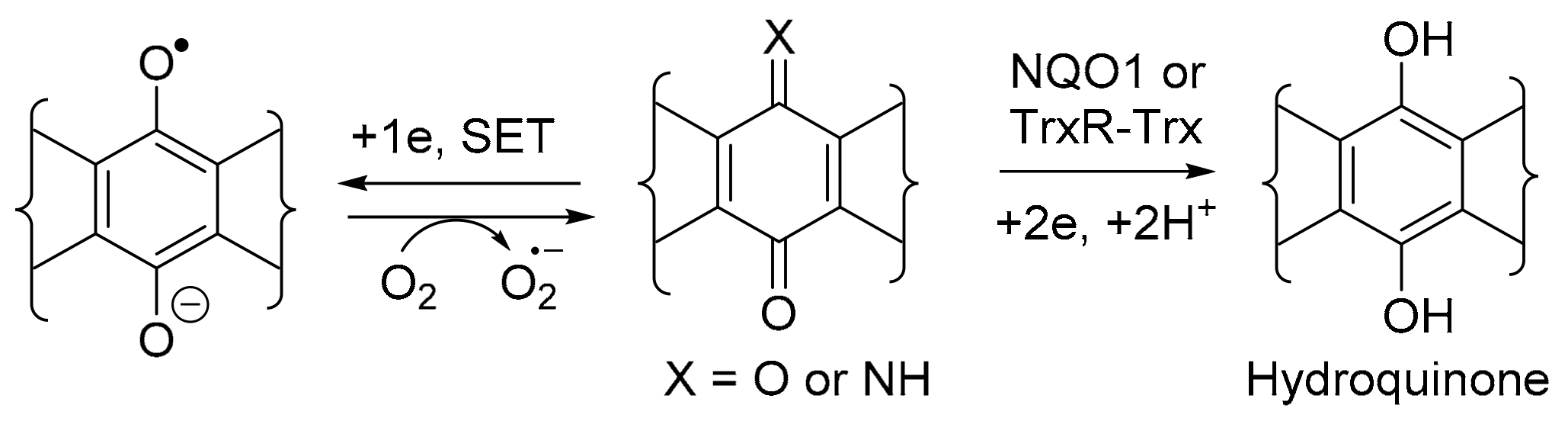
3.4. COMPARE Analysis Using Molecular Target Expression
3.4.1. Compound Correlations to NQO1 Expression
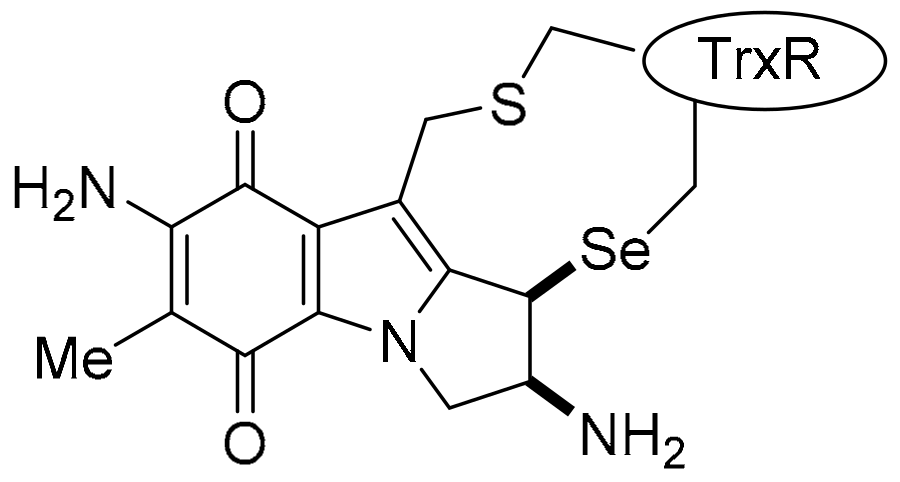
3.4.2. Compound Correlations to TrxR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Holbeck, S.L. Update on NCI in vitro drug screen utilities. Eur. J. Cancer 2004, 40, 785–793. [Google Scholar] [CrossRef]
- Sharma, S.V.; Haber, D.A.; Settleman, J. Cell line-based platforms to evaluate the therapeutic efficacy of candidate anticancer agents. Nat. Rev. Cancer 2010, 10, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Holbeck, S.L.; Camalier, R.; Crowell, J.A.; Govindharajulu, J.P.; Hollingshead, M.; Anderson, L.W.; Polley, E.; Rubinstein, L.; Srivastava, A.; Wilsker, D.; et al. The National Cancer Institute ALMANAC: A Comprehensive screening resource for the detection of anticancer drug pairs with enhanced therapeutic activity. Cancer Res. 2017, 77, 3564–3576. [Google Scholar] [CrossRef] [PubMed]
- Paull, K.D.; Shoemaker, R.H.; Hodes, L.; Monks, A.; Scudiero, D.A.; Rubinstein, L.; Plowman, J.; Boyd, M.R. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: Development of mean graph and COMPARE algorithm. J. Natl. Cancer Inst. 1989, 81, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Fagan, V. Synthesis and Anti-Cancer Activity of Novel Imidazo[5,4-f]benzimidazolequinones. Ph.D. Thesis, National University of Ireland Galway, Galway, Ireland, 2011. [Google Scholar]
- Fagan, V.; Bonham, S.; Carty, M.P.; Saenz-Méndez, P.; Eriksson, L.A.; Aldabbagh, F. COMPARE analysis of the toxicity of an iminoquinone derivative of the imidazo[5,4-f]benzimidazoles with NAD(P)H:quinone oxidoreductase 1 (NQO1) activity and computational docking of quinones as NQO1 substrates. Bioorganic Med. Chem. 2012, 20, 3223–3232. [Google Scholar] [CrossRef]
- Fagan, V.; Bonham, S.; McArdle, P.; Carty, M.P.; Aldabbagh, F. Synthesis and toxicity of new ring-fused imidazo[5,4-f]benzimidazolequinones and mechanism using amine N-oxide cyclizations. Eur. J. Org. Chem. 2012, 2012, 1967–1975. [Google Scholar] [CrossRef]
- Fagan, V.; Bonham, S.; Carty, M.P.; Aldabbagh, F. One-pot double intramolecular homolytic aromatic substitution routes to dialicyclic ring fused imidazobenzimidazolequinones and preliminary analysis of anticancer activity. Org. Biomol. Chem. 2010, 8, 3149–3156. [Google Scholar] [CrossRef]
- Sweeney, M.; Coyle, R.; Kavanagh, P.; Berezin, A.A.; Re, D.L.; Zissimou, G.A.; Koutentis, P.A.; Carty, M.P.; Aldabbagh, F. Discovery of anti-cancer activity for benzo[1,2,4]triazin-7-ones: Very strong correlation to pleurotin and thioredoxin reductase inhibition. Bioorganic Med. Chem. 2016, 24, 3565–3570. [Google Scholar] [CrossRef]
- Keane, L.-A.J.; Mirallai, S.I.; Sweeney, M.; Carty, M.P.; Zissimou, G.A.; Berezin, A.A.; Koutentis, P.A.; Aldabbagh, F. Anti-cancer activity of phenyl and pyrid-2-yl 1,3-substituted benzo[1,2,4]triazin-7-ones and stable free radical precursors. Molecules 2018, 23, 574. [Google Scholar] [CrossRef]
- Conboy, D.; Aldabbagh, F. 6-Imino-1,2,3,4,8,9,10,11-octahydropyrido[1,2-a]pyridoimidazo[4,5-f]benzimidazole-13-one: Synthesis and cytotoxicity evaluation. Molbank 2020, 2020, M1118. [Google Scholar] [CrossRef]
- Fitzsimmons, S.A.; Workman, P.; Grever, M.; Paull, K.; Camalier, R.; Lewis, A.D. Reductase enzyme expression across the National Cancer Institute tumor cell line panel: Correlation with sensitivity to mitomycin C and EO9. J. Natl. Cancer Inst. 1996, 88, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Catto, M.; Berezin, A.A.; Re, D.L.; Loizou, G.; Demetriades, M.; De Stradis, A.; Campagna, F.; Koutentis, P.A.; Carotti, A. Design, synthesis and biological evaluation of benzo[e][1,2,4]triazin-7(1H)-one and [1,2,4]-triazino[5,6,1-jk]carbazol-6-one derivatives as dual inhibitors of beta-amyloid aggregation and acetyl/butyryl cholinesterase. Eur. J. Med. Chem. 2012, 58, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Welsh, S.J.; Williams, R.R.; Birmingham, A.; Newman, D.J.; Kirkpatrick, D.L.; Powis, G. The thioredoxin redox inhibitors 1-methylpropyl 2-imidazolyl disulfide and pleurotin inhibit hypoxia-induced factor 1α and vascular endothelial growth factor formation. Mol. Cancer Ther. 2003, 2, 235–243. [Google Scholar] [PubMed]
- Saikolappan, S.; Kumar, B.; Shishodia, G.; Koul, S.; Koul, H.K. Reactive oxygen species and cancer: A complex interaction. Cancer Lett. 2019, 452, 132–143. [Google Scholar] [CrossRef]
- Bolton, J.L.; Dunlap, T. Formation and biological targets of quinones: Cytotoxic versus cytoprotective effects. Chem. Res. Toxicol. 2017, 30, 13–37. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, D.; Ma, K.; Wu, X.; Hao, H.; Jiang, S. NAD(P)H:quinone oxidoreductase 1 (NQO1) as a therapeutic and diagnostic target in cancer. J. Med. Chem. 2018, 61, 6983–7003. [Google Scholar] [CrossRef]
- Bian, M.; Fan, R.; Zhao, S.; Liu, W. Targeting the thioredoxin system as a strategy for cancer therapy. J. Med. Chem. 2019, 62, 7309–7321. [Google Scholar] [CrossRef]
- Tomasz, M.; Lipman, R.; Chowdary, D.; Pawlak, J.; Verdine, G.L.; Nakanishi, K. Isolation and structure of a covalent cross-link adduct between mitomycin C and DNA. Science 1987, 235, 1204–1208. [Google Scholar] [CrossRef]
- Parkinson, E.I.; Bair, J.S.; Cismesia, M.; Hergenrother, P.J. Efficient NQO1 substrates are potent and selective anticancer agents. ACS Chem. Biol. 2013, 8, 2173–2183. [Google Scholar] [CrossRef]
- Li, R.; Bianchet, M.A.; Talalay, P.; Amzel, L.M. The three-dimensional structure of NAD(P)H:quinone reductase, a flavoprotein involved in cancer chemoprotection and chemotherapy: Mechanism of the two-electron reduction. Proc. Natl. Acad. Sci. USA 1995, 92, 8846–8850. [Google Scholar] [CrossRef] [PubMed]
- Coyle, R. Barton Esters for Initiator-Free Radical Cyclisation with Heteroaromatic Substitution and Anti-Cancer Evaluation of Benzo[e][1,2,4]triazin-7-ones. Ph.D. Thesis, National University of Ireland Galway, Galway, Ireland, 2014. [Google Scholar]
- Dai, B.; Augustine, J.J.; Kang, Y.; Roife, D.; Li, X.; Deng, J.; Tan, L.; Rusling, L.A.; Weinstein, J.N.; Lorenzi, P.L.; et al. Compound NSC84167 selectively targets NRF2-activated pancreatic cancer by inhibiting asparagine synthesis pathway. Cell Death Dis. 2021, 12, 693. [Google Scholar] [CrossRef] [PubMed]
- Lister, A.; NedjadI, T.; Kitteringham, N.R.; Campbell, F.; Costello, E.; Lloyd, B.; Copple, I.M.; Williams, S.; Owen, A.; Neoptolemos, J.P.; et al. Nrf2 is overexpressed in pancreatic cancer: Implications for cell proliferation and therapy. Mol. Cancer 2011, 10, 37. [Google Scholar] [CrossRef]
- Stefañska, B.; Dzieduszycka, M.; Martelli, S.; Tarasiuk, J.; Bontemps-Gracz, M.; Borowski, E. 6-[(Aminoalkyl)amino]-substituted 7H-benzo[e]perimidin-7-ones as novel antineoplastic agents. Synthesis and biological evaluation. J. Med. Chem. 1993, 36, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Stefañska, B.; Dzieduszycka, M.; Bontemps-Gracz, M.M.; Borowski, E.; Martelli, S.; Supino, R.; Pratesi, G.; De Cesare, M.; Zunino, F.; Kuśnierczyk, H.; et al. 8,11-Dihydroxy-6-[(aminoalkyl)amino]-7H-benzo[e]perimidin-7-ones with activity in multidrug-resistant cell lines: Synthesis and antitumor evaluation. J. Med. Chem. 1999, 42, 3494–3501. [Google Scholar] [CrossRef]
- Dzieduszycka, M.; Martelli, S.; Arciemiuk, M.; Bontemps-Gracz, M.M.; Kupiec, A.; Borowski, E. Effect of modification of 6-[(Aminoalkyl)amino]-7H-benzo[e]perimidin-7-ones on their cytotoxic activity toward sensitive and multidrug resistant tumor cell lines. Synthesis and biological evaluation. Bioorganic Med. Chem. 2002, 10, 1025–1035. [Google Scholar] [CrossRef]
- Anzai, K.; Isono, K.; Okuma, K.; Suzuki, S. The new antibiotics, questiomycins A and B. J. Antibiot. 1960, 13, 125–132. [Google Scholar]
- Kozlovsky, A.G.; Zhelifonova, V.P.; Antipova, T.V.; Adanin, V.M.; Novikova, N.D.; Deshevaya, E.A.; Schlegel, B.; Dahse, H.M.; Gollmik, F.; Grafe, U. Penicillium expansum, a resident fungal strain of the orbital complex Mir, producing xanthocyllin X and questiomycin A. Appl. Biochem. Microbiol. 2004, 40, 344–349. [Google Scholar] [CrossRef]
- Bitzer, J.; Große, T.; Wang, L.; Lang, S.; Beil, W.; Zeeck, A. New aminophenoxazinones from a marine Halomonas sp.: Fermentation, structure elucidation, and biological activity. J. Antibiot. 2006, 59, 86–92. [Google Scholar] [CrossRef]
- Hanawa, T.; Osaki, T.; Manzoku, T.; Fukuda, M.; Kawakami, H.; Tomoda, A.; Kamiya, S. In vitro antibacterial activity of Phx-3 against Helicobacter pylori. Biol. Pharm. Bull. 2010, 33, 188–191. [Google Scholar] [CrossRef]
- Guo, S.; Hu, H.; Bilal, M.; Zhang, X. Production of antibacterial questiomycin A in metabolically engineered Pseudomonas chlororaphis HT66. J. Agric. Food Chem. 2022, 70, 7742–7750. [Google Scholar] [CrossRef]
- Barry, C.E., III; Nayar, P.G.; Begley, T.P. Phenoxazinone synthase: Mechanism for the formation of the phenoxazinone chromophore of actinomycin. Biochemistry 1989, 28, 6323–6333. [Google Scholar] [CrossRef] [PubMed]
- Le Roes-Hill, M.; Goodwin, C.; Burton, S. Phenoxazinone synthase: What’s in a name? Trends Biotechnol. 2009, 27, 248–258. [Google Scholar] [CrossRef] [PubMed]
- Nakachi, T.; Tabuchi, T.; Takasaki, A.; Arai, S.; Miyazawa, K.; Tomoda, A. Anticancer activity of phenoxazines produced by bovine erythrocytes on colon cancer cells. Oncol. Rep. 2010, 23, 1517–1522. [Google Scholar] [PubMed]
- Hongo, T.; Miyano-Kurosaki, N.; Kurosaki, K.; Hata, A.; Harigae, S.; Tomoda, A. 2-Aminophenoxazine-3-one prevents pulmonary metastasis of mouse B16 melanoma cells in mice. J. Pharmacol. Sci. 2010, 114, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Kohno, K.; Miyake, M.; Sano, O.; Tanaka-Kataoka, M.; Yamamoto, S.; Koya-Miyata, S.; Arai, N.; Fujii, M.; Watanabe, H.; Ushio, S.; et al. Anti-inflammatory and immunomodulatory properties of 2-amino-3H-phenoxazin-3-one. Biol. Pharm. Bull. 2008, 31, 1938–1945. [Google Scholar] [CrossRef]
- Baindara, P.; Mandal, S.M. Bacteria and bacterial anticancer agents as a promising alternative for cancer therapeutics. Biochimie 2020, 177, 164–189. [Google Scholar] [CrossRef]
- Bhansali, K.G.; Kook, A.M. Synthesis and characterization of a series of 5H-benzo[a]phenoxazin-5-one derivatives as potential antiviral/antitumor agents. Heterocycles 1993, 36, 1239–1251. [Google Scholar] [CrossRef]
- Chau, N.-M.; Rogers, P.; Aherne, W.; Carroll, V.; Collins, I.; McDonald, E.; Workman, P.; Ashcroft, M. Identification of novel small molecule inhibitors of hypoxia-inducible factor-1 that differentially block hypoxia-inducible factor-1 activity and hypoxia-inducible factor-1α induction in response to hypoxic stress and growth factors. Cancer Res. 2005, 65, 4918–4928. [Google Scholar] [CrossRef]
- Lv, X.; Li, J.; Zhang, C.; Hu, T.; Li, S.; He, S.; Yan, H.; Tan, Y.; Lei, M.; Wen, M.; et al. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017, 4, 19–24. [Google Scholar] [CrossRef]
- Li, Z.; You, Q.; Zhang, X. Small-Molecule modulators of the hypoxia-inducible factor pathway: Development and therapeutic applications. J. Med. Chem. 2019, 62, 5725–5749. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Arambula, J.F.; Koo, S.; Kumar, R.; Singh, H.; Sessler, J.L.; Kim, J.-S. Hypoxia-targeted drug delivery. Chem. Soc. Rev. 2019, 48, 771–813. [Google Scholar] [CrossRef] [PubMed]
- Faig, M.; Bianchet, M.A.; Talalay, P.; Chen, S.; Winski, S.; Ross, D.; Amzel, L.M. Structures of recombinant human and mouseNAD(P)H:quinone oxidoreductases: Species comparison and structural changes with substrate binding and release. Proc. Natl. Acad. Sci. USA 2000, 97, 3177–3182. [Google Scholar] [CrossRef]
- Song, Y.; Wang, S.; Zhao, M.; Yang, X.; Yu, B. Strategies targeting protein tyrosine phosphatase SHP2 for cancer therapy. J. Med. Chem. 2022, 65, 3066–3079. [Google Scholar] [CrossRef]
- Homan, K.T.; Balasubramaniam, D.; Zabell, A.P.R.; Wiest, O.; Helquist, P.; Stauffacher, C.V. Identification of novel inhibitors for a low molecular weight protein tyrosine phosphatase via virtual screening. Bioorganic Med. Chem. 2010, 18, 5449–5456. [Google Scholar] [CrossRef] [PubMed]
- Defant, A.; Guella, G.; Mancini, I. Synthesis and in vitro cytotoxicity evaluation of novel naphthindolizinedione derivatives. Arch. Pharm. Chem. Life Sci. 2007, 340, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Chen, Y.; Pan, L.; Yang, Y.; Zheng, Q.; Hu, Y.; Wang, Y.; Zhang, L.; Sun, Y.; Li, Z.; et al. Design, synthesis and structure-activity relationship study of novel naphthoindolizine and indolizinoquinoline-5,12-dione derivatives as IDO1 inhibitors. Bioorganic Med. Chem. 2018, 26, 4886–4897. [Google Scholar] [CrossRef]
- Feng, X.; Liao, D.; Liu, D.; Ping, A.; Li, Z.; Bian, J. Development of indoleamine 2,3-dioxygenase 1 inhibitors for cancer therapy and beyond: A recent perspective. J. Med. Chem. 2020, 63, 15115–15139. [Google Scholar] [CrossRef]
- Ryu, C.-K.; Yoon, J.-H.; Song, A.L.; Im, H.A.; Kim, J.Y.; Kim, A. Synthesis and antifungal evaluation of pyrido[1,2-a]indole-1,4-diones and benzo[f]pyrido[1,2-a]indole-6,11-diones. Bioorganic Med. Chem. 2012, 22, 497–499. [Google Scholar] [CrossRef]
- Székely, R.; Rengifo-Gonzalez, M.; Singh, V.; Riabova, O.; Benjak, A.; Piton, J.; Gimino, M.; Kornobus, E.; Mizrahi, V.; Johnsson, K.; et al. 6,11-Dioxobenzo[f]pyrido[1,2-a]indoles kill mycobacterium tuberculosis by targeting iron−sulfur protein Rv0338c (IspQ), a putative redox sensor. ACS Infect. Dis. 2020, 6, 3015–3025. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, J.-W. Copper(II)-catalyzed synthesis of benzo[f]pyrido[1,2-a]indole-6,11-dione derivatives via naphthoquinone difunctionalization reaction. J. Org. Chem. 2012, 77, 1191–1197. [Google Scholar] [CrossRef]
- Sun, J.; Wang, F.; Hu, H.; Wang, X.; Wu, H.; Liu, Y. Copper(II)-catalyzed carbon−carbon triple bond cleavage of internal alkynes for the synthesis of annulated indolizines. J. Org. Chem. 2014, 79, 3992–3998. [Google Scholar] [CrossRef]
- Matsuoka, M.; Iwamoto, A.; Kitao, T. Reaction of 2,3-dichloro-1,4-naphthoquinone with dithiooxamide. Synthesis of dibenzo[b,i]thianthrene-5,7,12,14-tetrone. J. Heterocycl. Chem. 1991, 28, 1445–1447. [Google Scholar] [CrossRef]
- Trzoss, L.; Fukuda, T.; Costa-Lotufo, L.V.; Jimenez, P.; La Clair, J.J.; Fenical, W. Seriniquinone, a selective anticancer agent, induces cell death by autophagocytosis, targeting the cancer-protective protein dermcidin. Proc. Natl. Acad. Sci. USA 2014, 111, 14687–14692. [Google Scholar] [CrossRef] [PubMed]
- Hammons, J.C.; Trzoss, L.; Jimenez, P.C.; Hirata, A.S.; Costa-Lotufo, L.V.; La Clair, J.J.; Fenical, W. Advance of seriniquinone analogues as melanoma agents. ACS Med. Chem. Lett. 2019, 10, 186–190. [Google Scholar] [CrossRef]
- Burian, M.; Schittek, B. The secrets of dermcidin action. Int. J. Med. Microbiol. 2015, 305, 283–286. [Google Scholar] [CrossRef]
- Nagao, H.; Ninomiya, M.; Sugiyama, H.; Itabashi, A.; Uno, K.; Tanaka, K.; Koketsu, M. Comparative analysis of p-terphenylquinone and seriniquinone derivatives as reactive oxygen species-modulating agents. Bioorganic Med. Chem. Lett. 2022, 76, 128992. [Google Scholar] [CrossRef]
- Tuchinda, P.; Pohmakotr, M.; Munyoo, B.; Reutrakul, V.; Santisuk, T. An azaanthracene alkaloid from Polyalthia suberosa. Phytochemistry 2000, 53, 1079–1082. [Google Scholar] [CrossRef] [PubMed]
- Gandy, M.N.; Piggott, M.J. Synthesis of kalasinamide. A putative plant defense photoxin. J. Nat. Prod. 2008, 71, 866–868. [Google Scholar] [CrossRef]
- Lang, S.; Groth, U. Total syntheses of cytotoxic, naturally occurring kalasinamide, geovanine, and marcanine A. Angew. Chem. Int. Ed. 2009, 48, 911–913. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Kong, M.; Chen, R.-Y.; Yu, D.Q. Alkaloids from the roots of Goniothalamus griffithii. J. Nat. Prod. 1999, 62, 1050–1052. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Landward, M.B.; Rainier, J.D. Photoelectrocyclization reactions of amidonaphthoquinones. J. Org. Chem. 2020, 85, 4298–4311. [Google Scholar] [CrossRef] [PubMed]
- Soonthornchareonnon, N.; Suwanborirux, K.; Bavovada, R.; Patarapanich, C.; Cassady, J.M. New cytotoxic 1-azaanthraquinones and 3-aminonaphthoquinone from the stem bark of Goniothalamus marcanii. J. Nat. Prod. 1999, 62, 1390–1394. [Google Scholar] [CrossRef] [PubMed]
- Sedmera, P.; Pospisil, S.; Novak, J. New furanonaphthoquinone from Streptomyces cinnamonensis. J. Nat. Prod. 1991, 54, 870–872. [Google Scholar] [CrossRef]
- Bringmann, G.; Haagen, Y.; Gulder, T.A.M.; Gulder, T.; Heide, L. Biosynthesis of the isoprenoid moieties of furanonaphthoquinone I and endophenazine A in Streptomyces cinnamonensis DSM 1042. J. Org. Chem. 2007, 72, 4198–4204. [Google Scholar] [CrossRef]
- Simamura, E.; Hirai, K.-I.; Shimada, H.; Koyama, J.; Niwa, Y.; Shimizu, S. Furanonaphthoquinones cause apoptosis of cancer cells by inducing the production of reactive oxygen species by the mitochondrial voltage-dependent anion channel. Cancer Biol. Ther. 2006, 5, 1523–1529. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Watanabe, A.; Yutani, M.; Yamano, Y.; Sagara, M.; Nagai, S.; Saito, K.; Yamashita, M.; Ihara, M.; Iida, A. STAT3 inhibitory activity of naphthoquinones isolated from Tabebuia avellanedae. Bioorganic Med. Chem. 2020, 28, 115347. [Google Scholar] [CrossRef]
- Feng, X.; Qiu, X.; Huang, H.; Wang, J.; Xu, X.; Xu, P.; Ge, R.; Liu, X.; Li, Z.; Bian, J. Palladium(II)-catalyzed reaction of Lawsones and propargyl carbonates: Construction of 2,3-furanonaphthoquinones and evaluation as potential indoleamine 2,3-dioxygenase inhibitors. J. Org. Chem. 2018, 83, 8003–8010. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Wu, Z.-Z.; Jang, Y.-J.; Lee, C.-J.; Lin, W. A versatile and practical method for the regioselective synthesis of polysubstituted furanonapthoquinones. Org. Biomol. Chem. 2013, 11, 828–834. [Google Scholar] [CrossRef]
- Lin, C.-Y.; Wu, H.-Y.; Hsu, Y.-L.; Cheng, T.-J.R.; Liu, J.-H.; Huang, R.-J.; Hsiao, T.-H.; Wang, C.-J.; Hung, P.-F.; Lan, A.; et al. Suppression of drug-resistant non-small-cell lung cancer with inhibitors targeting minichromosomal maintenance protein. J. Med. Chem. 2020, 63, 3172–3187. [Google Scholar] [CrossRef]
- Tsai, C.-J.; Chen, C.-C.; Tsai, C.-W.; Wu, M.J. Base-mediated cyclization reaction of 2-(5-hydroxy-1-pentynyl)benzonitriles to 4-amino-2,3-dihydronaphtho[2,3-b]furanes and synthesis of furanonaphthoquinones. J. Org. Chem. 2016, 81, 3882–3889. [Google Scholar] [CrossRef] [PubMed]
- El-Feraly, F.S. Melampolides from Magnolia grandiflora. Phytochemistry 1984, 23, 2372–2374. [Google Scholar] [CrossRef]
- Nasim, S.; Pei, S.; Hagen, F.K.; Jordan, C.T.; Crooks, P.A. Melampomagnolide B: A new antileukemic sesquiterpene. Bioorganic Med. Chem. 2011, 19, 1515–1519. [Google Scholar] [CrossRef]
- Janganati, V.; Ponder, J.; Jordan, C.T.; Borrelli, M.J.; Penthala, N.R.; Crooks, P.A. Dimers of melampomagnolide B exhibit potent anticancer activity against hematological and solid tumor cells. J. Med. Chem. 2015, 58, 8896–8906. [Google Scholar] [CrossRef] [PubMed]
- Penthala, N.R.; Balasubramaniam, M.; Dachavaram, S.S.; Morris, E.J.; Bhat-Nakshatri, P.; Ponder, J.; Jordan, C.T.; Nakshatri, H.; Crooks, P.A. Antitumor properties of novel sesquiterpene lactone analogs as NFκB inhibitors that bind to the IKKβ ubiquitin-like domain (ULD). Eur. J. Med. Chem. 2021, 224, 113675. [Google Scholar] [CrossRef] [PubMed]
- Mischenko, N.P.; Fedoreyev, S.A.; Pokhilo, N.D.; Anufriev, V.P.; Denisenko, V.A.; Glazunov, V.P. Echinamines A and B, first aminated hydroxynaphthazarins from the sea urchin Scaphechinus mirabilis. J. Nat. Prod. 2005, 68, 1390–1393. [Google Scholar] [CrossRef]
- Mishchenko, N.P.; Krylova, N.V.; Iunikhina, O.V.; Vasileva, E.A.; Likhatskaya, G.N.; Pislyagin, E.A.; Tarbeeva, D.V.; Dmitrenok, P.S.; Fedoreyev, S.A. Antiviral potential of sea urchin aminated spinochromes against herpes simplex virus type 1. Mar. Drugs 2020, 18, 550. [Google Scholar] [CrossRef]
- You, H.; Vegi, S.R.; Lagishetti, C.; Chen, S.; Reddy, R.S.; Yang, X.; Guo, J.; Wang, C.; He, Y. Synthesis of bioactive 3,4-dihydro-2H-naphtho[2,3-b][1,4]oxazine-5,10-dione and 2,3,4,5-tetrahydro-1H-naphtho[2,3-b]azepine-6,11-dione derivatives via the copper-catalyzed intramolecular coupling reaction. J. Org. Chem. 2018, 83, 4119–4130. [Google Scholar] [CrossRef]
- Kirkpatrick, D.L.; Powis, G. Inhibitors of Redox Signaling for Restoration of Apoptosis and Inhibition of Abnormal Cell Proliferation. WO2000006088 A2, 2 October 2000. [Google Scholar]
- Liu, L.; Richard, J.; Kim, S.; Wojcik, E.J. Small molecule screen for candidate antimalarials targeting plasmodium kinesin-5. J. Biol. Chem. 2014, 289, 16601–16614. [Google Scholar] [CrossRef]
- Tomek, P.; Palmer, B.D.; Flanagan, J.U.; Sun, C.; Raven, E.L.; Ching, L.-M. Discovery and evaluation of inhibitors to the immunosuppressive enzyme indoleamine 2,3-dioxygenase 1 (IDO1): Probing the active site-inhibitor interactions. Eur. J. Med. Chem. 2017, 126, 983–996. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Soni, I.; Kelkar, D.S.; Dharmaraja, A.T.; Sankar, R.K.; Beniwal, G.; Rajendran, A.; Tamhankar, S.; Chopra, S.; Kamat, S.S.; et al. Chemoproteomics of an indole-based quinone epoxide identifies druggable vulnerabilities in vancomycin-resistant Staphylococcus aureus. J. Med. Chem. 2019, 62, 6785–6795. [Google Scholar] [CrossRef] [PubMed]
- Bergamasco, R.; Porter, Q.N.; Yap, C. Vinylindenes and some heteroanalogues in the Diels-Alder reaction. V. 3-Vinylindoles and quinones. Aust. J. Chem. 1978, 31, 1841–1844. [Google Scholar] [CrossRef]
- Kuo, C.-W.; Konala, A.; Lin, L.; Chiang, T.-T.; Huang, C.-Y.; Yang, T.-H.; Kavala, V.; Yao, C.-F. Synthesis of benzo[a]carbazole derivatives from 3-ethylindoles by exploiting the dual character of benzoquinone as an oxidizing agent and dienophile. Chem. Commun. 2016, 52, 7870–7873. [Google Scholar] [CrossRef] [PubMed]
- Brimble, M.A.; Duncalf, L.J.; Nairn, M.R. Pyranonaphthoquinone antibiotics—Isolation, structure and biological activity. Nat. Prod. Rep. 1999, 16, 267–281. [Google Scholar] [CrossRef]
- Naysmith, B.J.; Hume, P.A.; Sperry, J.; Brimble, M.A. Pyranonaphthoquinones—Isolation, biology and synthesis: An update. Nat. Prod. Rep. 2017, 34, 25–61. [Google Scholar] [CrossRef]
- Bergy, M.E. Kalafungin, a new broad spectrum antibiotic: Isolation and characterization. J. Antibiot. 1968, 21, 454–457. [Google Scholar] [CrossRef]
- Kakinuma, S.; Ikeda, H.; Omura, S.; Hopwood, D.A. Biosynthesis of kalafungin in Streptomyces tanashiensis. J. Antibiot. 1990, 43, 391–396. [Google Scholar] [CrossRef]
- Omura, S.; Tanaka, H.; Okada, Y.; Marumo, H. Isolation and structure of nanaomycin D, an enantiomer of the antibiotic kalafungin. J. Chem. Soc. Chem. Commun. 1976, 1976, 320–321. [Google Scholar] [CrossRef]
- Heapy, A.M.; Patterson, A.V.; Smaill, J.B.; Jamieson, S.M.F.; Guise, C.P.; Sperry, J.; Hume, P.A.; Rathwell, K.; Brimble, M.A. Synthesis and cytotoxicity of pyranonaphthoquinone natural product analogues under bioreductive conditions. Bioorganic Med. Chem. 2013, 21, 7971–7980. [Google Scholar] [CrossRef]
- Tanaka, H.; Minami-Kakinuma, S.; Omura, S. Biosynthesis of nanaomycin. III. Nanaomycin A formation from nanaomycin D by nanaomycin D reductase via a hydroquinone. J. Antibiot. 1982, 35, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Salaski, E.J.; Krishnamurthy, G.; Ding, W.-D.; Yu, K.; Insaf, S.S.; Eid, C.; Shim, J.; Levin, J.I.; Tabei, K.; Toral-Barza, L.; et al. Pyranonaphthoquinone lactones: A new class of AKT selective kinase inhibitors alkylate a regulatory loop cysteine. J. Med. Chem. 2009, 52, 2181–2184. [Google Scholar] [CrossRef]
- Kuck, D.; Caulfield, T.; Lyko, F.; Medina-Franco, J.L. Nanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cells. Mol. Cancer Ther. 2010, 9, 3015–3023. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, N.; Kobayashi, M.; Wakisaka, Y.; Kawamura, Y.; Mayama, M.; Matsumoto, K. New antibiotics, griseusins A and B. Isolation and characterization. J. Antibiot. 1976, 29, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Masamoto, K.; Takeuchi, Y.; Takeda, K.; Yoshii, E. Synthesis of pentacyclic structure of griseusin A. Heterocycles 1981, 16, 1659–1664. [Google Scholar]
- Brimble, M.A.; Nairn, M.R. Synthesis of the Griseusin A ring system. J. Chem. Soc. Perkin Trans. 1 1990, 1990, 169–171. [Google Scholar] [CrossRef]
- Brimble, M.A.; Nairn, M.R. Synthesis of a pyranonaphthoquinone-spiroacetal. J. Chem. Soc. Perkin Trans. 1 1992, 1992, 579–583. [Google Scholar] [CrossRef]
- Brimble, M.A.; Nairn, M.R.; Park, J. Synthesis of Analogues of Griseusin, A. Org. Lett. 1999, 1, 1459–1462. [Google Scholar] [CrossRef]
- Perry, N.B.; Blunt, J.W.; McCombs, J.D.; Munro, M.H.G. Discorhabdin C, a highly cytotoxic pigment from a sponge of the genus Latrunculia. J. Org. Chem. 1986, 51, 5476–5478. [Google Scholar] [CrossRef]
- Hu, J.-F.; Fan, H.; Xiong, J.; Wu, S.-B. Discorhabdins and pyrroloiminoquinone-related alkaloids. Chem. Rev. 2011, 111, 5465–5491. [Google Scholar] [CrossRef]
- Copp, B.R.; Fulton, K.F.; Perry, N.B.; Blunt, J.W.; Munro, M.H.G. Natural and synthetic derivatives of discorhabdin C, a cytotoxic pigment from the New Zealand sponge Latrunculia cf. bocagei. J. Org. Chem. 1994, 59, 8233–8238. [Google Scholar] [CrossRef]
- Na, M.K.; Ding, Y.; Wang, B.; Tekwani, B.L.; Schinazi, R.F.; Franzblau, S.; Kelly, M.; Stone, R.; Li, X.-C.; Ferreira, D.; et al. Anti-infective discorhabdins from a deep-water Alaskan sponge of the genus Latrunculia. J. Nat. Prod. 2010, 73, 383–387. [Google Scholar] [CrossRef]
- Goey, A.K.L.; Chau, C.H.; Sissung, T.M.; Cook, K.M.; Venzon, D.J.; Castro, A.; Ransom, T.R.; Henrich, C.J.; McKee, T.C.; McMahon, J.B.; et al. Screening and biological effects of marine pyrroloiminoquinone alkaloids: Potential inhibitors of the HIF-1α/p300 interaction. J. Nat. Prod. 2016, 79, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Mąkosza, M.; Stalewski, J.; Maslennikova, O.S. Synthesis of 7,8-dimethoxy-4-oxo-1,3,4,5-tetrahydropyrrolo[4,3,2-de]quinoline. A key intermediate en route to makaluvamines, discorhabdin C and other marine alkaloids of this group via vicarious nucleophilic substitution of hydrogen. Synthesis 1997, 1997, 1131–1133. [Google Scholar] [CrossRef]
- Roberts, D.; Joule, J.A.; Bros, M.A.; Alvarez, M. Synthesis of pyrrolo[4,3,2-de]quinolines from 6,7-dimethoxy-4-methylquinoline. Formal total syntheses of damirones A and B, batzelline C, isobatzelline C, discorhabdin C, and makaluvamines A–D. J. Org. Chem. 1997, 62, 568–577. [Google Scholar] [CrossRef]
- Kita, Y.; Tohma, H.; Inagaki, M.; Hatanaka, K.; Yakura, T. Total synthesis of discorhabdin C: A general aza spiro dienone formation from O-silylated phenol derivatives using a hypervalent iodine reagent. J. Am. Chem. Soc. 1992, 114, 2175–2180. [Google Scholar] [CrossRef]
- Sadanandan, E.V.; Pillai, S.K.; Lakshmikantham, M.V.; Billimoria, A.D.; Culpepper, J.S.; Cava, M.P. Efficient syntheses of the marine alkaloids makaluvamine D and discorhabdin C: The 4,6,7-trimethoxyindole approach. J. Org. Chem. 1995, 60, 1800–1805. [Google Scholar] [CrossRef]
- Aubart, K.M.; Heathcock, C.H. A Biomimetic approach to the discorhabdin alkaloids: Total syntheses of discorhabdins C and E and dethiadiscorhabdin D. J. Org. Chem. 1999, 64, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Kublak, G.G.; Confalone, P.N. The preparation of the aza-spirobicyclic system of discorhabdin C via an intramolecular phenolate alkylation. Tetrahedron Lett. 1990, 31, 3845–3848. [Google Scholar] [CrossRef]
- Kadela-Tomanek, M.; Bebenek, E.; Chrobak, E.; Boryczka, S. 5,8-Quinolinedione scaffold as a promising moiety of bioactive agents. Molecules 2019, 24, 4115. [Google Scholar] [CrossRef]
- Bolzán, A.D.; Bianchi, M.S. Genotoxicity of streptonigrin: A review. Mutat. Res. 2001, 488, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Rao, K.V.; Cullen, W.P. Streptonigrin, an antitumor substance. In Antibiotics Annual: 1959–1960; Welch, H., Martilhanez, F., Eds.; Medical Encyclopedia, Inc.: New York, NY, USA, 1960; pp. 950–953. [Google Scholar]
- Doyle, T.W.; Balitz, D.M.; Grulich, R.E.; Nettleton, D.E.; Gould, S.J.; Tann, C.-H.; Moews, A.E. Structure determination of lavendamycin- a new antitumor antibiotic from Streptomyces lavendulae. Tetrahedron Lett. 1981, 22, 4595–4598. [Google Scholar] [CrossRef]
- Behforouz, M.; Cai, W.; Stocksdale, M.G.; Lucas, J.S.; Jung, J.; Briere, D.; Wang, A.; Katen, K.S.; Behforouz, N.C. Novel lavendamycin analogues as potent HIV-reverse transcriptase inhibitors: Synthesis and evaluation of anti-reverse transcriptase activity of amide and ester analogues of lavendamycin. J. Med. Chem. 2003, 46, 5773–5780. [Google Scholar] [CrossRef] [PubMed]
- Hassani, M.; Cai, W.; Koelsch, K.H.; Holley, D.C.; Rose, A.S.; Olang, F.; Lineswala, J.P.; Holloway, W.G.; Gerdes, J.M.; Behforouz, M.; et al. Lavendamycin antitumor agents: Structure-based design, synthesis, and NAD(P)H:quinone oxidoreductase 1 (NQO1) model validation with molecular docking and biological studies. J. Med. Chem. 2008, 51, 3104–3115. [Google Scholar] [CrossRef] [PubMed]
- Porter, T.H.; Skelton, F.S.; Folkers, K. Synthesis of new alkylamino- and alkylaminomethyl-5,8-quinolinequinones as inhibitors of Coenzyme Q and as antimalarials. J. Med. Chem. 1971, 14, 1029–1033. [Google Scholar] [CrossRef]
- Cossy, J.; Belotti, D.; Brisson, M.; Skoko, J.J.; Wipf, P.; Lazo, J.S. Biological evaluation of newly synthesized quinoline-5,8-quinones as Cdc25B inhibitors. Bioorganic Med. Chem. 2006, 14, 6283–6287. [Google Scholar] [CrossRef] [PubMed]
- Santoso, K.T.; Menorca, A.; Cheung, C.-Y.; Cook, G.M.; Stocker, B.L.; Timmer, M.S.M. The synthesis and evaluation of quinolinequinones as anti-mycobacterial agents. Bioorganic Med. Chem. 2019, 27, 3532–3545. [Google Scholar] [CrossRef]
- Egu, S.A.; Ibezim, A.; Onoabedje, E.A.; Okoro, U.C. Biological and in silico evaluation of quinolinedione and naphthoquinone derivatives as potent antibacterial agents. ChemistrySelect 2017, 2, 9222–9226. [Google Scholar] [CrossRef]
- Ciftci, H.I.; Bayrak, N.; Yıldız, M.; Yıldırım, H.; Sever, B.; Tateishi, H.; Otsuka, M.; Fujita, M.; Tuyun, A.F. Design, synthesis and investigation of the mechanism of action underlying anti-leukemic effects of the quinolinequinones as LY83583 analogs. Bioorganic Chem. 2021, 114, 105160. [Google Scholar] [CrossRef]
- Ryu, C.-K.; Jeong, H.-J.; Lee, S.K.; You, H.J.; Choi, K.U.; Shim, J.Y.; Heo, Y.H.; Lee, C.-O. Effects of 6-arylamino-5,8-quinolinediones and 6-chloro-7-arylamino-5,8-isoquinolinediones on NAD(P)H:quinone oxidoreductase (NQO1) activity and their cytotoxic potential. Arch. Pharm. Res. 2001, 24, 390–396. [Google Scholar] [CrossRef]
- Fryatt, T.; Pettersson, H.I.; Gardipee, W.T.; Bray, K.C.; Green, S.J.; Slawin, A.M.Z.; Beall, H.D.; Moody, C.J. Novel quinolinequinone antitumor agents: Structure-metabolism studies with NAD(P)H:quinone oxidoreductase (NQO1). Bioorganic Med. Chem. 2004, 12, 1667–1687. [Google Scholar] [CrossRef] [PubMed]
- Schlager, J.J.; Powis, G. Cytosolic NAD(P)H:(quinone-acceptor)oxidoreductase in human normal and tumor tissue: Effects of cigarette smoking and alcohol. Int. J. Cancer 1990, 45, 403–409. [Google Scholar] [CrossRef]
- Cerecetto, H.; González, M.; Lavaggi, M.L.; Azqueta, A.; de Cerain, A.L.; Monge, A. Phenazine 5,10-dioxide derivatives as hypoxic selective cytotoxins. J. Med. Chem. 2005, 48, 21–23. [Google Scholar] [CrossRef]
- Lavaggi, M.L.; Cabrera, M.; Pintos, C.; Arredondo, C.; Pachon, G.; Rodriguez, J.; Raymondo, S.; Pacheco, J.P.; Cascante, M.; Olea-Azar, C.; et al. Novel phenazine 5,10-dioxides release •OH in simulated hypoxia and induce reduction of tumour volume in vivo. ISRN Pharmacol. 2011, 2011, 314209. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.B.; Williamson, S.K. Tirapazamine: A novel agent targeting hypoxic tumor cells. Expert Opin. Investig. Drugs 2009, 18, 77–87. [Google Scholar] [CrossRef]
- Abi-Jaoudeh, N.; Dayyani, F.; Chen, P.J.; Fernando, D.; Fidelman, N.; Javan, H.; Liang, P.-C.; Hwang, J.-I.; Imagawa, D.K. Phase I trial on arterial embolization with hypoxia activated tirapazamine for unresectable hepatocellular carcinoma. J. Hepatocell. Carcinoma 2021, 8, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.H.; Tanabe, K.; Saggiomo, A.J.; Nodiff, E.A. Modifications of primaquine as antimalarials. 4. 5-Alkoxy derivatives of primaquine. J. Med. Chem. 1987, 30, 1193–1199. [Google Scholar] [CrossRef]
- Shelke, Y.G.; Hande, P.E.; Gharpure, S.J. Recent advances in the synthesis of pyrrolo[1,2-a]indoles and their derivatives. Org. Biomol. Chem. 2021, 19, 7544–7574. [Google Scholar] [CrossRef]
- Bass, P.D.; Gubler, D.A.; Judd, T.C.; Williams, R.M. Mitomycinoid alkaloids: Mechanism of action, biosynthesis, total syntheses, and synthetic approaches. Chem. Rev. 2013, 113, 6816–6863. [Google Scholar] [CrossRef]
- Liu, L.; Wang, Y.; Zhang, L. Formal synthesis of 7-methoxymitosene and synthesis of its analog via a key PtCl2-catalyzed cycloisomerization. Org. Lett. 2012, 14, 3736–3739. [Google Scholar] [CrossRef]
- Zheng, Z.; Touve, M.; Barnes, J.; Reich, N.; Zhang, L. Synthesis-enabled probing of mitosene structural space leads to improved IC50 over mitomycin C. Angew. Chem. Int. Ed. 2014, 53, 9302–9305. [Google Scholar] [CrossRef]
- Ayala, C.E.; Villalpando, A.; Nguyen, A.L.; McCandless, G.T.; Kartika, R. Chlorination of aliphatic primary alcohols via triphosgene–triethylamine activation. Org. Lett. 2012, 14, 3676–3679. [Google Scholar] [CrossRef]
- Sweeney, M. Hydrogen Peroxide and Hydrohalic acid Mediated Synthesis of Halogenated Benzimidazolequinones and Anti-cancer Evaluation of Benzotriazinones. Ph.D. Thesis, National University of Ireland Galway, Galway, Ireland, 2019. [Google Scholar]
- Lee, S.-R.; Yang, K.-S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.L.; Pitt, A.R.; Spickett, C.M. Approaches to investigating the protein interactome of PTEN. J. Proteome Res. 2021, 20, 60–77. [Google Scholar] [CrossRef] [PubMed]
- Meuillet, E.J.; Mahadevan, D.; Berggren, M.; Coon, A.; Powis, G. Thioredoxin-1 binds to the C2 domain of PTEN inhibiting PTEN’s lipid phosphatase activity and membrane binding: A mechanism for the functional loss of PTENs tumor suppressor activity. Arch. Biochem. Biophys. 2004, 429, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.; Zhang, J.; Yao, J.; Liu, Y.; Fang, J. Targeting thioredoxin reductase by parthenolide contributes to inducing apoptosis of HeLa Cells. J. Biol. Chem. 2016, 291, 10021–10031. [Google Scholar] [CrossRef]
- Madkour, M.M.; Anbar, H.S.; El-Gamal, M.I. Current status and future prospects of p38a/MAPK14 kinase and its inhibitors. Eur. J. Med. Chem. 2021, 213, 113216. [Google Scholar] [CrossRef]
- Paz, M.M.; Zhang, X.; Lu, J.; Holmgren, A. A new mechanism of action for the anticancer drug mitomycin C: Mechanism-based inhibition of thioredoxin reductase. Chem. Res. Toxicol. 2012, 25, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Newsome, J.J.; Swann, E.; Hassani, M.; Bray, K.C.; Slawin, A.M.Z.; Beall, H.D.; Moody, C.J. Indolequinone antitumour agents: Correlation between quinone structure and rate of metabolism by recombinant human NAD(P)H:quinone oxidoreductase. Org. Biomol. Chem. 2007, 5, 1629–1640. [Google Scholar] [CrossRef]
- Yan, C.; Siegel, D.; Newsome, J.J.; Chilloux, A.; Moody, C.J.; Ross, D. Antitumor indolequinones induced apoptosis in human pancreatic cancer cells via inhibition of thioredoxin reductase and activation of redox signaling. Mol. Pharmacol. 2012, 81, 401–410. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Soriga, S.G.; Socea, L.I.; Olaru, O.T.; Plesu, V. Structure-activity relationships and chemoinformatic analysis of the anticancer profile of an aminopyrazole derivative. Rev. Chim. 2016, 67, 162–165. [Google Scholar]


































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
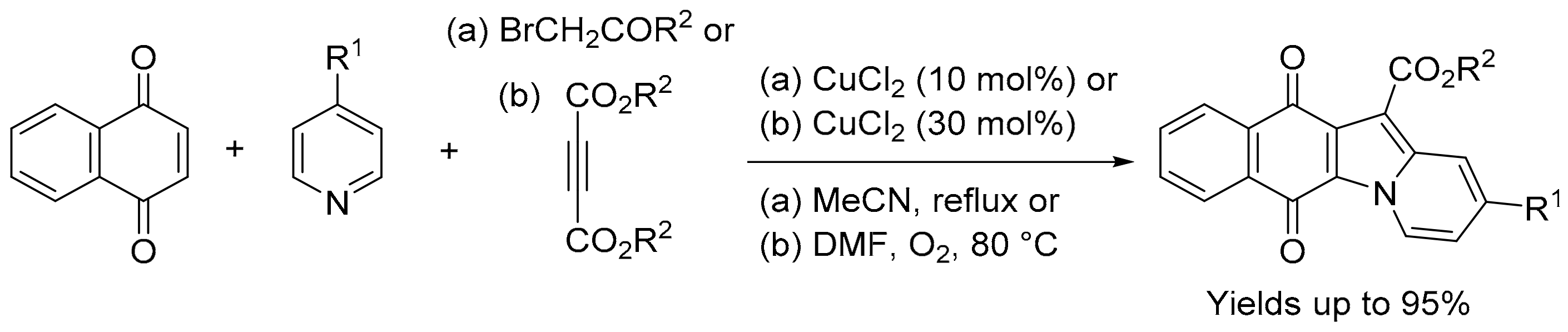{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | NQO1 | TrxR |
|---|---|---|
| 5a | −0.37 | −0.24 |
| 5b | −0.30 | −0.12 |
| Pleurotin | −0.27 | −0.21 |
| 14b | −0.48 | −0.22 |
| Kalafungin | −0.27 | −0.24 |
| Discorhabdin C | −0.29 | −0.37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haji, N.; Faizi, M.; Koutentis, P.A.; Carty, M.P.; Aldabbagh, F. Heterocyclic Iminoquinones and Quinones from the National Cancer Institute (NCI, USA) COMPARE Analysis. Molecules 2023, 28, 5202. https://doi.org/10.3390/molecules28135202
Haji N, Faizi M, Koutentis PA, Carty MP, Aldabbagh F. Heterocyclic Iminoquinones and Quinones from the National Cancer Institute (NCI, USA) COMPARE Analysis. Molecules. 2023; 28(13):5202. https://doi.org/10.3390/molecules28135202
Chicago/Turabian StyleHaji, Naemah, Masoma Faizi, Panayiotis A. Koutentis, Michael P. Carty, and Fawaz Aldabbagh. 2023. "Heterocyclic Iminoquinones and Quinones from the National Cancer Institute (NCI, USA) COMPARE Analysis" Molecules 28, no. 13: 5202. https://doi.org/10.3390/molecules28135202
APA StyleHaji, N., Faizi, M., Koutentis, P. A., Carty, M. P., & Aldabbagh, F. (2023). Heterocyclic Iminoquinones and Quinones from the National Cancer Institute (NCI, USA) COMPARE Analysis. Molecules, 28(13), 5202. https://doi.org/10.3390/molecules28135202