Abstract
Natural products are an excellent source of inspiration for the development of new drugs. Among them, betalains have been extensively studied for their antioxidant properties and potential application as natural food dyes. Herein, we describe the seven-step synthesis of new betalamic acid analogs without carboxy groups in the 2- and 6-position with an overall yield of ~70%. The Folin–Ciocalteu assay was used to determine the antioxidant properties of protected intermediate 21. Additionally, the five-step synthesis of betalamic acid analog 35 with three ester moieties was performed. Using NMR techniques, the stability of the obtained compounds towards oxygen was analyzed.
1. Introduction
Natural colors in fruits and plants are essential for photosynthesis, pollination, and seed dissemination [1,2]. Colors in plants are caused by three chemically distinct pigment types, anthocyanins (1), betalains (2), and carotenoids (3) (Figure 1). Anthocyanins are water-soluble pigments that give blue, red, and purple hues. The chemistry, production, and distribution of these compounds have been extensively investigated in the past [3,4,5,6,7].
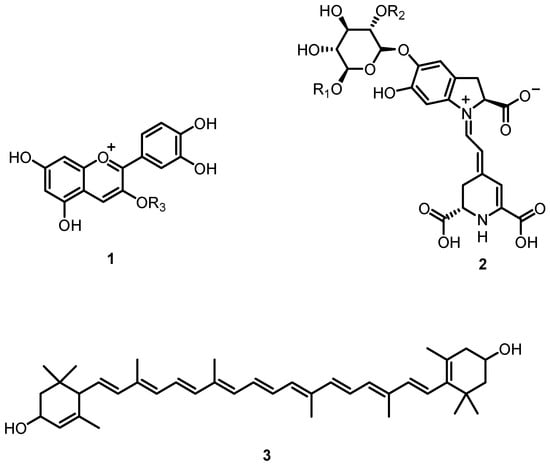
Figure 1.
Natural pigments present in various plants; anthocyanins (1), betalains (2) and carotenoids (3).
In the last fifty years, there has been a growing interest in betalains. With few exceptions, plants and fruits of the order Caryophyllales exhibit a range of colors from red/purple to orange/yellow, due to the presence of these hydrophilic pigments. Initially, betalains were classified as anthocyanins. However, it was later discovered that the main enzymes required for the formation of anthocyanins are not present in betalain-producing plants [8,9].
Betalains are nitrogen-containing water-soluble pigments. Their biosynthesis in plants starts with L-tyrosine (4), which is converted into L-3,4-dihydroxyphenylalanine L-DOPA (5). The enzyme tyrosinase was thought to be responsible for the hydroxylation of L-tyrosine [10]. Recently, it has been observed that cytochrome P-450 monooxygenases are also able to catalyze this reaction [11]. Through the action of the enzyme 4,5-DOPA-extradiol dioxygenase, L-DOPA is converted into 4,5-seco-DOPA (7). Spontaneous cyclization of 4,5-seco-DOPA leads to betalamic acid (9), the key intermediate in the biosynthesis of all betalains. Moreover, tyrosinase is also involved in the oxidation of L-DOPA to o-DOPA-quinone (6), which undergoes cyclization to form cyclo-DOPA (8). Spontaneous condensation between cyclo-DOPA (8) and betalamic acid (9) leads to red and violet pigments named betacyanins (e.g., betanidin (11)). Alternatively, the reaction of betalamic acid (9) with amino acids or amino acid derivatives provides yellow-colored betaxanthins (e.g., indicaxanthin (12)) (Figure 2).
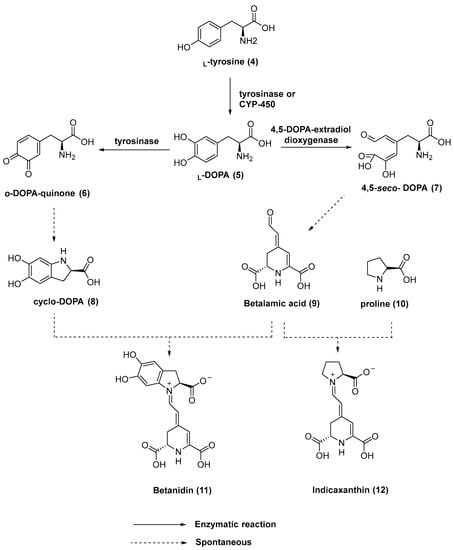
Figure 2.
Biosynthetic pathway of betalains 11 and 12.
Betaxanthins are yellow, regardless of the amino acid or amine condensed with betalamic acid. Betaxanthins have a maximum absorption wavelength of 480 nm, while betacyanins show a maximum absorption wavelength of 536 nm. Additionally, a sugar moiety is linked to one of the phenolic OH moieties in betacyanin’s cyclo-DOPA portion [10,12,13].
The food industry has demonstrated a growing interest in these pigments as food colorants [14,15]. Moreover, betalains exhibit antioxidant and radical scavenging activity [16,17,18,19,20,21,22,23,24,25]. Numerous investigations have demonstrated that indicaxanthin (12) possesses both antiproliferative and chemoprotective properties [26,27,28].
The majority of betalains employed in biological research are extracted directly from plants by solid–liquid extraction. Maceration of vegetables facilitates the diffusion of the substances. Additional cellular components are released after tissue breakdown, which makes further purification necessary. Although betalains are typically extracted with H2O, other solvents such as MeOH and EtOH are frequently added to aid the extraction process. Unfortunately, this approach requires longer extraction time, additional purification procedures, and provides limited yields. As a result, innovative extraction methods were used to increase the efficiency of the isolation process of betalains, such as diffusion extraction, ultrafiltration, reverse osmosis, and cryogenic freezing [25,29,30,31,32]. Another significant issue encountered during the extraction and purification of these pigments is their chemical instability when exposed to oxygen, acids, bases, light, and heat. These parameters have a considerable impact on the extraction and purification procedures’ efficiency [33]. Several strategies for increasing the stability of betalains have been implemented, most notably in the food industry [34].
Betalamic acid (9) is a critical intermediary in the formation of both kinds of betalains. Although two syntheses of this compound have already been reported [35,36,37], the first synthesis developed by Dreiding et al. [35,37] started with chelidamic acid (I). Hydrogenation of 19 in the presence of rhodium on activated alumina afforded an all-cis-configured piperidine derivative, which was converted into the dimethyl ester II upon treatment with methanol and HCl. Oxidation of the secondary alcohol led to the formation of piperidin-4-one III. To avoid overoxidation to the corresponding pyridine derivative, a polymeric carbodiimide was used for the Pfitzner–Moffatt oxidation and the transformation was carefully monitored. For the introduction of the side chain, a fully methyl-protected semicarbazide was employed as the Horner–Wittig reagent. This reagent led to the formation of hydrazone IV as a pure €-configured diastereomer (C=N bond). Dehydrogenation of IV with t-butyl hypochlorite and triethylamine (NEt3) provided dihydropyridine V as a 7:3 mixture of (E)- and (Z)-configured diastereomers. In this case, (E) and (Z) configuration refers to the exocyclic C=C double bond, whereas the C=N double bond is still (E)-configured. Recrystallization from t-butanol provided the pure (E,E)-configured betalamic acid derivative (E,E)-23 (Scheme 1).
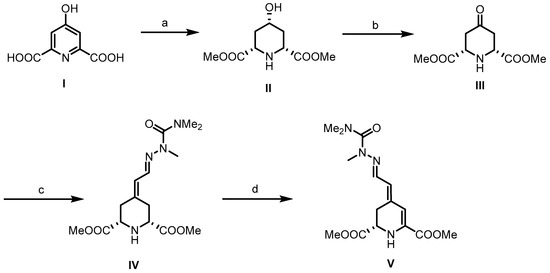
Scheme 1.
Synthesis of betalamic acid derivative 23 according to Dreiding et al. [35,37]. Reagents and reaction conditions were as follows: (a) 1. 5% Rh/Al2O3, H2O, H2 (4 atm), 75 °C; 2. HCl, MeOH, 42%. (b) Polimeric carbodiimide, pyridinum trifluoroacetate, DMSO, r.t., 33 h, 90%. (c) (EtO)2OPCH2CH=NN(Me)CONMe2, NaH, DME, r.t., 15 h, 44%. (d) t-BuOCl/t-BuOH, Et3N, C6H6, r.t., 1 h, 16%.
In Scheme 2, the second strategy for the synthesis of betalamic acid (9), developed by Bϋchi et al. [36], is displayed. This approach started from benzylnorteleoidine VI obtained by Robinson–Schöpf condensation. The first reaction includes the protection of the diol by formation of a cyclic ortho ester. Hydrogenolytic cleavage of the N-benzyl protective group provided the secondary amine VII. Reaction of the aminoketone VII with allyl magnesium chloride yielded the tertiary alcohol VIII with high diastereoselectivity. The secondary amine VIII was then converted into O-benzoylhydroxylamine IX. First, amine IX was neutralized with K2CO3 and reacted with dibenzoyl peroxide in DMF, leading to formation of the protected amine. Subsequently, acetylation of the alcohol provided O-benzoylhydroxylamine IX. Next, the ortho ester was cleaved with oxalic acid in water to obtain diol X. This latter compound was then oxidized with N-chlorosuccinimide (NCS) and dimethylsulfide to achieve the diketone XI. Ozonolysis of XI led to the formation of aldehyde XII. Treatment of XII with lead tetraacetate in benzene and methanol converted the diketone moiety into an unstable dicarboxylic acid, which, upon loss of HOAc and BzOH, yielded (±)-betalamic acid (9) as a mixture of (E)- and (Z)-configured diastereomers after silica gel chromatography [36].
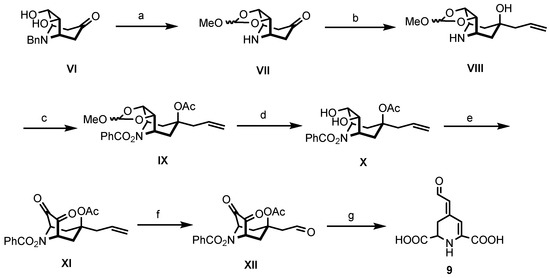
Scheme 2.
Synthesis of betalamic acid (9) according to Bϋchi et al. [36]. Reagents and reaction conditions were as follows: (a) 1. CF3COOH, DMF, (MeO)3CH, CH2Cl2, reflux, 5 h. 2. CF3COOH, H2, 5% Pd/C, MeOH, rt, 1 h, 96%. (b) H2C=CH-CH2MgCl, THF, Ar, 0 °C, 30 min, 71%. (c) 1. Bz2O2, K2CO3, DMF, RT, 30 h. 2. Ac2O, Et3N, DMAP, Et2O, reflux, 5d, 92%. (d) 5% oxalic acid, aceton, rt, 30 min, 81%. (e) NCS, Me2S, Et3N, PhMe, 0–40 °C, 30 min–1.5 h, 75%. (f) 1. O3, EtOAc:MeOH (30:7), −78 °C. 2. Me2S, −78 °C, 2.5 h, 68%. (g) 1. Pb(OAc)4, PhH:MeOH (1:1), Ar, 0 °C, 45 min. 2. H2NCONHNH2-HCl, NaOAc, EtOAc, rt, 2 h, 33%.
Despite the fact that two methods for the synthesis of betalamic acid (9) and its derivatives have been reported in the literature, the majority of betalamic acid (9) is produced through extraction from pigments, followed by basic hydrolysis.
To investigate relationships between the chemical structure and biological properties of indicaxanthin derivatives in further detail, analogs 13 of 12 that lack the two carboxy moieties in positions C-2- and C-6 were considered first. Herein, we describe the design and synthesis of betalamic acid analog 13 that is devoid of carboxy groups in positions C-2 and C-6. Additionally, experiments were conducted to synthesize the betalamic acid derivative 14 in a simpler and more cost-effective manner and to evaluate its reactivity toward oxygen (Scheme 3).
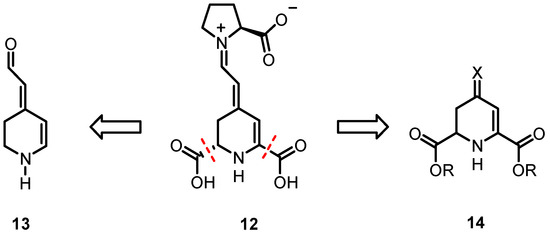
Scheme 3.
Development of indicaxanthin (12) analogs without carboxy moieties in positions C-2 and C-6 (13) and without side chain in position C-4 (14).
2. Results and Discussion
2.1. Chemistry
The plan for the synthesis of 13, the analog of betalamic acid without carboxy groups in positions C-2- and C-6, is outlined in Scheme 4.
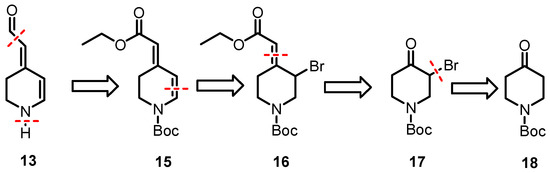
Scheme 4.
Retrosynthetic analysis of the key intermediate 13.
We planned to synthesize 13 from the α,β-unsaturated ester 15 that bears a Boc-protective group at the piperidine ring. At first, the ester must be reduced to afford an aldehyde and finally, the Boc-protective group must be removed. The α,β-unsaturated ester 15 can be obtained by a Wittig reaction of α-bromoketone 17 and the subsequent β-elimination of 16. The α-bromoketone should be available by α-bromination of an appropriate piperidone derivative, e.g., 18.
The synthesis started with piperidine 19 (Scheme 5), which was protected with (Boc)2O to afford Boc-protected piperidone 18. In order to introduce a double bond in positions C-5 and C-6 of the piperidine ring, piperidone 18 was brominated in the α-position using Br2 and AlCl3 to generate the α-bromoketone 17 in a 46% yield [38]. The conjugated double bond system is a characteristic feature of the class of betalains. Thus, the first double bond was introduced by a Wittig reaction of the α-bromoketone 17 with Ph3P=CHCO2Et to give the α,β-unsaturated ester 16 in a 95% yield [39]. Although the formation of (E)/(Z)-diastereomers was expected, the 1H and 13C NMR spectra reveal only one set of signals, indicating a single diastereomer, presumably (E)-16. LiBr and Li2CO3 were used to induce dehydrobromination (β-elimination), resulting in the formation of completely conjugated compound 15, which was isolated in a 88% yield [40]. The 1H NMR spectrum of 15 reveals two distinct sets of signals, indicating the presence of (E)- and (Z)-configured esters 15 in the ratio 9:1. Since diastereomeric (E)- and (Z)-configured esters 15 could not be separated by flash column chromatography, the mixture was used to prepare the aldehyde 21. According to the first theory, aldehyde 21 should be obtained directly by the reduction of the ester 15 with DIBAL-H. However, even at −78 °C in toluene, only the primary alcohol 20 was formed and isolated in a 94% yield. Alternatively, the primary alcohol 20 was synthesized by the reduction of the ester 15 with LiAlH4. Several methods have been reported in the literature for the oxidation of primary alcohols to aldehydes [41]. A method with broad applicability and high yields is the Dess–Martin periodinane (DMP) oxidation method. Unexpectedly, the oxidation of allyl alcohol 20 with DMP resulted in low yields of the product, which was difficult to purify. Therefore, the alcohol 20 was oxidized via radical oxidation with TEMPO [41] and CuCl to provide the aldehyde 21 in a 76% yield. To obtain the aldehyde 13 as an analog of betalamic acid (9), the Boc-protective group of 21 was removed. Unfortunately, removing the Boc-protective group under typical conditions with F3CCO2H did not result in the desired aldehyde 13. Several methods were investigated to remove the Boc-protective group from 21 to achieve 13. In the end, a rather unusual method, i.e., heating the Boc-protected compound 21 in a mixture of water and dioxane under neutral conditions [42], was successful. Due to the instability of the secondary amine 13, the isolated yield of 13 was rather low. In particular, condensation and polymerization reactions, as well as oxidation processes, were observed during the purification process. Despite the instability, 1H and 13C NMR spectra could be recorded to identify and characterize 13.
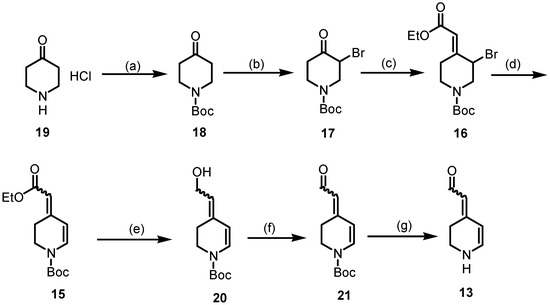
Scheme 5.
Synthesis of the key intermediate 13. Reagents and reaction conditions were as follows: (a) Boc2O, NaHCO3, THF/H2O, r.t., 16 h 96%. (b) Br2, AlCl3, THF/Et2O, 0 °C, 16 h, 46%. (c) Ph3P=CHCO2Et, CH2Cl2, 40 °C, 2 h, 95%. (d) Li2CO3, LiBr, DMF, 75 °C, 3 h, 88%. (e) DIBAL-H, toluene, −78 °C, 1 h, 94% or LiAlH4, THF, −10 °C, 1 h, 70%, (f) TEMPO, CuCl, DMF, r.t, 16 h, 90%. (g) H2O/dioxane, 90 °C, 2 h, 5%.
In addition to betalamic acid analog 13, 1,2,3,4-tetrahydropyridine derivatives 22 and 23 were designed and synthesized (Scheme 6). The reactivity of these 1,2,3,4-tetrahydropyridines 22 and 23 and further analogs towards oxygen should be investigated. The key intermediate for the synthesis of 22 and 23 is 4-methylenepiperidine 24, which can be obtained by double allylation of iminodiacetic acid diester 25 with dichloride 26, as reported by Einhorn et al. [43]. Transformation of the methylene moiety of 24 into a ketone and subsequent introduction of a double bond in the ring result in the formation of 23. The α,β-unsaturated ester 22 can prepared by an additional Wittig reaction of a ketone intermediate.
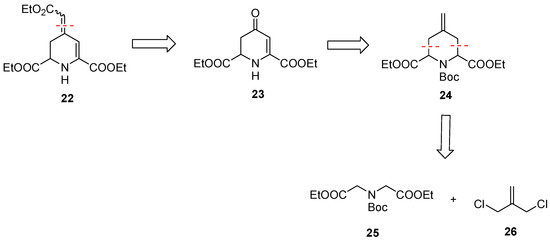
Scheme 6.
Retrosynthetic analysis of α,β-unsaturated ketone 22 and ester 23 with two ester moieties in positions C-2- and C-6.
For the synthesis of methylenepiperidine 24, the diester 25 and the diiodide 29 were prepared (Scheme 7). The diester HCl salt 28 was obtained by esterification of iminodiacetic acid (27) with SOCl2 in refluxing ethanol. The secondary amine of 27 was protected with Boc2O to afford the carbamate 25 in a 76% yield. The diester 25 was initially treated with dichloride 26, which, however, did not lead to the desired 4-methylenepiperidine 24. To obtain the desired methylenepiperidine 24, the more reactive diiodide 29 should be employed instead of the dichloride 26. Allyl diiodide 29 was freshly prepared by a Finkelstein reaction of commercially available 3-chloro-2-(chloromethyl)prop-1-ene (26) with NaI in acetone [44]. After a reaction time of 16 h in refluxing acetone, the diiodide 29 was obtained in a 99% yield.
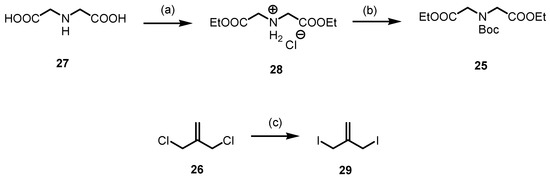
Scheme 7.
Synthesis of diester 25 and diiodide 29 for the designed preparation of methylenepiperidine 24. Reagents and reaction conditions were as follows: (a) SOCl2, EtOH, 0 °C to reflux, 16 h, 98%. (b) Boc2O, NaHCO3, THF, H2O, r.t., 16 h, 76%. (c) NaI, acetone, reflux, 16 h, 99%.
For the double allylation of diester 25, LDA was generated in situ from n-BuLi and i-Pr2NH. Deprotonation of diester 25 with freshly prepared LDA and subsequent treatment with diiodide 29 provided the methylenepiperidine 24 in a 77% yield. The IR and 1H NMR spectra of piperidine 24 demonstrate the successful synthesis of the piperidine ring. A band at 1655 cm−1 in the IR spectrum originates from the C=C stretching vibration. Two sets of signals can be found in the 1H NMR spectrum, as illustrated by two singlets for the protons of the exocyclic methylene moiety (R2C=CH2) at 4.83 and 4.92 ppm and two singlets for the Boc group at 1.42 and 1.47 ppm. These signal pairs confirm the formation of trans- and cis-configured diastereomers trans-24 and cis-24, which are present in the ratio 9:1. Lemieux–Johnson oxidation using catalytic amounts of OsO4 and an excess of NaIO4 transformed the 4-methylenepiperidine 24 into piperidinone 30 [45]. Despite the fact that compound 24 was used as a mixture of diastereomers, only one diastereomer could be observed for compound 30. The subsequent Wittig reaction of ketone 30 provided the α,β-unsaturated ester 31, which shows an even higher structural similarity to betalamic acid than methylenepiperidine 24 and piperidinone 30 (Scheme 8).
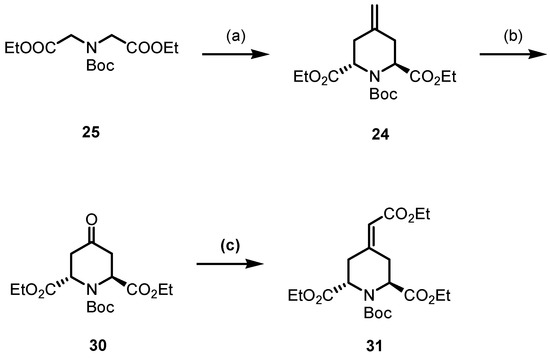
Scheme 8.
Synthesis of α,β-unsaturated ester 31. Reagents and reaction conditions were as follows: (a) 1. LDA, THF, −78 °C, 1 h. 2. Allyl diiodide 29, THF, −78 °C to r.t., 16 h, 77%; (b) OsO4 (0.05 m in H2SO4), NaIO4, pyridine, H2O, t-BuOH, r.t., 48 h, 73%; (c) Ph3P=CHCO2Et, toluene, reflux, 48 h, 75%.
Since the piperidines 24, 30, and 31 do not contain a halogen atom for elimination, another method for the introduction of a double bond into the piperidine ring was required. For this purpose, the Boc-protective group was removed, yielding the secondary amines 32, 33 and 34. The secondary amines 32–34 were reacted with in situ prepared t-BuOCl followed by base-induced HCl elimination, according to the method reported by Zhong et al. [46] (Scheme 9). For compound 32, isolation of the expected product 35 was not possible due to the fast oxidation to its pyridine form 38, isolated in a 6% yield. For compound 33, the formation of the conjugate system was successful, leading to the desired product 36 in a 39% yield. For this product, we did not observe the formation of the pyridine form 39. With compound 34, the conjugate derivative 37 was obtained. Although, a slow conversion to the pyridine form 40 was observed.
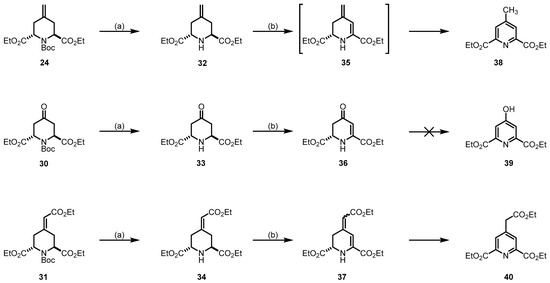
Scheme 9.
Removal of the Boc-protective group and introduction of a double bond in the 5,6-position of the piperidine ring. Reagents and reaction conditions were as follows: (a) CF3COOH, CH2Cl2, r.t., 16 h, 11% (32), 71% (33), 90% (34); (b) 1. NaOCl (0.75 m in H2O), AcOH, t-BuOH, t-butyl methyl ether, r.t., 1 h; 2. ethyldiisopropylamine, r.t., 16 h, 6% (38), 39% (36), 30% (37).
2.2. Antioxidant Activity and Stability
Due to the instability of aldehyde 13, we decided to evaluate the total antioxidant activity (TAC) of the protected aldehyde 21. For this purpose, the Folin–Ciocalteu assay was employed [47]. This method can be classified among the protocols used to evaluate the TAC in the electron transfer (ET) group [48]. Reduction of the oxidant leads to a change in its properties, such as light absorption or fluorescence, which are measured using spectroscopy techniques [49]. In the Folin–Ciocalteu assay, a molybdotungstophosphate heteropolyanion (3H2O-P2O5-14WO3-4MoO3-10H2O) is used for the oxidation of phenolic compounds in basic solution (carbonate buffer). The reduction leads to a colored product with an absorption maximum (λmax) at 765 nm. The molibdenum center in the complex is reduced from Mo(VI) to Mo(V) by an e− donated from the antioxidant, leading to a blue solution [49].
Unfortunately, during the test of the protected aldehyde 21 in the Folin–Ciocalteu assay, a change in the color of the solution could not be recorded as reduction of the molybdenum complex did not take place. All information were provided in Supplementary Materials (Page S2).
The stability towards oxygen of the 1,2,3,4-tetrahydropyridines 35–37 was observed spectroscopically using 1H NMR spectra. After oxidizing the 4-methylenepiperidine 32 with t-BuOCl, only pyridine 38 was detected, indicating the fast oxidation of intermediate tetrahydropyridine 35 by O2. In contrast to the fast oxidation of methylenetetrahydropyridine 35, tetrahydropyridone 36 did not show any potential to be further oxidized. The most promising properties were observed for the ester 37. Although it could be isolated in its pure form, recording of 1H NMR spectra over a period of several days revealed the slow oxidation of ester 37 to pyridine 40 (Figure 3).
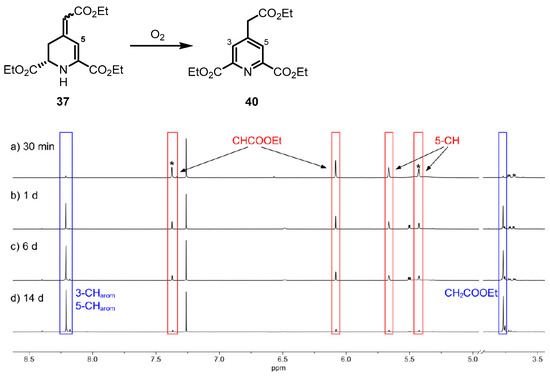
Figure 3.
1H NMR spectra of 1,2,3,4-tetrahydropyridine 37 after different time intervals. Ratio of diastereomers of cis-37 and trans-37 is 55:45. The signals for the minor diastereomer are marked with an asterisk (*). Increasing amounts of aromatic pyridine 40 can be detected. Signals of 37 are marked with red boxes and signals for pyridine 40 with blue boxes.
3. Materials and Methods
3.1. Chemistry
Moisture and oxygen sensitive reactions were carried out under nitrogen, dried with molecular sieves (3 or 4 Å, 8 to 12 mesh, Acros Organics), in dry glassware (Schlenk flasks or Schlenk tubes, sealed with rubber septa). All solvents were of analytical grade quality. Flash chromatography (FC): silica gel 60, 40–63 µm (Machery Nagel); parentheses include: diameter of the column (Ø), length of the stationary phase (h), fraction size (V) and eluent. Melting point: melting point system MP50 (Mettler Toledo, gießen, Germany), open capillary, uncorrected. MS: MicroTOFQII mass spectrometer (Bruker Daltonics, Bremen, Germany); deviations of the found exact masses from the calculated exact masses were 5 ppm or less; the data were analyzed with DataAnalysis® (Bruker Daltonics). NMR: NMR spectra were recorded in deuterated solvents on Agilent DD2 400 MHz and 600 MHz NMR spectrometers (Agilent, Santa Clara, CA, USA); chemical shifts (δ) are reported in parts per million (ppm) against the reference substance tetramethylsilane and calculated using the solvent residual peak of the undeuterated solvent; coupling constants are given with 0.5 Hz resolution; assignment of 1H and 13C NMR signals was supported by 2D NMR techniques where necessary. IR: FT/IR IR Affinity®-1 spectrometer (Shimadzu, Düsseldorf, Germany) using ATR technique. Characterization data including 1H and 13C NMR spectra for synthesized compounds are reported in Supplementary Materials (Page S3).
3.1.1. Synthesis of Tert-Butyl 4-Oxopiperidine-1-Carboxylate (18)
Piperidin-4-one monohydrate hydrochloride 19 (5.0 g, 32.5 mmol, 1.0 eq.) was dissolved in a 1:1 mixture of THF:H2O (100 mL) at room temperature. NaHCO3 (5.47 g, 65 mmol, 2.0 eq.) was added and the mixture was stirred for 15 min at rt. Afterwards, Boc2O (8.52 g, 39 mmol, 1.2 eq.) was added and the mixture was stirred for 16 h at room temperature. The mixture was diluted with Et2O (50 mL) and washed with aqueous solution of KHSO4 5% (3 × 50 mL), H2O (3 × 50 mL) and brine (3 × 50 mL). The combined organic layers were concentrated in vacuo. The residue was purified by flash column chromatography (petroleum ether/EtOAc 9/1 → 5/5, Rf = 0.34 (cHex/EtOAc 7:3)). Colorless solid, mp 73–76 °C, yield 6.22 g (31 mmol, 96%). Exact mass (APCI): m/z = 200.1279 (calcd.200.1287 for C10H18NO3+ [M+H+]).1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.42–1.44 (m, 9H, C(CH3)3), 2.34 (t, J = 6.2 Hz, 4H, 3-(CH2)2), 3.60 (t, J = 6.2 Hz, 4H, 2-(CH2)2). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 28.0 (3C, C(CH3)3), 40.0 (2C, C-3), 40.3 (2C, C-2), 79.2 (1C, OC(CH3)3), 153.8 (1C, C(=O)OC(CH3)3), 207.4 (1C, R2C=O). FT-IR (neat): ṽ (cm−1) = 2985, 2870 (C-H, aliph.), 1724 (C=Ocarb.), 1678 (C=Oketone), 1161 (C-O).
3.1.2. Synthesis of Tert-Butyl 3-Bromo-4-Oxopiperidine-1-Carboxylate (17)
1-Boc-piperidin-4-one 18 (10 g, 50 mmol, 1.0 eq.) was dissolved in THF (30 mL) and Et2O (30 mL). AlCl3 (0.67 g, 5.0 mmol, 0.1 eq.) was added and at 0 °C, Br2 (2.6 mL, 50 mmol, 1.0 eq.) was added slowly over a period of 30 min. Afterwards, the solution was stirred at 0 °C for 18 h. Afterwards, the formed solid was filtered off and washed with Et2O. The organic layer was dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (petroleum ether/EtOAc 9/1 → 5/5, Rf = 0.41 (cHex/EtOAc 7:3)). Colorless solid, mp 90–93 °C, yield 6.42 g (23 mmol, 46%). Exact mass (APCI): m/z = 278.0329 (calcd.278.0392 for C10H17BrNO3+ [M+H+]). 1H NMR (200 MHz, DMSO-d6): δ (ppm) = 1.44 (s, 9H, C(CH3)3), 2.50–2.52 (m, 1H, 5-H), 2.70–2.78 (m, 1H, 5-H), 3.58–3.68 (m, 3H, 2 × 6-H, 2-H), 3.98–4.08 (m, 1H, 2-H), 4.77 (s, 1H, 3-H). 13C NMR (50 MHz, DMSO-d6): δ (ppm) = 27.9 (3C, C(CH3)3), 35.8 (1C, C-5), 42.7 (1C, C-6), 47.7 (1C, C-2), 49.0 (1C, C-3), 79.8 (1C, OC(CH3)3), 153.8 (1C, C(=O)-OC(CH3)3), 199.7 (1C, R2C=Oketone). FT-IR (neat): ṽ (cm−1) = 2978, 2931 (C-H. aliph.), 1724 (C=Oketone), 1674 (C=Ocarbamate), 1157 (C-O), 648 (C-Br).
3.1.3. Synthesis of Tert-Butyl (E)-3-Bromo-4-(Ethoxycarbonylmethylene)Piperidine-1-CarBoxylate (16)
4-(Ethoxycarbonylmethylene)triphenylphosphorane (6.9 g, 20 mmol, 1.1 eq.) was added to a solution of tert-butyl 3-bromo-4-oxopiperidine-1-carboxylate 17 (5.03 g, 18 mmol, 1.0 eq.) in CH2Cl2 (450 mL) and the reaction mixture was stirred at reflux for 2 h. Then, the mixture was cooled to room temperature and concentrated in vacuo. The residue was purified by flash column chromatography (petroleum ether/EtOAc 9/1 → 7/3, Rf = 0.57 (cHex/EtOAc 7:3)). Colorless solid, mp 114–115 °C, yield 5.98 g (17 mmol, 95%). Exact mass (APCI): m/z = 348.0777 (calcd.348.0810 for C14H23BrNO4+ [M+H+]). 1H NMR (200 MHz, DMSO-d6): δ (ppm) = 1.21 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.42 (s, 9H, C(CH3)3), 2.56–2.88 (m, 2H, 5-CH2, 6-CH2), 3.34–3.51 (m, 2H, 2-CH2, 5-CH2), 4.01–4.29 (m, 4H, OCH2CH3, 6-CH2, 2-CH2), 5.04–5.12 (m, 1H, 3-CH), 6.12 (s, 1H, R2C=CH). 13C NMR (50 MHz, DMSO-d6): δ (ppm) = 14.0 (1C, OCH2CH3), 24.1 (1C, C-5), 27.9 (3C, C(CH3)3), 42.5 (1C, C-6), 51.0 (1C, C-2), 53.3 (1C, C-3), 59.9 (1C, OCH2CH3), 79.2 (1C, C(CH3)3), 113.8 (1C, R2C=CH), 153.7 (1C, C(=O)OC(CH3)3), 154.0 (1C, C-4), 165.1 (1C, CO2Et). Only one set of signals can be observed in the spectra. FT-IR (neat): ṽ (cm−1) = 2985, 2920 (C-H, aliph.), 1712 (C=O), 1670 (C=O), 1654 (C=C), 1161 (C-O), 641 (C-Br).
3.1.4. Synthesis of Tert-Butyl (E)- and (Z)-Ethoxycarbonylmethylene)-3,4-Dihydropyridine-1(2H)-Carboxylate (15)
Ester 16 (5.74 g, 16 mmol, 1.0 eq) was dissolved in dry DMF (165 mL). LiBr (8.6 g, 99 mmol, 6.0 eq.) and Li2CO3 (7.31 g, 99 mmol, 6.0 eq.) were added and the solution was stirred at 75 °C for 3 h. Then, the mixture was cooled to room temperature and extracted with EtOAc (3 × 100 mL). The combined organic layers were concentrated in vacuo. The residue was purified by flash column chromatography (petroleum ether/EtOAc 9/1 → 7/3, Rf = 0.72 (cHex/EtOAc 7:3)). Yellow oil, yield 3.88 g (14 mmol, 88%). Exact mass (APCI): m/z = 268.1497 (calcd. 268.1549 for C14H22NO4+ [M+H+]). Compound 15 was isolated as a mixture of ((E):(Z)) isomers. In the NMR spectra, a ratio of 9:1 is observed. 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.19 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.46 (s, 9H, C(CH3)3), 2.49–2.53 (m, 0.2H, 5-CH2), 3.00–3.07 (m, 1.8H, 5-CH2), 3.52–3.60 (m, 2H, 6-CH2), 4.06 (q, J = 7.1 Hz, 2H, OCH2CH3), 5.32–5.34 (m, 0.1H, R2C=CH), 5.47–5.53 (m, 0.9H, 3-CH) 5.55–5.59 (m, 0.9H, R2C=CH), 6.54–6.61 (m, 0.1H, 3-CH) 7.00–7.13 (m, 0.9H, 2-CH), 7.14–7.16 (m, 0.1H, 2-CH). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 14.2 (1C, OCH2CH3), 24.8 (0.9C, C-5), 27.7 (3C, C(CH3)3), 30.0 (0.1C, C-5), 40.0 (1C, C-6), 59.0 (1C, OCH2CH3), 81.5 (1C, C(CH3)3), 103.4 (0.1C, C-3), 108.1 (0.9C, C-3), 109.6 (0.1C, R2C=CH) 110.6 (0.9C, R2C=CH), 132.5 (0.9C, C-2), 133.0 (0.1C, C-2), 147.5 (1C, C(=O)OC(CH3)3), 147.9 (0.1C, R2C=CH) 149.1 (0.9C, R2C=CH), 166.0 (1C, CO2Et) FT-IR (neat): ṽ (cm−1) = 2978, 2931, 2900 (C-H, aliph.), 1708 (C=O), 1700 (C=O), 1608 (C=C), 1145 (C-O), 1111 (C-O).
3.1.5. Synthesis of Tert-Butyl (E)- and (Z)-4-(2-Hydroxyethylidene)-3,4-Dihydropyridine-1(2H)-Carboxylate (20)
Procedure 1
Under N2, 15 (2.80 g, 10.5 mmol, 1.0 eq.) was dissolved in dry toluene (25 mL). The solution was cooled to −78 °C, DIBAL-H (1 m solution in hexane, 31.5 mL, 31.5 mmol, 3.0 eq.) was added dropwise within 15 min, and the reaction mixture was stirred at −78 °C for 40 min. Then, at −78 °C, CH3OH (2 mL) was added carefully, and the mixture was warmed to room temperature. The mixture was filtered, the solid was washed with EtOAc (3 × 30 mL) and the combined organic layers were concentrated in vacuo. Yellow oil, yield 2.22 g (9.9 mmol, 94%).
Procedure 2
Under N2, 15 (570 mg, 2.1 mmol, 1.0 eq.) was dissolved in dry THF (5 mL) and at −10 °C, LiAlH4 (160 mg, 4.3 mmol, 2.0 eq.) was added and the mixture was stirred for 40 min. A saturated solution of potassium sodium tartrate (5 mL) was added, and the mixture was extracted with EtOAc (3 × 15 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (cHex/EtOAc 9/1 → 6/4, Rf = 0.45 (petroleum ether/EtOAc 7:3)). Yellow oil, yield 0.33 g (1.5 mmol, 70%). Exact mass (APCI): m/z = 226.1383 (calcd. 226.1443 for C12H20NO3+ [M+H+]). Compound 20 was isolated as a mixture of ((E):(Z)) isomers. In the NMR spectra, a ratio of 8.5:1.5 is observed. Peaks of minor isomer are not all clearly visible in the spectra. 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.44 (s, 9H, C(CH3)3), 2.36–2.44 (m, 2H, 5-CH2), 3.48–3.55 (m, 2H, 6-CH2), 3.96–4.01 (m, 2H, CH2OH), 4.53–4.58 (m, 1H, OH), 5.13 (t, J = 6.8 Hz, 0.15H, R2C=CH), 5.35 (t, J = 6.8 Hz, 0.85H, R2C=CH), 5.40–5.46 (m, 1H, 3-CH), 6.65–6.78 (m, 1H, 2-CH). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 26.9 (1C, C-5), 30.9 (3C, C(CH3)3), 43.1 (1C, C-6), 59.9 (1C, CH2OH), 83.6 (1C, C(CH3)3), 112.7 (1C, C-3), 127.8 (1C, R2C=CH), 128.4 (1C, C-2), 133.2 (1C, R2C=CH), 154.3 (1C, C(=O)OC(CH3)3). FT-IR (neat): ṽ (cm−1) = 3398 (O-H), 2974, 2931, 2873 (C-H, aliph.), 1701 (C=O), 1643 (C=C), 1612 (C=C), 1161, 1141 (C-O).
3.1.6. Synthesis of Tert-Butyl (E)- and (Z)-4-(Formylmethylene)-3,4-Dihydropyridine-1(2H)-Carboxylate (21)
CuCl (11 mg, 0.12 mmol, 0.1 eq.) and TEMPO (17 mg, 0.12 mmol, 0.1 eq.) were added to a solution of racemic mixture of allylic alcohol 20 (270 mg, 1.20 mmol, 1.0 eq.) in dry DMF (3 mL). The solution was stirred at room temperature for 16 h. Afterwards, the solution was poured into water/ice slowly and the mixture was warmed to room temperature. The solid was filtered, washed with H2O and dried. Purification by flash column chromatography (petroleum ether/EtOAc 9/1 → 7/3 Rfa = 0.38, Rfb = 0.30 (cHex/EtOAc 7:3)). Yellow oil, yield 240 mg (1.07 mmol, 90%). Exact mass (APCI): m/z = 224.1208 (calcd. 224.2495 for C24H35N2O6+ [M+H+]). Compound 21 was isolated as a mixture of ((E):(Z)) isomers. In the NMR spectra, a ratio of 6:4 is observed. Peaks of minor isomer are not all clearly visible in the spectra. 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.52 (s, 3.6H, C(CH3)3), 1.53 (s, 5.4H, C(CH3)3), 2.64 (t, J = 6.8 Hz, 0.8H, 5-CH2), 2.91–3.14 (m, 1.2H, 5-CH2), 3.74 (t, J = 6.8 Hz, 2H, 6-CH2), 5.44–5.53 (m, 0.6H, 3-CH), 5.57 (d, J = 7.8 Hz, 0.4H, R2C=CH), 5.78 (d, J = 7.7 Hz, 0.6H, R2C=CH), 6.23–6.36 (m, 0.4H, 3-CH), 7.13–7.19 (m, 0.6H, 2-CH), 7.27–7.41 (m, 0.4H, 2-CH), 9.94 (d, J = 7.9 Hz, 0.6H, CHO), 10.05 (d, J = 7.9 Hz, 0.4H, CHO). 13C NMR (151 MHz, CDCl3): δ (ppm) = 24.7 (0.6C, C-5), 28.2 (3C, C(CH3)3), 30.9 (0.4C, C-5), 40.4 (1C, C-6), 82.8 (1C, C(CH3)3), 101.2 (0.4C, C-3), 108.1 (0.6C, C-3), 121.5 (1C, R2C=CH), 134.0 (0.6C, C-2), 134.5 (0.4C, C-2), 151.1 (1C, R2C=CH), 151.3 (1C, C(=O)OC(CH3)3), 189.6 (0.4C, CHO), 190.0 (0.6C, CHO). FT-IR (neat): ṽ (cm−1) = 3062, 2966, 2877 (C-H, aliph.), 1712 (C=O), 1647 (C=C), 1593 (C=C), 1141, 1122 (C-O).
3.1.7. Synthesis of (E)- and (Z)-2-[2,3-Dihydropyridin-4(1H)-Ylidene]Acetaldehyde (13)
A solution of 21 (110 mg, 0.49 mmol, 1.0 eq.) in water (9 mL) and 1,4-dioxane (1 mL) was heated to 85 °C for 2 h. Then, the solution was cooled to room temperature and the mixture was extracted with EtOAc (6 × 15 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (petroleum ether/EtOAc 9/1 → 1/9 Rfa = 0.15 (EtOAc)). Yellow/orange oil, yield 12 mg (0.10 mmol, 20%). The compound is highly unstable and quickly decomposed during purification. Aldehyde 13 was isolated as a mixture of ((E):(Z)) isomers. In the NMR spectra, a ratio of 6:4 is observed. Peaks of minor isomer are not all clearly visible in the spectra. 1H NMR (200 MHz, CDCl3): δ (ppm) = 2.61 (t, J = 7.0 Hz, 0.8H, 5-CH2), 3.00–3.12 (m, 1.2H, 5-CH2), 3.31–3.46 (m, 2H, 6-CH2), 4.7–4.81 (m, 1H, NH), 5.19 (d, J = 7.2 Hz, 0.6H, R2C=CH), 5.29 (d, J = 7.9 Hz, 0.4H, R2C=CH), 5.60 (d, J = 8.3 Hz, 0.6H, 3-CH), 6.00 (d, J = 7.5 Hz, 0.4H, 3-CH), 6.66–6.77 (m, 1H, 2-CH), 9.79 (d, J = 8.4 Hz, 0.6H, CHO), 9.93 (d, J = 8.0 Hz, 0.4H, CHO). 13C NMR (50 MHz, CDCl3): δ (ppm) = 25.3 (0.6C, C-5), 31.75 (0.4C, C-5), 40.5 (0.6C, C-6), 40.9 (0.4C, C-6), 93.9 (0.4C, C-3), 100.3 (0.6C, C-3), 116.2 (0.4C, R2C=CH), 117.1 (0.6C, R2C=CH), 142.6 (1C, C-2), 143.4 (1C, C-2), 155.3 (0.4C, R2C=CH), 171.3 (0.6C, R2C=CH), 189.2 (0.4C, CHO), 189.3 (0.6C, CHO).
3.1.8. Synthesis of Diethyl 2,2′-Iminodiacetate∙HCl (28)
At 0 °C, SOCl2 (20.0 mL, 275 mmol, 1.5 eq) was added dropwise to a suspension of iminodiacetic acid 27 (24.4 g, 184 mmol, 1.0 eq) in EtOH abs. (200 mL). Afterwards, the reaction mixture was heated to reflux for 16 h. The solution was cooled down to room temperature and concentrated in vacuo. Colorless solid, mp 88–89 °C, yield 40.6 g (98%). C8H16ClNO4 (225.7 g/mol). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.24 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3), 3.70 (brs, 2H, NH2+), 3.99 (s, 4H, 2 × CH2), 4.21 (q, J = 7.1 Hz, 4H, 2 × OCH2CH3). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 13.9 (2C, 2 × OCH2CH3), 46.4 (2C, 2 × CH2), 61.8 (2C, 2 × OCH2CH3), 166.4 (2C, 2 × O=COEt). IR (neat): ṽ (cm−1) = 2936 (C-Halip), 1736 (C=Oester), 1204, 1076, 1015 (C-N, C-O). Exact mass (APCI): m/z = 190.1073 (calcd. 190.1074 for C8H16NO4 [M-Cl]+).
3.1.9. Synthesis of Diethyl 2,2′-[N-(Tert-Butoxycarboxyl)Imino]Diacetate (25)
NaHCO3 (22.8 g, 271 mmol, 3.0 eq) was added to a solution of 28 (20.4 g, 90.4 mmol, 1.0 eq) in THF (80 mL) and H2O (20 mL). The reaction mixture was stirred for 30 min at room temperature. After the addition of Boc2O (19.3 mL, 90.4 mmol, 1.0 eq), the mixture was stirred at room temperature for 16 h. Then, it was extracted with EtOAc (3 × 50 mL) and the combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (ø = 8 cm, h = 16 cm, cHex/EtOAc 8:2, V = 80 mL). Colorless oil, yield 19.9 g (76%). C13H23NO6 (289.3 g/mol). TLC: Rf = 0.36 (cHex/EtOAc 8:2). 1H NMR (400 MHz, DMSO-d6): δ (ppm) = 1.19 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.20 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.35 (s, 9H, C(CH3)3), 3.98 (s, 2H, NCH2), 4.01 (s, 2H, NCH2), 4.10 (q, J = 7.1 Hz, 2H, OCH2CH3), 4.12 (q, J = 7.1 Hz, 2H, OCH2CH3). Ratio of rotamers is 1:1. 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 14.0 (1C, OCH2CH3), 14.1 (1C, OCH2CH3), 27.7 (3C, C(CH3)3), 49.2 (1C, CH2), 49.7 (1C, CH2), 60.4 (2C, OCH2CH3), 79.9 (1C, C(CH3)3), 154.6 (1C, N(C=O)O), 169.45 (1C, O=COEt), 169.53 (1C, O=COEt). Ratio of rotamers is 1:1. IR (neat): ṽ (cm−1) = 2978 (CHaliph), 1747 (C=Oester), 1700 (C=Ocarbamate), 1185, 1159, 1026 (C-N, C-O). Exact mass (APCI): m/z = 290.1587 (calcd. 290.1598 for C13H24NO6+ [M+H]+).
3.1.10. Synthesis of 3-Iodo-2-(Iodomethyl)Prop-1-Ene (29) [44]
NaI (17.8 g, 119 mmol, 2.5 eq) was added to a solution of 3-chloro-2-(chloromethyl)prop-1-ene 26 (5.50 mL, 47.5 mmol, 1.0 eq) in acetone (100 mL) and the mixture was stirred at reflux for 16 h. The suspension was cooled to room temperature and concentrated in vacuo. The residue was dissolved in H2O (75 mL) and cHex (75 mL). After separation of the two layers, the organic layer was washed with Na2SO3 (2 × 50 mL) and H2O (50 mL), dried (Na2SO4), filtered and concentrated in vacuo. Light green solid, mp 28–29 °C, yield 14.5 g (99%). C4H6I2 (307.9 g/mol). 1H NMR (600 MHz, CD3OD): δ (ppm) = 4.21 (s, 4H, 2 × CH2I), 5.40 (s, 2H, R2C=CH2). 13C NMR (151 MHz, CD3OD): δ (ppm) = 6.6 (2C, 2 × CH2I), 116.4 (1C, R2C=CH2), 146.0 (1C, R2C=CH2). Exact mass (APCI): m/z = 308.8637 (calcd. 308.8632 for C4H7I2+ [M+H]+).
3.1.11. Synthesis of 1-Tert-Butyl 2,6-Diethyl Cis- and Trans-4-Methylenepiperidine-1,2,6-Tricarboxylate (24)
At −78 °C, n-BuLi (1.6 m in n-hexane, 74.4 mL, 119 mmol, 2.1 eq) was added dropwise to a solution of i-Pr2NH (16.7 mL, 119 mmol, 2.1 eq) in dry THF (170 mL). After the mixture was stirred for 1 h, a solution of 25 (16.4 g, 56.7 mmol, 1.0 eq) in dry THF (15 mL) was added and the mixture was stirred for 1 h at −78 °C. Then, a solution of 29 (22.1 g, 68.1 mmol, 1.2 eq) in dry THF (15 mL) was added and the reaction mixture was stirred for 30 min at −78 °C and warmed up to room temperature over 16 h. At 0 °C, H2O (150 mL) was added and the mixture was extracted with EtOAc (3 × 80 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified twice by flash column chromatography (1. ø = 8 cm, h = 25 cm, cHex/EtOAc 9:1 → 8:2, V = 80 mL, 2. ø = 8 cm, h = 25 cm, cHex/EtOAc 9:1 → 8:2, V = 80 mL). Yellow oil, yield 14.8 g (77%). C17H27NO6 (341.4 g/mol). TLC: Rf = 0.26 (cHex/EtOAc 8:2). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.26 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.42 (s, 9 × 0.9H, C(CH3)3), 1.47 (s, 9 × 0.1H, C(CH3)3*), 2.43–2.51 (m, 2 × 0.1H, 3/5-CH2*), 2.65 (dd, J = 15.9/2.7 Hz, 1H, 3/5-CH2), 2.74 (dd, J = 16.0/3.6 Hz, 1H, 3/5-CH2), 2.79–2.87 (m, 2 × 0.9H, 3/5-CH2), 4.09–4.24 (m, 4H, 2 × OCH2CH3), 4.60 (dd, J = 3.5/3.0 Hz, 1H, 2/6-CH), 4.69 (dd, J = 6.2/4.1 Hz, 1H, 2/6-CH), 4.81–4.83 (m, 2 × 0.9H, R2C=CH2), 4.90–4.93 (m, 2 × 0.1H, R2C=CH2*). Ratio of isomers is 9:1 (trans:cis). Signals for the cis diastereomer are marked with an asterisk (*). 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.3 (1C, OCH2CH3), 14.5 (1C, OCH2CH3), 28.3 (3 × 0.9C, C(CH3)3), 28.4 (3 × 0.1C, C(CH3)3*), 33.25 (0.9C, C-3/5), 33.34 (0.9C, C-3/5), 33.6 (2 × 0.1C, C-3*, C-5*), 54.6 (1C, C-2/6), 55.7 (1C, C-2/6), 61.0 (2 × 0.1C, OCH2CH3*), 61.2 (0.9C, OCH2CH3), 61.3 (0.9C, OCH2CH3), 81.17 (0.9C, C(CH3)3), 81.21 (0.1C, C(CH3)3*) 112.3 (0.9C, R2C=CH2), 112.7 (0.1C, R2C=CH2*), 137.5 (0.9C, R2C=CH2), 138.1 (0.1C, R2C=CH2*), 155.2 (0.1C, N(C=O)O*), 155.5 (0.9C, N(C=O)O), 171.3 (2 × 0.1C, O=COEt)*), 172.8 (2 × 0.9C, O=COEt). Ratio of isomers is 9:1 (trans:cis). Signals for the cis diastereomer are marked with an asterisk (*). Purity (HPLC, method A): 94.4% (tR = 21.5 min). IR (neat): ṽ (cm−1) = 2978 (CHaliph), 1740 (C=Oester), 1701 (C=Ocarbamate), 1655 (C=C), 1180, 1165, 1022 (C-N, C-O). Exact mass (APCI): m/z = 342.1913 (calcd. 342.1911 for C17H28NO6 [M+H]+).
3.1.12. Synthesis of 1-Tert-Butyl 2,6-Diethyl Trans-4-Oxopiperidine-1,2,6-Tricarboxylate (30)
OsO4 (0.05 m in H2SO4, 3.60 mL, 0.18 mmol, 0.01 eq), pyridine (0.70 mL, 9.10 mmol, 0.5 eq) and NaIO4 (15.6 g, 72.8 mmol, 4.0 eq) were added to a solution of 24 (6.20 g, 18.2 mmol, 1.0 eq) in t-BuOH (80 mL) and H2O (120 mL) and the suspension was stirred at room temperature for 48 h. Then, the mixture was filtered and Na2SO3 (50 mL) and EtOAc (50 mL) were added to the solution. The aqueous layer was extracted with EtOAc (3 × 50 mL) and the combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (ø = 8 cm, h = 16 cm, cHex/EtOAc 9:1 → 8:2, V = 80 mL). Colorless solid, mp 49–50 °C, yield 4.55 g (73%). C16H25NO7 (343.4 g/mol). TLC: Rf = 0.33 (cHex/EtOAc 8:2). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.27 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.45 (s, 9H, C(CH3)3), 2.72 (dd, J = 18.0/1.4 Hz, 1H, 3/5-CHeq), 2.86 (dd, J = 18.0/1.4 Hz, 1H, 3/5-CHeq), 3.01–3.08 (m, 2H, 3-CHax, 5-CHax), 4.12–4.26 (m, 4H, 2 × OCH2CH3), 4.83 (d, J = 7.8 Hz, 1H, 2/6-CH), 5.06 (d, J = 7.8 Hz, 1H, 2/6-CH). 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.2 (1C, OCH2CH3), 14.3 (1C, OCH2CH3), 28.3 (3C, C(CH3)3), 40.6 (1C, C-3/5), 41.0 (1C, C-3/5), 53.0 (1C, C-2/6), 54.2 (1C, C-2/6), 61.9 (1C, OCH2CH3), 62.1 (1C, OCH2CH3), 81.9 (1C, C(CH3)3), 154.5 (1C, N(C=O)O), 172.3 (1C, O=COEt), 172.4 (1C,O=COEt), 203.8 (1C, C-4). IR (neat): ṽ (cm−1) = 2978 (CHaliph), 1736 (C=Oester), 1701 (C=Oketone, carbamate), 1188, 1115, 1026 (C-N, C-O). Exact mass (APCI): m/z = 344.1690 (calcd. 344.1704 for C16H26NO7+ [M+H]+).
3.1.13. Synthesis of 1-Tert-Butyl 2,6-Diethyl Trans-(Ethoxycarbonylmethylene)-Piperidine-1,2,6-Tricarboxylate (31)
4-(Ethoxycarbonylmethylene)triphenylphosphorane (7.60 g, 21.7 mmol, 1.75 eq) was added to a solution of 30 (4.30 g, 12.4 mmol, 1.0 eq) in toluene (50 mL) and the mixture was heated at reflux for 48 h. After concentrating the mixture in vacuo, the residue was purified by flash column chromatography (ø = 6 cm, h = 17 cm, cHex/EtOAc 9:1, V = 80 mL). Colorless oil, yield 3.9 g (75%). C20H31NO8 (413.5 g/mol). TLC: Rf = 0.35 (cHex/EtOAc 8:2). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 1.11–1.22 (m, 9H, 3 × OCH2CH3), 1.35 (s, 9 × 0.54H, C(CH3)3), 1.36 (s, 9 × 0.46H, C(CH3)3*), 2.71 (dd, J = 16.8/2.2 Hz, 0.54H, 3/5-CH2), 2.78 (dd, J = 17.1/3.2 Hz, 0.46H, 3/5-CH2*), 2.83–2.95 (m, 1.46H, 3/5-CH2, 3-CH2*, 5-CH2*), 3.01 (ddm, J = 18.9/7.3 Hz, 0.54H, 3/5-CH2), 3.61 (dm, J = 18.9 Hz, 0.54H, 3/5-CH2), 3.68 (dm, J = 18.5 Hz, 0.46H, 3/5-CH2*), 4.02–4.17 (m, 6H, 3 × OCH2CH3), 4.57 (dd, J = 6.7/2.3 Hz, 0.54H, 2/6-CH), 4.62 (dd, J = 6.4/3.1 Hz, 0.46H, 2/6-CH*), 4.67 (dd, J = 7.2/1.9 Hz, 0.46H, 2/6-CH*), 4.75 (dd, J = 7.3/2.3 Hz, 0.54H, 2/6-CH), 5.80 (s, 0.54H, R2C=CH), 5.81 (s, 0.46H, R2C=CH*). Ratio of rotamers is 54:46. Signals for the minor rotamer are marked with an asterisk (*). 13C NMR (151 MHz, DMSO-d6): δ (ppm) = 13.9 (0.54C, OCH2CH3), 14.0 (0.46C, OCH2CH3*), 14.0 (0.46C, OCH2CH3*), 14.1 (0.54C, OCH2CH3), 14.11 (1C, OCH2CH3), 27.7 (s, 3C, C(CH3)3), 29.8 (0.46C, C-3/5*), 30.4 (0.54C, C-3/5), 33.8 (0.54C, C-3/5), 34.0 (0.46C, C-3/5*), 51.6 (0.54C, C-2/6), 53.0 (0.46C, C-2/6*), 53.1 (0.46C, C-2/6*), 54.1 (0.54C, C-2/6), 59.5 (1C, OCH2CH3), 60.8 (0.46C, OCH2CH3*), 60.9 (0.54C, OCH2CH3), 61.0 (0.46C, OCH2CH3*), 61.1 (0.54C, OCH2CH3), 80.3 (0.54C, C(CH3)3), 80.4 (0.46C, C(CH3)3*), 116.9 (0.54C, R2C=CH2), 117.0 (0.46C, R2C=CH2*), 151.4 (0.46C, R2C=CH2*), 151.6 (0.54C, R2C=CH2), 154.0 (0.54C, N(C=O)O), 154.2 (0.46C, N(C=O)O*), 165.0 (1C, O=COEt), 171.5 (0.46C, O=COEt*), 171.7 (0.54C, O=COEt), 172.0 (0.54C, O=COEt), 172.2 (0.46C, O=COEt*). Ratio of rotamers is 54:46. Signals for the minor rotamer are marked with an asterisk (*). Purity (HPLC, method A): 98.9% (tR = 22.1 min). IR (neat): ṽ (cm−1) = 2978 (C-Haliph), 1744 (C=Oester), 1701 (C=Ocarbamate), 1651 (C=C), 1184, 1142, 1026 (C-N, C-O). Exact mass (APCI): m/z = 414.2144 (calcd. 414.2122 for C20H32NO8+ [M+H]+).
3.1.14. Synthesis of Diethyl Cis/Trans-4-Methylenepiperidine-2,6-Dicarboxylate (32)
TFA (6.5 mL, 87.9 mmol, 30 eq) was added to a solution of 24 (1.0 g, 2.9 mmol, 1.0 eq) in dry CH2Cl2 (50 mL) and the mixture was stirred at room temperature for 16 h. The next day, Na2CO3 was added, the layers were separated and the aqueous layer was extracted with CH2Cl2 (2 × 40 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The diastereomers were separated twice by flash column chromatography (1.50 g cartridge, cHex/EtOAc 8:2 → 6:4; 2. 25 g cartridge, cHex/EtOAc 8:2). C12H19NO4 (241.3 g/mol). cis-32: Yellow oil, yield 79 mg (11%). TLC: Rf = 0.31 (cHex/EtOAc 1:1). 1H NMR (600 MHz CDCl3) δ = 1.29 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3), 2.10–2.17 (m, 2H, 3-CHax, 5-CHax), 2.62 (dd, J = 13.5/2.7 Hz, 2H, 3-CHeq, 5-CHeq), 3.37 (dd, J = 11.8/3.0 Hz, 2H, 2-CHax, 6-CHax), 4.22 (dq, J = 7.1/1.4 Hz, 4H, 2 × OCH2CH3), 4.87 (t, J = 1.7 Hz, 2H, R2C=CH2). NH signal is missing. 13C NMR (151 MHz, CDCl3) δ = 14.3 (2C, 2 × OCH2CH3), 37.7 (2C, C-3, C-5), 58.9 (2C, C-2, C-6), 61.4 (2C, 2 × OCH2CH3), 111.4 (1C, R2C=CH2), 142.5 (1C, C-4), 171.9 (2C, 2 × O=COEt)). IR (neat): ṽ (cm−1) = 2982 (C-Haliph), 1732 (C=Oester), 1651 (C=C), 1180, 1026 (C-N, C-O). Exact mass (APCI): m/z = 242.1360 (calcd. 242.1387 for C12H20NO4 [M+H]+). trans-32: Yellow oil, yield 623 mg (89%). TLC: Rf = 0.20 (cHex/EtOAc 1:1). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.29 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3), 2.46 (dd, J = 13.2/7.0 Hz, 2H, 3-CH2, 5-CH2), 2.56 (dd, J = 13.2/5.0 Hz, 2H, 3-CH2, 5-CH2), 3.84 (dd, J = 7.0/5.0 Hz, 2H, 2-CH, 6-CH), 4.14–4.24 (m, 4H, 2 × OCH2CH3), 4.87 (s, 2H, R2C=CH2). NH signal is missing. 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.4 (2C, 2 × OCH2CH3), 36.3 (2C, C-3, C-5), 56.1 (2C, C-2, C-6), 61.2 (2C, 2 × OCH2CH3), 111.7 (1C, R2C=CH2), 141.3 (1C, C-4), 172.7 (2C, 2 × O=COEt). IR (neat): ṽ (cm−1) = 3356 (N-H), 2978 (C-Halip), 1728 (C=Oester), 1655 (C=C), 1200, 1165, 1026 (C-N, C-O). Exact mass (APCI): m/z = 242.1373 (calcd. 242.1387 for C12H20NO4+ [M+H]+).
3.1.15. Synthesis of Diethyl Trans-4-Oxopiperidine-2,6-Dicarboxylate (33)
TFA (3.60 mL, 47.0 mmol, 30 eq) was added to a solution of 30 (0.54 g, 1.57 mmol, 1 eq) in CH2Cl2 (15 mL) and the mixture was stirred at room temperature for 16 h. Then, the mixture was washed with NaHCO3 (30 mL). The aqueous phase was extracted with CH2Cl2 (3 × 20 mL) and the combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. Yellow oil, yield 0.27 g (71%). C11H17NO5 (243.3 g/mol). TLC: Rf = 0.22 (cHex/EtOAc 1:1). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.28 (t, J = 7.1 Hz, 6H, 2 × OCH2CH3), 2.61 (ddd, J = 15.1/7.0/1.4 Hz, 2H, 3-CHax, 5-CHax), 2.71 (ddd, J = 15.0/5.5/1.3 Hz, 2H, 3-CHeq, 5-CHeq), 4.05 (dd, J = 7.0/5.5 Hz, 2H, 2-CH, 6-CH), 4.21 (q, J = 7.1 Hz, 4H, 2 × OCH2CH3). The signal for NH is not observed in the spectrum. 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.3 (2C, 2 × OCH2CH3), 42.7 (2C, C-3, C-5), 54.8 (2C, C-2, C-6), 61.8 (2C, 2 × OCH2CH3), 171.7 (2C, 2 × O=COEt), 204.7 (1C, C-4).
3.1.16. Synthesis of Diethyl Trans-4-(2-Ethoxy-2-Oxoethylidene)Piperidine-2,6-Dicarboxylate (34)
TFA (2.10 mL, 28.4 mmol, 30 eq) was added to a solution of 31 (0.39 g, 0.95 mmol, 1 eq) in CH2Cl2 (10 mL) and the mixture was stirred at room temperature for 16 h. Then, the mixture was washed with NaHCO3 (20 mL). The aqueous phase was extracted with CH2Cl2 (3 × 15 mL) and the combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. Yellow oil, yield 0.27 g (90%). C15H23NO6 (313.4 g/mol). TLC: Rf = 0.35 (cHex/EtOAc 1:1). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.25 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.27 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.28 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.52 (dd, J = 13.3/7.1 Hz, 1H, 3/5-CHax), 2.61 (dd, J = 13.3,/5.0 Hz, 1H, 3/5-CHeq), 3.21 (dd, J = 13.8, 6.9 Hz, 1H, 3/5-CHax), 3.26 (dd, J = 13.8/5.3 Hz, 1H, 3/5-CHeq), 3.86 (dd, J = 6.9/5.3 Hz, 1H, 2/6-CH), 3.92 (dd, J = 7.1/5.0 Hz, 1H, 2/6-CH), 4.12–4.24 (m, 6H, 3 × OCH2CH3), 5.75 (s, 1H, R2C=CH). 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.3 (1C, OCH2CH3), 14.4 (2C, 2 × OCH2CH3), 31.2 (1C, C-3/5), 38.2 (1C, C-3/5), 55.6 (1C, C-2/6), 56.0 (1C, C-2/6), 60.0 (1C, OCH2CH3), 61.3 (1C, OCH2CH3), 61.4 (1C, OCH2CH3), 117.5 (1C, R2C=CH), 153.4 (1C, R2C=CH), 166.0 (1C, O=COEt), 172.2 (1C, O=COEt), 172.6. (1C, O=COEt).
3.1.17. Synthesis of Diethyl 4-Methylpyridine-2,6-Dicarboxylate (38)
At 0 °C, NaOCl (0.75 m in H2O, 3.90 mL, 2.90 mmol, 2 eq) was added to a solution of 32 (350 mg, 1.45 mmol, 1 eq) and AcOH (0.17 mL, 2.90 mmol, 2 eq) in t-BuOH (0.20 mL, 1.74 mmol, 1.2 eq) and methyl t-butyl ether (8 mL) and the mixture was stirred for 1 h. Then, DIPEA (2.05 mL, 11.6 mmol, 8 eq) was added and the mixture was stirred at room temperature for 16 h. After the addition of H2O (10 mL), the mixture was extracted with EtOAc (3 × 15 mL). The combined organic layers were dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (25 g cartridge, cHex/EtOAc 95:5 → 9:1). Yellow oil, yield 20 mg (6%). C12H15NO4 (237.3 g/mol). TLC: Rf = 0.59 (cHex/EtOAc 9:1). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.45 (t, J = 7.2 Hz, 6H, 2 × OCH2CH3), 2.51 (s, 3H, CH3), 4.47 (q, J = Hz, 4H, 2 × OCH2CH3), 8.10 (s, 2H, 3-CHarom, 5-CHarom). 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.4 (2C, 2 × OCH2CH3), 21.3 (1C, CH3), 62.4 (2C, 2 × OCH2CH3), 128.8 (2C, C-3arom, C-5arom), 148.6 (2C, C-2arom, C-6arom), 150.2 (1C, C-4arom), 165.1 (2C, 2 × O=COEt).
3.1.18. Synthesis of Diethyl (RS)-4-Oxo-1,2,3,4-Tetrahydropyridine-2,6-Dicarboxylate (36)
At 0 °C, NaOCl (0.75 m in H2O, 1.70 mL, 1.28 mmol, 1.2 eq) was added to a solution of 33 (260 mg, 1.07 mmol, 1 eq) and AcOH (0.07 mL, 1.28 mmol, 1.2 eq) in t-BuOH (0.12 mL, 1.28 mmol, 1.2 eq) and methyl t-butyl ether (10 mL) and the mixture was stirred for 1 h. Then, DIPEA (0.90 mL, 5.35 mmol, 5 eq) was added and the mixture was stirred at room temperature for 16 h. H2O (15 mL) was added and the mixture was extracted with EtOAc (3 × 15 mL) and dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (25 g cartridge, cHex/EtOAc 7:3 → 1:1). Colorless oil, yield 101 mg (39%). C11H15NO5 (241.2 g/mol). TLC: Rf = 0.40 (cHex/EtOAc 1:1). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.30 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.35 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.70 (dd, J = 16.5/12.3 Hz, 1H, 3-CH2), 2.80 (dd J = 16.5/5.8 Hz, 1H, 3-CH2), 4.23–4.29 (m, 2H, OCH2CH3), 4.32–4.37 (m, 3H, 2-CH, OCH2CH3), 5.77 (s, 1H, 5-CH), 6.07 (s, 1H, NH). 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.1 (1C, OCH2CH3), 14.2 (1C, OCH2CH3), 38.2 (1C, C-3), 54.8 (1C, C-2), 62.4 (1C, OCH2CH3), 63.0 (1C, OCH2CH3), 102.2 (1C, C-5), 147.7 (1C, C-6), 162.9 (1C, O=COEt), 169.9 (1C, O=COEt), 192.5 (1C, C-4).
3.1.19. Synthesis of Diethyl (E)- and (Z)-(RS)-4-(2-Ethoxy-2-Oxoethylidene)-1,2,3,4-Tetrahydropyridine-2,6-Dicarboxylate (37)
At 0 °C, NaOCl (0.75 m in H2O, 2.20 mL, 1.65 mmol, 2 eq) was added to a solution of 34 (260 mg, 0.82 mmol, 1 eq) and AcOH (0.10 mL, 1.65 mmol, 2 eq) in t-BuOH (0.10 mL, 1.00 mmol, 1.2 eq) and methyl t-butyl ether (8 mL) and the mixture was stirred for 1 h. Then, DIPEA (1.15 mL, 6.61 mmol, 8 eq) was added and the mixture was stirred at room temperature for 16 h. H2O (10 mL) was added and the mixture was extracted with EtOAc (3 × 15 mL) and dried (Na2SO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (25 g cartridge, cHex/EtOAc 8:2 → 6:4). Yellow oil, yield 77 mg (30%). C15H21NO6 (311.3 g/mol). TLC: Rf = 0.49 (cHex/EtOAc 7:3). 1H NMR (600 MHz, CDCl3): δ (ppm) = 1.24–1.32 (m, 6H, 2 × OCH2CH3, 2 × OCH2CH3*), 1.34 (t, J = 7.1 Hz, 3 × 0.55H, OCH2CH3), 1.35 (t, J = 7.1 Hz, 3 × 0.45H, OCH2CH2*), 2.76 (ddd, J = 15.5/9.6/1.5 Hz, 0.45H, 3-CH2*), 2.83 (ddd, J = 15.5/5.0/1.2 Hz, 0.45H, 3-CH2*), 3.10 (ddd, J = 17.0/10.2/2.0 Hz, 0.55H, 3-CH2), 3.70 (ddd, J = 17.0/4.8/1.6 Hz, 0.55H, 3-CH2), 4.00 (dd, J = 10.2/4.8 Hz, 0.55H, 2-CH), 4.06 (dd, J = 9.6/5.0 Hz, 0.45H, 2-CH*), 4.12–4.27 (m, 4H, 2 × OCH2CH3, 2 × OCH2CH3*), 4.30 (q, J = 7.1 Hz, 1.10H, OCH2CH3), 4.31 (q, J = 7.1 Hz, 0.9H, OCH2CH3*), 5.43 (s, 0.45H, 5-CH*), 5.66 (s, 0.55H, 5-CH), 6.08 (s, 0.55H, CHCOOEt), 7.37 (s, 0.45H, CHCOOEt*). Ratio of diastereomers is 55:45. The signals for the minor diastereomer are marked with an asterisk (*). 13C NMR (151 MHz, CDCl3): δ (ppm) = 14.25, 14.28, 14.20, 14.31 (2C, 2 × OCH2CH3, 2 × OCH2CH3*), 14.50, 14.51 (1C, OCH2CH3, OCH2CH3*), 28.4 (0.55C, 3-C), 33.7 (0.45C, C-3*), 53.5 (0.55C, C-2), 55.8 (0.45C, C-2*), 59.82 (0.55, OCH2CH3), 59.84 (0.45C, OCH2CH3*), 61.8 (0.45C, OCH2CH3*), 61.9 (0.55C, OCH2CH3), 62.06 (0.55C, OCH2CH3), 62.13 (0.45C, OCH2CH3*), 102.4 (0.45C, CHCOOEt*), 107.1 (0.55C, CHCOOEt), 112.3 (0.45C, C-5*), 113.2 (0.55C, C-5), 137.9 (0.55C, C-6), 138.1 (0.45C, C-6*), 146.1 (0.45C, C-4*), 148.0 (0.55C, C-4), 163.2 (0.55C, O=COEt), 163.9 (0.45C, O=COEt*), 166.7 (0.45C, O=COEt*), 167.0 (0.55C, O=COEt), 170.8 (0.45C, O=COEt*), 171.2 (0.55C, O=COEt). Ratio of diastereomers is 55:45. The signals for the minor diastereomer are marked with an asterisk (*).
4. Conclusions
In order to learn more about the relevance of the carboxy moieties of betalamic acid (9), the seven-step synthesis of the betalamic acid analog 13 without carboxy groups in positions C-2-and C-6 was designed and carried out. Due to low stability, in particular against O2, the free amine 13 could be characterized only by NMR spectroscopy. However, the Boc-protected precursor 21 could be isolated. In the Folin–Ciocalteu assay, 21 did not show any antioxidative properties, indicating that a free amine within the piperidine ring is essential for its antioxidative activity. Analogous 1,2,3,4-tetrahydropyridines 35–37 with two ester moieties in positions C-2 and C-6 and different substituents in position C-4 showed different levels of stability, i.e., different antioxidative properties in NMR studies.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules27217423/s1, Page S2: Folin–Cicalteau Assay; Page S3: NMR spectra.
Author Contributions
Synthesis, analytical data and preparation of the manuscript, D.A.; preparation of some intermediates, analytical data and preparation of the manuscript, H.J.; preparation of some intermediates, A.C.; preparation of the manuscript, D.C. and C.P.; biological evaluation, L.T.; NMR spectroscopy, J.K.; supervision of the project and supervising Daniele Aiello and Hendrik Jonas during the writing of the manuscript, B.W.; responsible for the idea of the project and Supervisor of Daniele Aiello and Hendrik Jonas during their PhD, P.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by a European Union 2014–2020 PON Ricerca e Innovazione grant from the Italian Ministry of Education, University and Research, entitled “PROGEMA-Processi Green per l’Estrazione di Principi Attivi e la Depurazione di Matrici di Scarto e Non” (ARS01_00432) (to Patrizia Diana).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Sample of the compounds and all data are available from the authors.
Acknowledgments
This work was supported by the Ministero dell’istruzione, dell’università e della ricerca (MIUR), which is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Grotewold, E. The Genetics and Biochemistry of Floral Pigments. Annu. Rev. Plant Biol. 2006, 57, 761–780. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.R. Floral Colour Changes as Cues for Pollinators. Nature 1991, 354, 227–229. [Google Scholar] [CrossRef]
- Forkmann, G. Flavonoids as Flower Pigments: The Formation of the Natural Spectrum and Its Extension by Genetic Engineering. Plant Breed. 1991, 106, 1–26. [Google Scholar] [CrossRef]
- Harborne, J.B. The Flavonoids: Advances in Research Since 1986. J. Chem. Educ. 1995, 72, A73. [Google Scholar] [CrossRef]
- Harborne, J.B.; Williams, C.A. Advances in Flavonoid Research since 1992. Phytochemistry 2000, 55, 481–504. [Google Scholar] [CrossRef]
- Mol, J.; Grotewold, E.; Koes, R. How Genes Paint Flowers and Seeds. Trends Plant Sci. 1998, 3, 212–217. [Google Scholar] [CrossRef]
- Winkel-Shirley, B. Flavonoid Biosynthesis. A Colorful Model for Genetics, Biochemistry, Cell Biology, and Biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef]
- Brockington, S.F.; Walker, R.H.; Glover, B.J.; Soltis, P.S.; Soltis, D.E. Complex Pigment Evolution in the Caryophyllales. New Phytol. 2011, 190, 854–864. [Google Scholar] [CrossRef]
- Shimada, S.; Otsuki, H.; Sakuta, M. Transcriptional Control of Anthocyanin Biosynthetic Genes in the Caryophyllales. J. Exp. Bot. 2007, 58, 957–967. [Google Scholar] [CrossRef]
- Gandía-Herrero, F.; García-Carmona, F. Biosynthesis of Betalains: Yellow and Violet Plant Pigments. Trends Plant Sci. 2013, 18, 334–343. [Google Scholar] [CrossRef]
- Sunnadeniya, R.; Bean, A.; Brown, M.; Akhavan, N.; Hatlestad, G.; Gonzalez, A.; Symonds, V.V.; Lloyd, A. Tyrosine Hydroxylation in Betalain Pigment Biosynthesis Is Performed by Cytochrome P450 Enzymes in Beets (Beta Vulgaris). PLoS ONE 2016, 11, e0149417. [Google Scholar] [CrossRef] [PubMed]
- Gandía-Herrero, F.; García-Carmona, F.; Escribano, J. Floral Fluorescence Effect. Nature 2005, 437, 334. [Google Scholar] [CrossRef] [PubMed]
- Felker, P.; Stintzing, F.C.; Müssig, E.; Leitenberger, M.; Carle, R.; Vogt, T.; Bunch, R. Colour Inheritance in Cactus Pear (Opuntia Ficus-Indica) Fruits. Ann. Appl. Biol. 2008, 152, 307–318. [Google Scholar] [CrossRef]
- Moreno, D.A.; García-Viguera, C.; Gil, J.I.; Gil-Izquierdo, A. Betalains in the Era of Global Agri-Food Science, Technology and Nutritional Health. Phytochem. Rev. 2008, 2, 261–280. [Google Scholar] [CrossRef]
- Henry, B.S. Natural Food Colours. In Natural Food Colorants; Hendry, G.A.F., Houghton, J.D., Eds.; Springer: New York, NY, USA, 1996; pp. 40–79. [Google Scholar] [CrossRef]
- Gandía-Herrero, F.; Escribano, J.; García-Carmona, F. Structural Implications on Color, Fluorescence, and Antiradical Activity in Betalains. Planta 2010, 232, 449–460. [Google Scholar] [CrossRef]
- Livrea, M.A.; Tesoriere, L. Antioxidant Activities of Prickly Pear (Opuntia Ficus Indica) Fruit and Its Betalains. In Herbal and Traditional Medicines; Packer, L., Ong, C.N., Halliwell, B., Eds.; CRC Press: Boca Raton, FL, USA; Taylor & Francis: Abingdon, UK, 2004; p. 537. [Google Scholar]
- Wybraniec, S.; Stalica, P.; Spòrna, A.; Nemzer, B.; Pietrzkowski, Z.; Michalowski, T. Antioxidant Activity of Betanidin: Electrochemical Study in Aqueous Media. J. Agric. Food Chem. 2011, 59, 12163–12170. [Google Scholar] [CrossRef]
- Tesoriere, L.; Allegra, M.; Gentile, C.; Livrea, M.A. Betacyanins as Phenol Antioxidants. Chemistry and Mechanistic Aspects of the Lipoperoxyl Radical-Scavenging Activity in Solution and Liposomes. Free Radic. Res. 2009, 43, 706–717. [Google Scholar] [CrossRef]
- Wybraniec, S.; Starzak, K.; Skopińska, A.; Nemzer, B.; Pietrzkowski, Z.; Michałowski, T. Studies on Nonenzymatic Oxidation Mechanisms in Neobetanin, Betanin, and Decarboxylated Betanins. J. Agric. Food Chem. 2013, 61, 6465–6476. [Google Scholar] [CrossRef]
- Wybraniec, S.; Michalowski, T. New Pathways of Betanidin and Betanin Enzymatic Oxidation. J. Agric. Food Chem. 2011, 59, 9612–9622. [Google Scholar] [CrossRef]
- Gandía-Herrero, F.; Escribano, J.; García-Carmona, F. The Role of Phenolic Hydroxy Groups in the Free Radical Scavenging Activity of Betalains. J. Nat. Prod. 2009, 72, 1142–1146. [Google Scholar] [CrossRef]
- Butera, D.; Tesoriere, L.; Di Gaudio, F.; Bongiorno, A.; Allegra, M.; Pintaudi, A.M.; Kohen, R.; Livrea, M.A. Antioxidant Activities of Sicilian Prickly Pear (Opuntia Ficus Indica) Fruit Extracts and Reducing Properties of Its Betalains: Betanin and Indicaxanthin. J. Agric. Food Chem. 2002, 50, 6895–6901. [Google Scholar] [CrossRef]
- Vidal, P.J.; López-Nicolás, J.M.; Gandía-Herrero, F.; García-Carmona, F. Inactivation of Lipoxygenase and Cyclooxygenase by Natural Betalains and Semi-Synthetic Analogues. Food Chem. 2014, 154, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Gandía-Herrero, F.; Escribano, J.; García-Carmona, F. Purification and Antiradical Properties of the Structural Unit of Betalains. J. Nat. Prod. 2012, 75, 1030–1036. [Google Scholar] [CrossRef]
- Sreekanth, D.; Arunasree, M.K.; Roy, K.R.; Chandramohan Reddy, T.; Reddy, G.V.; Reddanna, P. Betanin a Betacyanin Pigment Purified from Fruits of Opuntia Ficus-Indica Induces Apoptosis in Human Chronic Myeloid Leukemia Cell Line-K562. Phytomedicine 2007, 14, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Lechner, J.F.; Wang, L.-S.; Rocha, C.M.; Larue, B.; Henry, C.; McIntyre, C.M.; Riedl, K.M.; Schwartz, S.J.; Stoner, G.D. Drinking Water with Red Beetroot Food Color Antagonizes Esophageal Carcinogenesis in N-Nitrosomethylbenzylamine-Treated Rats. J. Med. Food 2010, 13, 733–739. [Google Scholar] [CrossRef]
- Naselli, F.; Tesoriere, L.; Caradonna, F.; Bellavia, D.; Attanzio, A.; Gentile, C.; Livrea, M.A. Anti-Proliferative and pro-Apoptotic Activity of Whole Extract and Isolated Indicaxanthin from Opuntia Ficus-Indica Associated with Re-Activation of the Onco-Suppressor P16INK4a Gene in Human Colorectal Carcinoma (Caco-2) Cells. Biochem. Biophys. Res. Commun. 2014, 450, 652–658. [Google Scholar] [CrossRef]
- Cardoso-Ugarte, G.A.; Sosa-Morales, M.E.; Ballard, T.; Liceaga, A.; San Martín-González, M.F. Microwave-Assisted Extraction of Betalains from Red Beet (Beta Vulgaris). LWT Food Sci. Technol. 2014, 59, 276–282. [Google Scholar] [CrossRef]
- De Azeredo, H.M.C.; Pereira, A.C.; De Souza, A.C.R.; Gouveia, S.T.; Mendes, K.C.B. Study on Efficiency of Betacyanin Extraction from Red Beetroots. Int. J. Food Sci. Technol. 2009, 44, 2464–2469. [Google Scholar] [CrossRef]
- Gandía-Herrero, F.; García-Carmona, F.; Escribano, J. Development of a Protocol for the Semi-Synthesis and Purification of Betaxanthins. Phytochem. Anal. 2006, 17, 262–269. [Google Scholar] [CrossRef]
- Chandrasekhar, J.; Sonika, G.; Madhusudhan, M.C.; Raghavarao, K.S.M.S. Differential Partitioning of Betacyanins and Betaxanthins Employing Aqueous Two Phase Extraction. J. Food Eng. 2015, 144, 156–163. [Google Scholar] [CrossRef]
- Herbach, K.M.; Stintzing, F.C.; Carle, R. Betalain Stability and Degradation—Structural and Chromatic Aspects. J. Food Sci. 2006, 71, 41–50. [Google Scholar] [CrossRef]
- Robert, P.; Torres, V.; García, P.; Vergara, C.; Sáenz, C. The Encapsulation of Purple Cactus Pear (Opuntia Ficus-Indica) Pulp by Using Polysaccharide-Proteins as Encapsulating Agents. LWT Food Sci. Technol. 2015, 60, 1039–1045. [Google Scholar] [CrossRef]
- Hermann, K. Totalsynthese von Betalainen. Helv. Chim. Acta 1977, 60, 673–683. [Google Scholar] [CrossRef]
- Buechi, G.H.; Fliri, H.; Shapiro, R. Synthesis of Betalamic Acid. J. Org. Chem. 1977, 42, 2192–2194. [Google Scholar] [CrossRef]
- Hilpert, H.; Dreiding, A.S. Über Die Totalsynthese vo Betalainen. Helv. Chim. Acta 1984, 67, 1547–1561. [Google Scholar] [CrossRef]
- Nirogi, R.; Shinde, A.K.; Kambhampati, R.; Namala, R.; Dwarampudi, A.R.; Kota, L.; Gampa, M.; Kodru, P.; Tiriveedhi, T.N.V.; Kandikere, V.N.; et al. Heterocyclyl Compounds as Histamine h3 Receptor Ligands. U.S. Patent No 8,912,179, 16 December 2014. [Google Scholar]
- Robins, M.J.; Yang, H.; Miranda, K.; Peterson, M.A.; De Clercq, E.; Balzarini, J. Synthesis and Biological Evaluation of 3,3-Difluoropyridine-2,4(1H,3H)-Dione and 3-Deaza-3-Fluorouracil Base and Nucleoside Derivatives. J. Med. Chem. 2009, 52, 3018–3027. [Google Scholar] [CrossRef]
- Miyamoto, K.; Kubodera, N.; Murayama, E.; Ochi, K.; Mori, T.; Matsunaga, I. Synthetic Studies on Vitamin D Analogues VI. A New Synthesis of 25-Hydroxycholesterol from Lithocholic Acid. Synth. Commun. 1986, 16, 513–521. [Google Scholar] [CrossRef]
- Semmelhack, M.F.; Schmid, C.R.; Cortes, D.A.; Chou, C.S. Oxidation of Alcohols to Aldehydes with Oxygen and Cupric Ion, Mediated by Nitrosonium Ion. J. Am. Chem. Soc. 1984, 106, 3374–3376. [Google Scholar] [CrossRef]
- Wang, G.; Li, C.; Li, J.; Jia, X. Catalyst-Free Water-Mediated N-Boc Deprotection. Tetrahedron Lett. 2009, 50, 1438–1440. [Google Scholar] [CrossRef]
- Einhorn, J.; Einhorn, C.; Pierre, J.-L. Dianions of N-Protected Iminodiacetic Acid Diesters: New Synthons with Broad Synthetic Potentialities. Synlett 1994, 1994, 1023. [Google Scholar] [CrossRef]
- John, J.P.; Jost, J.; Novikov, A.V. Synthesis of Plakortethers F and G. J. Org. Chem. 2009, 74, 6083–6091. [Google Scholar] [CrossRef] [PubMed]
- Pappo, R.; Allen, D.S.; Lemieux, R.U.; Johnson, W.S. Osmium Tetroxide-Catalyzed Periodate Oxidation of Olefinic Bonds. J. Org. Chem. 1956, 21, 478. [Google Scholar] [CrossRef]
- Zhong, Y.L.; Zhou, H.; Gauthier, D.R.; Lee, J.; Askin, D.; Dolling, U.H.; Volante, R.P. Practical and Efficient Synthesis of N-Halo Compounds. Tetrahedron Lett. 2005, 46, 1099–1101. [Google Scholar] [CrossRef]
- Singleton, V.L.; Orthofer, R.; Lamuela-Raventós, R.M. [14] Analysis of Total Phenols and Other Oxidation Substrates and Antioxidants by Means of Folin-Ciocalteu Reagent. In Oxidants and Antioxidants Part A; Packer, L., Ed.; Academic Press: Cambridge, MA, USA, 1999; Volume 299, pp. 152–178. [Google Scholar] [CrossRef]
- Huang, D.; Ou, B.; Prior, R.L. The Chemistry behind Antioxidant Capacity Assays. J. Agric. Food Chem. 2005, 53, 1841–1856. [Google Scholar] [CrossRef]
- Berker, K.I.; Ozdemir Olgun, F.A.; Ozyurt, D.; Demirata, B.; Apak, R. Modified Folin-Ciocalteu Antioxidant Capacity Assay for Measuring Lipophilic Antioxidants. J. Agric. Food Chem. 2013, 61, 4783–4791. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).