Recent Advances in the Discovery of Novel Antiprotozoal Agents

Abstract

1. Introduction
2. Recent Progress of Antiprotozoan Agents
2.1. Anti-Giardiasis
Important Highlights of Table 1 Compounds
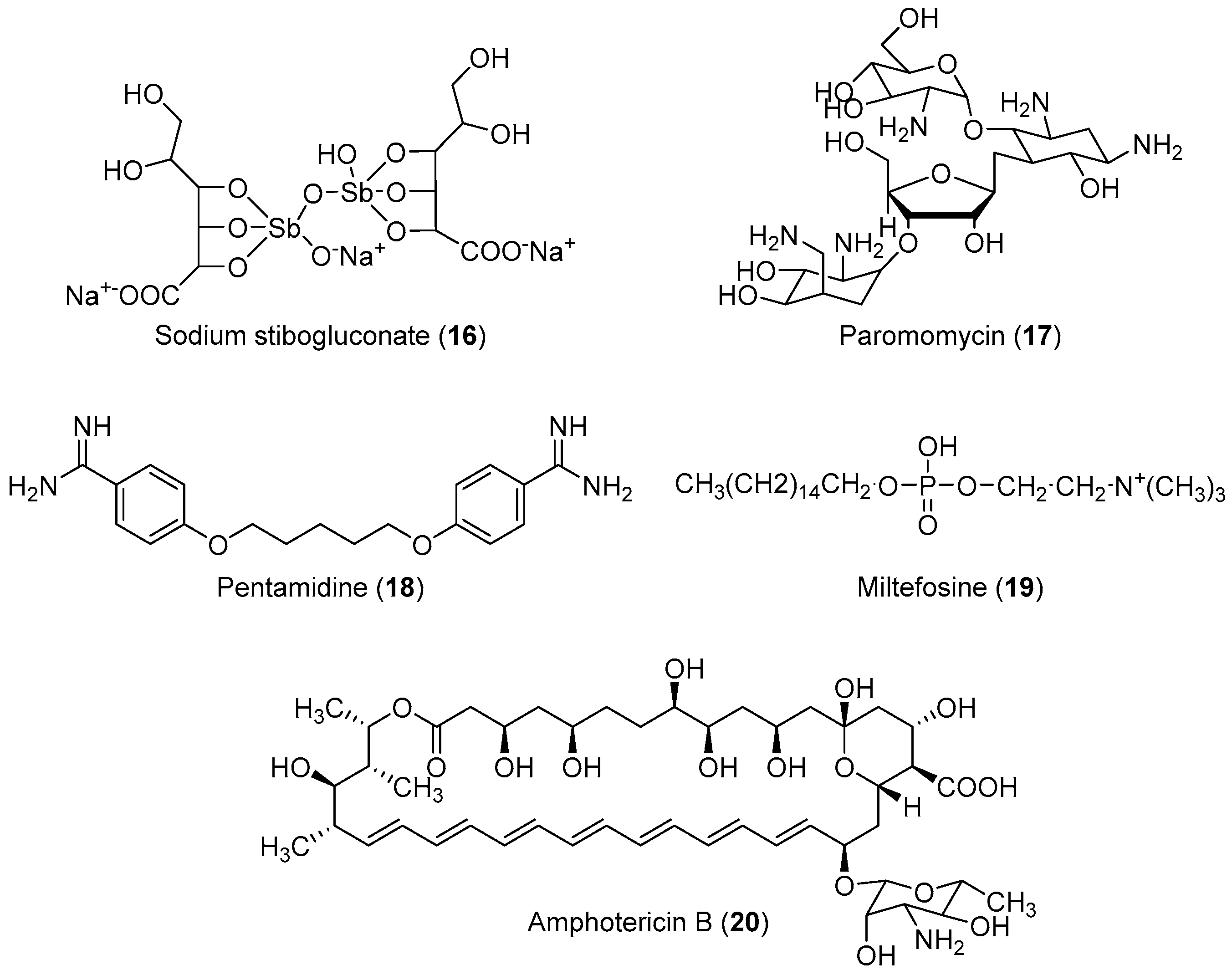
2.2. Anti-Leishmaniasis
Important Highlights of Table 2 Compounds
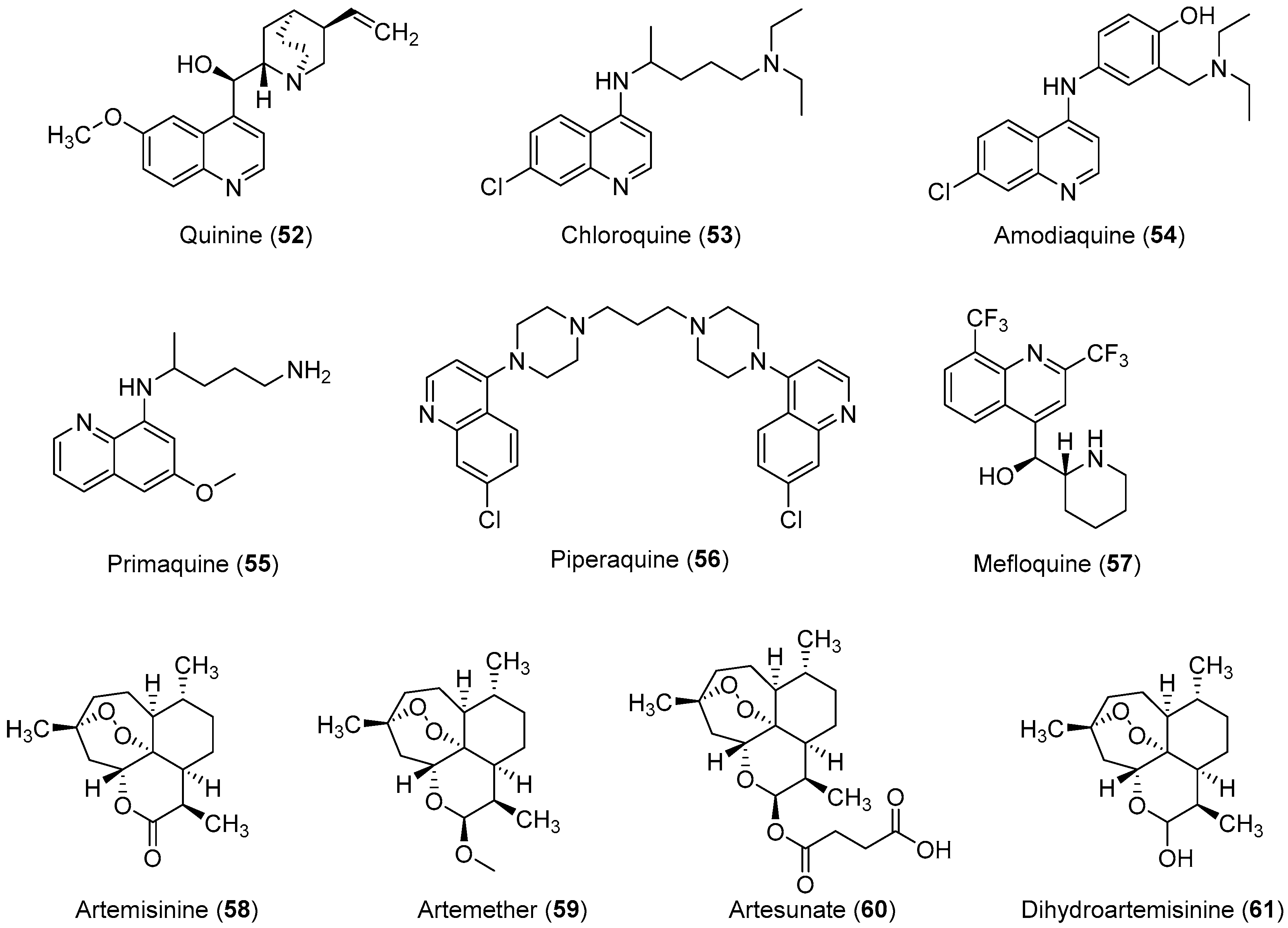
2.3. Anti-Malaria
Important Highlights of Table 3 Compounds
2.4. Anti-Trichomoniasis
Important Highlights of Table 4 Compounds
2.5. Anti-Trypanosomiasis
2.5.1. HAT/African Sleeping Sickness
2.5.2. Chagas Disease
Important Highlights of Table 5 Compounds
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| HAT | Human African trypanosomiasis |
| MTZ | Metronidazole |
| ML | Mucosal leishmaniasis |
| SSG | Sodium gluconate |
| AmB | Amphotericin-B |
| CFM | Clofazimine |
| EEF | Exoerythrocytic form |
| RBC | Red blood cell |
| pf-DHFR-TS | P.falciparum dihydrofolate reductase thymidylate synthase |
| CP | Cysteine peptidase |
| NTD | Neglected tropical disease |
| CNS | Central nervous system |
References
- Nash, T.E. Parasitic Diseases that Cause Seizures. Epilepsy Curr. 2014, 14 (Suppl. 1), 29–34. [Google Scholar] [CrossRef]
- Renslo, A.R.; McKerrow, J.H. Drug discovery and development for neglected parasitic diseases. Nat. Chem. Biol. 2006, 2, 701–710. [Google Scholar] [CrossRef]
- Pozio, E. World distribution of Trichinella spp. Infections in animals and humans. Vet. Parasitol. 2007, 149, 3–21. [Google Scholar] [CrossRef] [PubMed]
- Garcia, H.H.; Gonzalez, A.E.; Gilman, R.H. Diagnosis, treatment and control of Taenia solium cysticercosis. Curr. Opin. Infect. Dis. 2003, 16, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Fennell, B. Microtubules as antiparasitic drug targets. Expert Opin. Drug Discov. 2008, 3, 501–518. [Google Scholar] [CrossRef] [PubMed]
- Pink, R. Opportunities and challenges in antiparasitic drug discovery. Nat. Rev. Drug Discov. 2005, 4, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Tekwani, B.L.; Walker, L.A. 8-Aminoquinolines: Future role as antiprotozoal drugs. Curr. Opin. Infect. Dis. 2006, 19, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Watkins, B.M. Drugs for the control of parasitic diseases: Current status and development. Trends Parasitol. 2003, 19, 477–478. [Google Scholar] [CrossRef]
- Woodhall, D. Neglected parasitic infections: What every family physician needs to know. Am. Fam. Physician 2014, 89, 803–811. [Google Scholar]
- Mathers, C. The Global Burden of Disease: 2004 Update; World Health Organization: Geneva, Switzerland, 2008. [Google Scholar]
- Hotez, P.J. Control of neglected tropical diseases. N. Engl. J. Med. 2007, 357, 1018–1027. [Google Scholar] [CrossRef]
- Utzinger, J. Neglected tropical diseases: Diagnosis, clinical management, treatment and control. Swiss Med. Wkly. 2012, 142, w13727. [Google Scholar] [CrossRef] [PubMed]
- Secor, W.E. Neglected parasitic infections in the United States: Trichomoniasis. Am. J. Trop. Med. Hyg. 2014, 90, 800–804. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.S. Targeting Amoebiasis: Status and Developments. Curr. Bioact. Compd. 2007, 3, 121–133. [Google Scholar]
- Ordaz-Pichardo, C.; Shibayama, M.; Villa-Treviño, S.; Arriaga-Alba, M.; Angeles, E.; de la Garza, M. Antiamoebic and toxicity studies of a carbamic acid derivative and its therapeutic effect in a hamster model of hepatic amoebiasis. Antimicrob. Agents Chemother. 2005, 49, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- El-Nahas, A.F.; el-Ashmawy, I.M. Reproductive and cytogenetic toxicity of metronidazole in male mice. Basic Clin. Pharmacol. Toxicol. 2004, 94, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Basu, A.K. Mutagenicity of nitroaromatic compounds. Chem. Res. Toxicol. 2000, 13, 673–692. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.; Levesque, D.; Knowles, K.; Longshore, R.; Plummer, S. Diazepam as a treatment for metronidazole toxicosis in dogs: A retrospective study of 21 cases. J. Vet. Intern. Med. 2003, 17, 304–310. [Google Scholar] [CrossRef]
- Jha, T.K. Drug unresponsiveness & combination therapy for kala-azar. Indian J. Med. Res. 2006, 123, 389–398. [Google Scholar]
- Rudrapaul, P.; Sarma, I.S.; Das, N.; De, U.C.; Bhattacharjee, S.; Dinda, B. New flavonol methyl ether from the leaves of Vitex peduncularis exhibitspotential inhibitory activity against Leishmania donovani through activation of iNOS expression. Eur. J. Med. Chem. 2014, 87, 328–335. [Google Scholar] [CrossRef]
- Guimarães, T.T.; Pinto Mdo, C.; Lanza, J.S.; Melo, M.N.; do Monte-Neto, R.L.; de Melo, I.M.; Diogo, E.B.; Ferreira, V.F.; Camara, C.A.; Valença, W.O.; et al. Potent naphthoquinones against antimony-sensitive and -resistant Leishmania parasites: Synthesis of novel α-and nor-α-lapachone-based 1,2,3-triazoles by copper-catalyzed azide–alkyne cycloaddition. Eur. J. Med. Chem. 2013, 63, 523–530. [Google Scholar] [CrossRef]
- Oh, S.; Kim, S.; Kong, S.; Yang, G.; Lee, N.; Han, D.; Goo, J.; Siqueira-Neto, J.L.; Freitas-Junior, L.H.; Song, R. Synthesis and biological evaluation of 2,3-dihydroimidazo[1,2-a]benzimidazole derivatives against Leishmania donovani and Trypanosoma cruzi. Eur. J. Med. Chem. 2014, 84, 395–403. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. The World Health Report 1997—Conquering suffering, enriching humanity. World Health Forum 1997, 18, 248–260. [Google Scholar]
- Watkins, W.M.; Sixsmith, D.G.; Spencer, H.C.; Boriga, D.A.; Kariuki, D.M.; Kipingor, T.; Koech, D.K. Effectiveness of amodiaquine as treatment for chloroquine-resistant Plasmodium falciparum infections in Kenya. Lancet 1984, 1, 357–359. [Google Scholar] [CrossRef]
- White, N.J. Can amodiaquine be resurrected? Lancet 1996, 348, 1184–1185. [Google Scholar] [CrossRef]
- Olliaro, P.; Nevill, C.; LeBras, J.; Ringwald, P.; Mussano, P.; Garner, P.; Brasseur, P. Systematic review of amodiaquine treatment in uncomplicated malaria. Lancet 1996, 348, 1196–1201. [Google Scholar] [CrossRef]
- O’Neill, P.M.; Mukhtar, A.; Stocks, P.A.; Randle, L.E.; Hindley, S.; Ward, S.A.; Storr, R.C.; Bickley, J.F.; O’Neil, I.A.; Maggs, J.L.; et al. Isoquine and related amodiaquine analogues: A new generation of improved 4-aminoquinoline antimalarials. J. Med. Chem. 2003, 46, 4933–4945. [Google Scholar] [CrossRef]
- Neftel, K.A.; Woodtly, W.; Schmid, M.; Frick, P.G.; Fehr, J. Amodiaquine induced agranulocytosis and liver damage. Br. Med. J. (Clin. Res. Ed.) 1986, 292, 721–723. [Google Scholar] [CrossRef]
- Lind, D.E.; Levi, J.A.; Vincent, P.C. Amodiaquine-induced agranulocytosis: Toxic effect of amodiaquine in bone marrow cultures in vitro. Br. Med. J. 1973, 1, 458–460. [Google Scholar] [CrossRef]
- Zuckerman, J. Principles and Practice of Travel Medicine. J. Travel Med. 2003, 10, 313–314. [Google Scholar] [CrossRef]
- Gelb, M.H.; Hol, W.G. Drugs to combat tropical protozoan parasites. Science 2002, 297, 343–344. [Google Scholar] [CrossRef]
- Njogu, P.M.; Guantai, E.M.; Pavadai, E.; Chibale, K. Computer-Aided Drug Discovery Approaches against the Tropical Infectious Diseases Malaria, Tuberculosis, Trypanosomiasis, and Leishmaniasis. ACS Infect. Dis. 2016, 2, 8–31. [Google Scholar] [CrossRef] [PubMed]
- Torresa, F.A.E.; Passalacquaa, T.G.; Velásqueza, A.M.A.; de Souzab, R.A.; Colepicolod, P.; Graminha, M.A.S. New drugs with antiprotozoal activity from marine algae: A review. Rev. Bras. Farmacogn. 2014, 24, 265–276. [Google Scholar] [CrossRef]
- Andrews, K.T.; Fisher, G.; Skinner-Adams, T.S. Drug repurposing and human parasitic protozoan diseases. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 95–111. [Google Scholar] [CrossRef]
- Sangshetti, J.N.; Kalam Khan, F.A.; Kulkarni, A.A.; Arote, R.; Patil, R.H. Antileishmanial Drug Discovery: Comprehensive Review of the Last 10 Years. RSC Adv. 2015, 5, 32376–32415. [Google Scholar] [CrossRef]
- Nagle, A.S.; Khare, S.; Kumar, A.B.; Supek, F.; Buchynskyy, A.; Mathison, C.J.; Chennamaneni, N.K.; Pendem, N.; Buckner, F.S.; Gelb, M.H.; et al. Recent Developments in Drug Discovery for Leishmaniasis and Human African Trypanosomiasis. Chem. Rev. 2014, 114, 11305–11347. [Google Scholar]
- Biamonte, M.A.; Wanner, J.; Le Roch, K.G. Recent advances in malaria drug discovery. Bioorg. Med. Chem. Lett. 2013, 23, 2829–2843. [Google Scholar] [CrossRef]
- Chen, C. Development of antimalarial drugs and their application in China: A historical review. Infect. Dis. Poverty 2014, 3, 9. [Google Scholar] [CrossRef]
- Lawal, B.; Shittu, O.K.; Kabiru, A.Y.; Jigam, A.A.; Umar, M.B.; Berinyuy, E.B.; Alozieuwa, B.U. Potential antimalarials from African natural products: A reviw. J. Intercult. Ethnopharmacol. 2015, 4, 318–343. [Google Scholar] [CrossRef]
- Barnett, D.S.; Guy, R.K. Antimalarials in Development in 2014. Chem. Rev. 2014, 114, 11221–11241. [Google Scholar] [CrossRef]
- Sinha, S.; Singh, A.; Medhi, B.; Sehgal, R. Systematic Review: Insight into Antimalarial Peptide. Int. J. Pept. Res. Ther. 2016, 2, 325–340. [Google Scholar] [CrossRef]
- Mushtaque, M. Reemergence of chloroquine (CQ) analogs as multi-targeting antimalarial agents: A review. Eur. J. Med. Chem. 2015, 90, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Vandekerckhove, S.; D’Hooghe, M. Quinoline-based antimalarial hybrid compounds. Bioorg. Med. Chem. 2015, 23, 5098–5119. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Paliwal, D.; Saini, D.; Thakur, A.; Aggarwal, S.; Kaushik, D. A comprehensive review on synthetic approach for antimalarial agents. Eur. J. Med. Chem. 2014, 85, 147–178. [Google Scholar] [CrossRef] [PubMed]
- Mishra, M.; Mishra, V.K.; Kashaw, V.; Iyer, A.K.; Kashaw, S.K. Comprehensive review on various strategies for antimalarial drug discovery. Eur. J. Med. Chem. 2017, 125, 1300–1320. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.J.; Grkovic, T.; Sykes, M.L.; Avery, V.M. Trypanocidal activity of marine natural products. Mar. Drugs 2013, 11, 4058–4082. [Google Scholar] [CrossRef] [PubMed]
- de Brum Vieira, P.; Giordani, R.B.; Macedo, A.J.; Tasca, T. Natural and synthetic compound anti-Trichomonas vaginalis: An update review. Parasitol. Res. 2015, 114, 1249–1261. [Google Scholar] [CrossRef] [PubMed]
- Eckmann, L.; Gillin, F.D. Microbes and microbial toxins: Paradigms for microbial-mucosal interactions I. Pathophysiological aspects of enteric infections with the lumen-dwelling protozoan pathogen Giardia lamblia. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, G1–G6. [Google Scholar] [CrossRef]
- Tripathi, D.M.; Gupta, N.; Lakshmi, V.; Saxena, K.C.; Agrawal, A.K. Antigiardial and immunostimulatory effect of Piper longum on giardiasis due to Giardia lamblia. Phytother. Res. 1999, 13, 561–565. [Google Scholar] [CrossRef]
- Upcroft, P.; Upcroft, J.A. Drug targets and mechanisms of resistance in the anaerobic protozoa. Clin. Microbiol. Rev. 2001, 14, 150–164. [Google Scholar] [CrossRef]
- Kfir, R.; Hilner, C.; du Preez, M.; Bateman, B. Studies on the prevalence of Giardia cysts and Cryptosporidium oocysts in South African water. Water Sci. Technol. 1995, 31, 435–438. [Google Scholar] [CrossRef]
- Nash, T.E. Surface antigenic variation in Giardia lamblia. Mol. Microbiol. 2002, 45, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Halliez, M.C.; Buret, A.G. Extra-intestinal and long term consequences of Giardia duodenalis infections. World J. Gastroenterol. 2013, 19, 8974–8985. [Google Scholar] [CrossRef] [PubMed]
- Wensaas, K.A.; Langeland, N.; Hanevik, K.; Morch, K.; Eide, G.E.; Rortveit, G. Irritable bowel syndrome and chronic fatigue 3 years after acute giardiasis: Historic cohort study. Gut 2012, 61, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Monis, P.T.; Caccio, S.M.; Thompson, R.C. Variation in Giardia: Towards a taxonomic revision of the genus. Trends Parasitol. 2009, 25, 93–100. [Google Scholar] [CrossRef]
- Lasek-Nesselquist, E.; Welch, D.M.; Sogin, M.L. The identification of a new Giardia duodenalis assemblage in marine vertebrates and a preliminary analysis of G. duodenalis population biology in marine systems. Int. J. Parasitol. 2010, 40, 1063–1074. [Google Scholar] [CrossRef]
- Anderson, V.R.; Curran, M.P. Nitazoxanide: A review of its use in the treatment of gastrointestinal infections. Drugs 2007, 67, 1947–1967. [Google Scholar] [CrossRef]
- Chen, C.Z.; Kulakova, L.; Southall, N.; Marugan, J.J.; Galkin, A.; Austin, C.P.; Herzberg, O.; Zheng, W. High-throughput Giardia lamblia viability assay using bioluminescent ATP content measurements. Antimicrob. Agents Chemother. 2011, 55, 667–675. [Google Scholar] [CrossRef]
- Upcroft, J.A.; Upcroft, P.; Boreham, P.F. Drug resistance in Giardia intestinalis. Int. J. Parasitol. 1990, 20, 489–496. [Google Scholar] [CrossRef]
- Wright, J.M. Efficacy of antigiardial drugs. Expert Opin. Drug Saf. 2003, 2, 529–541. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Xue, M.Q.; Bai, Y.C.; Yuan, H.H.; Zhao, H.L.; Lan, M.B. 3,5-Dicaffeoylquinic acid isolated from Artemisia argyi and its ester derivatives exert anti-leucyl-tRNA synthetase of Giardia lamblia (GlLeuRS) and potential anti-giardial effects. Fitoterapia 2012, 83, 1281–1285. [Google Scholar] [CrossRef]
- Cano, P.A.; Islas-Jacome, A.; Gonzalez-Marrero, J.; Yepez-Mulia, L.; Calzada, F.; Gamez-Montano, R. Synthesis of 3-tetrazolylmethyl-4H-chromen-4-ones via Ugi-azide and biological evaluation against Entamoeba histolytica, Giardia lamblia and Trichomona vaginalis. Bioorg. Med. Chem. 2014, 22, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Vazquez, G.; Chavez-Silva, F.; Colin-Lozano, B.; Estrada-Soto, S.; Hidalgo-Figueroa, S.; Guerrero-Alvarez, J.; Mendez, S.T.; Reyes-Vivas, H.; Oria-Hernandez, J.; Canul-Canche, J.; et al. Synthesis of nitro(benzo)thiazole acetamides and in vitro antiprotozoal effect against amitochondriate parasites Giardia intestinalis and Trichomonas vaginalis. Bioorg. Med. Chem. 2015, 23, 2204–2210. [Google Scholar] [CrossRef] [PubMed]
- Singh, G.; Arora, A.; Mangat, S.S.; Rani, S.; Kaur, H.; Goyal, K.; Sehgal, R.; Maurya, I.K.; Tewari, R.; Choquesillo-Lazarte, D.; et al. Design, synthesis and biological evaluation of chalconyl blended triazole allied organosilatranes as giardicidal and trichomonacidal agents. Eur. J. Med. Chem. 2016, 108, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Bautista, E.; Calzada, F.; Lopez-Huerta, F.A.; Yepez-Mulia, L.; Ortega, A. Antiprotozoal activity of 8-acyl and 8-alkyl incomptine A analogs. Bioorg. Med. Chem. Lett. 2014, 24, 3260–3262. [Google Scholar] [CrossRef] [PubMed]
- Soria-Arteche, O.; Hernandez-Campos, A.; Yepez-Mulia, L.; Trejo-Soto, P.J.; Hernandez-Luis, F.; Gres-Molina, J.; Maldonado, L.A.; Castillo, R. Synthesis and antiprotozoal activity of nitazoxanide-N-methylbenzimidazole hybrids. Bioorg. Med. Chem. Lett. 2013, 23, 6838–6841. [Google Scholar] [CrossRef] [PubMed]
- Leboho, T.C.; Giri, S.; Popova, I.; Cock, I.; Michael, J.P.; de Koning, C.B. Double Sonogashira reactions on dihalogenated aminopyridines for the assembly of an array of 7-azaindoles bearing triazole and quinoxaline substituents at C-5: Inhibitory bioactivity against Giardia duodenalis trophozoites. Bioorg. Med. Chem. 2015, 23, 4943–4951. [Google Scholar] [CrossRef]
- Mocelo-Castell, R.; Villanueva-Novelo, C.; Cáceres-Castillo, D.; Carballo, R.M.; Quijano-Quiñones, R.F.; Quesadas-Rojas, M.; Cantillo-Ciau, Z.; Cedillo-Rivera, R.; Moo-Puc, R.E.; Moujir, L.M.; et al. 2-Amino-4-arylthiazole Derivatives as Anti-giardial Agents: Synthesis, Biological Evaluation and QSAR Studies. Open Chem. 2015, 13, 1127–1136. [Google Scholar] [CrossRef]
- Castillo-Villanueva, A.; Rufino-Gonzalez, Y.; Mendez, S.T.; Torres-Arroyo, A.; Ponce-Macotela, M.; Martinez-Gordillo, M.N.; Reyes-Vivas, H.; Oria-Hernandez, J. Disulfiram as a novel inactivator of Giardia lamblia triosephosphate isomerase with antigiardial potential. Int. J. Parasitol. Drugs Drug Resist. 2017, 7, 425–432. [Google Scholar] [CrossRef]
- Hayani, K.; Dandashli, A.; Weisshaar, E. Cutaneous leishmaniasis in Syria: Clinical features, current status and the effects of war. Acta Derm. Venereol. 2015, 95, 62–66. [Google Scholar] [CrossRef]
- Herwaldt, B.L. Leishmaniasis. Lancet 1999, 354, 1191–1199. [Google Scholar] [CrossRef]
- Desjeux, P. The increase in risk factors for leishmaniasis worldwide. Trans. R. Soc. Trop. Med. Hyg. 2001, 95, 239–243. [Google Scholar] [CrossRef]
- Chappuis, F.; Sundar, S.; Hailu, A.; Ghalib, H.; Rijal, S.; Peeling, R.W.; Alvar, J.; Boelaert, M. Visceral leishmaniasis: What are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 2007, 5, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Murray, H.W.; Berman, J.D.; Davies, C.R.; Saravia, N.G. Advances in leishmaniasis. Lancet 2005, 366, 1561–1577. [Google Scholar] [CrossRef]
- Reithinger, R.; Dujardin, J.C.; Louzir, H.; Pirmez, C.; Alexander, B.; Brooker, S. Cutaneous leishmaniasis. Lancet Infect. Dis. 2007, 7, 581–596. [Google Scholar] [CrossRef]
- Scope, A.; Trau, H.; Bakon, M.; Yarom, N.; Nasereddin, A.; Schwartz, E. Imported mucosal leishmaniasis in a traveler. Clin. Infect. Dis. 2003, 37, e83–e87. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Marsden, P.D. Mucosal leishmaniasis (“espundia” Escomel, 1911). Trans. R. Soc. Trop. Med. Hyg. 1986, 80, 859–876. [Google Scholar] [CrossRef]
- Desjeux, P. Leishmaniasis: Public health aspects and control. Clin. Dermatol. 1996, 14, 417–423. [Google Scholar] [CrossRef]
- Habib, Z.H.; Yusuf, M.A.; Ahmed, I.; Jhora, S.T. Clinical Burden of Kala-azar in Bangladesh: A Review Update. J. Sci. Found. 2012, 10, 70–79. [Google Scholar] [CrossRef]
- Sharlow, E.R.; Grogl, M.; Johnson, J.; Lazo, J.S. Anti-leishmanial drug discovery: Rising to the challenges of a highly neglected disease. Mol. Interv. 2010, 10, 72–75. [Google Scholar] [CrossRef]
- WHO Expert Committee on the Control of the Leishmaniases & World Health Organizatio. Conotrol of the Leishmaniases: Report of a Meeting of the WHO Expert Commitee on the Control of Leishmaniases; WHO Expert Committee on the Control of the Leishmaniases & World Health Organizatio: Geneva, Switzerland, 2010. [Google Scholar]
- Mathers, C.D.; Ezzati, M.; Lopez, A.D. Measuring the burden of neglected tropical diseases: The global burden of disease framework. PLoS Negl. Trop. Dis. 2007, 1, e114. [Google Scholar] [CrossRef]
- den Boer, M.; Argaw, D.; Jannin, J.; Alvar, J. Leishmaniasis impact and treatment access. Clin. Microbiol. Infect. 2011, 17, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Fuertes, M.A.; Nguewa, P.A.; Castilla, J.; Alonso, C.; Perez, J.M. Anticancer compounds as leishmanicidal drugs: Challenges in chemotherapy and future perspectives. Curr. Med. Chem. 2008, 15, 433–439. [Google Scholar] [PubMed]
- Bern, C.; Maguire, J.H.; Alvar, J. Complexities of assessing the disease burden attributable to leishmaniasis. PLoS Negl. Trop. Dis. 2008, 2, e313. [Google Scholar] [CrossRef] [PubMed]
- Vannier-Santos, M.A.; Martiny, A.; de Souza, W. Cell biology of Leishmania spp.: Invading and evading. Curr. Pharm. Des. 2002, 8, 297–318. [Google Scholar] [CrossRef]
- Croft, S.L.; Coombs, G.H. Leishmaniasis--current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol. 2003, 19, 502–508. [Google Scholar] [CrossRef]
- Hussain, H.; Al-Harrasi, A.; Al-Rawahi, A.; Green, I.R.; Gibbons, S. Fruitful decade for antileishmanial compounds from 2002 to late 2011. Chem. Rev. 2014, 114, 10369–10428. [Google Scholar] [CrossRef]
- Alvar, J.; Croft, S.; Olliaro, P. Chemotherapy in the Treatment and Control of Leishmaniasis. Adv. Parasitol. 2006, 61, 223–274. [Google Scholar]
- Plano, D.; Baquedano, Y.; Moreno-Mateos, D.; Font, M.; Jimenez-Ruiz, A.; Palop, J.A.; Sanmartin, C. Selenocyanates and diselenides: A new class of potent antileishmanial agents. Eur. J. Med. Chem. 2011, 46, 3315–3323. [Google Scholar] [CrossRef]
- Rodrigues, R.F.; Castro-Pinto, D.; Echevarria, A.; dos Reis, C.M.; Del Cistia, C.N.; Sant’Anna, C.M.; Teixeira, F.; Castro, H.; Canto-Cavalheiro, M.; Leon, L.L.; et al. Investigation of trypanothione reductase inhibitory activity by 1,3,4-thiadiazolium-2-aminide derivatives and molecular docking studies. Bioorg. Med. Chem. 2012, 20, 1760–1766. [Google Scholar] [CrossRef]
- Maarouf, M.; Adeline, M.T.; Solignac, M.; Vautrin, D.; Robert-Gero, M. Development and characterization of paromomycin-resistant Leishmania donovani promastigotes. Parasite 1998, 5, 167–173. [Google Scholar] [CrossRef]
- Croft, S.L.; Sundar, S.; Fairlamb, A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [Google Scholar] [CrossRef] [PubMed]
- de Melos, J.L.R.; Torres-Santos, E.C.; Faioes, V.D.S.; Del Cistia, C.D.N.; Sant’Anna, C.M.R.; Rodrigues-Santos, C.E.; Echevarria, A. Novel 3,4-methylenedioxyde-6-X-benzaldehyde-thiosemicarbazones: Synthesis and antileishmanial effects against Leishmania amazonensis. Eur. J. Med. Chem. 2015, 103, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Rashid, U.; Sultana, R.; Shaheen, N.; Hassan, S.F.; Yaqoob, F.; Ahmad, M.J.; Iftikhar, F.; Sultana, N.; Asghar, S.; Yasinzai, M.; et al. Structure based medicinal chemistry-driven strategy to design substituted dihydropyrimidines as potential antileishmanial agents. Eur. J. Med. Chem. 2016, 115, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Sangshetti, J.N.; Shaikh, R.I.; Khan, F.A.; Patil, R.H.; Marathe, S.D.; Gade, W.N.; Shinde, D.B. Synthesis, antileishmanial activity and docking study of N′-substitutedbenzylidene-2-(6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)acetohydraz ides. Bioorg. Med. Chem. Lett. 2014, 24, 1605–1610. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Shivahare, R.; Korthikunta, V.; Singh, R.; Gupta, S.; Tadigoppula, N. Synthesis and biological evaluation of chalcones as potential antileishmanial agents. Eur. J. Med. Chem. 2014, 81, 359–366. [Google Scholar] [CrossRef]
- Manda, S.; Khan, S.I.; Jain, S.K.; Mohammed, S.; Tekwani, B.L.; Khan, I.A.; Vishwakarma, R.A.; Bharate, S.B. Synthesis, antileishmanial and antitrypanosomal activities of N-substituted tetrahydro-beta–carbolines. Bioorg. Med. Chem. Lett. 2014, 24, 3247–3250. [Google Scholar] [CrossRef]
- Zhu, X.; Farahat, A.A.; Mattamana, M.; Joice, A.; Pandharkar, T.; Holt, E.; Banerjee, M.; Gragg, J.L.; Hu, L.; Kumar, A.; et al. Synthesis and pharmacological evaluation of mono-arylimidamides as antileishmanial agents. Bioorg. Med. Chem. Lett. 2016, 26, 2551–2556. [Google Scholar] [CrossRef]
- Ronga, L.; Del Favero, M.; Cohen, A.; Soum, C.; Le Pape, P.; Savrimoutou, S.; Pinaud, N.; Mullie, C.; Daulouede, S.; Vincendeau, P.; et al. Design, synthesis and biological evaluation of novel 4-alkapolyenylpyrrolo[1,2-a]quinoxalines as antileishmanial agents—Part III. Eur. J. Med. Chem. 2014, 81, 378–393. [Google Scholar] [CrossRef]
- Bekhit, A.A.; Hassan, A.M.; Abd El Razik, H.A.; El-Miligy, M.M.; El-Agroudy, E.J.; Bekhit Ael, D. New heterocyclic hybrids of pyrazole and its bioisosteres: Design, synthesis and biological evaluation as dual acting antimalarial-antileishmanial agents. Eur. J. Med. Chem. 2015, 94, 30–44. [Google Scholar] [CrossRef]
- Dea-Ayuela, M.A.; Bilbao-Ramos, P.; Bolas-Fernandez, F.; Gonzalez-Cardenete, M.A. Synthesis and antileishmanial activity of C7-and C12-functionalized dehydroabietylamine derivatives. Eur. J. Med. Chem. 2016, 121, 445–450. [Google Scholar] [CrossRef]
- Marchand, P.; Bazin, M.-A.; Pagniez, F.; Rivière, G.; Bodero, L.; Marhadour, S.; Nourrisson, M.-R.; Picot, C.; Ruchaud, S.; Bach, S.; et al. Synthesis, antileishmanial activity and cytotoxicity of 2,3-diaryl- and 2,3,8-trisubstituted imidazo[1,2-a]pyrazines. Eur. J. Med. Chem. 2015, 103, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Sangshetti, J.N.; Kalam Khan, F.A.; Kulkarni, A.A.; Patil, R.H.; Pachpinde, A.M.; Lohar, K.S.; Shinde, D.B. Antileishmanial activity of novel indolyl-coumarin hybrids: Design, synthesis, biological evaluation, molecular docking study and in silico ADME prediction. Bioorg. Med. Chem. Lett. 2016, 26, 829–835. [Google Scholar] [CrossRef] [PubMed]
- Lunagariya, N.A.; Gohil, V.M.; Kushwah, V.; Neelagiri, S.; Jain, S.; Singh, S.; Bhutani, K.K. Design, synthesis and biological evaluation of 1,3,6-trisubstituted beta–carboline derivatives for cytotoxic and anti-leishmanial potential. Bioorg. Med. Chem. Lett. 2016, 26, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Tiwari, A.; Suryawanshi, S.N.; Mittal, M.; Vishwakarma, P.; Gupta, S. Chemotherapy of leishmaniasis. Part IX: Synthesis and bioevaluation of aryl substituted ketene dithioacetals as antileishmanial agents. Bioorg. Med. Chem. Lett. 2012, 22, 6728–6730. [Google Scholar] [CrossRef] [PubMed]
- Saad, S.M.; Ghouri, N.; Perveen, S.; Khan, K.M.; Choudhary, M.I. 4-Arylamino-6-nitroquinazolines: Synthesis and their activities against neglected disease leishmaniasis. Eur. J. Med. Chem. 2016, 108, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Gopinath, V.S.; Rao, M.; Shivahare, R.; Vishwakarma, P.; Ghose, S.; Pradhan, A.; Hindupur, R.; Sarma, K.D.; Gupta, S.; Puri, S.K.; et al. Design, synthesis, ADME characterization and antileishmanial evaluation of novel substituted quinoline analogs. Bioorg. Med. Chem. Lett. 2014, 24, 2046–2052. [Google Scholar] [CrossRef] [PubMed]
- Baquedano, Y.; Moreno, E.; Espuelas, S.; Nguewa, P.; Font, M.; Gutierrez, K.J.; Jimenez-Ruiz, A.; Palop, J.A.; Sanmartin, C. Novel hybrid selenosulfonamides as potent antileishmanial agents. Eur. J. Med. Chem. 2014, 74, 116–123. [Google Scholar] [CrossRef]
- Adam, R.; Bilbao-Ramos, P.; Lopez-Molina, S.; Abarca, B.; Ballesteros, R.; Gonzalez-Rosende, M.E.; Dea-Ayuela, M.A.; Alzuet-Pina, G. Triazolopyridyl ketones as a novel class of antileishmanial agents. DNA binding and BSA interaction. Bioorg. Med. Chem. 2014, 22, 4018–4027. [Google Scholar] [CrossRef]
- Sharma, R.; Pandey, A.K.; Shivahare, R.; Srivastava, K.; Gupta, S.; Chauhan, P.M. Triazino indole-quinoline hybrid: A novel approach to antileishmanial agents. Bioorg. Med. Chem. Lett. 2014, 24, 298–301. [Google Scholar] [CrossRef]
- Tiwari, A.; Kumar, S.; Shivahare, R.; Kant, P.; Gupta, S.; Suryawanshi, S.N. Chemotherapy of leishmaniasis part XIII: Design and synthesis of novel heteroretinoid-bisbenzylidine ketone hybrids as antileishmanial agents. Bioorg. Med. Chem. Lett. 2015, 25, 410–413. [Google Scholar] [CrossRef]
- Pandey, S.; Chauhan, S.S.; Shivahare, R.; Sharma, A.; Jaiswal, S.; Gupta, S.; Lal, J.; Chauhan, P.M. Identification of a diverse indole-2-carboxamides as a potent antileishmanial chemotypes. Eur. J. Med. Chem. 2016, 110, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Barteselli, A.; Casagrande, M.; Basilico, N.; Parapini, S.; Rusconi, C.M.; Tonelli, M.; Boido, V.; Taramelli, D.; Sparatore, F.; Sparatore, A. Clofazimine analogs with antileishmanial and antiplasmodial activity. Bioorg. Med. Chem. 2015, 23, 55–65. [Google Scholar] [CrossRef] [PubMed]
- De Vita, D.; Moraca, F.; Zamperini, C.; Pandolfi, F.; Di Santo, R.; Matheeussen, A.; Maes, L.; Tortorella, S.; Scipione, L. In vitro screening of 2-(1H-imidazol-1-yl)-1-phenylethanol derivatives as antiprotozoal agents and docking studies on Trypanosoma cruzi CYP51. Eur. J. Med. Chem. 2016, 113, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Patrick, D.A.; Bakunov, S.A.; Bakunova, S.M.; Jones, S.K.; Wenzler, T.; Barszcz, T.; Kumar, A.; Boykin, D.W.; Werbovetz, K.A.; Brun, R.; et al. Synthesis and antiprotozoal activities of benzyl phenyl ether diamidine derivatives. Eur. J. Med. Chem. 2013, 67, 310–324. [Google Scholar] [CrossRef] [PubMed]
- Rye, C.E.; Barker, D. Asymmetric synthesis and anti-protozoal activity of the 8,4′-oxyneolignans virolin, surinamensin and analogues. Eur. J. Med. Chem. 2013, 60, 240–248. [Google Scholar] [CrossRef]
- Durust, Y.; Karakus, H.; Kaiser, M.; Tasdemir, D. Synthesis and anti-protozoal activity of novel dihydropyrrolo[3,4-d][1,2,3]triazoles. Eur. J. Med. Chem. 2012, 48, 296–304. [Google Scholar] [CrossRef]
- Pierson, J.T.; Dumetre, A.; Hutter, S.; Delmas, F.; Laget, M.; Finet, J.P.; Azas, N.; Combes, S. Synthesis and antiprotozoal activity of 4-arylcoumarins. Eur. J. Med. Chem. 2010, 45, 864–869. [Google Scholar] [CrossRef]
- Patrick, D.A.; Ismail, M.A.; Arafa, R.K.; Wenzler, T.; Zhu, X.; Pandharkar, T.; Jones, S.K.; Werbovetz, K.A.; Brun, R.; Boykin, D.W.; et al. Synthesis and antiprotozoal activity of dicationic m-terphenyl and 1,3-dipyridylbenzene derivatives. J. Med. Chem. 2013, 56, 5473–5494. [Google Scholar] [CrossRef]
- Saccolitia, F.; Angiullib, G.; Pupoa, G.; Pescatoria, L.; Madiaa, V.N.; Messorea, A.; Colottib, G.; Fiorillob, A.; Scipionea, L.; Santoa, R.D.; et al. Inhibition of Leishmania infantum trypanothione reductase by diaryl sulfide derivatives. J. Enzyme Inhit. Med. Chem. 2017, 32, 304–310. [Google Scholar] [CrossRef]
- Vishwakarma, P.; Parmar, N.; Chandrakar, P.; Sharma, T.; Kathuria, M.; Agnihotri, P.K.; Siddiqi, M.I.; Mitra, K.; Kar, S. Ammonium trichloro [1,2-ethanediolato-O,O′]-tellurate cures experimental visceral leishmaniasis by redox modulation of Leishmania donovani trypanothione reductase and inhibiting host integrin linked PI3K/Akt pathway. Cell. Mol Life Sci. 2018, 75, 563–588. [Google Scholar] [CrossRef]
- Rodríguez-Hernández, D.; Barbosa, L.C.A.; Demuner, A.J.; Nain-Perez, A.; Ferreira, S.R.; Fujiwara, R.T.; de Almeida, R.M.; Heller, L.; Csuk, R. Leishmanicidal and cytotoxic activity of hederagenin-bistriazolyl derivatives. Eur. J. Med. Chem. 2017, 140, 624–635. [Google Scholar] [CrossRef] [PubMed]
- Tahghighi, A.; Babalouei, F. Thiadiazoles: The appropriate pharmacological scaffolds with leishmanicidal and antimalarial activities: A review. Iran. J. Basic Med. Sci. 2017, 20, 613–622. [Google Scholar] [PubMed]
- Njoroge, M.; Njuguna, N.M.; Mutai, P.; Ongarora, D.S.; Smith, P.W.; Chibale, K. Recent Approaches to Chemical Discovery and Development against Malaria and the Neglected Tropical Diseases Human African Trypanosomiasis and Schistosomiasis. Chem. Rev. 2014, 114, 11138–11163. [Google Scholar] [CrossRef] [PubMed]
- Caraballo, H.; King, K. Emergency department management of mosquito-borne illness: Malaria, dengue, and West Nile virus. Emerg. Med. Pract. 2014, 16, 1–23. [Google Scholar] [PubMed]
- World Health Organization. World Malaria Report 2014; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- World Health Organization. World Malaria Report 2012; World Health Organization: Geneva, Switzerland, 2012. [Google Scholar]
- Murray, C.J.; Rosenfeld, L.C.; Lim, S.S.; Andrews, K.G.; Foreman, K.J.; Haring, D.; Fullman, N.; Naghavi, M.; Lozano, R.; Lopez, A.D. Global malaria mortality between 1980 and 2010: A systematic analysis. Lancet 2012, 379, 413–431. [Google Scholar] [CrossRef]
- Sachs, J.; Malaney, P. The economic and social burden of malaria. Nature 2002, 415, 680–685. [Google Scholar] [CrossRef]
- Brieger, W. The World Malaria Report 2015: Prospects for Malaria Elimination. Africa Health 2016, 14–16. [Google Scholar]
- Baird, J.K. Neglect of Plasmodium vivax malaria. Trends Parasitol. 2007, 23, 533–539. [Google Scholar] [CrossRef]
- Prudencio, M.; Rodriguez, A.; Mota, M.M. The silent path to thousands of merozoites: The Plasmodium liver stage. Nat. Rev. Microbiol. 2006, 4, 849–856. [Google Scholar] [CrossRef]
- Wells, T.N.; Burrows, J.N.; Baird, J.K. Targeting the hypnozoite reservoir of Plasmodium vivax: The hidden obstacle to malaria elimination. Trends Parasitol. 2010, 26, 145–151. [Google Scholar] [CrossRef]
- Clark, I.A.; al Yaman, F.M.; Jacobson, L.S. The biological basis of malarial disease. Int. J. Parasitol. 1997, 27, 1237–1249. [Google Scholar] [CrossRef]
- Baker, D.A. Malaria gametocytogenesis. Mol. Biochem. Parasitol. 2010, 172, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.C. Quinine and fever: The development of the effective dosage. J. Hist. Med. Allied Sci. 1976, 31, 343–367. [Google Scholar] [CrossRef] [PubMed]
- Croft, S. Antimalarial Chemotherapy: Mechanisms of Action, Resistance and New Directions in Drug Discovery. Drug Discov. Today 2001, 6, 1151. [Google Scholar] [CrossRef]
- Salas, P.F.; Herrmann, C.; Orvig, C. Metalloantimalarials. Chem. Rev. 2013, 113, 3450–3492. [Google Scholar] [CrossRef]
- Bloland, P.B.; Ettling, M.; Meek, S. Combination therapy for malaria in Africa: Hype or hope? Bull. World Health Organ. 2000, 78, 1378–1388. [Google Scholar]
- Mutabingwa, T.K. Artemisinin-based combination therapies (ACTs): Best hope for malaria treatment but inaccessible to the needy! Acta Trop. 2005, 95, 305–315. [Google Scholar] [CrossRef]
- Jambou, R. Resistance of Plasmodium falciparum field isolates to in-vitro artemether and point mutations of the SERCA-type PfATPase6. Lancet 2005, 366, 1960–1963. [Google Scholar] [CrossRef]
- Pingaew, R.; Saekee, A.; Mandi, P.; Nantasenamat, C.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Synthesis, biological evaluation and molecular docking of novel chalcone-coumarin hybrids as anticancer and antimalarial agents. Eur. J. Med. Chem. 2014, 85, 65–76. [Google Scholar] [CrossRef]
- Gut, J.; Rosenthal, P.J.; Kumar, V. β-beta-amino-alcohol tethered 4-aminoquinoline-isatin conjugates: Synthesis and antimalarial evaluation. Eur. J. Med. Chem. 2014, 84, 566–573. [Google Scholar]
- Kumar, K.; Pradines, B.; Madamet, M.; Amalvict, R.; Benoit, N.; Kumar, V. 1H-1,2,3-triazole tethered isatin-ferrocene conjugates: Synthesis and in vitro antimalarial evaluation. Eur. J. Med. Chem. 2014, 87, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Helgren, T.R.; Sciotti, R.J.; Lee, P.; Duffy, S.; Avery, V.M.; Igbinoba, O.; Akoto, M.; Hagen, T.J. The synthesis, antimalarial activity and CoMFA analysis of novel aminoalkylated quercetin analogs. Bioorg. Med. Chem. Lett. 2015, 25, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Faisca Phillips, A.M.; Nogueira, F.; Murtinheira, F. Synthesis and antimalarial evaluation of prodrugs of novel fosmidomycin analogues. Bioorg. Med. Chem. Lett. 2015, 25, 2112–2116. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.R.; Khan, S.I.; Singh, S.; Khan, I.A.; Vishwakarma, R.A.; Bharate, S.B. Synthesis, antimalarial and antitubercular activities of meridianin derivatives. Eur. J. Med. Chem. 2015, 98, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Nguyen Le, T.; De Borggraeve, W.M.; Grellier, P.; Pham, V.C.; Dehaen, W.; Nguyen, V.H. Synthesis of 11-aza-artemisinin derivatives using the Ugi reaction and an evaluation of their antimalarial activity. Tetrahedron Lett. 2014, 55, 4892–4894. [Google Scholar] [CrossRef]
- Reiter, C.; Frohlich, T.; Gruber, L.; Hutterer, C.; Marschall, M.; Voigtlander, C.; Friedrich, O.; Kappes, B.; Efferth, T.; Tsogoeva, S.B. Highly potent artemisinin-derived dimers and trimers: Synthesis and evaluation of their antimalarial, antileukemia and antiviral activities. Bioorg. Med. Chem. 2015, 23, 5452–5458. [Google Scholar] [CrossRef]
- Parthiban, A.; Muthukumaran, J.; Manhas, A.; Srivastava, K.; Krishna, R.; Rao, H.S. Synthesis, in vitro and in silico antimalarial activity of 7-chloroquinoline and 4H-chromene conjugates. Bioorg. Med. Chem. Lett. 2015, 25, 4657–4663. [Google Scholar] [CrossRef]
- Bhat, H.R.; Singh, U.P.; Thakur, A.; Kumar Ghosh, S.; Gogoi, K.; Prakash, A.; Singh, R.K. Synthesis, antimalarial activity and molecular docking of hybrid 4-aminoquinoline-1,3,5-triazine derivatives. Exp. Parasitol. 2015, 157, 59–67. [Google Scholar] [CrossRef]
- Carneiro, P.F.; Pinto, M.C.R.F.; Marra, R.K.F.; da Silva, F.C.; Resende, J.A.L.C.; Rocha, E.; Silva, L.F.; Alves, H.G.; Barbosa, G.S.; de Vasconcellos, M.C.; et al. Synthesis and antimalarial activity of quinones and structurally-related oxirane derivatives. Eur. J. Med. Chem. 2016, 108, 134–140. [Google Scholar] [CrossRef]
- Karad, S.C.; Purohit, V.B.; Thakor, P.; Thakkar, V.R.; Raval, D.K. Novel morpholinoquinoline nucleus clubbed with pyrazoline scaffolds: Synthesis, antibacterial, antitubercular and antimalarial activities. Eur. J. Med. Chem. 2016, 112, 270–279. [Google Scholar] [CrossRef]
- Devender, N.; Gunjan, S.; Chhabra, S.; Singh, K.; Pasam, V.R.; Shukla, S.K.; Sharma, A.; Jaiswal, S.; Singh, S.K.; Kumar, Y.; et al. Identification of beta-Amino alcohol grafted 1,4,5 trisubstituted 1,2,3-triazoles as potent antimalarial agents. Eur. J. Med. Chem. 2016, 109, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Svogie, A.L.; Isaacs, M.; Hoppe, H.C.; Khanye, S.D.; Veale, C.G. Indolyl-3-ethanone-alpha-thioethers: A promising new class of non-toxic antimalarial agents. Eur. J. Med. Chem. 2016, 114, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.R.; Sun, W.; Kim, M.; Huang, X.; Sanderson, P.E.; Tanaka, T.Q.; McKew, J.C.; Simeonov, A.; Williamson, K.C.; Zheng, W.; et al. In vitro evaluation of imidazo[4,5-c]quinolin-2-ones as gametocytocidal antimalarial agents. Bioorg. Med. Chem. Lett. 2016, 26, 2907–2911. [Google Scholar] [CrossRef][Green Version]
- Seebacher, W.; Wolkinger, V.; Faist, J.; Kaiser, M.; Brun, R.; Saf, R.; Bucar, F.; Groblacher, B.; Brantner, A.; Merino, V.; et al. Synthesis of 3-azabicyclo[3.2.2]nonanes and their antiprotozoal activities. Bioorg. Med. Chem. Lett. 2015, 25, 1390–1393. [Google Scholar] [CrossRef] [PubMed]
- Inam, A.; Siddiqui, S.M.; Macedo, T.S.; Moreira, D.R.; Leite, A.C.; Soares, M.B.; Azam, A. Design, synthesis and biological evaluation of 3-[4-(7-chloro-quinolin-4-yl)-piperazin-1-yl]-propionic acid hydrazones as antiprotozoal agents. Eur. J. Med. Chem. 2014, 75, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Juneja, A.; Macedo, T.S.; Magalhaes Moreira, D.R.; Pereira Soares, M.B.; Lima Leite, A.C.; Kelle de Andrade Lemoine Neves, J.; Alves Pereira, V.R.; Avecilla, F.; Azam, A. Synthesis of 4′-(2-ferrocenyl)-2,2′:6′2″-terpyridine: Characterization and antiprotozoal activity of Mn(II), Co(II), Ni(II), Cu(II) and Zn(II) complexes. Eur. J. Med. Chem. 2014, 75, 203–210. [Google Scholar] [CrossRef] [PubMed]
- McKeever, C.; Kaiser, M.; Rozas, I. Aminoalkyl derivatives of guanidine diaromatic minor groove binders with antiprotozoal activity. J. Med. Chem. 2013, 56, 700–711. [Google Scholar] [CrossRef]
- Opsenica, I.M.; Tot, M.; Gomba, L.; Nuss, J.E.; Sciotti, R.J.; Bavari, S.; Burnett, J.C.; Solaja, B.A. 4-Amino-7-chloroquinolines: Probing ligand efficiency provides botulinum neurotoxin serotype A light chain inhibitors with significant antiprotozoal activity. J. Med. Chem. 2013, 56, 5860–5871. [Google Scholar] [CrossRef]
- Hanessian, S.; Vakiti, R.R.; Chattopadhyay, A.K.; Dorich, S.; Lavallee, C. Probing functional diversity in pactamycin toward antibiotic, antitumor, and antiprotozoal activity. Bioorg. Med. Chem. 2013, 21, 1775–1786. [Google Scholar] [CrossRef]
- Abada, Z.; Cojean, S.; Pomel, S.; Ferrie, L.; Akagah, B.; Lormier, A.T.; Loiseau, P.M.; Figadere, B. Synthesis and antiprotozoal activity of original porphyrin precursors and derivatives. Eur. J. Med. Chem. 2013, 67, 158–165. [Google Scholar] [CrossRef]
- Yeo, S.J.; Liu, D.X.; Kim, H.S.; Park, H. Anti-malarial effect of novel chloroquine derivatives as agents for the treatment of malaria. Malar. J. 2017, 16, 80. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Gut, J.; Rosenthal, P.J.; Kumar, V. 4-Aminoquinoline-ferrocenyl-chalcone conjugates: Synthesis and anti-plasmodial evaluation. Eur. J. Med. Chem. 2017, 125, 269–277. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization, Department of Reproductive Health and Research. Global Incidence and Prevalence of Selected Curable Sexually Transmitted Infections; World Health Organization: Geneva, Switzerland, 2008. [Google Scholar]
- Petrin, D.; Delgaty, K.; Bhatt, R.; Garber, G. Clinical and microbiological aspects of Trichomonas vaginalis. Clin. Microbiol. Rev. 1998, 11, 300–317. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, M.A.; Carey, J.C.; Hauth, J.C.; Hillier, S.L.; Nugent, R.P.; Thom, E.A.; Ernest, J.M.; Heine, R.P.; Wapner, R.J.; Trout, W.; et al. Failure of metronidazole to prevent preterm delivery among pregnant women with asymptomatic Trichomonas vaginalis infection. N. Engl. J. Med. 2001, 345, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Goldman, M.B.; Cramer, D.W. Relation of tubal infertility to history of sexually transmitted diseases. Am. J. Epidemiol. 1993, 137, 577–584. [Google Scholar]
- Viikki, M.; Pukkala, E.; Nieminen, P.; Hakama, M. Gynaecological infections as risk determinants of subsequent cervical neoplasia. Acta Oncol. 2000, 39, 71–75. [Google Scholar] [CrossRef]
- Cherpes, T.L.; Wiesenfeld, H.C.; Melan, M.A.; Kant, J.A.; Cosentino, L.A.; Meyn, L.A.; Hillier, S.L. The associations between pelvic inflammatory disease, Trichomonas vaginalis infection, and positive herpes simplex virus type 2 serology. Sex. Transm. Dis. 2006, 33, 747–752. [Google Scholar] [CrossRef]
- Laga, M.; Manoka, A.; Kivuvu, M.; Malele, B.; Tuliza, M.; Nzila, N.; Goeman, J.; Behets, F.; Batter, V.; Alary, M.; et al. Non-ulcerative sexually transmitted diseases as risk factors for HIV-1 transmission in women: Results from a cohort study. AIDS 1993, 7, 95–102. [Google Scholar] [CrossRef]
- Cu-Uvin, S.; Ko, H.; Jamieson, D.J.; Hogan, J.W.; Schuman, P.; Anderson, J.; Klein, R.S. Prevalence, incidence, and persistence or recurrence of trichomoniasis among human immunodeficiency virus (HIV)-positive women and among HIV-negative women at high risk for HIV infection. Clin. Infect. Dis. 2002, 34, 1406–1411. [Google Scholar] [CrossRef][Green Version]
- Shethwala, N.D.; Mulla, S.A.; Kosambiya, J.K.; Desai, V.K. Sexually transmitted infections and reproductive tract infections in female sex workers. Indian J. Pathol. Microbiol. 2009, 52, 198–199. [Google Scholar]
- Mavedzenge, S.N.; Pol, B.V.; Cheng, H.; Montgomery, E.T.; Blanchard, K.; de Bruyn, G.; Ramjee, G.; Straten, A.V. Epidemiological synergy of Trichomonas vaginalis and HIV in Zimbabwean and South African women. Sex. Transm. Dis. 2010, 37, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Sorvillo, F.; Smith, L.; Kerndt, P.; Ash, L. Trichomonas vaginalis, HIV, and African-Americans. Emerg. Infect. Dis. 2001, 7, 927–932. [Google Scholar] [CrossRef] [PubMed]
- van Der Pol, B.; Kwok, C.; Pierre-Louis, B.; Rinaldi, A.; Salata, R.A.; Chen, P.L.; van de Wijgert, J.; Mmiro, F.; Mugerwa, R.; Chipato, T.; et al. Trichomonas vaginalis infection and human immunodeficiency virus acquisition in African women. J. Infect. Dis. 2008, 197, 548–554. [Google Scholar] [CrossRef]
- Wolner-Hanssen, P.; Krieger, J.N.; Stevens, C.E.; Kiviat, N.B.; Koutsky, L.; Critchlow, C.; DeRouen, T.; Hillier, S.; Holmes, K.K. Clinical manifestations of vaginal trichomoniasis. JAMA 1989, 261, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.S.; Chung, H.L.; Min, D.Y.; Cho, Y.H.; Ro, Y.S.; Kim, S.R. Diagnosis of trichomoniasis by polymerase chain reaction. Yonsei Med. J. 1999, 40, 56–60. [Google Scholar] [CrossRef]
- Coombs, G.H.; North, M.J. An analysis of the proteinases of Trichomonas vaginalis by polyacrylamide gel electrophoresis. Parasitology 1983, 86 Pt 1, 1–6. [Google Scholar] [CrossRef]
- Lockwood, B.C.; North, M.J.; Scott, K.I.; Bremner, A.F.; Coombs, G.H. The use of a highly sensitive electrophoretic method to compare the proteinases of trichomonads. Mol. Biochem. Parasitol. 1987, 24, 89–95. [Google Scholar] [CrossRef]
- Arroyo, R.; Alderete, J.F. Trichomonas vaginalis surface proteinase activity is necessary for parasite adherence to epithelial cells. Infect. Immun. 1989, 57, 2991–2997. [Google Scholar]
- Neale, K.A.; Alderete, J.F. Analysis of the proteinases of representative Trichomonas vaginalis isolates. Infect. Immun. 1990, 58, 157–162. [Google Scholar]
- Mallinson, D.J.; Lockwood, B.C.; Coombs, G.H.; North, M.J. Identification and molecular cloning of four cysteine proteinase genes from the pathogenic protozoon Trichomonas vaginalis. Microbiology 1994, 140 Pt 10, 2725–2735. [Google Scholar] [CrossRef][Green Version]
- Garber, G.E.; Lemchuk-Favel, L.T. Analysis of the extracellular proteases of Trichomonas vaginalis. Parasitol. Res. 1994, 80, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.A.; North, M.J.; Coombs, G.H. The pathway of secretion of proteinases in Trichomonas vaginalis. Int. J. Parasitol. 1995, 25, 657–666. [Google Scholar] [CrossRef]
- Wendel, K.A.; Workowski, K.A. Trichomoniasis: Challenges to appropriate management. Clin. Infect. Dis. 2007, 44 (Suppl. 3), S123–S129. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.M.; Webb, R.I.; O’Donoghue, P.; Upcroft, P.; Upcroft, J.A. Hydrogenosomes of laboratory-induced metronidazole-resistant Trichomonas vaginalis lines are downsized while those from clinically metronidazole-resistant isolates are not. J. Eukaryot. Microbiol. 2010, 57, 171–176. [Google Scholar] [CrossRef]
- Upcroft, J.A.; Dunn, L.A.; Wal, T.; Tabrizi, S.; Delgadillo-Correa, M.G.; Johnson, P.J.; Garland, S.; Siba, P.; Upcroft, P. Metronidazole resistance in Trichomonas vaginalis from highland women in Papua New Guinea. Sex. Health 2009, 6, 334–338. [Google Scholar] [CrossRef]
- Shokar, A.; Au, A.; An, S.H.; Tong, E.; Garza, G.; Zayas, J.; Wnuk, S.F.; Land, K.M. S-Adenosylhomocysteine hydrolase of the protozoan parasite Trichomonas vaginalis: Potent inhibitory activity of 9-(2-deoxy-2-fluoro-beta,D-arabinofuranosyl)adenine. Bioorg. Med. Chem. Lett. 2012, 22, 4203–4205. [Google Scholar] [CrossRef]
- Raj, R.; Singh, P.; Haberkern, N.T.; Faucher, R.M.; Patel, N.; Land, K.M.; Kumar, V. Synthesis of 1H-1,2,3-triazole linked beta-lactam-isatin bi-functional hybrids and preliminary analysis of in vitro activity against the protozoal parasite Trichomonas vaginalis. Eur. J. Med. Chem. 2013, 63, 897–906. [Google Scholar] [CrossRef]
- Anthwal, A.; Rajesh, U.C.; Rawat, M.S.; Kushwaha, B.; Maikhuri, J.P.; Sharma, V.L.; Gupta, G.; Rawat, D.S. Novel metronidazole-chalcone conjugates with potential to counter drug resistance in Trichomonas vaginalis. Eur. J. Med. Chem. 2014, 79, 89–94. [Google Scholar] [CrossRef]
- Kumar, K.; Bhargava, G.; Land, K.M.; Chang, K.H.; Arora, R.; Sen, S.; Kumar, V. N-Propargylated isatin-Mannich mono-and bis-adducts: Synthesis and preliminary analysis of in vitro activity against Tritrichomonas foetus. Eur. J. Med. Chem. 2014, 74, 657–663. [Google Scholar]
- Kumar, K.; Liu, N.; Yang, D.; Na, D.; Thompson, J.; Wrischnik, L.A.; Land, K.M.; Kumar, V. Synthesis and antiprotozoal activity of mono-and bis-uracil isatin conjugates against the human pathogen Trichomonas vaginalis. Bioorg. Med. Chem. 2015, 23, 5190–5197. [Google Scholar] [CrossRef]
- Nisha, N.; Tran, R.; Yang, D.; Hall, D.; Hopper, M.J.; Wrischnik, L.A.; Land, K.M.; Kumar, V. Cu(I)Cl-promoted synthesis of novel N-alkylated isatin analogs with an extension toward isatin-4-aminoquinoline conjugates: In vitro analysis against Trichomonas vaginalis. Med. Chem. Res. 2014, 23, 4570–4578. [Google Scholar] [CrossRef]
- Raj, R.; Sharma, V.; Hopper, M.J.; Patel, N.; Hall, D.; Wrischnik, L.A.; Land, K.M.; Kumar, V. Synthesis and preliminary in vitro activity of mono- And bis-1H-1,2,3-triazole-tethered-βlactam-isatin conjugates against the human protozoal pathogen Trichomonas vaginalis. Med. Chem. Res. 2014, 23, 3671–3680. [Google Scholar] [CrossRef]
- Saleh, Y.R.H.; Saadeh, H.A.; Kaur, H.; Goyal, K.; Sehgal, R.; Mubarak, M.S. The synthesis of novel hybrid compounds containing 5-nitrothiazole moiety as potential antiparasitic agents. Monatshefte für Chemie 2015, 146, 2087–2095. [Google Scholar] [CrossRef]
- Adams, M.; Li, Y.; Khot, H.; De Kock, C.; Smith, P.J.; Land, K.; Chibale, K.; Smith, G.S. The synthesis and antiparasitic activity of aryl-and ferrocenyl-derived thiosemicarbazone ruthenium(II)-arene complexes. Dalton Trans. 2013, 42, 4677–4685. [Google Scholar] [CrossRef] [PubMed]
- Nisha; Mehra, V.; Hopper, M.; Patel, N.; Hall, D.; Wrischnik, L.A.; Land, K.M.; Kumar, V. Design and synthesis of β-amino alcohol based β-lactam-isatin chimeras and preliminary analysis of in vitro activity against the protozoal pathogen Trichomonas vaginalis. MedChemComm 2013, 4, 1018–1024. [Google Scholar] [CrossRef]
- Stringer, T.; Taylor, D.; Guzgay, H.; Shokar, A.; Au, A.; Smith, P.J.; Hendricks, D.T.; Land, K.M.; Egan, T.J.; Smith, G.S. Polyamine quinoline rhodium complexes: Synthesis and pharmacological evaluation as antiparasitic agents against Plasmodium falciparum and Trichomonas vaginalis. Dalton Trans. 2015, 44, 14906–14917. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kushwaha, B.; Srivastava, A.; Maikhuri, J.P.; Sankhwar, S.N.; Gupta, G.; Dwivedi, A.K. Design and synthesis of coumarin-glyoxal hybrids for spermicidal and antimicrobial actions: A dual approach to contraception. RSC Adv. 2016, 6, 76288–76297. [Google Scholar] [CrossRef]
- Deborggraeve, S.; Koffi, M.; Jamonneau, V.; Bonsu, F.A.; Queyson, R.; Simarro, P.P.; Herdewijn, P.; Buscher, P. Molecular analysis of archived blood slides reveals an atypical human Trypanosoma infection. Diagn. Microbiol. Infect. Dis. 2008, 61, 428–433. [Google Scholar] [CrossRef]
- Truc, P.; Jamonneau, V.; N’Guessan, P.; N’Dri, L.; Diallo, P.B.; Cuny, G. Trypanosoma brucei ssp. and T congolense: Mixed human infection in Cote d’Ivoire. Trans. R. Soc. Trop. Med. Hyg. 1998, 92, 537–538. [Google Scholar] [CrossRef]
- Joshi, P.P.; Shegokar, V.R.; Powar, R.M.; Herder, S.; Katti, R.; Salkar, H.R.; Dani, V.S.; Bhargava, A.; Jannin, J.; Truc, P. Human trypanosomiasis caused by Trypanosoma evansi in India: The first case report. Am. J. Trop. Med. Hyg. 2005, 73, 491–495. [Google Scholar] [CrossRef]
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human African trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef]
- Ferguson, M.A.J.; Homans, S.W.; Dwek, R.A.; Rademacher, T.W. Glycosyl-phosphatidylinositol moiety that anchors Trypanosoma brucei variant surface glycoprotein to the membrane. Science 1988, 239, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Mehlert, A.; Richardson, J.M.; Ferguson, M.A.J. Structure of the glycosylphosphatidylinositol membrane anchor glycan of a class-2 variant surface glycoprotein from Trypanosoma brucei 11Edited by I. B. Holland. J. Mol. Biol. 1998, 277, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Mehlert, A.; Sullivan, L.; Ferguson, M.A. Glycotyping of Trypanosoma brucei variant surface glycoprotein MITat1.8. Mol. Biochem. Parasitol. 2010, 174, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Mehlert, A.; Zitzmann, N.; Richardson, J.M.; Treumann, A.; Ferguson, M.A. The glycosylation of the variant surface glycoproteins and procyclic acidic repetitive proteins of Trypanosoma brucei. Mol. Biochem. Parasitol. 1998, 91, 145–152. [Google Scholar] [CrossRef]
- Zamze, S.E.; Ashford, D.A.; Wooten, E.W.; Rademacher, T.W.; Dwek, R.A. Structural characterization of the asparagine-linked oligosaccharides from Trypanosoma brucei type II and type III variant surface glycoproteins. J. Biol. Chem. 1991, 266, 20244–20261. [Google Scholar]
- Zamze, S.E.; Wooten, E.W.; Ashford, D.A.; Ferguson, M.A.; Dwek, R.A.; Rademacher, T.W. Characterisation of the asparagine-linked oligosaccharides from Trypanosoma brucei type-I variant surface glycoproteins. Eur. J. Biochem. 1990, 187, 657–663. [Google Scholar] [CrossRef]
- Alirol, E.; Schrumpf, D.; Amici Heradi, J.; Riedel, A.; de Patoul, C.; Quere, M.; Chappuis, F. Nifurtimox-eflornithine combination therapy for second-stage gambiense human African trypanosomiasis: Medecins Sans Frontieres experience in the Democratic Republic of the Congo. Clin. Infect. Dis. 2013, 56, 195–203. [Google Scholar] [CrossRef]
- Andrade, L.O.; Andrews, N.W. The Trypanosoma cruzi-host-cell interplay: Location, invasion, retention. Nat. Rev. Microbiol. 2005, 3, 819–823. [Google Scholar] [CrossRef]
- Clayton, J. Chagas disease: Pushing through the pipeline. Nature 2010, 465, S12–S15. [Google Scholar] [CrossRef]
- Rassi, A., Jr.; Dias, J.C.; Marin-Neto, J.A.; Rassi, A. Challenges and opportunities for primary, secondary, and tertiary prevention of Chagas’ disease. Heart 2009, 95, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Bhambra, A.S.; Edgar, M.; Elsegood, M.R.J.; Li, Y.; Weaver, G.W.; Arroo, R.R.J.; Yardley, V.; Burrell-Saward, H.; Krystof, V. Design, synthesis and antitrypanosomal activities of 2,6-disubstituted-4,5,7-trifluorobenzothiophenes. Eur. J. Med. Chem. 2016, 108, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Trunz, B.B.; Jedrysiak, R.; Tweats, D.; Brun, R.; Kaiser, M.; Suwinski, J.; Torreele, E. 1-Aryl-4-nitro-1H-imidazoles, a new promising series for the treatment of human African trypanosomiasis. Eur. J. Med. Chem. 2011, 46, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- Bouchikhi, F.; Anizon, F.; Brun, R.; Moreau, P. Biological evaluation of glycosyl-isoindigo derivatives against the pathogenic agents of tropical diseases (malaria, Chagas disease, leishmaniasis and human African trypanosomiasis). Bioorg. Med. Chem. Lett. 2011, 21, 6319–6321. [Google Scholar] [CrossRef]
- Hwang, J.Y.; Smithson, D.; Connelly, M.; Maier, J.; Zhu, F.; Guy, K.R. Discovery of halo-nitrobenzamides with potential application against human African trypanosomiasis. Bioorg. Med. Chem. Lett. 2010, 20, 149–152. [Google Scholar] [CrossRef]
- Samant, B.S.; Sukhthankar, M.G. Compounds containing 2-substituted imidazole ring for treatment against human African trypanosomiasis. Bioorg. Med. Chem. Lett. 2011, 21, 1015–1018. [Google Scholar] [CrossRef]
- Ferrins, L.; Rahmani, R.; Sykes, M.L.; Jones, A.J.; Avery, V.M.; Teston, E.; Almohaywi, B.; Yin, J.; Smith, J.; Hyland, C.; et al. 3-(Oxazolo[4,5-b]pyridin-2-yl)anilides as a novel class of potent inhibitors for the kinetoplastid Trypanosoma brucei, the causative agent for human African trypanosomiasis. Eur. J. Med. Chem. 2013, 66, 450–465. [Google Scholar] [CrossRef]
- Pham, N.B.; Deydier, S.; Labaied, M.; Monnerat, S.; Stuart, K.; Quinn, R.J. N1,N1-Dimethyl-N3-(3-(trifluoromethyl)phenethyl)propane-1,3-diamine, a new lead for the treatment of human African trypanosomiasis. Eur. J. Med. Chem. 2014, 74, 541–551. [Google Scholar] [CrossRef]
- Samant, B.S.; Chakaingesu, C. Novel naphthoquinone derivatives: Synthesis and activity against human African trypanosomiasis. Bioorg. Med. Chem. Lett. 2013, 23, 1420–1423. [Google Scholar] [CrossRef]
- Papadopoulou, M.V.; Bloomer, W.D.; Rosenzweig, H.S.; Wilkinson, S.R.; Kaiser, M. Novel nitro(triazole/imidazole)-based heteroarylamides/sulfonamides as potential antitrypanosomal agents. Eur. J. Med. Chem. 2014, 87, 79–88. [Google Scholar] [CrossRef]
- Papadopoulou, M.V.; Bloomer, W.D.; Rosenzweig, H.S.; O’Shea, I.P.; Wilkinson, S.R.; Kaiser, M.; Chatelain, E.; Ioset, J.R. Discovery of potent nitrotriazole-based antitrypanosomal agents: In vitro and in vivo evaluation. Bioorg. Med. Chem. 2015, 23, 6467–6476. [Google Scholar] [CrossRef] [PubMed]
- Zelisko, N.; Atamanyuk, D.; Vasylenko, O.; Grellier, P.; Lesyk, R. Synthesis and antitrypanosomal activity of new 6,6,7-trisubstituted thiopyrano[2,3-d][1,3]thiazoles. Bioorg. Med. Chem. Lett. 2012, 22, 7071–7074. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.A.; de Queiroz, A.C.; Alexandre-Moreira, M.S.; Varela, J.; Cerecetto, H.; González, M.; Doriguetto, A.C.; Landre, I.M.; Barreiro, E.J.; Lima, L.M. Design, synthesis and in vitro trypanocidal and leishmanicidal activities of novel semicarbazone derivatives. Eur. J. Med. Chem. 2015, 100, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Upadhayaya, R.S.; Dixit, S.S.; Foldesi, A.; Chattopadhyaya, J. New antiprotozoal agents: Their synthesis and biological evaluations. Bioorg. Med. Chem. Lett. 2013, 23, 2750–2758. [Google Scholar] [CrossRef]
- Rios Martinez, C.H.; Lagartera, L.; Kaiser, M.; Dardonville, C. Antiprotozoal activity and DNA binding of N-substituted N-phenylbenzamide and 1,3-diphenylurea bisguanidines. Eur. J. Med. Chem. 2014, 81, 481–491. [Google Scholar] [CrossRef]
- Sola, I.; Artigas, A.; Taylor, M.C.; Gbedema, S.Y.; Pérez, B.; Clos, M.V.; Wright, C.W.; Kelly, J.M.; Muñoz-Torrero, D. Synthesis and antiprotozoal activity of oligomethylene-and p-phenylene-bis(methylene)-linked bis(+)-huprines. Bioorg. Med. Chem. Lett. 2014, 24, 5435–5438. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Activity | Ref. | |
|---|---|---|---|
 | G. lamblia [IC50 (µg/mL)] | [61] | |
| 7 | 4.62 ± 0.12 | ||
| MTZ | 0.86 ± 0.03 | ||
 | G. lamblia [IC50 (µg/mL)] | [62] | |
| 8 | 84.2 | ||
| MTZ | 1.22 | ||
 | G. intestinalis [IC50 (µM)] | [63] | |
| 9a | 0.122 | ||
| 9b | 0.151 | ||
| Aminitrozole | 0.490 | ||
| MTZ | 5.360 | ||
 | G. lamblia [IC50 (µM)] | [64] | |
| 10 | 19.58 | ||
| MTZ | 62.48 | ||
 | G. lamblia [IC50 (µg/mL)] | [65] | |
| 11 | 30.6 | ||
| MTZ | 0.21 | ||
 | G. intestinalis [IC50 (µM)] | [66] | |
| 12a | 0.027 ± 0.002 | ||
| 12b | 0.022 ± 0.001 | ||
| 12c | 0.027 ± 0.002 | ||
| 12d | 0.021 ± 0.005 | ||
| Nitazoxamide | 0.015 ± 0.002 | ||
| Mebendazole | 0.046 ± 0.009 | ||
| MTZ | 1.224 ± 0.021 | ||
 | G. duodenalis [IC50 (µg/mL)] | [67] | |
| 13a | 14.3 | ||
| 13b | 8.2 | ||
| MTZ | 11.4 | ||
 | G. intestinalis [IC50 (µM)] | [68] | |
| 14a | 0.87 | ||
| 14b | 0.39 | ||
| MTZ | 1.40 | ||
 | G. lamblia [IC50 (µM)] | [69] | |
| Disulfiram (15) | 6.6 | ||
| Compound | Activity | Ref. | ||
|---|---|---|---|---|
 | L. amazonensis (Promastigotes) [IC50 (µM)] | L. amazonensis (Amastigotes) [IC50 (µM)] | [94] | |
| 21a | 20.74 ± 0.5 | 22.0 ± 0.1 | ||
| 22b | 16.4 ± 1.1 | 17.0 ± 1.1 | ||
| Pentamidine | 4.8 ± 0.1 | 1.9 ± 0.1 | ||
 | L. major [IC50 (µg/mL)] | L. donovani [IC50 (µg/mL)] | [95] | |
| 22 | 0.47 ± 0.02 | 1.5 ± 0.17 | ||
| AmB | 0.56 ± 0.01 | - | ||
| SSG | - | 2.98 | ||
 | L. donovani [IC50 (µg/mL)] | [96] | ||
| 23a | 98.75 | |||
| 23b | 93.75 | |||
| SSG | 490 | |||
| Pentamidine | 5.5 | |||
 | L. donovani (Amastigotes) [IC50 (µM)] | [97] | ||
| 24a | 2.8 | |||
| 24b | 2.0 | |||
| SSG | 49.7 | |||
| Miltefosine | 8.4 | |||
 | L. donovani [IC50 (µM)] | [98] | ||
| Promastigotes | ||||
| 25a | 12.7 | |||
| 25b | 9.1 | |||
| Pentamidine | 4.8 | |||
| AmB | 0.3 | |||
 | L. amazonensis [IC50 (µM)] | L. donovani [IC50 (µM)] | [99] | |
| 26a | 0.13 | 0.31 | ||
| 26b | 0.14 | 0.17 | ||
 | L. major [IC50 (µM)] | L. donovani [IC50 (µM)] | [100] | |
| 27a | 1.2 ± 0.7 | 10.5 ± 0.6 | ||
| 27b | 1.5 ± 1.1 | 4.0 ± 0.6 | ||
| Pentamidine | 4.3 ± 0.8 | 5.5 ± 0.8 | ||
 | L. aethiopica [IC50 (µg/mL)] | [101] | ||
| Promastigotes | Amastigotes | |||
| 28 | 0.0142 ± 0.004 | 0.13 ± 0.02 | ||
| Miltefosine | 3.19 ± 14 | 0.3 ± 0.04 | ||
| AmB | 0.0472 ± 0.002 | 0.2 ± 0.02 | ||
 | L. infantum [IC50 (µM)] | [102] | ||
| 29 | 1.5 ± 0.1 | |||
| Miltefosine | 3.4 ± 0.6 | |||
 | L. major [IC50 (µM)] | [103] | ||
| Promastigotes | Amastigotes | |||
| 30a | 2.8 ± 0.4 | 0.2 ± 0.1 | ||
| 30b | 6.4 ± 0.2 | 0.8 | ||
| Pentamidine | 4.6 ± 1.1 | 3 ± 1 | ||
 | L. major [IC50 (µg/mL)] | [104] | ||
| 31a | 95.00 | |||
| 31b | 99.00 | |||
| SSG | 490.00 | |||
| Pentamidine | 5.50 | |||
 | L. donovani (Promastigotes) [IC50 (µM)] | [105] | ||
| 32 | 9.0 ± 2.80 | |||
| Miltefosine | 11.9 ± 2.70 | |||
 | L. donovani (Antiamastigotes) [IC50 (µM)] | [106] | ||
| 33a | 5.12 | |||
| 33b | 3.56 | |||
| Miltefosine | 12.50 | |||
 | L. major [IC50 (µM)] | [107] | ||
| 34a | 1.87 ± 0.31 | |||
| 34b | 4.37 ± 0.02 | |||
| Pentamidine | 5.09 ± 0.09 | |||
 | L. donovani [IC50 (µM)] | [108] | ||
| 35 | 0.84 ± 0.12 | |||
| Miltefosine | 8.10 ± 0.60 | |||
 | L. infantum [IC50 (µM)] | [109] | ||
| 36a | 1.40 ± 0.09 | |||
| 36b | 1.47 ± 0.07 | |||
| 36c | 0.83 ± 0.04 | |||
| Miltefosine | 2.84 ± 0.10 | |||
| Edelfosine | 0.82 ± 0.13 | |||
 | L. infantum [IC50(µM)] | L. amazonesis [IC50(µM)] | [110] | |
| 37a | 19.5 | 114.6 | ||
| 37b | 38.0 | inactive | ||
| Miltefosine | 23.7 | 20.9 | ||
 | L. donovani [IC50 (µM)] | [111] | ||
| Promastogote | Amastogote | |||
| 38a | 8.57 ± 1.58 | 1.11 ± 0.19 | ||
| 38b | 6.27 ± 0.65 | 0.36 ± 0.10 | ||
| Miltefosine | 1.10 ± 0.26 | 8.10 ± 1.41 | ||
 | L. donovani [IC50 (µM)] | [112] | ||
| 39a | 3.75 ± 0.31 | |||
| 39b | 5.02 ± 0.49 | |||
| 39c | 1.83 ± 0.21 | |||
| Miltefosine | 8.10 ± 0.51 | |||
| SSG | 53.12 ± 4.56 | |||
 | L. donovani Amastigote [IC50 (µM)] | [113] | ||
| 40 | 0.6 ± 0.2 | |||
| Miltefosine | 8.4 ± 1.2 | |||
| SSG | 56.1 ± 3.2 | |||
 | L. infantum [IC50 (µM)] | L. tropica [IC50 (µM)] | [114] | |
| 41 | 0.23 ± 0.05 | 0.12 ± 0.03 | ||
| Clofazimine | 4.48 ± 1.06 | 2.96 ± 1.23 | ||
| AmB | 0.08 ± 0.02 | 0.09 ± 0.04 | ||
 | L. infantum [IC50 (µM)] | [115] | ||
| 42a | 12.7 | |||
| 42b | 8.0 | |||
| Miltefosine | 10.4 | |||
 | L. donovani [IC50 (µM)] | [116] | ||
| 43a | 1.94 | |||
| 43b | 1.6 | |||
| 43c | 1.86 | |||
| 43d | 1.27 | |||
| 43e | 1.39 | |||
| Pentamidine | 1.84 | |||
 | L. donovani [IC50 (µg/mL)] | [117] | ||
| 44a | 2.29 | |||
| 44b | 2.48 | |||
 | L. donovani [IC50 (µg/mL)] | [118] | ||
| 45a | 1.6 | |||
| 45b | 2.0 | |||
| Miltefosine | 0.171 | |||
 | L. donovani (Amastigotes) [IC50 (µM)] | [119] | ||
| 46a | 5.4 | |||
| 46b | 1.1 | |||
| 46c | 2.4 | |||
| AmB | 0.10 | |||
 | L. amazonensis [IC50 (µM)] | [120] | ||
| 47a | 0.095 | |||
| 47b | 0.123 | |||
| 46c | 0.211 | |||
| AmB | 0.124 | |||
 | L. infantum [IC50 (µM)] | [121] | ||
| 48 | 29.43 ± 1.34 | |||
| AmB | 0.52 ± 1.34 | |||
 | L. donovani [IC50 (µM)] | [122] | ||
| Promastigotes | Amastigotes | |||
| 49 | 26.9 | 10.6 | ||
| Miltefosine | 6.1 | 8.2 | ||
 | L. infantum [IC50 (µM)] | [123] | ||
| 50 | 5.6 ± 0.14 | |||
 | L. major [IC50 (µM)] | [124] | ||
| Promastigotes | Amastigotes | |||
| 51a | 9.35 | 2.7 | ||
| 51b | 0.08 | - | ||
| Compounds | Activity | Ref. | ||
|---|---|---|---|---|
 | P. falciparum [IC50 (µM)] | [143] | ||
| 62 | 1.60 | |||
| Dihydro-artemisinin | 0.0011 | |||
 | P. falciparum [IC50 (nM)] | [144] | ||
| 63a | 11.7 | |||
| 63b | 13.5 | |||
| Chloroquine | 36.37 | |||
| Artemisinin | 4.37 | |||
 | P. falciparum [IC50 (µM)] | [145] | ||
| 3D7 strain | W2 strain | |||
| 64a | 3.76 | 5.97 | ||
| 64b | 8.49 | 4.58 | ||
| Chloroquine | 0.021 | 0.49 | ||
 | P. falciparum [IC50 (µM)] | [146] | ||
| W2 strain | D6 strain | |||
| 65a | 0.071 | 0.065 | ||
| 65b | 0.079 | 0.069 | ||
| Artemisinin | 0.007 | 0.009 | ||
| Chloroquine | 0.63 | 0.014 | ||
 | P. falciparum [IC50 (nM)] | [147] | ||
| 66 | 27.4 | |||
| Chloroquine | 300 | |||
| Mefloquine | 92 | |||
 | P. falciparum [IC50 (µM)] | [148] | ||
| D6 strain | W2 strain | |||
| 67 | 2.56 | 3.41 | ||
| Artemisinin | <0.09 | <0.09 | ||
| Chloroquine | <0.08 | 0.72 | ||
 | P. falciparum [IC50 (nM)] | [149] | ||
| 68a | 0.3 | |||
| 68b | 0.7 | |||
| 68c | 1.5 | |||
| Artemisinin | 19 | |||
 | P. falciparum [IC50 (nM)] | [150] | ||
| 69 | 2.6 ± 0.4 | |||
| Chloroquine | 9.8 ± 2.8 | |||
| Dihydro-Artemisinin | 1.1 ± 0.5 | |||
 | P. falciparum [IC50 (µM)] | [151] | ||
| 3D7 | K1 | |||
| 70a | 0.62 | 1.78 | ||
| 70b | 0.29 | 0.496 | ||
| Chloroquine | 0.005 | 0.254 | ||
 | % Dead asexual parasites (P. falciparum) | [152] | ||
| 3D7 | RKL-2 | |||
| 71a | 25.7 | 5.0 | ||
| 71b | 20.0 | 1.0 | ||
 | P. falciparum [IC50 (µM)] 3D7 strain | [153] | ||
| 72a | 3.71 | |||
| 72b | 3.95 | |||
| Chloroquine | 0.18 | |||
 | P. falciparum [IC50 (µM)] | [154] | ||
| 73a | 0.018 | |||
| 73b | 0.015 | |||
| Chloroquine | 0.062 | |||
 | P. falciparum [IC50 (µM)] | [155] | ||
| 3D7 | K1 | |||
| 74a | 0.87 | 2.04 | ||
| 74b | 0.3 | 2.11 | ||
| Chloroquine | 0.011 | 1.2 | ||
 | P. falciparum [IC50 (µM)] 3D7 strain | [156] | ||
| 75a | 0.24 | |||
| 75b | 0.09 | |||
| Chloroquine | 0.028 | |||
 | P. falciparum [IC50 (µM)] 3D7 strain | [157] | ||
| 76a | 0.007 | |||
| 76b | 0.017 | |||
 | P. falciparum [IC50 (µM)] K1 strain | [158] | ||
| 77a | 0.28 | |||
| 77b | 0.095 | |||
| Chloroquine | 0.12 | |||
 | P. falciparum [IC50 (µM)] K1 strain | [115] | ||
| 78 | 0.6 | |||
| Chloroquine | 0.14 | |||
 | P. falciparum [(IC50 µM)] W2 strain | [159] | ||
| 79a | 0.33 ± 0.095 | |||
| 79b | 0.2 ± 0.05 | |||
| Mefloquine | 0.04 ± 0.01 | |||
 | P. falciparum [IC50 (µM)] | [116] | ||
| 80a | 0.006 | |||
| 80b | 0.004 | |||
| 80c | 0.006 | |||
| Chloroquine | 0.201 | |||
 | P. falciparum [IC50 (µg/mL)] | [117] | ||
| 81a | 0.608 | |||
| 81b | 2.50 | |||
| 81c | 1.71 | |||
 | P. falciparum [IC50 (µg/mL)] | [118] | ||
| 82a | 13.2 | |||
| 82b | 14.7 | |||
| Chloroquine | 0.073 | |||
 | P. falciparum [IC50 (µM)] | [160] | ||
| 83a | 1.1 ± 1.1 | |||
| 83b | 1.9 ± 0.9 | |||
| 83c | 1.5 ± 0.14 | |||
| Mefloquine | 0.004 ± 0.02 | |||
 | P. falciparum [IC50 (µM)] | [161] | ||
| 84a | 0.106 | |||
| 84b | 0.149 | |||
| Chloroquine | 0.0039 | |||
 | P. falciparum [IC50 (µM)] | [120] | ||
| 85a | 0.002 | |||
| 85b | 0.003 | |||
| Chloroquine | 0.125 | |||
| Artemisinin | 0.004 | |||
 | P. falciparum [IC50 (nM)] | [162] | ||
| W2 | D6 | |||
| 86a | 45.78 | 26.38 | ||
| 86b | 9.47 | 4.10 | ||
| 86c | 5.93 | 3.95 | ||
| Artemisinin | 6.7 | 9.00 | ||
 | P. falciparum [IC50 (nM)] | [163] | ||
| D6 | Dd2 | |||
| 87a | 6.5 | 7.4 | ||
| 87b | 6.7 | 3.5 | ||
 | P. falciparum [IC50 (µM)] | [164] | ||
| 88 | 0.02 | |||
| Chloroquine | 15~25 | |||
 | P. falciparum [IC50 (µM)] | [165] | ||
| 89a | 0.17 ± 0.01 | |||
| 89b | 0.23 ± 0.01 | |||
 | P. falciparum [IC50 (µM)] | [166] | ||
| 90a | 0.43 ± 0.02 | |||
| 90b | 0.53 ± 0.04 | |||
| 90c | 0.37 ± 0.03 | |||
| Chloroquine | 0.06 | |||
| Compound | Activity | Ref. | ||
|---|---|---|---|---|
 | T. vaginalis [IC50 (µM)] | [63] | ||
| 91a | 0.331 | |||
| 91b | 0.221 | |||
| MTZ | 0.290 | |||
 | T. vaginalis [IC50 (µM)] | [191] | ||
| 92 | 0.09 | |||
| MTZ | 0.72 | |||
 | T. vaginalis [IC50 (µM)] | [192] | ||
| 93 | 7.06 | |||
| MTZ | 0.72 | |||
 | T. vaginalis [IC50 (µg/mL)] | [193] | ||
| MTZ-susceptible | MTZ-resistant | |||
| 94a | 1.56 | 3.125 | ||
| 94b | 1.56 | 3.125 | ||
| MTZ | 1.56 | 12.5 | ||
 | T. vaginalis [IC50 (µM)] | [66] | ||
| 95 | 0.023± 0.005 | |||
| Nitazoxanide | 0.107 ± 0.009 | |||
| MTZ | 0.213 ± 0.004 | |||
 | T. vaginalis [IC50 (µg/mL)] | [62] | ||
| 96 | 83.9 | |||
| MTZ | 0.037 | |||
 | T.foetus [IC50 (µM)] | [194] | ||
| 97a | 22.2 | |||
| 97b | 11.3 | |||
| 97c | 24.5 | |||
| MTZ | 0.72 | |||
 | T. vaginalis [IC50 (µM)] | [195] | ||
| 98a | 9.86 | |||
| 98b | 9.79 | |||
| MTZ | 0.72 | |||
 | T. vaginalis [IC50 (µM)] | [196] | ||
| 99 | 23 | |||
| MTZ | 0.72 | |||
 | T. vaginalis [IC50 (µM)] | [197] | ||
| 100a | 10.49 ± 1.05 | |||
| 100b | 25.60 ± 1.08 | |||
| MTZ | 0.72 | |||
 | T. vaginalis [IC50 (µg/mL)] | [198] | ||
| 101 | 4.3 ± 1.2 | |||
| MTZ | 8.5 ± 0.9 | |||
 | T. vaginalis [IC50 (µM)] | [199] | ||
| 102a | 5.47 | |||
| 102b | 7.56 | |||
| MTZ | 0.72 | |||
 | T. vaginalis [IC50 (µM)] | [200] | ||
| 103 | 9.73 ± 1.13 | |||
| MTZ | 0.72 | |||
 | T. vaginalis [IC50 (µM)] | [201] | ||
| 104 | 4.80 ± 0.54 | |||
| MTZ | 0.72 | |||
 | T. vaginalis [IC50 (µg/mL)] | [202] | ||
| MTZ-susceptible | MTZ-resistant | |||
| 105a | 0.0294 | 0.2363 | ||
| 105b | 0.0140 | 0.1122 | ||
| 105c | 0.0136 | 0.1091 | ||
| MTZ | 0.0182 | 0.3655 | ||
| Compound | Activity | Ref. | ||
|---|---|---|---|---|
 | T. b. rhodesiense [IC50 (µM)] | [217] | ||
| 114a | 0.60 | |||
| 114b | 0.53 | |||
| Melarsoprol | 0.051 | |||
 | T. b. rhodesiense [IC50 (µM)] | [218] | ||
| 115a | 0.16 | |||
| 115b | 0.10 | |||
| Melarsoprol | 0.009 | |||
| Eflornithine | 3.80 | |||
 | T. brucei [IC50 (µM)] | [219] | ||
| 116a | 0.84 | |||
| 116b | 0.60 | |||
| 116c | 0.51 | |||
| Melarsoprol | 0.013 | |||
 | T. brucei [IC50 (µM)] | [220] | ||
| 117 | 1.5 ± 0.4 | |||
 | T. brucei [IC50 (µM)] | [221] | ||
| 118a | 0.25 ± 0.1 | |||
| 118b | 0.67 ± 10.1 | |||
 | T. b. rhodesiense [IC50 (µM)] | [222] | ||
| 119 | 0.091 | |||
 | T. b. brucei [IC50 (µM)] | [223] | ||
| 120a | 0.7 | |||
| 120b | 0.5 | |||
 | T. brucei [IC50 (µM)] | [224] | ||
| 121a | 0.07 ± 0.01 | |||
| 121b | 0.05 ± 0.01 | |||
 | T. b. rhodesiense [IC50 (µM)] | T. cruzi [IC50 (µM)] | [225] | |
| 122a | 0.218 | 0.373 | ||
| 122b | 0.187 | 0.239 | ||
 | T. cruzi [IC50 (µM)] | [226] | ||
| 123 | 0.008 | |||
| Benznidazole | 2.153 ± 0.176 | |||
 | T. b. brucei [IC50 (µM)] | [227] | ||
| 124 | 0.2624 ± 0.0139 | |||
| pentamidine | 0.0032 ± 0.0004 | |||
 | T. brucei [IC50 (µM)] | [98] | ||
| 125 | 1.01 | |||
| Pentamidine | 0.0041 | |||
 | T. cruzi [IC50 (µM)] | [228] | ||
| 126 | 8.5 | |||
| Nifurtimox | 7.7 | |||
 | T. b. rhodesiense [IC50 (µM)] | [158] | ||
| 127a | 0.061 | |||
| 127b | 0.065 | |||
| Melarsoprol | 0.0039 | |||
| Suramin | 0.0075 | |||
 | T. b. rhodesiense [IC50 (µM)] | T. cruzi [IC50 (µM)] | [229] | |
| 128a | 1.81 | 0.25 | ||
 | T. cruzi [IC50 (µM)] | [115] | ||
| 129 | 0.04 | |||
| Benznidazole | 1.95 | |||
 | T. b. rhodesiense [IC50 (µM)] | [230] | ||
| 130 | 0.009 | |||
| Melarsoprol | 0.008 | |||
 | T. b. rhodesiense [IC50 (µM)] | [116] | ||
| 131 | 0.003 | |||
| Pentamidine | 0.003 | |||
| Melarsoprol | 0.006 | |||
 | T. b. rhodesiense [IC50(µg/mL)] | [118] | ||
| 132 | 7.0 | |||
| Melarsoprol | 0.005 | |||
 | T. b. rhodesiense [IC50(µM)] | [161] | ||
| 133a | 13.1 | |||
| 133b | 20.2 | |||
| Pentamidine | 0.005 | |||
 | T. b. rhodesiense [IC50 (µM)] | T. cruzi [IC50 (µM)] | [120] | |
| 134a | 0.004 | 71.6 | ||
| 134b | 0.019 | 0.053 | ||
| Pentamidine | 0.003 | - | ||
| Melarsoprol | 0.004 | - | ||
| Benznidazole | - | 1.30 | ||
 | T. brucei [IC50 (µM)] | [231] | ||
| 135a | 0.52 ± 0.01 | |||
| 135b | 0.57 ± 0.02 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.-M.; Kim, M.-S.; Hayat, F.; Shin, D. Recent Advances in the Discovery of Novel Antiprotozoal Agents. Molecules 2019, 24, 3886. https://doi.org/10.3390/molecules24213886
Lee S-M, Kim M-S, Hayat F, Shin D. Recent Advances in the Discovery of Novel Antiprotozoal Agents. Molecules. 2019; 24(21):3886. https://doi.org/10.3390/molecules24213886
Chicago/Turabian StyleLee, Seong-Min, Min-Sun Kim, Faisal Hayat, and Dongyun Shin. 2019. "Recent Advances in the Discovery of Novel Antiprotozoal Agents" Molecules 24, no. 21: 3886. https://doi.org/10.3390/molecules24213886
APA StyleLee, S.-M., Kim, M.-S., Hayat, F., & Shin, D. (2019). Recent Advances in the Discovery of Novel Antiprotozoal Agents. Molecules, 24(21), 3886. https://doi.org/10.3390/molecules24213886