Abstract
The kinetics of adsorption phenomena are investigated in terms of local and non-local kinetic equations of the Langmuir type. The sample is assumed in the shape of a slab, limited by two homogeneous planar-parallel surfaces, in such a manner that the problem can be considered one-dimensional. The local kinetic equations in time are analyzed when both saturation and non-saturation regimes are considered. These effects result from an extra dependence of the adsorption coefficient on the density of adsorbed particles, which implies the consideration of nonlinear balance equations. Non-local kinetic equations, arising from the existence of a time delay characterizing a type of reaction occurring between a bulk particle and the surface, are analyzed and show the existence of adsorption effects accompanied by temporal oscillations.
1. Introduction
When a solid surface comes in contact with a liquid in which impurities are dissolved, according to the interaction energy between these impurities and the surface, they can either be attracted or repelled from the surface [1,2]. There is also the possibility that will be handled here in which the particles are attracted to the surface and remain stuck on it. In the equilibrium state, the number of particles passing from the liquid to the surface (adsorbed) is equal to that passing from the surface to the liquid (desorbed). A simple description of the phenomenon, in cases where the particles are neutral and long-range effects can be neglected, is based on the kinetic equation relating the rate of adsorbed particles with the adsorption phenomenon of particles from the bulk, and the desorption of adsorbed particles from the surface [3]. In the approximation whereby the adsorption energy is independent of the surface density of adsorbed particles and all adsorbing sites are equivalent [4], the kinetic equation reads
where is the surface density of adsorbed particles, is the bulk density of adsorbable particles, k and are the adsorption coefficient and the desorption time, respectively. Expression (1) has been widely used to analyze the time-dependence of the surface density of adsorbed particles under different conditions [5,6,7,8]. It has been also employed in the investigation of the adsorption phenomenon in the presence of an external electric field [9,10,11]. A generalization of Equation (1) is discussed in [12]. The adsorption parameters depend, clearly, from the adsorbing surface and the adsorbable particles. They depend also on the temperature and on the presence of an external electric field, as reported in Refs. [7,10,12,13,14,15,16].
Indicating the surface density of adsorbing sites by , in the case of , the adsorption coefficient is independent of [17]. On the contrary, for , the possibility of a multilayer adsorption has to be considered. In conditions where only the formation of a monolayer is possible, has to be such that for , , indicating that when the surface density overcomes the density of sites, the adsorption phenomenon is no longer possible. A proposed form for the dependence of on is [18]
with . When the formation of a multilayer is possible, and can depend on the number of layers already adsorbed.
The work focuses on the kinetic evolution of the system towards the equilibrium state, where and are time-independent. To solve the problem, in addition to the kinetic balance equation, one has to also consider conservation of the total particle number comprising the bulk plus the surface system.
The analysis deals with a sample limited by two homogeneous planar-parallel surfaces at a distance d apart. Thus, the bulk density of particles depends only on the distance of the considered point from the limiting surfaces, i.e., the mathematical problem is one-dimensional. This is the so-called slab approximation, which is assumed henceforth. It is also assumed that the surface forces responsible for the adsorption phenomenon are short range, in such a manner that in the bulk can be considered uniform. This approximation is valid if the diffusion time is extremely small with respect to the characteristic times of the adsorption phenomenon. Indicating the diffusion coefficient by D of the adsorbable particles, . If m and m/s, s. The analysis presented in the following, works well for samples of few μm in thickness, and for ordinary diffusion coefficient, such as Na in water [3]. In cases where the adsorbing surface is in contact with a reservoir, fixing the value of adsorbable particles, the quantity appearing in the kinetic equation, with or without saturation, coincides with , the bulk equilibrium density. In this case, since , it is no longer necessary to impose conservation of the number of particles. Several measurements reported in the literature correspond to this situation, and our analysis can be easily generalized and applied to these cases.
The goal of the paper is to develop a model capable of analyzing interesting data reported in the literature [5,6,7,8,9,10,11,12,13,14,15,16] relevant to the dependence on surface density of the adsorbed particles versus the concentration and the time evolution of the surface density. Furthermore, a model is proposed that is able to explain the time-oscillating behavior of the surface density of adsorbed particles [19,20,21,22,23,24,25,26] toward the equilibrium state.
The paper is organized as follows. Section 2 is dedicated to the investigation of local kinetic equations from three different perspectives. In Section 2.1, the dynamical evolution of the surface density of adsorbed particles when the kinetic equation is well described by Langmuir isotherm is analyzed. This limiting case works well when the surface density of adsorbed particles in the steady state is extremely small with respect to the surface density of adsorbing sites. The scenario for large adsorption, when saturation effects are important, is considered in Section 2.2, where the adsorption coefficient depends linearly on the surface density of adsorbed particles is investigated first. The relevant problem for which the dependence is quadratic is discussed in Section 2.3. Section 3 deals with non-local kinetic equations involving a time delay. The analysis shows that the adsorption–desorption process could present a combination of saturation accompanied by an oscillating behavior, when the time delay is comparable with the desorption time. Lastly, the conclusions are provided in Section 4.
2. Local Kinetic Equations
In this section, the adsorption–desorption phenomena are analyzed in the presence of local kinetic equation when saturation and non-saturation regimes are considered. These equations are local in time because the reaction characterizing the adsorption–desorption process at the surfaces are assumed to be instantaneous, i.e., there is no time delay characterizing the bulk-surface dynamics.
2.1. No Saturation:
Let us consider first the simple case in which the kinetic equation describing the adsorption phenomenon (1) involves an adsorption term proportional to the bulk density of adsorbable particles , related to the constant adsorption coefficient k, and a desorption term proportional to the surface density of adsorbed particles, related to the desorption time . Assuming a uniform bulk distribution of the adsorbable particles , the conservation of the number of particles (bulk plus surface) implies that
For the analysis presented in the following, it is useful to measure the time in units of , i.e., , and to introduce the dimensionless parameter defined as , where is an intrinsic length and is an intrinsic time, both connected to the adsorption phenomenon [17], and d is the thickness of the sample. The maximum surface density of adsorbed particles is . For this reason, in the following, the surface and the bulk densities of particles will be measured in units of and , i.e.,
in such a way that Equations (1) and (3) can be rewritten as
and
respectively. Substitution of Equation (6) into Equation (5) yields
In the equilibrium state, . It follows that in the steady state the dimensionless surface density of adsorbed particles and bulk density of adsorbable particles are given, respectively, by
In absolute units, the corresponding surface and bulk particles densities are
The kinetic evolution of is obtained by solving the differential Equation (7) with the boundary condition . Simple calculations give
The effective relaxation time, , of the surface density of adsorbed particles , in absolute units, is
Note that Equation (11) can be rewritten as
indicating that, in the case in which is very different from , is approximatively equal to the smallest between and . When , i.e., , . This case represents a surface in contact with a reservoir of particles. The opposite case, in which , i.e., , yields , which is not of experimental interest.
2.2. Linear Saturation:
A simple generalization of Equation (1) to take the saturation into account is obtained when one considers in Equation (2), for , namely:
where is the surface density of adsorbing sites. In terms of the dimensionless quantities defined in (4), Equation (13) can be rewritten as
where
is a dimensionless parameter comparing the maximum density of adsorbable particles with the density of sites on the surface. Note that for a fixed adsorbing surface in contact with a solution the parameter is fixed and different values of r correspond to different bulk equilibrium concentrations of adsorbable particles. In the present case, the conservation of particles is still represented by Equation (6). The saturation is important only in the case where . When , the analysis presented above has to be recovered.
As before, in the equilibrium state , and the surface density characterizing the steady state is the solution of the second-order equation
whose solutions are
and
Since
for all meaningful physical parameters, and are real quantities. From Equations (18) and (19), in the limit of , one obtains
and
Since, in the limit, one has to recover the result given by (8), it is possible to conclude that the actual surface density in the equilibrium state is . The dependence of the equilibrium surface density versus r is shown in Figure 1.
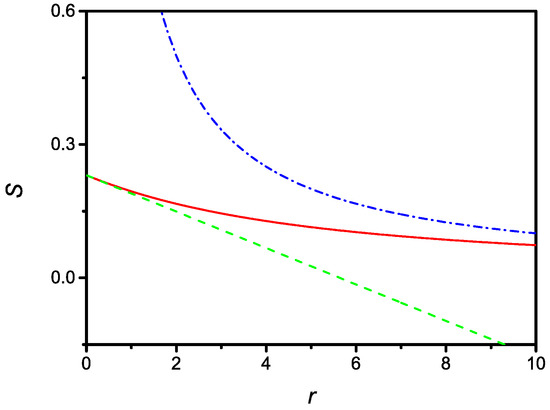
Figure 1.
Dependence of the dimensionless surface density of the steady state versus the dimensionless parameter r. Dashed and dash-dotted lines represent the limiting cases corresponding to small and large r, respectively, as given by Equations (21) and (23). The curves are drawn for or , i.e., the adsorption time is approximately three-times faster than the desorption time.
Notice that in the limit of , corresponding to a sample of infinite thickness, from Equation (18) one obtains
Taking into account that the actual surface density is given by , if one expresses in terms of , then one has . It follows that, from Equation (23), in the considered limit of large r
which states that, in an infinite sample, all adsorbing sites are occupied.
The evolution of toward to the equilibrium state is determined by solving Equation (16). A simple calculation gives
where
as it follows from Equation (20). The time dependence of given by Equation (25) is shown in Figure 2 for two different values of r.
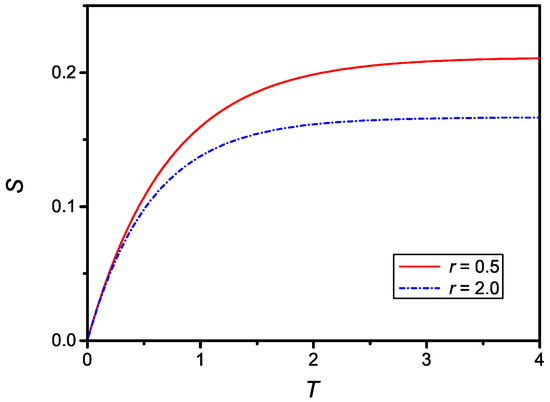
Figure 2.
Dependence on the dimensionless surface density of adsorbed particles, , versus the dimensionless time, T, for , , (dash-dotted line) and , (full line).
The time dependence of the dimensionless surface density of adsorbed particles compare qualitatively well with the experimental data reported in Refs. [8,10,14], relevant to the influence of the equilibrium concentration on the adsorption phenomenon. The relaxation time, in absolute units for , is then
One notices that the divergence of is only apparent because the quantity is always positive for all values of r and u physically meaningful, as stressed above. From Equation (27), in the limit, one gets
that, in the same limit, coincides with (11), as expected. The dependence of on r is shown in Figure 3, where are also reported the limiting cases corresponding to small and large values of r.
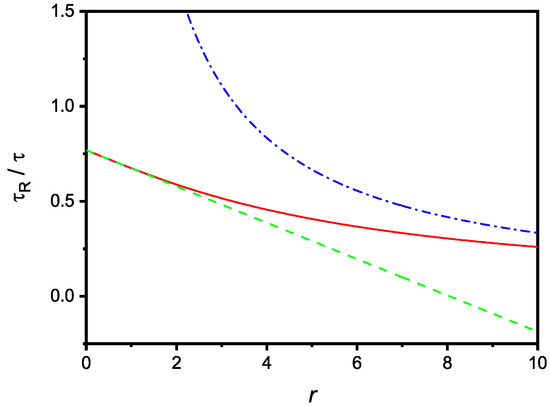
Figure 3.
Relaxation time, in units, versus the dimensionless parameter r. The dashed and dash-dotted lines are the limiting cases of large and small r, respectively. The figure is drawn for .
2.3. Quadratic Saturation:
Let us consider now the case in which the kinetic equation is represented by
where, as before, is the surface density of adsorbing sites. In terms of reduced quantities, it takes the form
and the equation stating the conservation of the number of particles is still represented by Equation (6). Differential Equation (29), taking into account Equation (6), can be rewritten as
In this way, the equilibrium value of S is obtained by solving the third-order equation
By inspection, one observes that for all physically meaningful values of r and u, Equation (32) always contains three distinct real solutions as shown in Figure 4. The searched solution to the kinetic Equation (29) is the one that, in the , reduces to Equation (8).
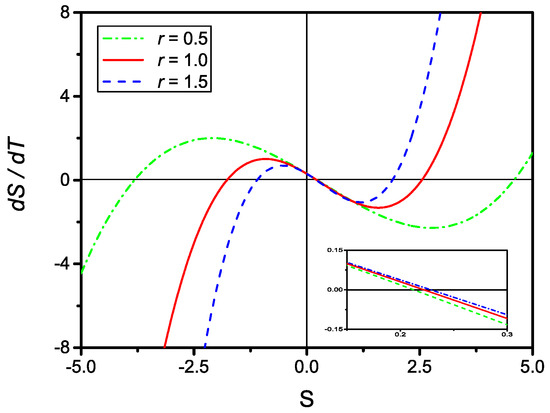
Figure 4.
Plot of dS/dT, given by Equation (31), versus S for and (dash-dotted), (full), and (dashed). The insert shows a closeup of the three curves around . Clearly, for all physically meaningful values of r and u, there are three distinct real solutions.
Indicating by , and the three solutions of Equation (32), a simple calculation shows that in the limit they tend respectively to
from which one deduces that solutions tends to the value given by (8), as expected. In the opposite limit of , the solutions tend to
at the second order in . As already mentioned, the surface density in units of , are and tend to
showing again that the solution of the problem also has the correct limit in the case of large r. The numerical solution of Equation (31) is shown in Figure 5, for and , , and .
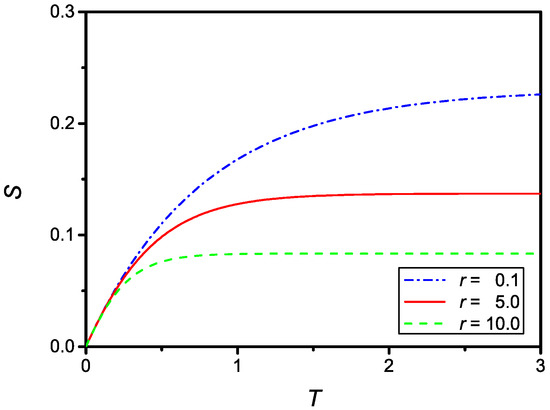
Figure 5.
Dependence on the dimensionless surface density of adsorbed particles, , versus the dimensionless time, T, in the case of a quadratic reduction of the adsorption coefficient. The curves were numerically obtained for and three values of r: (dash-dotted), (full), and (dashed).
Finally, Figure 6 compares the time evolution of deduced in the case of no saturation (dash-dotted line), linear saturation (full line), and quadratic saturation (dashed line).
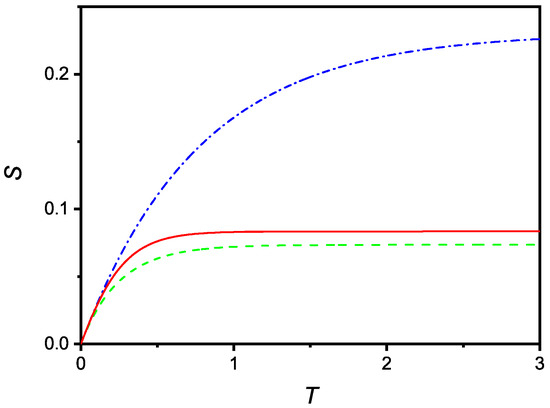
Figure 6.
Comparison (in dimensionless units) of the time dependencies of S relevant to cases where saturation is absent (dash-dotted), with saturation linear in S (full) and saturation quadratic in S (dashed). The curves were drawn for and .
3. Non-Local Kinetic Equations
In the previous sections, a few kinetic equations describing the adsorption phenomenon, in the absence of saturation, Equation (1), in the presence of linear, Equation (13) and quadratic, Equation (29), saturation have been considered. These equations were solved by taking into account conservation of the number of particles, that we assumed as represented by Equation (3). This is a rather rough approximation, but allows one to derive some general properties on the adsorption. These kinetic equations are of the type
i.e., they relate the time variation of at t with the bulk density of adsorbable particles, and with the surface density of adsorbed particles, at the same time t. However, since the adsorption phenomenon implies a form of chemical reaction occurring between a bulk particle and the surface, the time rate at t depends on and at an antecedent time , where depends on the particular reaction responsible for the adsorption. This particular time, , can differ from the desorption time, . In other words, usually, there is an intrinsic delay between and, and . This delay is related to in such a way that Equation (40) has to be written as
Assuming small with respect to the observation time t, from Equation (41) one gets
i.e., now the kinetic equation contains a second order derivative in t.
To investigate the effect of the delay on the evolution of the surface density of adsorbed particles, Equation (1) is now generalized, with the condition (3). In the presence of delay, Equation (1) becomes
that, by taking into account Equation (3) and using dimensionless quantities, assumes the form:
Equation (44) is the new version of Equation (7) in the presence of a time delay. The parameter compares the delay time with the desorption time .
To solve Equation (44), one has to impose the initial conditions on and S. It will be assumed, as before, that the adsorption phenomenon begins at , and hence and, furthermore, that . In this framework, as it is evident from (44), the case is singular, because the differential equation passes from the second to the first order. Solution of Equation (44) with the mentioned initial conditions, when , is
where
In the particular case in which , solution of Equation (44) with the above mentioned boundary conditions is
The exponents and depend critically on . If , then and are negative real numbers, and the solution tends uniformly to . In this case, the relaxation toward to the equilibrium value of the surface density of adsorbed particles is characterized by two different relaxation times and the explicit solution is
where . An expression of the type given by Equation (45) has been proposed to interpret the adsorption data by Vanegas et al. [27], and used more recently to analyze adsorption data in [15,16].
In the opposite case, where , and are complex conjugate numbers, with a negative real part. The solution tends to oscillating, and the explicit solution is
where .
Figure 7 shows the time evolution of S for and (dash-dotted line), (full line), and the solution for , i.e., assuming no delay (dashed line). As can be observed, all curves tend to the same limit . The inset of Figure 7 shows the evolution of S for small T. From this frame, it is clear that the case corresponding to does not satisfy the initial condition on .
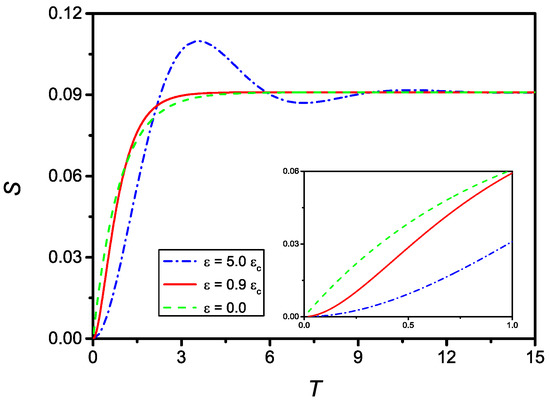
Figure 7.
Time evolution (in dimensionless units) of S for (dash-dotted), (full), and (dashed), with . From the inset, where it is reported the evolution for small T, it is evident that in the case the initial boundary condition on is not satisfied, as a consequence from the fact that in this case the differential equation passes from the second to the first order.
The simple analysis presented above to take into account the delay was possible because the governing differential equation is linear. The same generalization in the case where the adsorption phenomenon is described by Equation (16) or by Equation (31) gives rise to the differential equations
respectively. In presence of the saturation effect, it is no longer possible to obtain an analytical solution, because Equations (51) and (52) are no longer linear. However, a numerical solution can be easily obtained. Figure 8 compares the time evolution of S described by Equation (51), (dashed line), and by Equation (52), (dash-dotted line), for with and . Furthermore, in this case, the solutions are oscillating around the stable limiting value.
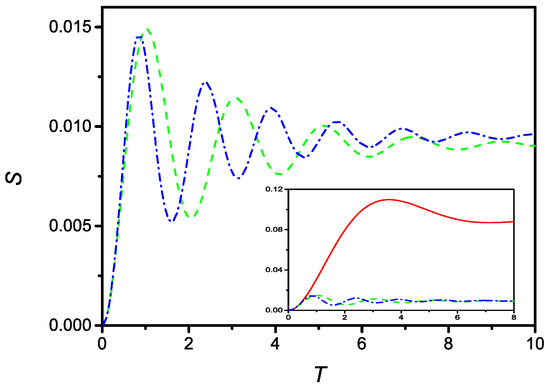
Figure 8.
Time evolution (in dimensionless units) of the surface density of adsorbed particles in the presence of linear (dashed), and quadratic (dash-dotted), saturation effect. The curve are drawn for , and . Observed in the inset, comparison between the time evolution of S in the absence of saturation effect (full), with linear (dashed), and quadratic (dash-dotted) saturation effect.
This oscillating behavior can be qualitatively compared with the ones exhibited by some acid monolayers whose adsorption is governed by physical forces, but in which the role of the head group has to be taken into account [19,20]. As pointed out in Ref. [19], an oscillating behavior such as the ones depicted in Figure 7 and Figure 8 is in agreement with the behavior found by Fourier transform IR spectroscopy for siloxane polymers chemisorbing to aluminia [20,21]. In these systems, measurements of the variation of physically adsorbed and chemically adsorbed segments show that the physisorption process is strong in the first instants, presenting a pronounced maximum and tending to a small value; the chemisorption process, on the contrary, tends to a saturation value. When these effects are combined, because they occur during the same process, the resulting curve may present oscillating behavior. The effect of the non-local term, represented by the second order time-derivative, may be connected with memory effects. Indeed, it may indicate that the adsorption process is much better described if one considers the memory of the preceding state for the molecules being adsorbed at the surface.
An effect of this kind is not unusual and can even be expected because, for instance, in the adsorption phenomenon by a solid surface, the collision of a molecule can be represented by different processes [22]. One of these is an elastic scattering, that occurs when there is no loss in translational energy during the collision; however, if the molecule is still in a weakly bound state, even if it is on the surface, the thermal motion of the surface atoms can cause the molecule to desorb and this represents another kind of process. Finally, when the molecule collides with a surface, it loses energy and is converted into a state where it remains on the surface for a reasonable length of time [23]. These phenomena indicate that the actual position of the molecule on the surface has a memory of its incoming state, eventually modifying the adsorption–desorption rates. An alternative analysis of this process has been carried out by considering modified linear kinetic equation in which some suitable choices for a kernel in the desorption rate accounted for the importance of the physisorption or of the chemisorption, according to the time scale governing the adsorption phenomena [19]. In the present analysis, the oscillating behavior is obtained with linear and nonlinear kinetic equations incorporating an expected time delay in the reaction of the molecules from the bulk at the surface, during the adsorption process and may be relevant to understand oscillation phenomena in polymers [24], multicomponent adsorption [25], surfactant ions [26], among many others.
4. Conclusions
The work approaches the kinetic equation in the Langmuir approximation from two different perspectives. The first deals with a balance equation that is local in time, but in which a phenomenological adsorption coefficient intervenes that may depend on the density of adsorbed particles according to a general power law. After formulating the problem in more general terms, the conventional case is considered and two other possibilities: a linear and a quadratic dependence. In such cases, the density of particles at equilibrium is described by algebraic equations and the time evolution by nonlinear differential equations. Thus, it is possible to analyze various adsorption regimes that are essentially governed by a saturation phenomenon dictated by the finite number of sites at the interface. In the second perspective, the adsorption–desorption phenomena are analyzed in a non-local picture in time, that is, it is assumed that there exists a time delay between the arrival of the particle in the bulk, but in the vicinity of the interface, and the adsorption–desorption itself, taking place over the interface. In this case, the entire kinetics are described by an equation involving a second derivative in time, which reduces to first-order when the time delay is negligible. In this context, it is found that the adsorption–desorption regimes could be accompanied by an oscillation phenomenon in the coverage ratio behavior. A similar phenomenon can be found using linear kinetic equations, but containing temporal kernels that take memory effects into account. There is, therefore, a very complete approach to the adsorption–desorption phenomena of neutral particles (although the extension to the case of charged particles can also be considered from the same perspective) at the liquid (bulk)–solid (surface) interface.
Author Contributions
Conceptualization, methodology, writing, and modification, G.B.; calculation, writing—original draft preparation, L.R.E.; writing—review and editing, A.M.S. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
Not applicable.
Acknowledgments
This work was partially supported by the MEPhI Program Priority 2030.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| d | Thickness of the cell [m]. |
| D | Diffusion coefficient [m/s]. |
| Delay time [s]. | |
| Dimensionless delay time. | |
| Adsorption coefficient [m/s]. | |
| Bulk density of adsorbable particles [1/m]. | |
| Bulk density of adsorbable particles in thermodynamic | |
| equilibrium in the absence of adsorption [1/m]. | |
| Equilibrium bulk density of particles | |
| in the presence of adsorption [1/m]. | |
| Reduced bulk density of particles. | |
| Reduced equilibrium bulk density of particles | |
| in the presence of adsorption | |
| Maximum density of adsorbed particles, , | |
| in units of surface density of adsorption sites, . | |
| Reduced surface density of particles. | |
| Reduced equilibrium surface density of particles | |
| in the presence of adsorption. | |
| Surface density of adsorbed particles [1/m]. | |
| Equilibrium value of the surface density of adsorbed | |
| particles [1/m]. | |
| Maximum density of adsorbable particles [1/m]. | |
| Surface density of adsorbing sites [1/m]. | |
| Desorption time [s]. | |
| Diffusion time [s]. | |
| Intrinsic adsorption time [s]. | |
| Effective relaxation time [s]. | |
| Dimensionless time. | |
| Desorption time, , in units of intrinsic adsorption time, . |
References
- Garrod, C. Statistical Mechanics and Thermodynamics; Oxford University Press: Oxford, UK, 1995. [Google Scholar]
- Lyklema, J. Fundamentals of Interface and Colloid Surfaces; Academic Press: London, UK, 1993. [Google Scholar]
- Atkins, P.W. Physical Chemistry; Oxford University Press: Oxford, UK, 1982. [Google Scholar]
- Evangelista, L.R.; Barbero, G. Statistical interpretation of the kinetic equation in the adsorption problem. Eur. Phys. J. E 2004, 15, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Umoren, S.A.; Etim, U.J.; Israel, A.U. Adsorption of methylene blue from industrial effluent using poly (vinyl alcohol). J. Mater. Environ. Sci. 2013, 4, 75–86. [Google Scholar]
- Chen, S.; Zhou, M.; Wang, H.-F.; Wang, T.; Wang, X.-S.; Hou, H.-B.; Song, B.-Y. Adsorption of Reactive Brilliant Red X-3B in Aqueous Solutions on Clay-Biochar Composites from Bagasse and Natural Attapulgite. Water 2018, 10, 703. [Google Scholar] [CrossRef]
- Darwish, A.A.A.; Rashada, M.; L-Aoh, H.A.A. Methyl orange adsorption comparison on nanoparticles: Isotherm, kinetics, and thermodynamic studies. Dyes Pigm. 2019, 160, 563–571. [Google Scholar] [CrossRef]
- Shahin, S.A.; Mossad, M.; Fouad, M. Evaluation of copper removal efficiency using water treatment sludge. Water Sci. Eng. 2019, 12, 37–44. [Google Scholar] [CrossRef]
- Mossad, M.; Zou, L. Evaluation of the salt removal efficiency of capacitive deionisation: Kinetics, isotherms and thermodynamics. Chem. Eng. J. 2013, 223, 704–713. [Google Scholar] [CrossRef]
- Wimalasiri, Y.; Mossad, M.; Zou, L. Thermodynamics and kinetics of adsorption of ammonium ions by graphene laminate electrodes in capacitive deionization. Desalination 2015, 357, 178–188. [Google Scholar] [CrossRef]
- Yang, W.; Han, H.; Zhou, M.; Yang, J. Simultaneous electricity generation and tetracycline removal in continuous flow electrosorption driven by microbial fuel cells. RSC Adv. 2015, 5, 49513–49520. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Q.; Wang, H.; Ni, Y.; Zhu, Z. Absorption behaviors study on doped Li4SiO4 under a humidified atmosphere with low CO2 concentration. Int. J. Hydrogen Energy 2014, 39, 17913–17920. [Google Scholar] [CrossRef]
- Wang, T.; Liang, H.; Bai, L.; Zhu, X.; Gan, Z.; Xing, J.; Li, G.; Aminabhavi, T.M. Adsorption behavior of powdered activated carbon to control capacitive deionization fouling of organic matter. Chem. Eng. J. 2020, 384, 123277. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, Q.; Zhang, J.; Chen, G.; Wang, Z.; Wu, Z. Development of novel ZnZr-COOH/CNT composite electrode for selectively removing phosphate by capacitive deionization. Chem. Eng. J. 2022, 439, 135527. [Google Scholar] [CrossRef]
- Wang, K.; Zhao, Y.; Clough, P.T.; Zhao, P.; Anthony, E.J. Structural and kinetic analysis of CO2 sorption on NaNO2-promoted MgO at moderate temperatures. Chem. Eng. J. 2019, 372, 886–895. [Google Scholar] [CrossRef]
- Cui, H.; Zhang, Q.; Hu, Y.; Peng, C.; Fang, X.; Cheng, Z.; Galvita, V.V.; Zhou, Z. Ultrafast and Stable CO2 Capture Using Alkali Metal Salt-Promoted MgO–CaCO3 Sorbents. ACS Appl. Mater. Interfaces 2018, 10, 20611–20620. [Google Scholar] [CrossRef] [PubMed]
- Barbero, G.; Evangelista, L.R. Adsorption Phenomena and Anchoring Energy in Nematic Liquid Crystals; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Barbero, G.; Evangelista, L.R.; Lelidis, I. Effective adsorption energy and generalization of the Frumkin-Fowler-Guggenheim isotherm. J. Mol. Liq. 2021, 327, 114795. [Google Scholar] [CrossRef]
- Zola, R.S.; Lenzi, E.K.; Evangelista, L.R.; Barbero, G. Memory effect in the adsorption phenomena of neutral particles. Phys. Rev. E 2007, 75, 042601. [Google Scholar] [CrossRef]
- Adamson, A.W.; Gast, A.P. Physical Chemistry of Surfaces; Wiley: New York, NY, USA, 1997. [Google Scholar]
- Cosgrove, T.; Prestidge, C.A.; Vincent, B. Chemisorption of linear and cyclic polymethylsiloxanes on alumina studied by Fourier-transform infrared spectroscopy. J. Chem. Soc. Faraday Trans. 1990, 86, 1377–1382. [Google Scholar] [CrossRef]
- Patrylak, K.I.; Taranooka, O.M. On the Phenomenon of Oscillations by Adsorption. Zeolites 1997, 18, 7–9. [Google Scholar] [CrossRef]
- Masel, R.I. Principles of Adsorption and Reaction on Solid Surfaces; Wiley: New York, NY, USA, 1996. [Google Scholar]
- Minko, S.; Voronov, A.; Pefferkorn, E. Oscillation Phenomenon at Polymer Adsorption. Langmuir 2000, 16, 7876–7878. [Google Scholar] [CrossRef]
- Jaksic, O.M.; Jaksic, K.; Rasljic, M.B.; Kolar-Anic, L.Z. On Oscillations and Noise in Multicomponent Adsorption: The Nature of Multiple Stationary States. Adv. Math. Phys. 2019, 2019, 7687643. [Google Scholar] [CrossRef]
- Ikezoe, Y.I.; Yui, S.I.H.; Fujinami, M.; Sawada, T. Chemical Oscillation with Periodic Adsorption and Desorption of Surfactant Ions at a Water/Nitrobenzene Interface. Anal. Sci. 2004, 20, 1509–1514. [Google Scholar] [CrossRef] [PubMed]
- Vanegas, M.J.; Fregoso-Israel, E.; Escamilla, P.; Pfeiffer, H. Kinetic and Reaction Mechanism of CO22 Sorption on Li4SiO4: Study of the Particle Size Effect. Ind. Eng. Chem. Res. 2007, 46, 2407–2412. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).