



Synthesis, Structure and Photochemistry of Dibenzylidenecyclobutanones
 , , , and
, , , and
Abstract
1. Introduction
2. Results and Discussion
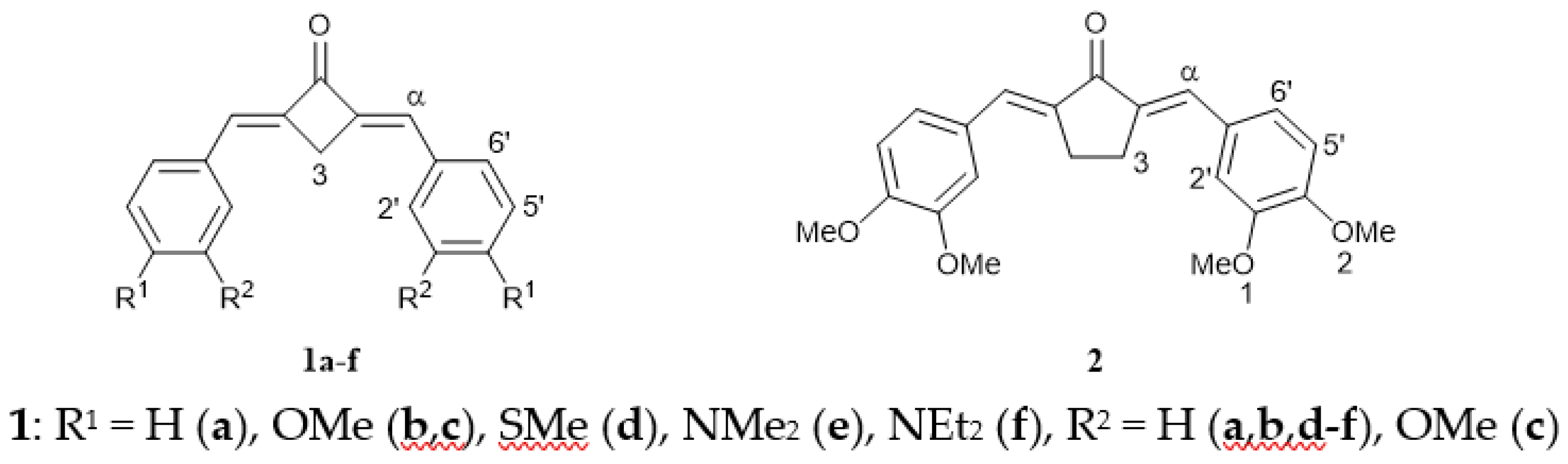
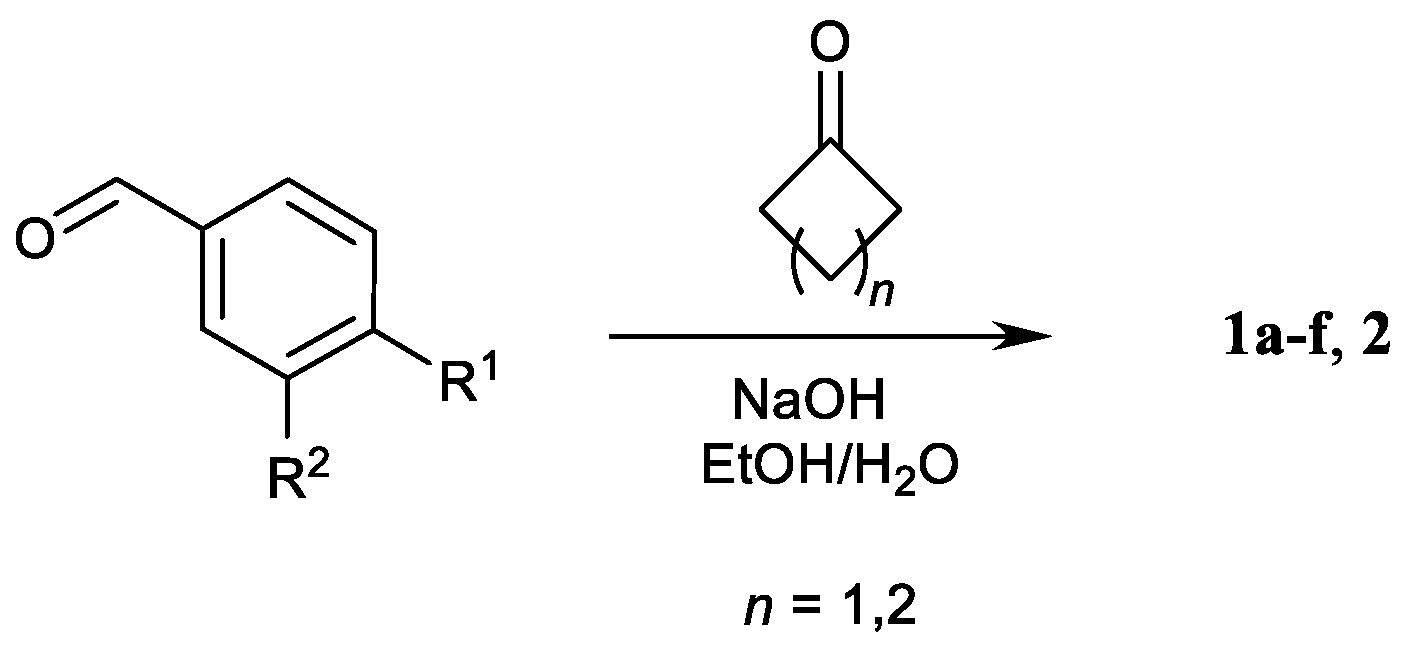
2.1. Synthesis of 1a–f
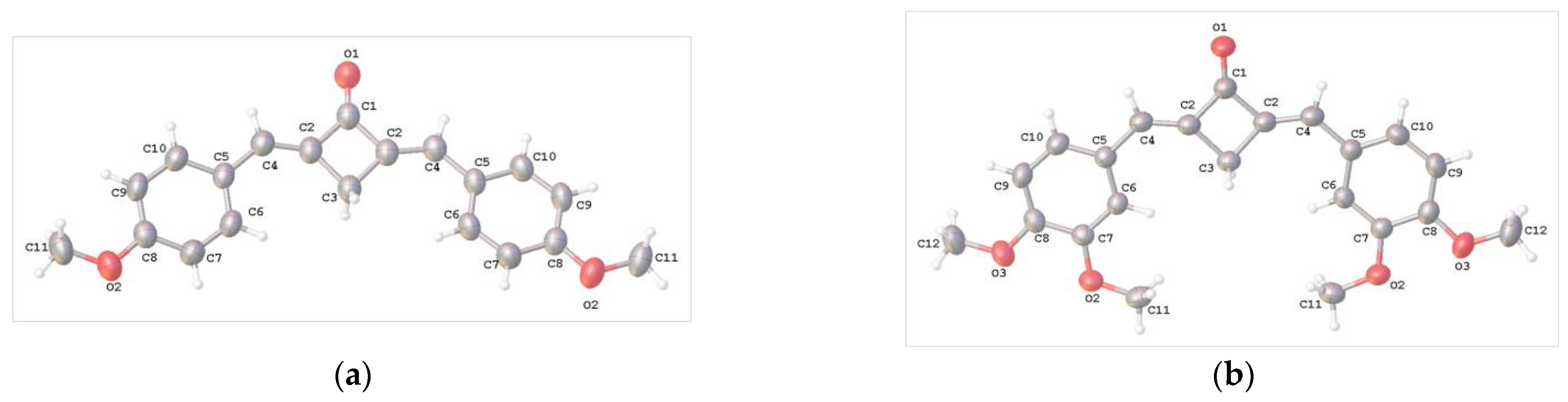
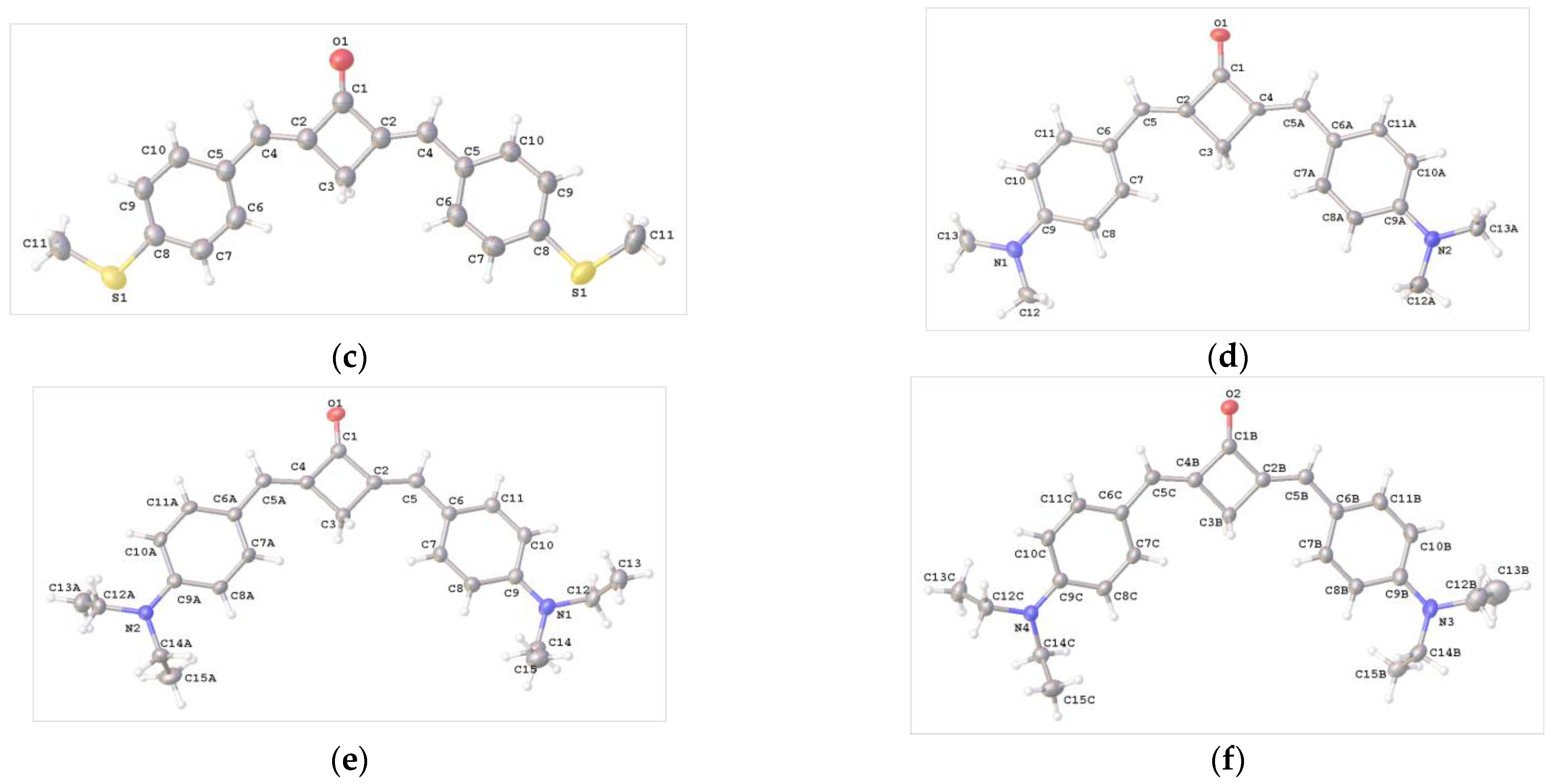
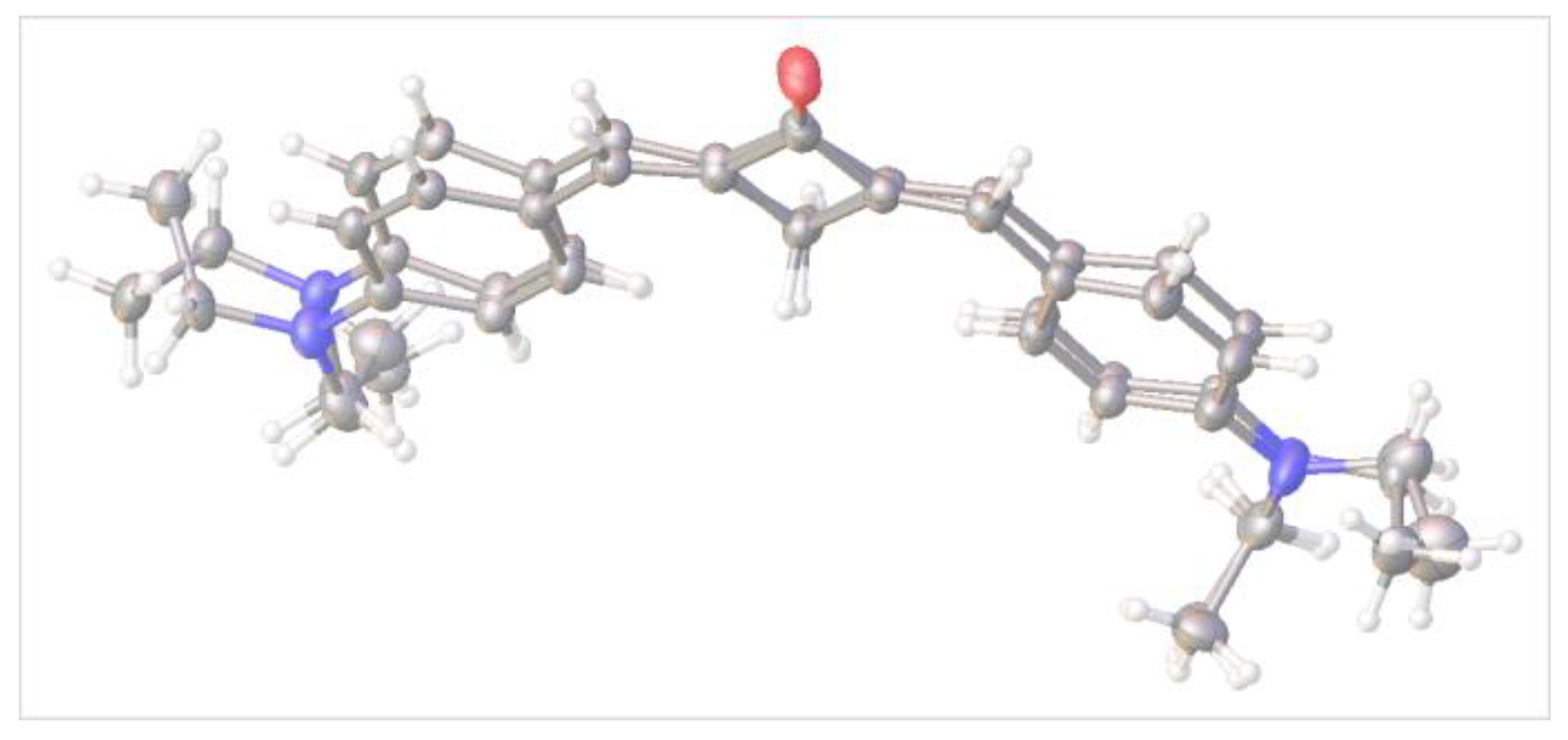
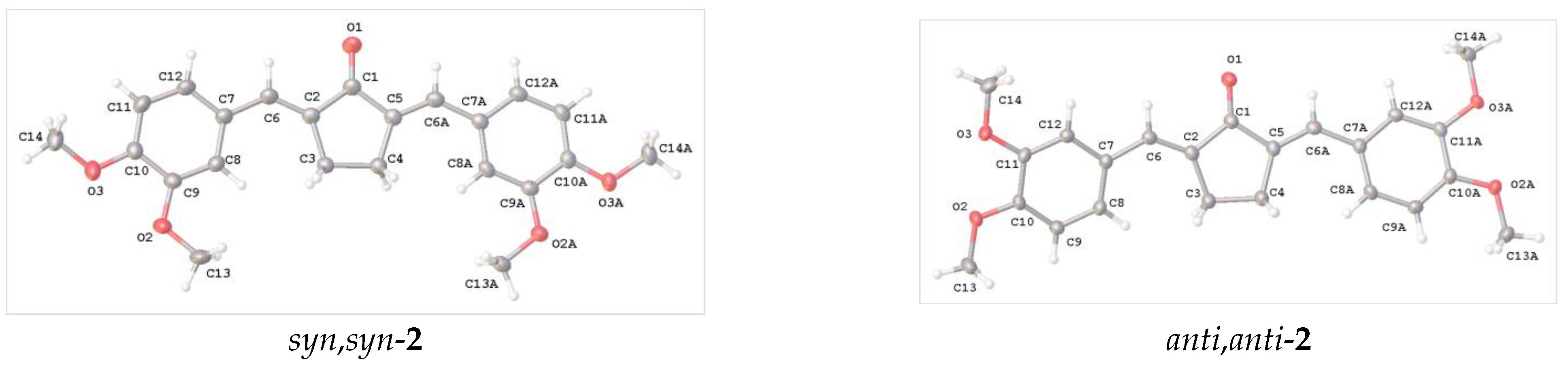
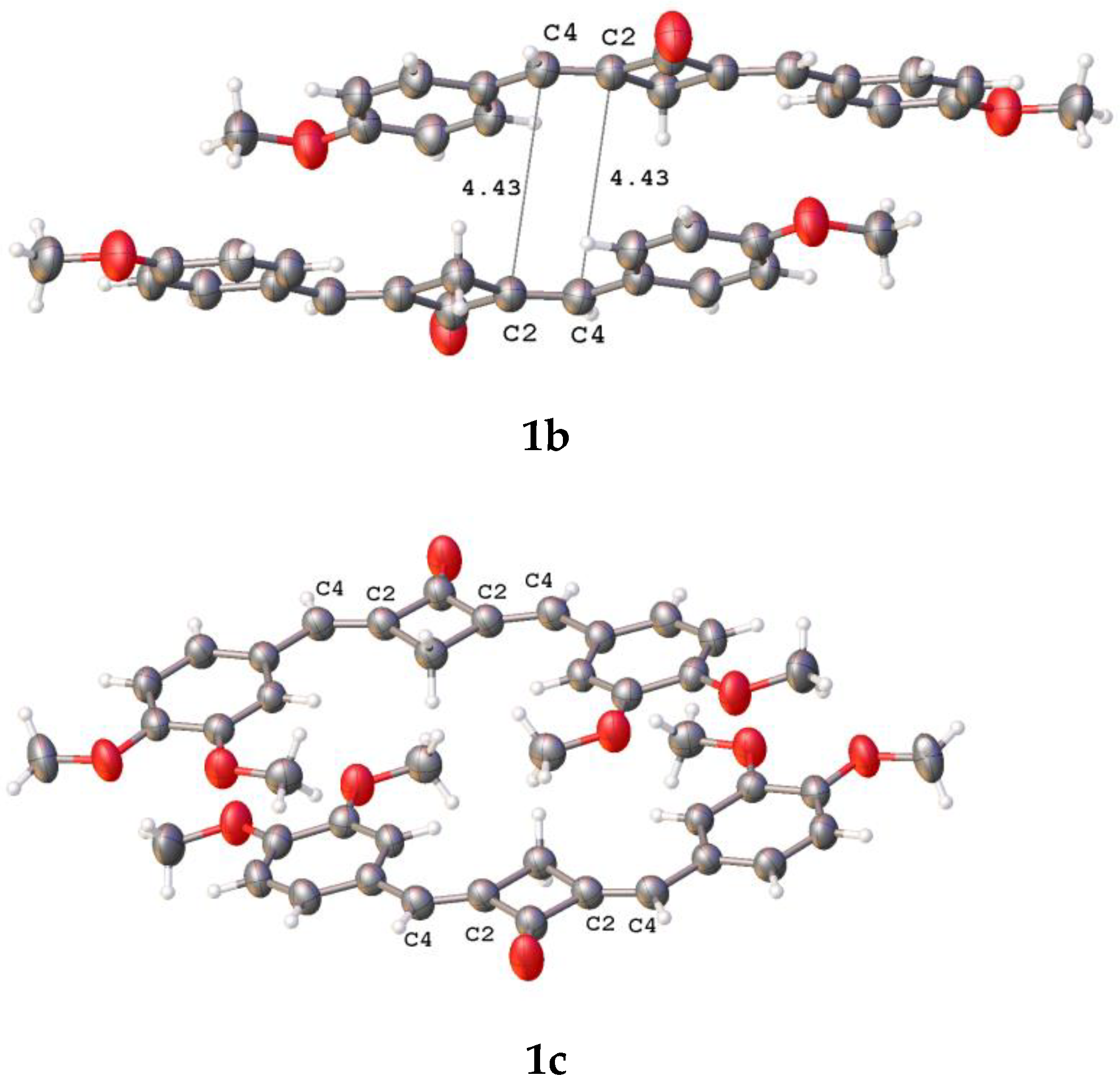
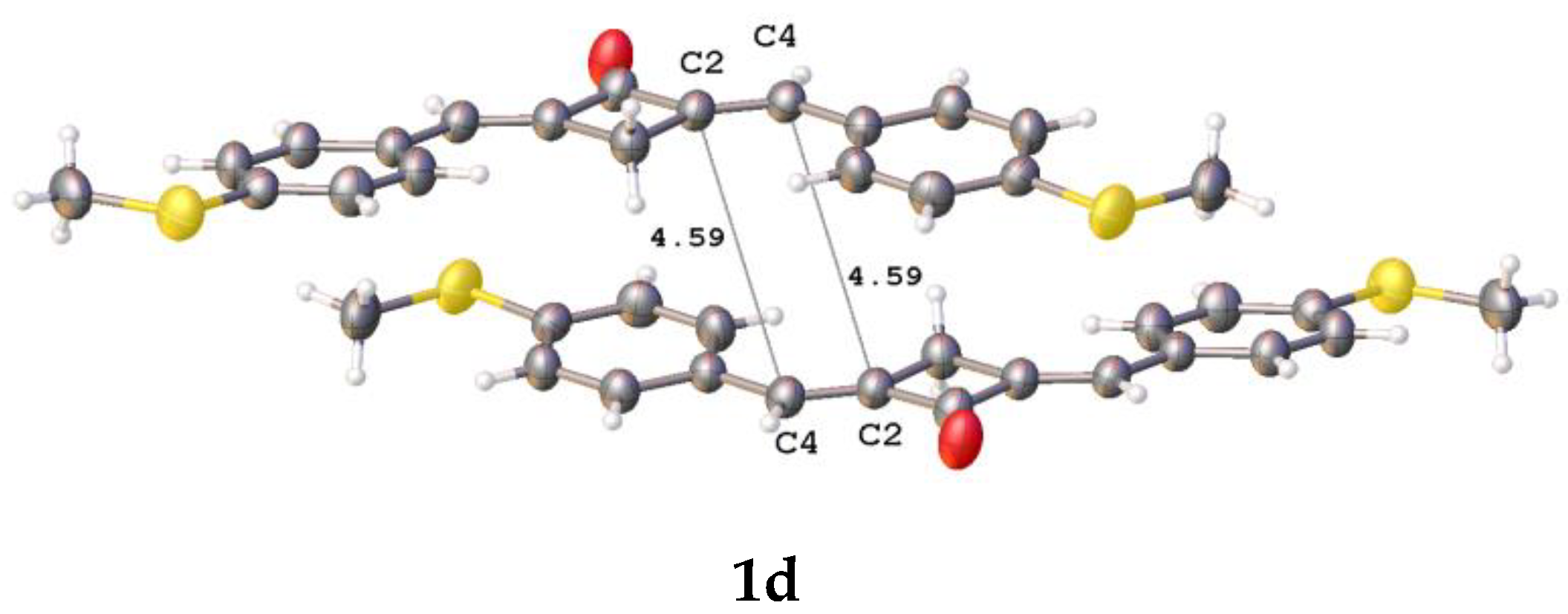
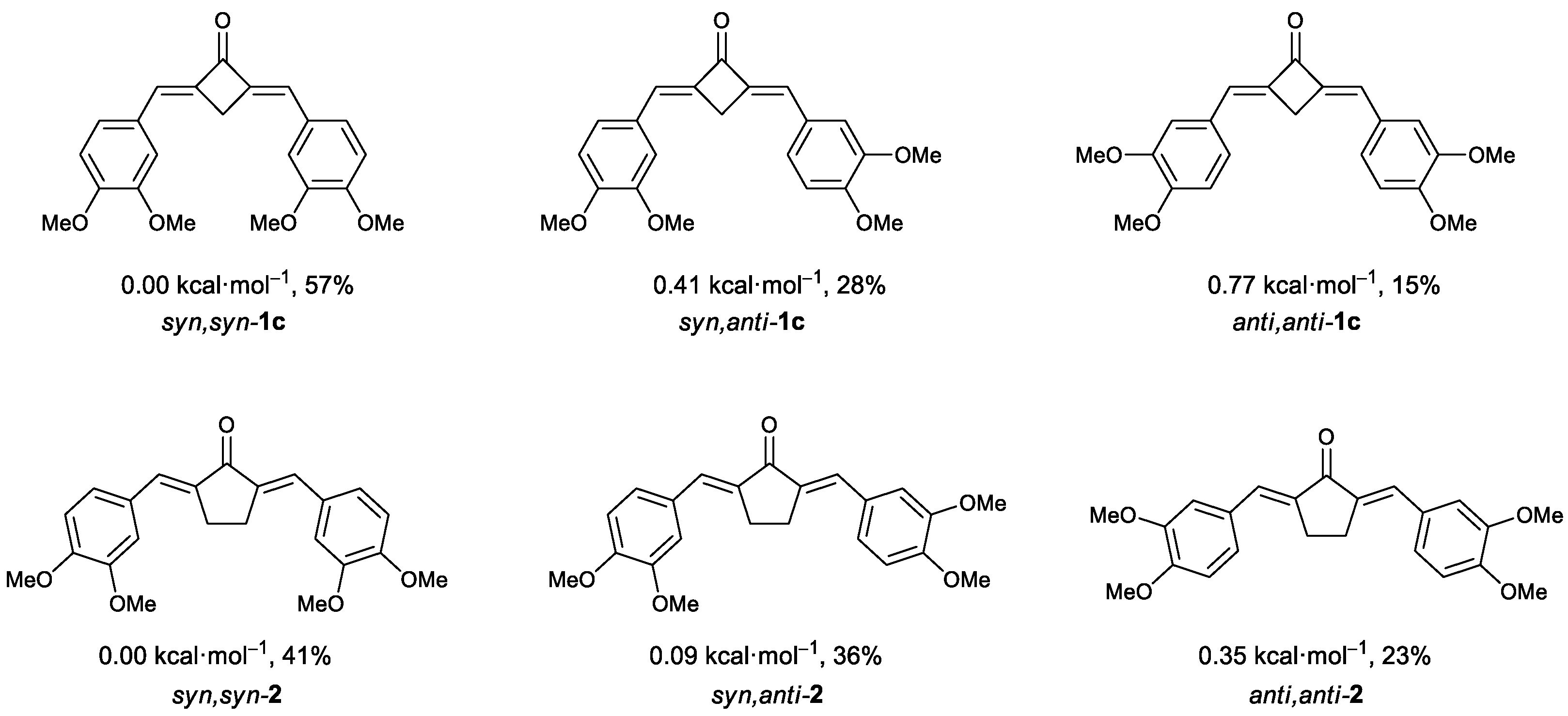
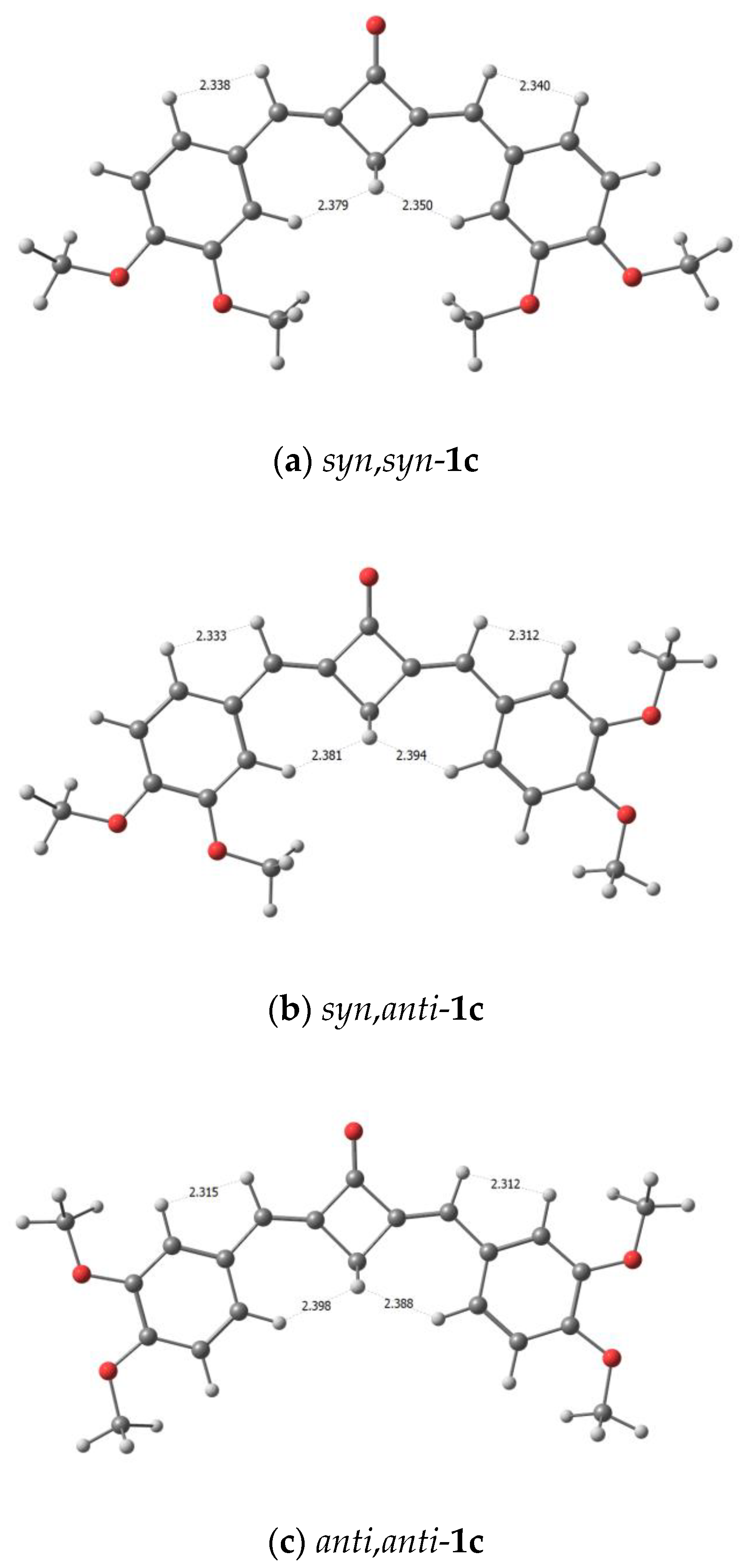
2.2. X-ray Diffraction Analysis
2.3. NMR Spectroscopy
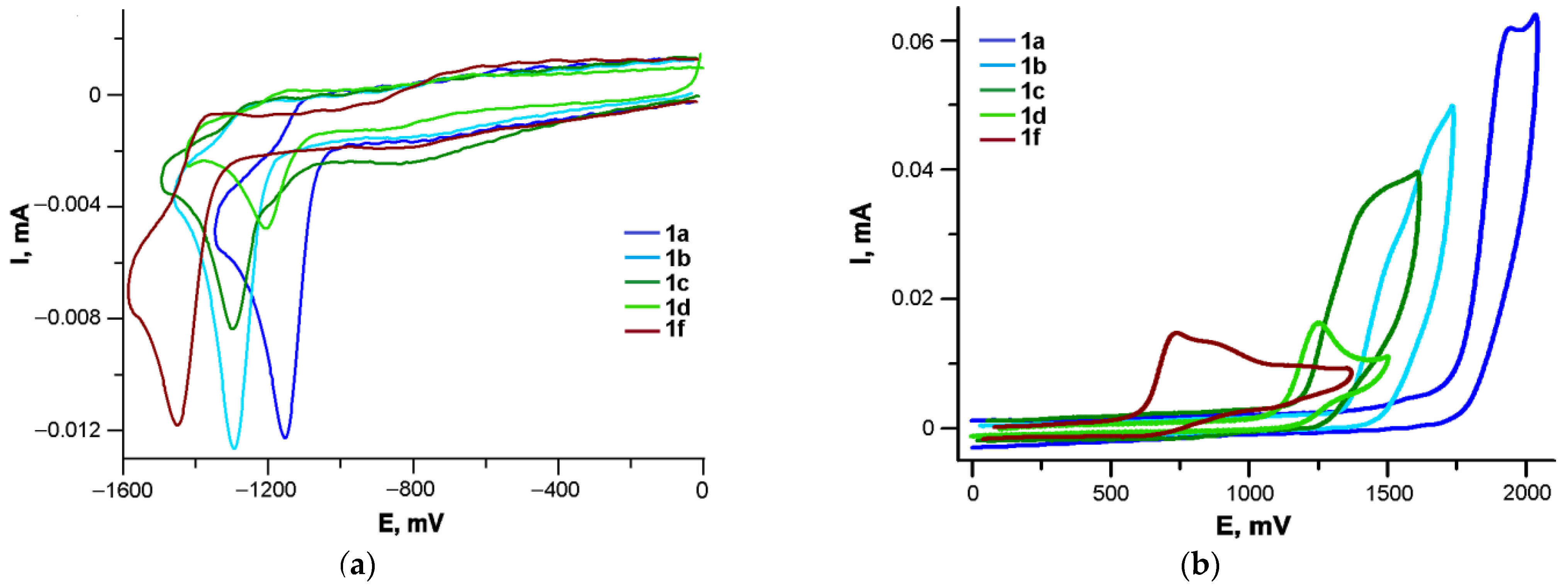
2.4. Electrochemistry
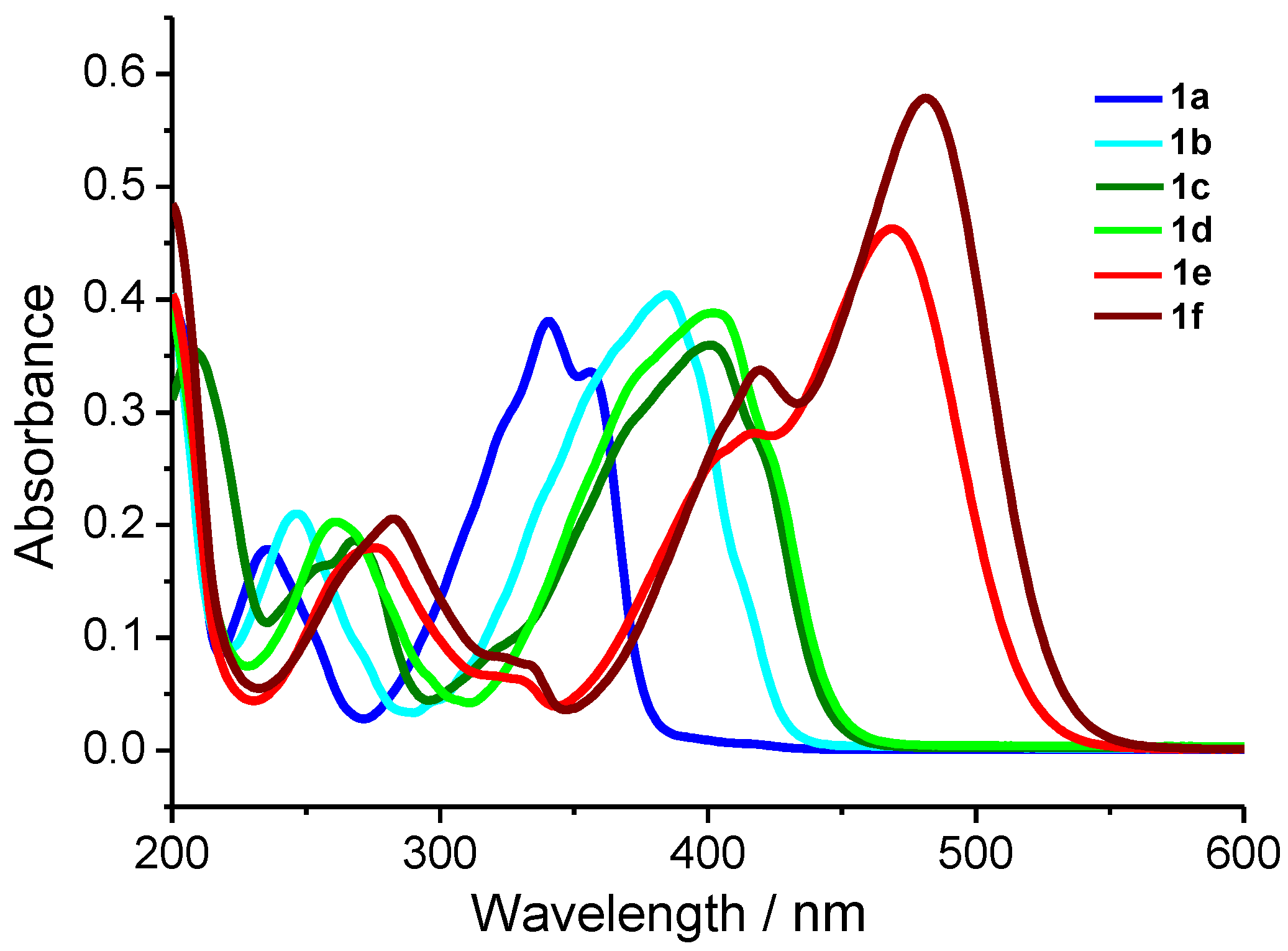
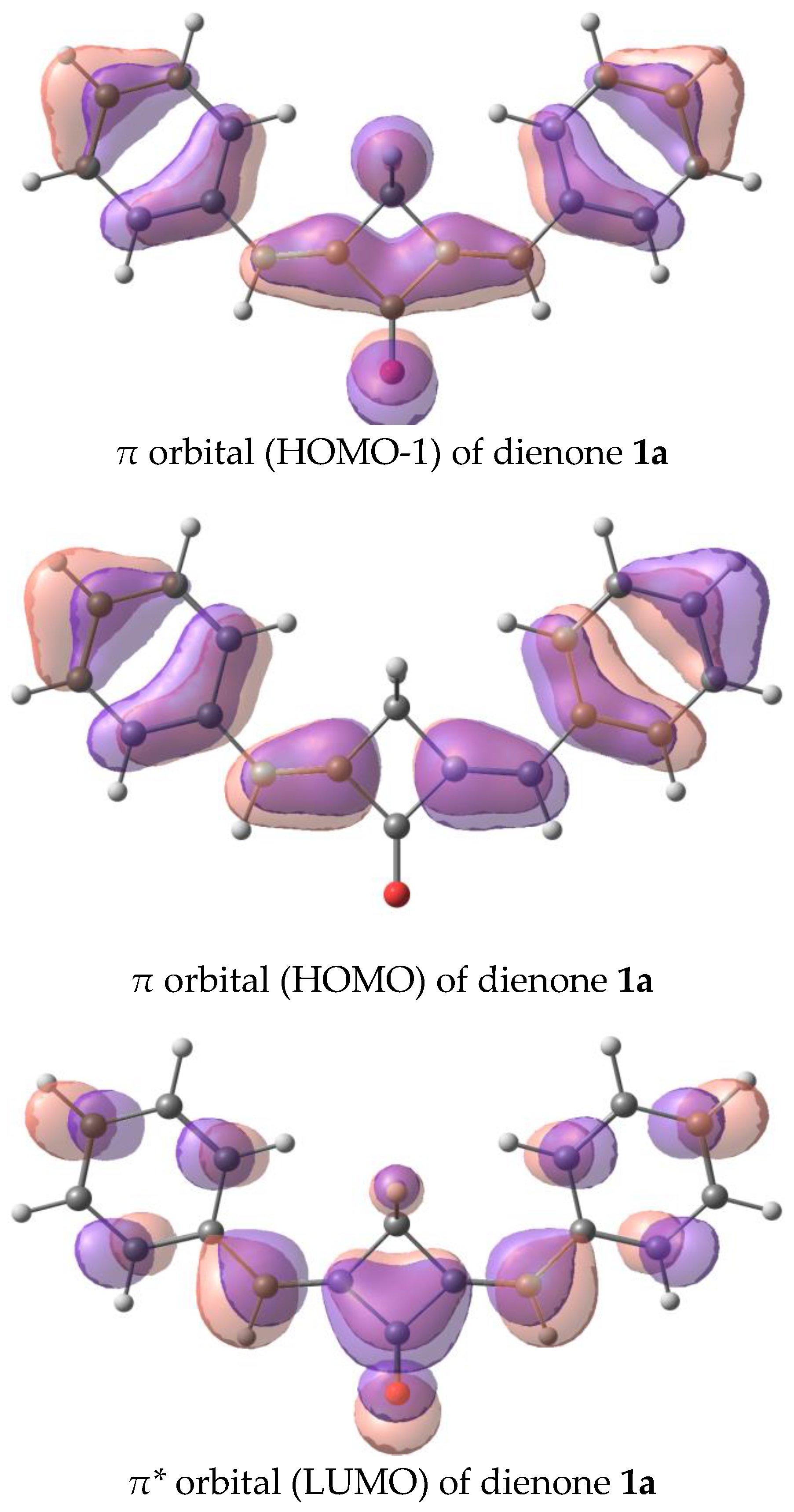
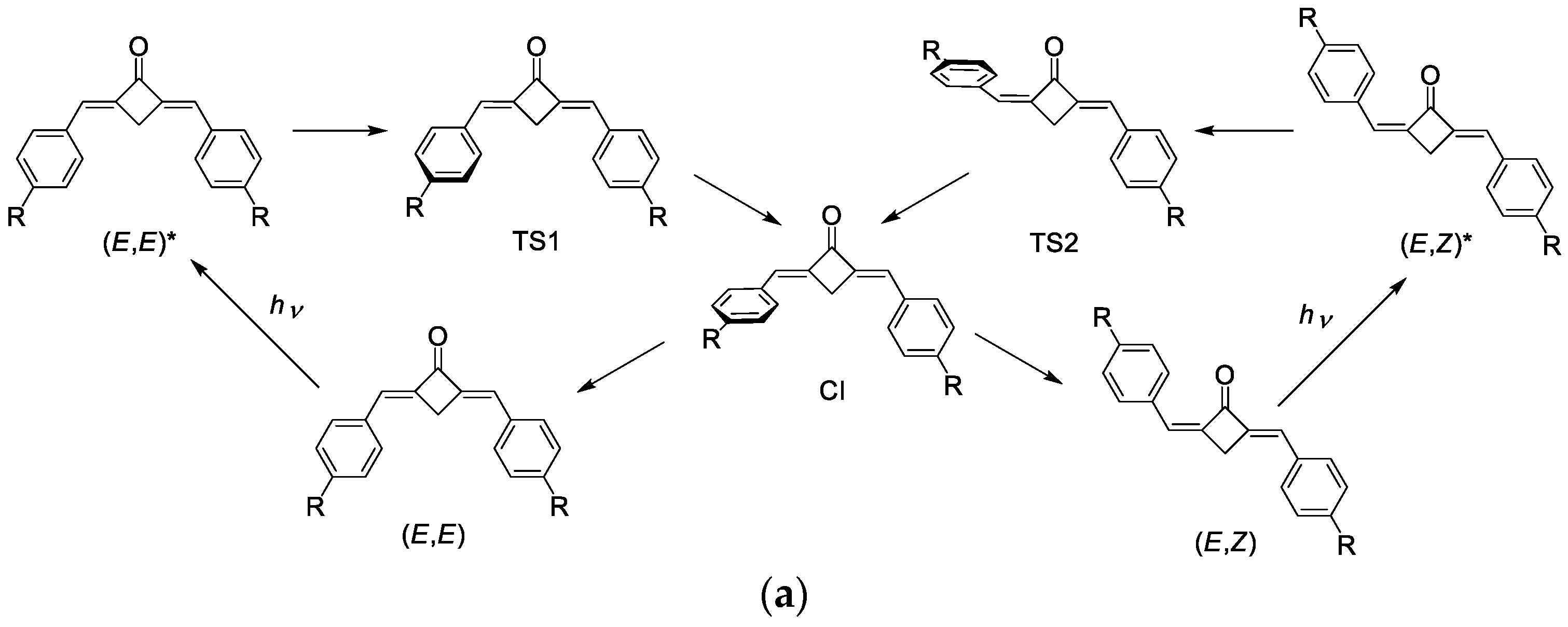
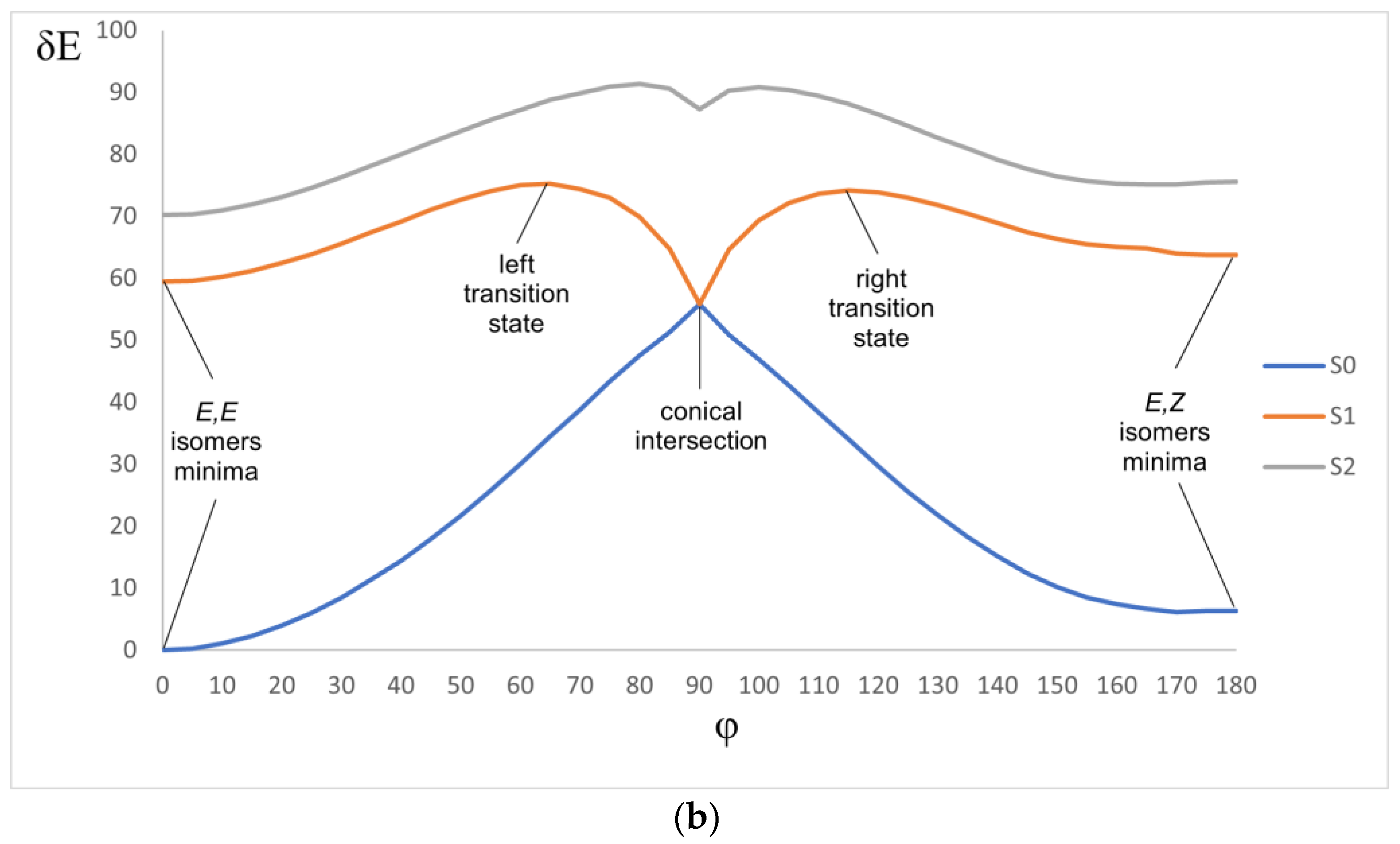
2.5. Photophysics
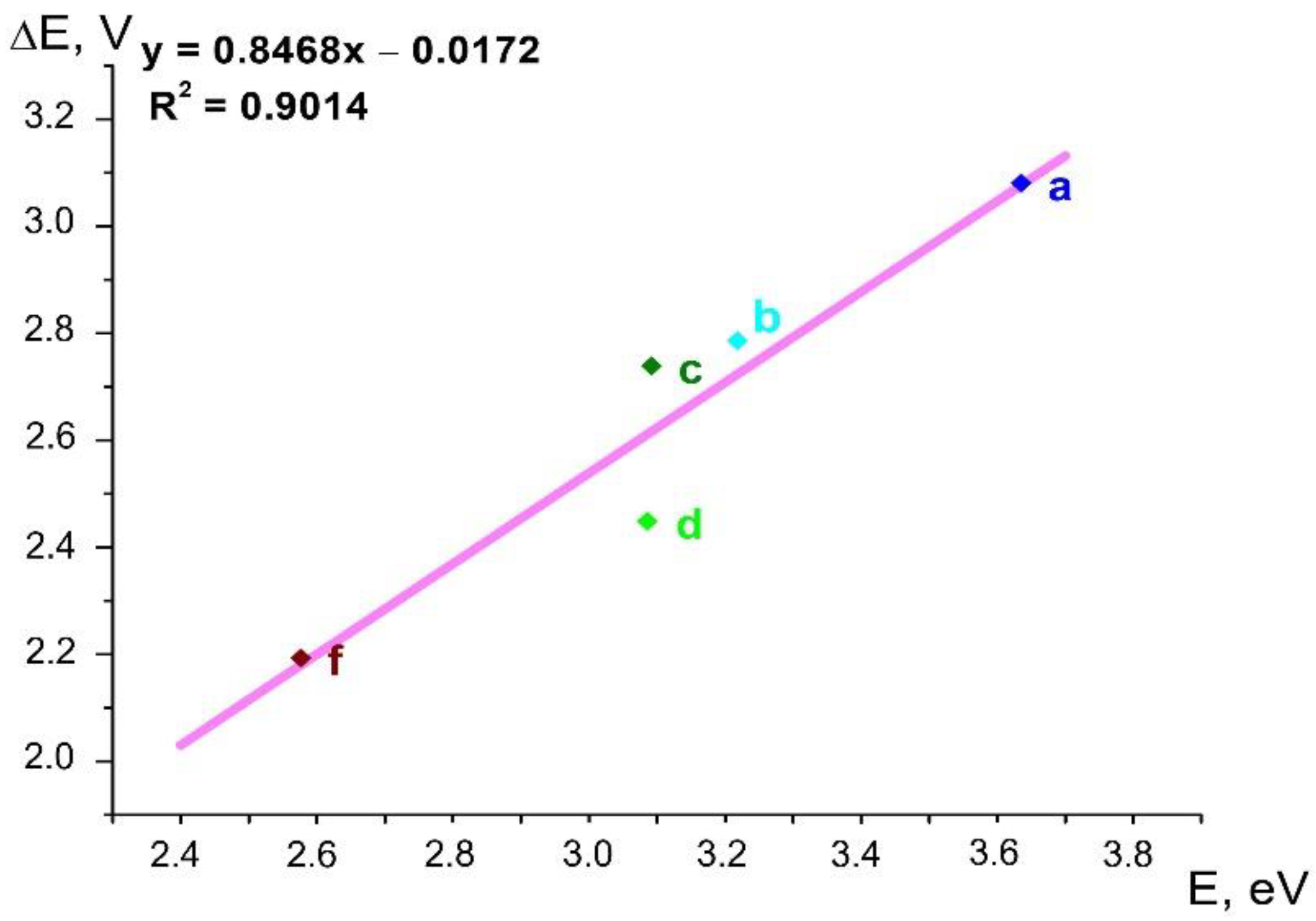
2.6. Correlations
3. Materials and Methods
3.1. Materials
Synthesis of 2,6-Dibenzylidenecyclobutanone Derivatives 1a-f
3.2. Methods
3.3. Cyclic Voltammetry
3.4. X-ray Diffraction Experiments
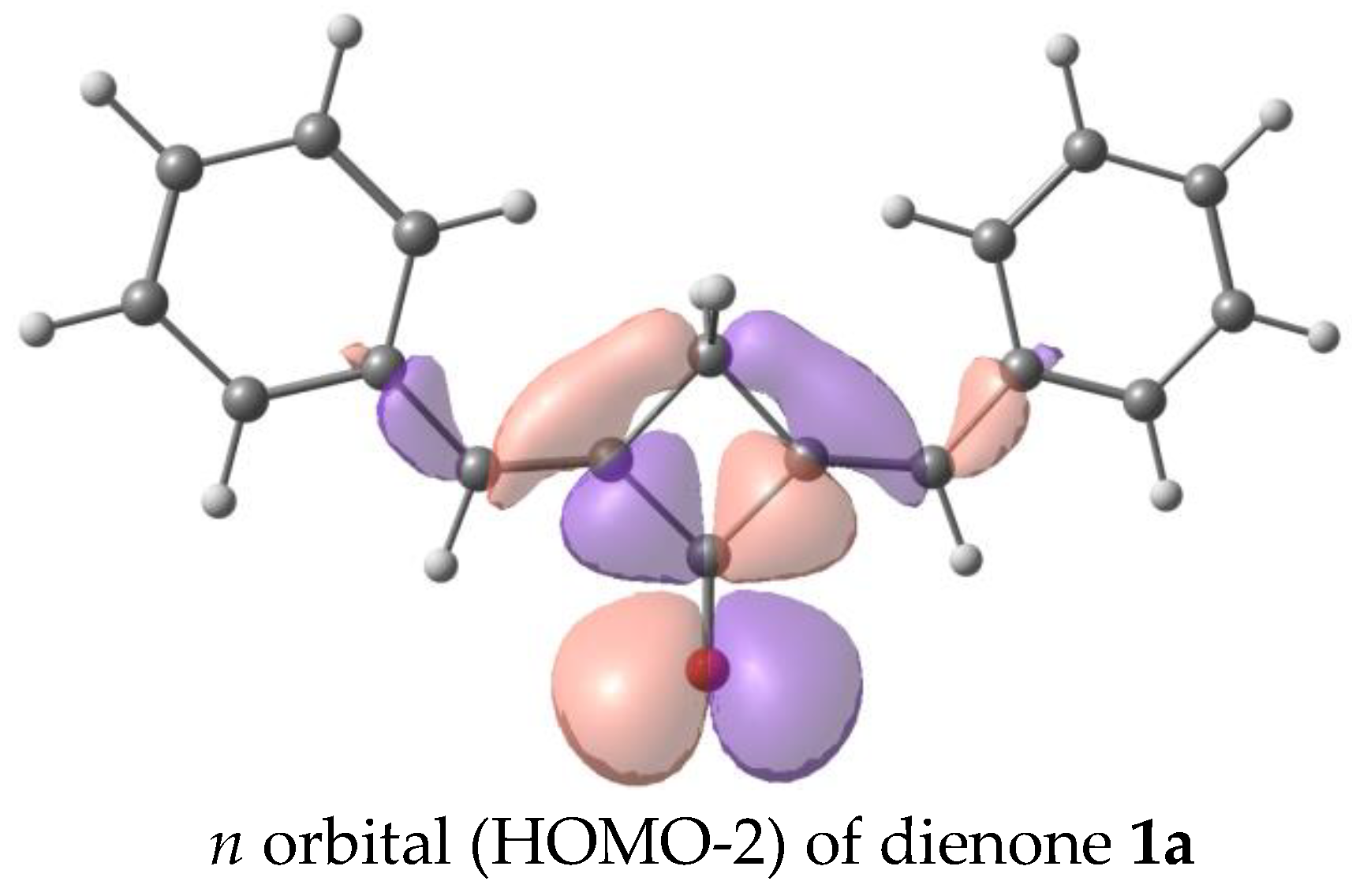
3.5. Density Functional Theory (DFT) Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Adams, R.V. Organic Reactions; Adams, R., Ed.; Wiley & Sons, Inc.: London, UK; New York, NY, USA, 1959; Volume 10, 576p. [Google Scholar]
- Vatsadze, S.Z.; Golikov, A.G.; Kriven’ko, A.P.; Zyk, N.V. Chemistry of cross-conjugated dienones. Russ. Chem. Rev. 2008, 77, 707–728. [Google Scholar] [CrossRef]
- Cui, J.; Crich, D.; Wink, D.; Lam, M.; Rheingold, A.L.; Case, D.A.; Fu, W.T.; Zhou, Y.; Rao, M.; Olson, A.J.; et al. Design and synthesis of highly constrained factor Xa inhibitors: Amidine-Substituted bis(benzoyl)-1,3-diazepan-2-ones and bis(benzylidene)-bis(gem-dimethyl)cycloketones. Bioorg. Med. Chem. 2003, 11, 3379–3392. [Google Scholar] [CrossRef]
- Jin, R.; Chen, Q.; Yao, S.; Bai, E.; Fu, W.; Wang, L.; Wang, J.; Du, X.; Wei, T.; Xu, H.; et al. Synthesis and anti-tumor activity of EF24 analogues as IKKβ inhibitors. Eur. J. Med. Chem. 2018, 144, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Cersosimo, U.; Sgorbissa, A.; Foti, C.; Drioli, S.; Angelica, R.; Tomasella, A.; Picco, R.; Semrau, M.S.; Storici, P.; Benedetti, F.; et al. Synthesis, Characterization, and Optimization for in Vivo Delivery of a Nonselective Isopeptidase Inhibitor as New Antineoplastic Agent. J. Med. Chem. 2015, 58, 1691–1704. [Google Scholar] [CrossRef] [PubMed]
- Gangadhara; Kishore, K. Novel photocrosslinkable liquid-crystalline polymers: Poly[bis(benzylidene)] esters. Macromolecules 1993, 26, 2995–3003. [Google Scholar] [CrossRef]
- Kannan, P.; Gangadhara; Kishore, K. Novel photocrosslinkable flame retardant polyvanillylidene arylphosphate esters. Polymer 1997, 38, 4349–4355. [Google Scholar] [CrossRef]
- Yakimansky, A.V.; Tenkovtsev, A.V.; Dudkina, M.M.; Voigt-Martin, I.G.; Kolb, U.; Lukoshkin, V.A.; Böhme, F. Studies of structures and properties of polymeric systems containing bis-(hydroxy-arylidene)alkanones as NLO-active chromophores. J. Non-Cryst. Solids 2002, 303, 237–245. [Google Scholar] [CrossRef]
- Doroshenko, A.O.; Sychevskaya, L.B.; Grygorovych, A.V.; Pivovarenko, V.G. Fluorescence Probing of Cell Membranes with Azacrown Substituted Ketocyanine Dyes. J. Fluoresc. 2002, 12, 455–464. [Google Scholar] [CrossRef]
- Demianov, M.J.; Dojarenko, M. Darstellung von Cyclobutanon durch pyrochemische Zersetzung der 1-Oxy-cyclobutan-1-carbonsäure. Ber. Dtsch. Chem. Ges. 1922, 55, 2737. [Google Scholar]
- Thieme, P. Notiz zur Darstellung von 2.4-Dibenzyliden-cyclobutanonen. Chem. Ber. B 1968, 101, 378–380. [Google Scholar] [CrossRef]
- Nielsen, A.T.; Weiss, R.C.; Moore, D.W. Base-Catalyzed Intermolecular Condensation of α,β-Unsaturated Ketones. Dimerization of 2,4-Diarylidenecyclobutanones to 2-Spiro(2 oxocyclobutyl)bicyclo[3.2.0]heptan-6-one Derivatives. J. Org. Chem. 1972, 37, 1086–1092. [Google Scholar] [CrossRef]
- Clark, G.R.; Lin, J.; Nikaido, M. Aldole reactions of a cyclobutanone enolate. Tetrahedron Lett. 1984, 25, 2645–2648. [Google Scholar] [CrossRef]
- Vidal, J.; Huet, F. Synthesis of α-Methylenecyclobutanones. The First Preparation of Norsarkomycin Methyl Ester. J. Org. Chem. 1988, 53, 611–616. [Google Scholar] [CrossRef]
- Zou, Q.; Zhao, Y.; Makarov, N.S.; Campo, J.; Yuan, H.; Fang, D.-C.; Perry, J.W.; Feipeng, W. Effect of alicyclic ring size on the photophysical and photochemical properties of bis(arylidene)cycloalkanone compounds. Phys. Chem. Chem. Phys. 2012, 14, 11743–11752. [Google Scholar] [CrossRef] [PubMed]
- Zou, Q.; Zhao, H.; Zhao, Y.; Fang, Y.; Chen, D.; Ren, J.; Wang, X.; Wang, Y.; Gu, Y.; Feipeng, W. Effective Two-Photon Excited Photodynamic Therapy of Xenograft Tumors Sensitized by Water-Soluble Bis(arylidene)cycloalkanone Photosensitizers. J. Med. Chem. 2015, 58, 7949–7958. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, X.-F.; Duan, X.; Zeng, F.; Wu, B.; Wu, S. Therapeutic Nanosystem Consisting of Singlet-Oxygen-Responsive Prodrug and Photosensitizer Excited by Two-Photon Light. ACS Med. Chem. Lett. 2018, 9, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Sanford, E.M.; Paulisse, K.W.; Reeves, J.T. A computational study of 2,5-dibenzylidenecyclopentanone and 2,6-dibenzylidenecyclohexanone, model compounds for poly(arylidenecycloalkanones). J. Appl. Polym. Sci. 1999, 74, 2255–2257. [Google Scholar] [CrossRef]
- Grandeury, A.; Petit, S.; Coste, S.; Coquerel, G.; Perrio, C.; Gouhier, G. New synthesis of (Z,E)-2,7-bis(4-cyanobenzylidene)cycloheptan-1-one under stereospecific constraints induced by host-guest interactions. Chem. Commun. 2005, 31, 4007–4009. [Google Scholar] [CrossRef]
- Vatsadze, S.Z.; Manaenkova, M.A.; Sviridenkova, N.V.; Zyk, N.V.; Krut’ko, D.P.; Churakov, A.V.; Antipin, M.Y.; Howard, J.A.K.; Lang, H. Synthesis and spectroscopic and structural studies of cross-conjugated dienones derived from cyclic ketones and aromatic aldehydes. Russ. Chem. Bull. 2006, 55, 1184–1194. [Google Scholar] [CrossRef]
- Aizenshtat, Z.; Hausman, M.; Pickholtz, Y.; Tal, D.; Blum, J. Chlorocarbonylbis(triphenylphosphine)iridium-catalyzed isomerization, isoaromatization, and disproportionation of some cycloalkanones having exocyclic double bonds. J. Org. Chem. 1977, 42, 2386–2394. [Google Scholar] [CrossRef]
- George, H.; Roth, H.J. Photoisomerisierung und Cyclo-1,2-Addition α,β-ungesättigter Cyclanone. Tetrahedron Lett. 1971, 12, 4057–4060. [Google Scholar] [CrossRef]
- Kaupp, G.; Zimmermann, I. First Detection of a π-Coupled 1,5-Diradical via Cycloaddition. Angew. Chem. 1981, 20, 1018–1019. [Google Scholar] [CrossRef]
- Ovchinnikova, I.G.; Nikulov, D.K.; Bartashevich, E.V.; Matochkina, E.G.; Kodess, M.I.; Slepukhin, P.A.; Druzhinin, A.V.; Fedorova, O.V.; Rusinov, G.L.; Charushin, V.N. Pre-organization of diarylideneacetonyl crownophanes in single crystals to photochemical transformations. Russ. Chem. Bull. 2011, 60, 824–840. [Google Scholar] [CrossRef]
- Alfimov, M.V.; Gromov, S.P.; Stanislavskii, O.B.; Ushakov, E.N.; Fedorova, O.A. Crown-containing styryl dyes. 8. Cation-dependent concerted [2 + 2]-autophotocycloaddition of photochromic 15-crown-5 ether betaines. Russ. Chem. Bull. 1993, 42, 1385–1389. [Google Scholar] [CrossRef]
- Gromov, S.P.; Vedernikov, A.I.; Lobova, N.A.; Kuz’mina, L.G.; Dmitrieva, S.N.; Strelenko, Y.A.; Howard, J.A.K. Synthesis, Structure, and Properties of Supramolecular Photoswitches Based on Ammonioalkyl Derivatives of Crown-Ether Styryl Dyes. J. Org. Chem. 2014, 79, 11416–11430. [Google Scholar] [CrossRef]
- Kuz’mina, L.G.; Vedernikov, A.I.; Gromov, S.P.; Alfimov, M.V. Crystallographic Approach to the [2 + 2] Photocycloaddition Topochemical Reactions of Unsaturated Compounds with Single Crystal Retention. Crystallogr. Rep. 2019, 64, 691–712. [Google Scholar] [CrossRef]
- Biradha, K.; Santra, R. Crystal engineering of topochemical solid state reactions. Chem. Soc. Rev. 2013, 42, 950–967. [Google Scholar] [CrossRef]
- Elacqua, E.; Kaushik, P.; Groeneman, R.H.; Sumrak, J.C.; Bučar, D.-K.; MacGillivray, L.R. A Supramolecular Protecting Group Strategy Introduced to the Organic Solid State: Enhanced Reactivity through Molecular Pedal Motion. Angew. Chem. Int. Ed. 2012, 51, 1037–1041. [Google Scholar] [CrossRef] [PubMed]
- Santra, R.; Garai, M.; Mondal, D.; Biradha, K. Anion Influence in Directing and Altering the Stereochemistry of the Double [2+2] Reaction of Bis-Pyridyl Dienes in their Silver Complexes: A Green Synthetic Route. Chem. Eur. J. 2013, 19, 489–493. [Google Scholar] [CrossRef]
- Santra, R.; Biradha, K. Solid state double [2 + 2] photochemical reactions in the co-crystal forms of 1,5-bis(4-pyridyl)-1,4-pentadiene-3-one: Establishing mechanism using single crystal X-ray, UV and H-1 NMR. CrystEngComm 2011, 13, 3246–3257. [Google Scholar] [CrossRef]
- Vatsadze, S.Z.; Gromov, S.P. Novel Linear Bis-Crown Receptors with Cross-Conjugated and Conjugated Central Cores. Macroheterocycles 2017, 10, 432–445. [Google Scholar] [CrossRef][Green Version]
- Fomina, M.V.; Kurchavov, N.A.; Freidzon, A.Y.; Nuriev, V.N.; Vedernikov, A.I.; Strelenko, Y.A.; Gromov, S.P. Self-assembly involving hydrogen bonds. Spectral properties and structure of supramolecular complexes of bis-aza-18-crown-6-containing dienones with alkanediammonium salts. J. Photochem. Photobiol. A 2020, 402, 112801. [Google Scholar] [CrossRef]
- Vatsadze, S.Z.; Gavrilova, G.V.; Zyuz’kevich, F.S.; Nuriev, V.N.; Krut’ko, D.P.; Moiseeva, A.A.; Shumyantsev, A.V.; Vedernikov, A.I.; Churakov, A.V.; Kuz’mina, L.G.; et al. Synthesis, structure, electrochemistry, and photophysics of 2,5-dibenzylidenecyclopentanones containing in benzene rings substituents different in polarity. Russ. Chem. Bull. 2016, 65, 1761–1772. [Google Scholar] [CrossRef]
- Fomina, M.V.; Vatsadze, S.Z.; Freidzon, A.Y.; Kuz’mina, L.G.; Moiseeva, A.A.; Starostin, R.O.; Nuriev, V.N.; Gromov, S.P. Structure–Property Relationships of dibenzylidenecyclohexanones. ACS Omega 2022, 7, 10087–10099. [Google Scholar] [CrossRef]
- Butcher, R.J.; Jasinski, J.P.; Narayana, B.; Sarojini, B.K.; Bindya, S.; Yathirajan, H.S. 2,5-Bis(3,4-dimethoxybenzylidene) cyclopentanone. Acta Cryst. 2007, E63, o3270–o3271. [Google Scholar] [CrossRef]
- Tsedilin, A.M.; Fakhrutdinov, A.N.; Eremin, D.B.; Zalesskiy, S.S.; Chizhov, A.O.; Kolotyrkina, N.G.; Ananikov, V.P. How sensitive and accurate are routine NMR and MS measurements? Mendeleev Commun. 2015, 25, 454–456. [Google Scholar] [CrossRef]
- Vedernikov, A.I.; Basok, S.S.; Gromov, S.P.; Kuz’mina, L.G.; Avakyan, V.G.; Lobova, N.A.; Kulygina, E.Y.; Titkov, T.V.; Strelenko, Y.A.; Ivanov, E.I.; et al. Synthesis and structure of bis-crown-containing stilbenes. Russ. J. Org. Chem. 2005, 41, 843–854. [Google Scholar] [CrossRef]
- Nuriev, V.N.; Fedorov, O.V.; Moiseeva, A.A.; Freidzon, A.Y.; Kurchavov, N.A.; Vedernikov, A.I.; Medved’ko, A.V.; Pod’yacheva, E.S.; Vatsadze, S.Z.; Gromov, S.P. Synthesis, structure, spectral properties, and electrochemistry of bis(crown ether) containing 1,3-distyrylbenzenes. Russ. J. Org. Chem. 2017, 53, 1726–1737. [Google Scholar] [CrossRef]
- Tsukerman, S.V.; Kutulya, L.A.; Lavrushin, V.F. Spectra and halo-chromism of dibenzylidenecycloalkanones and their thiophene and furan analogs. Zurn. Obshch. Khim. (Russ. J. Obshch. Chem.) 1964, 34, 3597. (In Russian) [Google Scholar]
- Issa, R.M.; Etaiw, S.H.; Issa, I.M.; El-Shafie, A.K. Electronic Absorption Spectra of some Diarylidene-Cyclopentanones and -Cyclohexanones. Acta Chim. Acad. Sceint. Hung. 1976, 89, 381–391. [Google Scholar]
- Gutrov, V.N.; Zakharova, G.V.; Fomina, M.V.; Nuriev, V.N.; Gromov, S.P.; Chibisov, A.K. Molecular photonics of dienones based on cycloalkanones and their derivatives. J. Photochem. Photobiol. A 2022, 425, 113678. [Google Scholar] [CrossRef]
- Freidzon, A.Y.; Bagatur’yants, A.A.; Gromov, S.P.; Alfimov, M.V. Recoordination of a metal ion in the cavity of a crown compound: A theoretical study 3. Absorption spectra and excited states of azacrown-containing styryl dyes and their complexes. Russ. Chem. Bull. 2008, 57, 2045–2055. [Google Scholar] [CrossRef]
- Al-Anber, M.; Vatsadze, S.Z.; Holze, R.; Lang, H.; Thiel, W.R. π-Conjugated N-heterocyclic compounds: Correlation of computational and electrochemical data. Dalton Trans. 2005, 22, 3632–3637. [Google Scholar] [CrossRef] [PubMed]
- Vatsadze, S.Z.; Al-Anber, M.; Thiel, W.R.; Lang, H.; Holze, R. Electrochemical studies and semiempirical calculations on pi-conjugated dienones and heterocyclic nitrogen containing donor ligand molecules. J. Solid. State. Electrochem. 2005, 9, 764–777. [Google Scholar] [CrossRef]
- Bruker. APEX2, SADABS and SAINT; Bruker AXS Inc.: Madison, WI, USA, 2008. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Pushman, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- SHELXTL-Plus, Version 5.10; Bruker AXS, Inc.: Madison, WI, USA, 1997.
- Granovsky, A.A. Firefly Version 8.2.0. Available online: http://classic.chem.msu.su/gran/firefly/index.html (accessed on 28 October 2022).
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.J.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Freidzon, A.Y.; Safonov, A.A.; Bagaturyants, A.A.; Alfimov, M.V. Solvatofluorochromism and Twisted Intramolecular Charge-Transfer State of the Nile Red Dye. Int. J. Quantum Chem. 2012, 112, 3059–3067. [Google Scholar] [CrossRef]
- Quentin, C.; Gerasimaitė, R.; Freidzon, A.; Atabekyan, L.S.; Lukinavičius, G.; Belov, V.N.; Mitronova, G.Y. Direct Visualization of Amlodipine Intervention into Living Cells by Means of Fluorescence Microscopy. Molecules 2021, 26, 2997. [Google Scholar] [CrossRef]
- Laikov, D.N. Fast evaluation of density functional exchange-correlation terms using the expansion of the electron density in auxiliary basis sets. Chem. Phys. Lett. 1997, 281, 151–156. [Google Scholar] [CrossRef]
- Laikov, D.N.; Ustynyuk, Y.A. PRIRODA-04: A quantum-chemical program suite. New possibilities in the study of molecular systems with the application of parallel computing. Russ. Chem. Bull. 2005, 54, 820–826. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protons | 1c | 2 | ||
|---|---|---|---|---|
| δ, ppm | ||||
| Experiment | Calculation | Experiment | Calculation | |
| C(3)H2 | 3.80 | 3.78 | 3.10 | 3.10 |
| H(5′) | 6.92 | 6.73 | 6.93 | 6.77 |
| H(2′) | 7.06 | 7.14 | 7.13 | 7.12 |
| H(6′) | 7.19 | 7.22 | 7.22 | 7.32 |
| CHAr (α) | 7.10 | 7.18 | 7.44 | 7.64 |
| Dienone | R1, R2 | Ered, V | EA, eV | ΔEred, mV (ΔEA) * | Eox, V | IP, eV | ΔEox, mV (ΔIP) ** | ΔE = Eox − Ered, V | EA–IP, eV |
|---|---|---|---|---|---|---|---|---|---|
| 1a | H, H | −1.15 | 2.878 | 1.92 | 6.36 | 3.07 | 3.49 | ||
| 1b | OMe, H | −1.29 | 2.716 | 140 | 1.49 | 5.87 | 430 | 2.78 | 3.16 |
| 1c | OMe, OMe | −1.30 | 2.736 | 150 | 1.44 | 5.70 | 480 | 2.73 | 2.96 |
| 1d | SMe, H | −1.20 | 2.827 | 50 | 1.24 | 5.75 | 680 | 2.44 | 2.92 |
| 1f | NEt2, H | −1.46 | 2.481 | 310 | 0.73 | 5.07 | 1190 | 2.19 | 2.98 |
| Dienone | R1, R2 | Ered, V | ||
|---|---|---|---|---|
| Cyclobutanones | Cyclopentanones [34] | Cyclohexanones [35] | ||
| 1a | H, H | −1.15 | −1.26 | −1.38 |
| 1b | OMe, H | −1.29 | −1.38 | −1.51 |
| 1c | OMe, OMe | −1.30 | −1.39 | −1.46 |
| 1d | SMe, H | −1.20 | −1.24 | −1.39 |
| 1f | NEt2, H | −1.46 | −1.53 | −1.63 |
| Dienone | λabs (ε)/nm (M−1 cm−1) | λfl/nm | ||
|---|---|---|---|---|
| Experiment (M−1 cm−1) | Calculation (Oscillator Strength) | Experiment | Calculation (Oscillator Strength, τrad, ns) | |
| 1a | 341 (38,000) 356 (33,600) | 359 (0.90) a 378 (0.00) | - | 411 (23.35 × 106) |
| 1b | 385 (40,400) | 394 (1.05) | - | 419 (2.28) c 446 (3.32) 446 (3.25) |
| 1c | 401 (36,000) | 411 (0.92) | 505 | 441 (3.05) b |
| 1d | 402 (39,000) | 433 (1.11) | 522 | 452 (2.62) c 474 (3.19) 475 (3.38) |
| 1e | 418 (28,000) 469 (46,000) | 402 (0.21) 471(1.21) | 576 | 481 (2.89) |
| 1f | 419 (33,800) 481 (58,000) | 459 (1.02) | 575 | 494 (3.07) d 517 (3.45) |
| Dienone | (E,E) | (E,Z) | (Z,Z) | |||
|---|---|---|---|---|---|---|
| Rel. Energy | Mole Fraction | Rel. Energy | Mole Fraction | Rel. Energy | Mole Fraction | |
| 1a | 0 | 0.78 | 0.99 | 0.15 | 1.44 | 0.07 |
| 1b | 0 | 0.43 | 0.08 | 0.38 | 0.48 | 0.19 |
| 1c | 0 | 0.79 | 1.04 | 0.14 | 1.45 | 0.7 |
| 1d | 0 | 0.57 | 0.76 | 0.16 | 0.43 | 0.28 |
| 1e | 0 | 0.75 | 1.08 | 0.12 | 1.03 | 0.13 |
| 1f | 0 | 0.81 | 2.03 | 0.03 | 0.93 | 0.17 |
| Dienone | Ea1, kcal/mol | Ea2, kcal/mol | CI, kcal/mol |
|---|---|---|---|
| 1a | 3.20 | 4.35 | −12.81 |
| 1b | 7.14 | 4.71 | −11.80 |
| 1c | 10.4 | 6.4 | −7.59 |
| 1d | 11.18 | 7.14 | −5.97 |
| 1e | 13.27 | 9.15 | −3.64 |
| 1f | 13.16 | 10.79 | −0.53 |
| Dienone | τr, ns | k, s−1 | ttc, ns |
|---|---|---|---|
| 1a | 23,350,824.12 | 7.04 × 1010 | 0.01 |
| 1b | 2.28 | 9.97 × 107 | 10.03 |
| 1c | 3.05 | 4.39 × 105 | 2.28 × 103 |
| 1d | 2.62 | 5.38 × 104 | 18.6 × 103 |
| 1e | 2.89 | 3.44 × 102 | 2.91 × 105 |
| 1f | 3.07 | 2.42 × 103 | 4.13 × 105 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fomina, M.V.; Freidzon, A.Y.; Kuz’mina, L.G.; Moiseeva, A.A.; Starostin, R.O.; Kurchavov, N.A.; Nuriev, V.N.; Gromov, S.P. Synthesis, Structure and Photochemistry of Dibenzylidenecyclobutanones. Molecules 2022, 27, 7602. https://doi.org/10.3390/molecules27217602
Fomina MV, Freidzon AY, Kuz’mina LG, Moiseeva AA, Starostin RO, Kurchavov NA, Nuriev VN, Gromov SP. Synthesis, Structure and Photochemistry of Dibenzylidenecyclobutanones. Molecules. 2022; 27(21):7602. https://doi.org/10.3390/molecules27217602
Chicago/Turabian StyleFomina, Marina V., Alexandra Y. Freidzon, Lyudmila G. Kuz’mina, Anna A. Moiseeva, Roman O. Starostin, Nikolai A. Kurchavov, Vyacheslav N. Nuriev, and Sergey P. Gromov. 2022. "Synthesis, Structure and Photochemistry of Dibenzylidenecyclobutanones" Molecules 27, no. 21: 7602. https://doi.org/10.3390/molecules27217602
APA StyleFomina, M. V., Freidzon, A. Y., Kuz’mina, L. G., Moiseeva, A. A., Starostin, R. O., Kurchavov, N. A., Nuriev, V. N., & Gromov, S. P. (2022). Synthesis, Structure and Photochemistry of Dibenzylidenecyclobutanones. Molecules, 27(21), 7602. https://doi.org/10.3390/molecules27217602