Enhanced Nanoencapsulation of Sepiapterin within PEG-PCL Nanoparticles by Complexation with Triacetyl-Beta Cyclodextrin



Abstract
1. Introduction
2. Results and Discussion
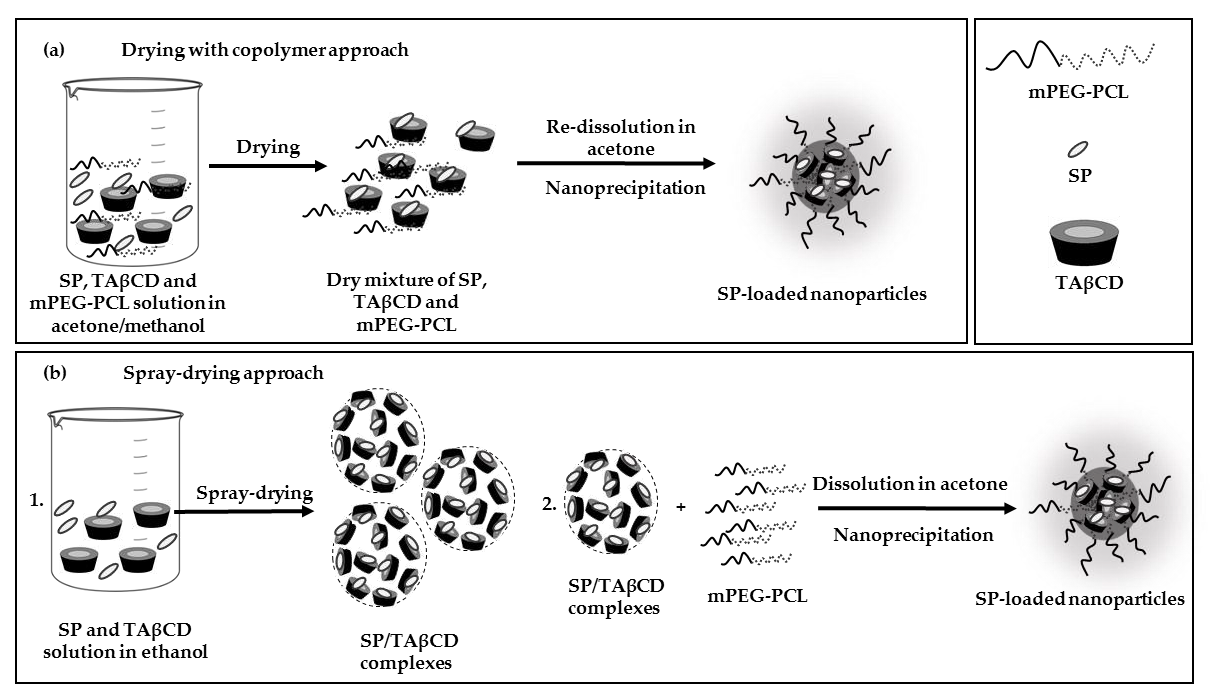
2.1. BH4 and SP Stability
2.2. Characterization of Spray-Dried SP/TAβCD Complexes
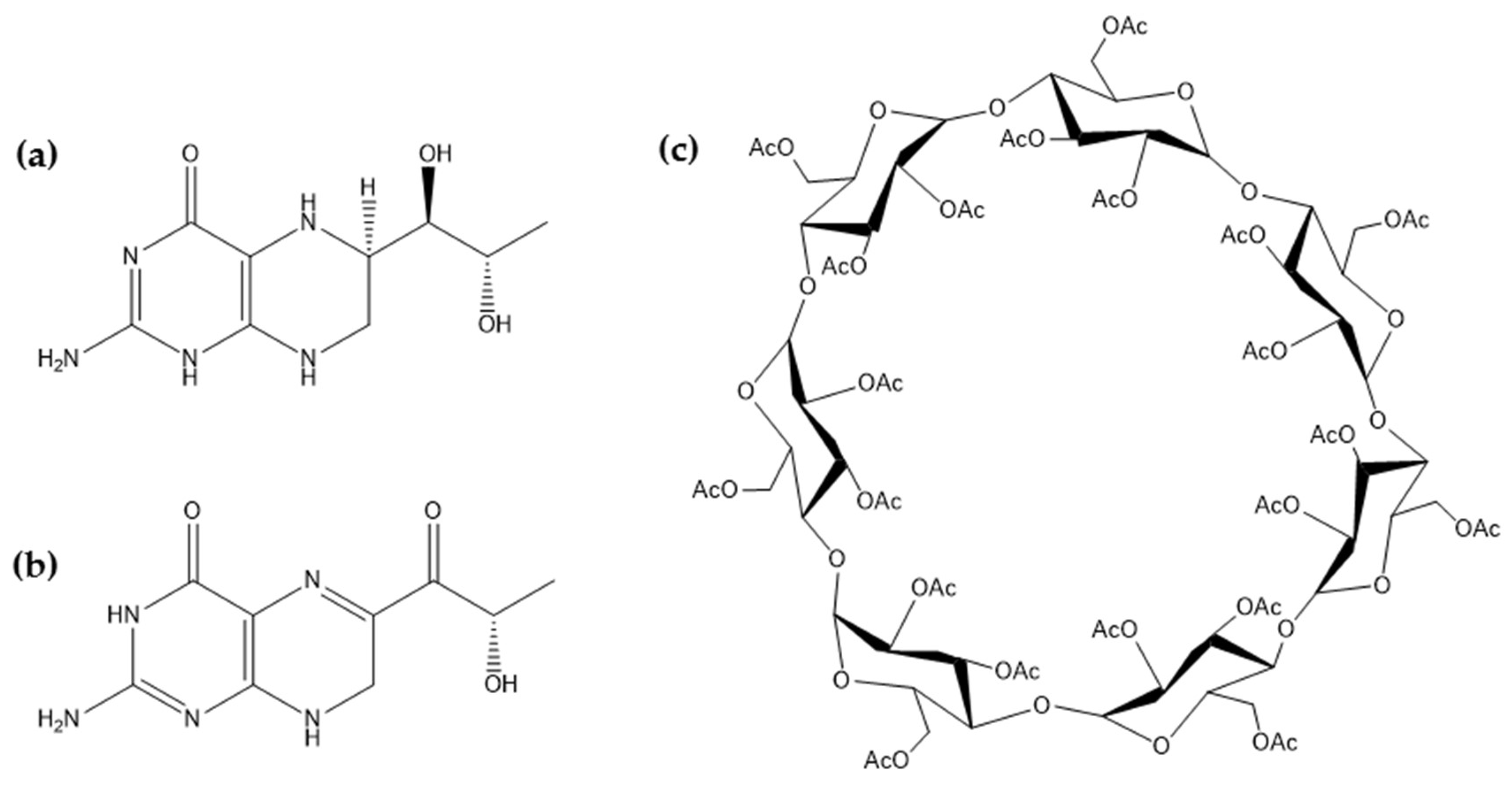
2.2.1. Fourier-Transform Infrared Spectroscopy
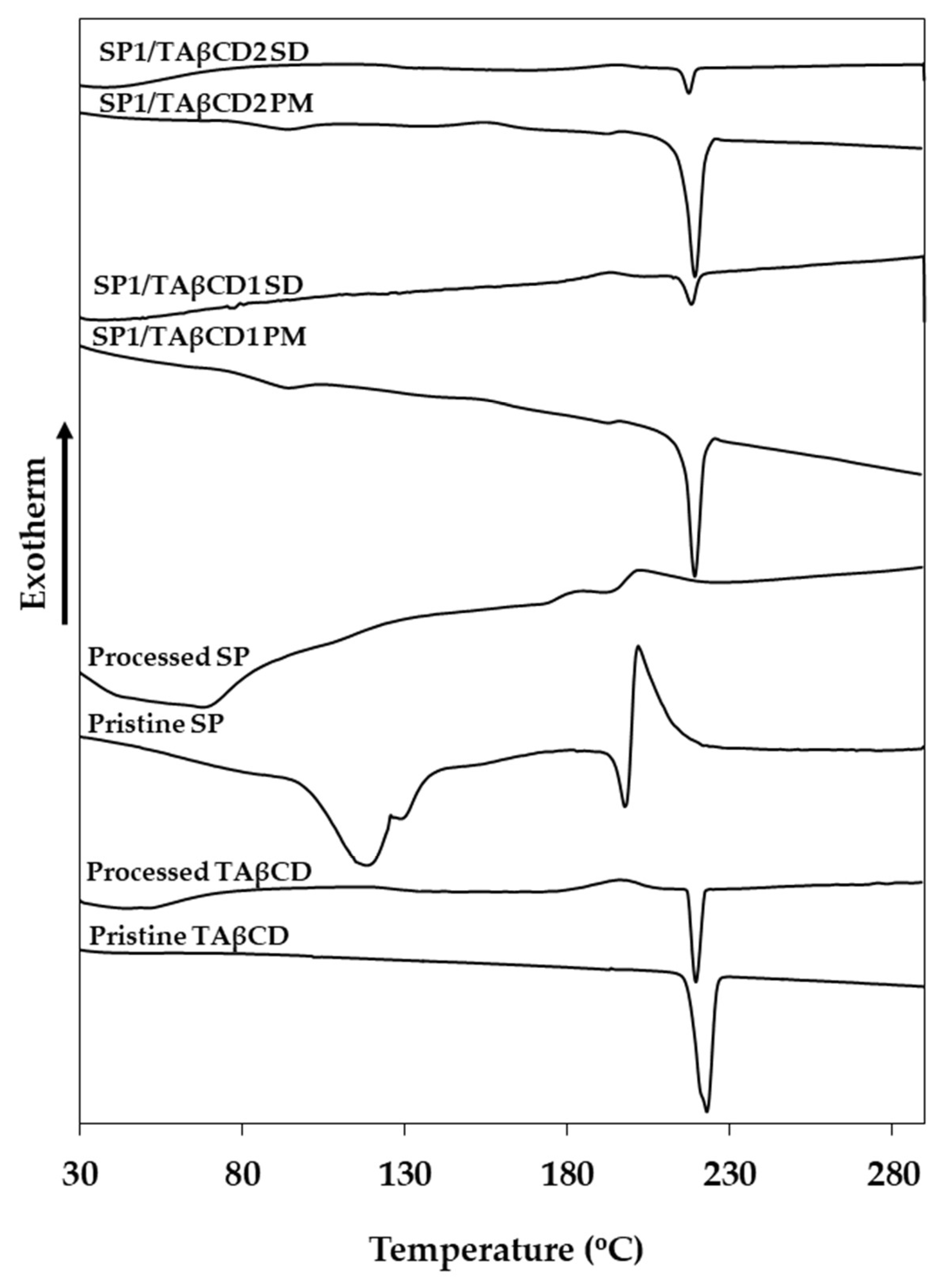
2.2.2. Differential Scanning Calorimetry (DSC)
2.2.3. Morphological Characterization of Spray-Dried Complexes
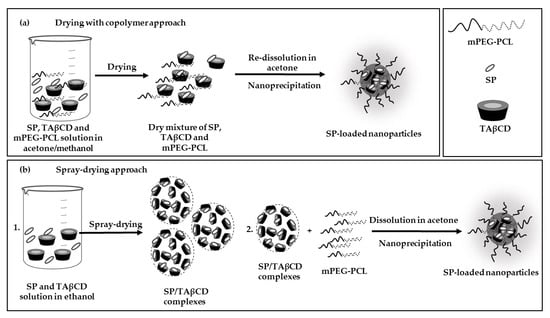
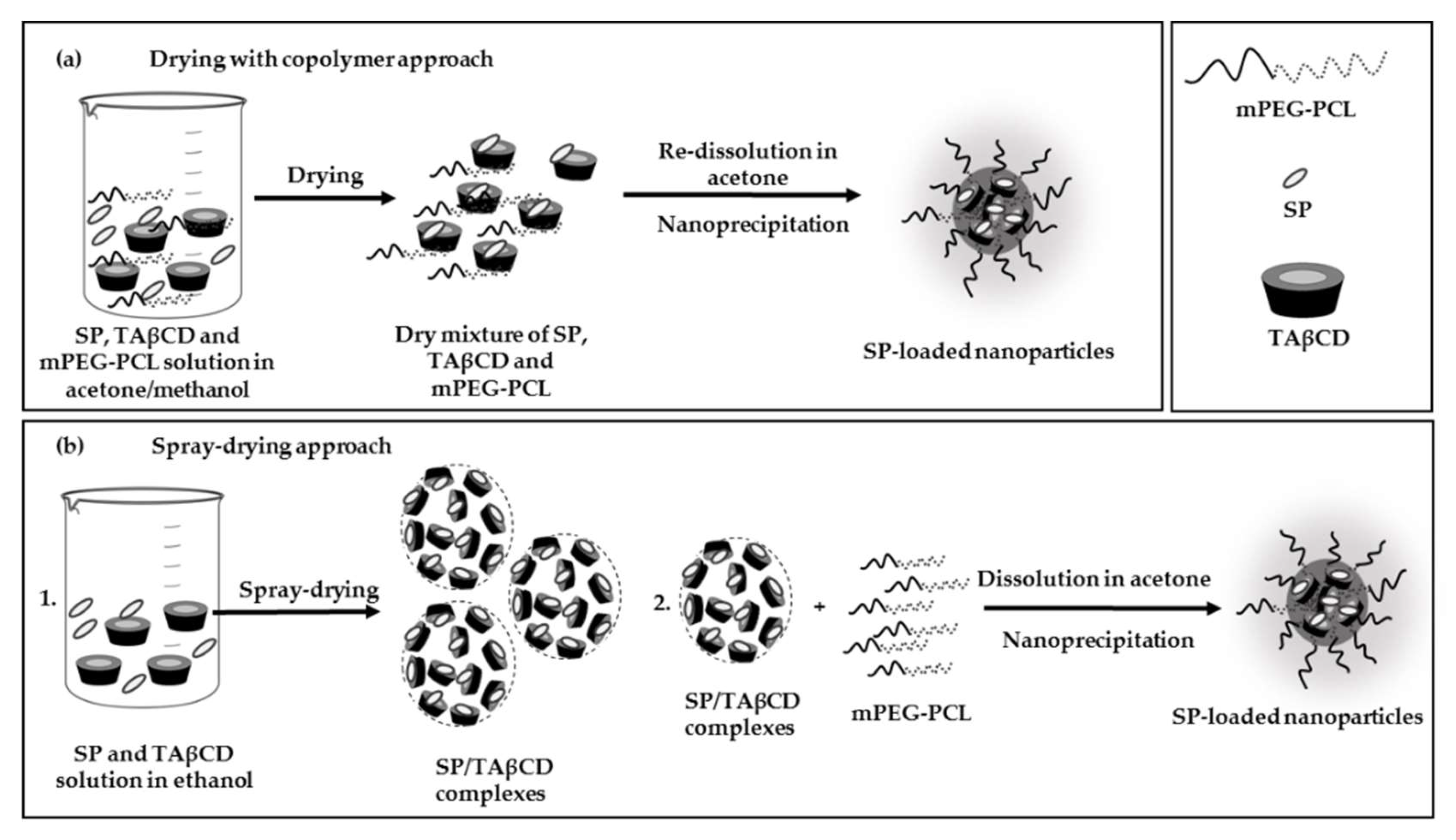
2.3. Production and Characterization of SP-Loaded NPs
2.3.1. mPEG-PCL Copolymer Synthesis
2.3.2. Nanoencapsulation of SP
2.3.3. Size and Size Distribution of SP-Loaded NPs
2.3.4. Encapsulation Efficiency and SP Loading
2.3.5. Morphological Characterization of SP-Loaded NPs
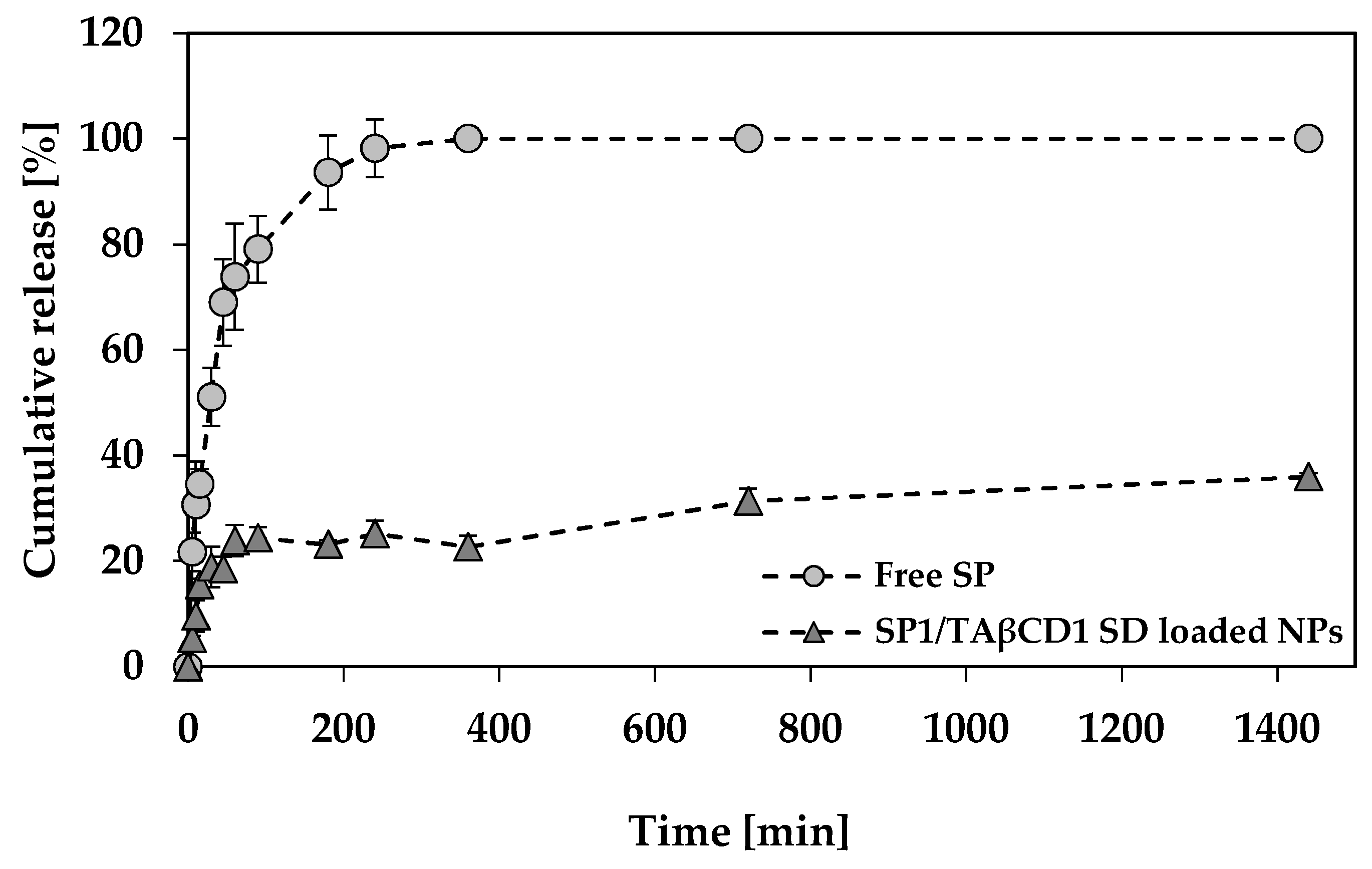
2.3.6. SP Release Study
3. Materials and Methods
3.1. Preparation of Spray-Dried SP/TAβCD Complexes
3.2. Preparation of PMs
3.3. Characterization of Spray-Dried SP/TAβCD Complexes
3.3.1. Differential scanning colorimetry
3.3.2. Fourier-Transform Infrared Spectroscopy
3.3.3. Scanning Electron Microscopy
3.3.4. Transmission Electron Microscopy
3.4. Preparation of SP-Loaded mPEG-PCL NPs
3.4.1. Synthesis of mPEG-PCL Copolymer
3.4.2. Drying of SP and TAβCD with mPEG-PCL Copolymer
3.4.3. Encapsulation of SP within mPEG-PCL NPs
3.5. Characterization of SP-Loaded NPs
3.5.1. Size and Size Distribution
3.5.2. SP Encapsulation Efficiency and Drug Loading
3.5.3. Morphological Analysis of SP-Loaded NPs
3.5.4. SP Release Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Thöny, B.; Auerbach, G.; Blau, N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem. J. 2000, 347, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Werner, E.R.; Blau, N.; Thöny, B. Tetrahydrobiopterin: Biochemistry and pathophysiology and its functions. Biochem. J. 2011, 438, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Alp, N.J.; Channon, K.M. Regulation of Endothelial Nitric Oxide Synthase by Tetrahydrobiopterin in Vascular Disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Hyland, K. Inherited Disorders Affecting Dopamine and Serotonin: Critical Neurotransmitters Derived from Aromatic Amino Acids. J. Nutr. 2007, 137, 1568S–1572S. [Google Scholar] [CrossRef] [PubMed]
- Shintaku, H. Disorders of tetrahydrobiopterin metabolism and their treatment. Curr. Drug Metab. 2002, 3, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.D.; Kaufman, S.; Milstein, S. The auto-oxidation of tetrahydrobiopterin. Eur. J. Biochem. 1988, 173, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Sosnik, A.; Carcaboso, A.; Chiappetta, D. Polymeric Nanocarriers: New Endeavors for the Optimization of the Technological Aspects of Drugs. Recent Patents Biomed. Eng. 2010, 1, 43–59. [Google Scholar] [CrossRef]
- Moretton, M.A.; Glisoni, R.J.; Chiappetta, D.A.; Sosnik, A. Molecular implications in the nanoencapsulation of the anti-tuberculosis drug rifampicin within flower-like polymeric micelles. Colloids Surf. B Biointerfaces 2010, 79, 467–479. [Google Scholar] [CrossRef]
- Maitz, M.F. Applications of synthetic polymers in clinical medicine. Biosurf. Biotribol. 2015, 1, 161–176. [Google Scholar] [CrossRef]
- Guo, J.; Gao, X.; Su, L.; Xia, H.; Gu, G.; Pang, Z.; Jiang, X.; Yao, L.; Chen, J.; Chen, H. Aptamer-functionalized PEG–PLGA nanoparticles for enhanced anti-glioma drug delivery. Biomaterials 2011, 32, 8010–8020. [Google Scholar] [CrossRef]
- Danafar, H.; Davaran, S.; Rostamizadeh, K.; Valizadeh, H.; Hamidi, M. Biodegradable m-PEG/PCL core-shell micelles: Preparation and characterization as a sustained release formulation for curcumin. Adv. Pharm. Bull. 2014, 4, 501–510. [Google Scholar] [PubMed]
- Peng, W.; Jiang, X.; Zhu, Y.; Deng, W.; Yu, J.; Xu, X.; Zhang, W. Oral delivery of capsaicin using MPEG-PCL nanoparticles. Acta Pharmacol. Sin. 2014, 36, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Riley, T.; Govender, T.; Stolnik, S.; Xiong, C.D.; Garnett, M.C.; Illum, L.; Davis, S.S. Colloidal stability and drug incorporation aspects of micellar-like PLA- PEG nanoparticles. Colloids Surf. B Biointerfaces 1999, 16, 147–159. [Google Scholar] [CrossRef]
- Danafar, H. MPEG–PCL copolymeric nanoparticles in drug delivery systems. Cogent Med. 2016, 3, 1142411. [Google Scholar] [CrossRef]
- Grossen, P.; Witzigmann, D.; Sieber, S.; Huwyler, J. PEG-PCL-based nanomedicines: A biodegradable drug delivery system and its application. J. Control. Release 2017, 260, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Pannirselvam, M.; Simon, V.; Verma, S.; Anderson, T.; Triggle, C.R. Chronic oral supplementation with sepiapterin prevents endothelial dysfunction and oxidative stress in small mesenteric arteries from diabetic (db/db) mice. Br. J. Pharmacol. 2003, 140, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Massella, D.; Celasco, E.; Salaün, F.; Ferri, A.; Barresi, A.A. Overcoming the limits of flash nanoprecipitation: Effective loading of hydrophilic drug into polymeric nanoparticles with controlled structure. Polymers (Basel) 2018, 10, 1092. [Google Scholar] [CrossRef] [PubMed]
- Barichello, J.M.; Morishita, M.; Takayama, K.; Nagai, T. Encapsulation of Hydrophilic and Lipophilic Drugs in PLGA Nanoparticles by the Nanoprecipitation Method. Drug Dev. Ind. Pharm. 1999, 25, 471–476. [Google Scholar] [CrossRef]
- Seremeta, K.P.; Martínez Pérez, S.; López Hernández, O.D.; Höcht, C.; Taira, C.; Reyes Tur, M.I.; Sosnik, A. Spray-dried didanosine-loaded polymeric particles for enhanced oral bioavailability. Colloids Surf. B Biointerfaces 2014, 123, 515–523. [Google Scholar] [CrossRef]
- Seremeta, K.P.; Höcht, C.; Taira, C.; Cortez Tornello, P.R.; Abraham, G.A.; Sosnik, A. Didanosine-loaded poly(epsilon-caprolactone) microparticles by a coaxial electrohydrodynamic atomization (CEHDA) technique. J. Mater. Chem. B 2015, 3, 102–111. [Google Scholar] [CrossRef]
- Sherje, A.; Londhe, V. Inclusion Complexes of Hydroxy Propyl-beta-Cyclodextrin and Paliperidone: Preparation and Characterization. Curr. Drug Discov. Technol. 2014, 11, 271–278. [Google Scholar]
- Yang, H.; Parniak, M.A.; Isaacs, C.E.; Hillier, S.L.; Rohan, L.C. Characterization of cyclodextrin inclusion complexes of the anti-HIV non-nucleoside reverse transcriptase inhibitor UC781. AAPS J. 2008, 10, 606–613. [Google Scholar] [CrossRef] [PubMed]
- Loftsson, T.; Ólafsdóttir, B.J.; Friðriksdóttir, H.; Jónsdóttir, S. Cyclodextrin complexation of NSAIDSs: Physicochemical characteristics. Eur. J. Pharm. Sci. 1993, 1, 95–101. [Google Scholar] [CrossRef]
- Loftsson, T.; Hreinsdóttir, D.; Másson, M. Evaluation of cyclodextrin solubilization of drugs. Int. J. Pharm. 2005, 302, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Masukawa, T.; Nadai, T.; Yoshii, K.; Okada, S.; Miyajima, K. Prolonged release of drug from triacetyl-β-CyD complex for oral and rectal administration. J. Incl. Phenom. Molecular Recognit. Chem. 1996, 1–3, 181–184. [Google Scholar] [CrossRef]
- Nakanishi, K.; Masukawa, T.; Nadai, T.; Yoshii, K.; Okada, S.; Miyajima, K. Sustained release of flufenamic acid from a drug-triacetyl-beta-cyclodextrin complex. Biol. Pharm. Bull. 1997, 20, 66–70. [Google Scholar] [CrossRef][Green Version]
- Fernandes, C.M.; Veiga, F.J.B. Effect of the hydrophobic nature of triacetyl-beta-cyclodextrin on the complexation with nicardipine hydrochloride: Physicochemical and dissolution properties of the kneaded and spray-dried complexes. Chem. Pharm. Bull. (Tokyo) 2002, 50, 1597–1602. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.M.; Ramos, P.; Jose, F. Hydrophilic and hydrophobic cyclodextrins in a new sustained release oral formulation of nicardipine: In vitro evaluation and bioavailability studies in rabbits. J. Control. Release 2003, 88, 127–134. [Google Scholar] [CrossRef]
- Corti, G.; Capasso, G.; Maestrelli, F.; Cirri, M.; Mura, P. Physical–chemical characterization of binary systems of metformin hydrochloride with triacetyl- β-cyclodextrin. J. Pharm. Biomed. Anal. 2007, 45, 480–486. [Google Scholar] [CrossRef]
- Nunes, A.V.M.; Almeida, A.P.C.; Marques, S.R.; De Sousa, A.R.S.; Casimiro, T.; Duarte, C.M.M. Processing triacetyl-β-cyclodextrin in the liquid phase using supercritical CO2. J. Supercrit. Fluids 2010, 54, 357–361. [Google Scholar] [CrossRef]
- Bratu, I.; Veiga, F.; Fernandes, C.; Hernanz, A.; Gavira, J.M. Infrared spectroscopic study of triacetyl-β-cyclodextrin and its inclusion complex with nicardipine. Spectroscopy 2004, 18, 459–467. [Google Scholar] [CrossRef]
- Ghorab, M.K.; Adeyeye, M.C. Enhancement of ibuprofen dissolution via wet granulation with β-cyclodextrin. Pharm. Dev. Technol. 2001, 6, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, S.B.; Duarte, F.Í.C.; Heimfarth, L.; Quintans, J.D.S.S.; Quintans-Júnior, L.J.; Júnior, V.F.D.V.; De Lima, Á.A.N. Cyclodextrin-drug inclusion complexes: In vivo and in vitro approaches. Int. J. Mol. Sci. 2019, 20, 642. [Google Scholar] [CrossRef] [PubMed]
- Bettinetti, G.; Sorrenti, M.; Catenacci, L.; Ferrari, F.; Rossi, S. Polymorphism, pseudopolymorphism, and amorphism of peracetylated α-, β-, and γ-cyclodextrins. J. Pharm. Biomed. Anal. 2006, 41, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Sapte, S.; Pore, Y. Inclusion complexes of cefuroxime axetil with β-cyclodextrin: Physicochemical characterization, molecular modeling and effect of l-arginine on complexation. J. Pharm. Anal. 2016, 6, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E. Polymorphs of sepiapterin and salts thereof. Patent Application WO2018102315.
- Sami, F.; Philip, B.; Pathak, K. Effect of auxiliary substances on complexation efficiency and intrinsic dissolution rate of gemfibrozil-beta-CD complexes. AAPS PharmSciTech 2009, 11, 27–35. [Google Scholar] [CrossRef]
- Sosnik, A.; Cohn, D. Poly(ethylene glycol)-poly(epsilon-caprolactone) block oligomers as injectable materials. Polymer 2003, 44, 7033–7042. [Google Scholar] [CrossRef]
- Glisoni, R.J.; Chiappetta, D.A.; Moglioni, A.G.; Sosnik, A. Novel 1-indanone thiosemicarbazone antiviral candidates: Aqueous solubilization and physical stabilization by means of cyclodextrins. Pharm. Res. 2012, 29, 739–755. [Google Scholar] [CrossRef]
- Daneshmand, S.; Golmohammadzadeh, S.; Jaafari, M.R.; Movaffagh, J.; Rezaee, M.; Sahebkar, A.; Malaekeh-Nikouei, B. Encapsulation challenges, the substantial issue in solid lipid nanoparticles characterization. J. Cell. Biochem. 2018, 119, 4251–4264. [Google Scholar] [CrossRef]
- Zhang, Y.; Huo, M.; Zhou, J.; Zou, A.; Li, W.; Yao, C.; Xie, S. DDSolver: An add-in program for modeling and comparison of drug dissolution profiles. AAPS J. 2010, 12, 263–271. [Google Scholar] [CrossRef]
- Moretton, M.A.; Hocht, C.; Taira, C.; Sosnik, A. Rifampicin-loaded “flower-like” polymeric micelles for enhanced oral bioavailability in an extemporaneous liquid fixed-dose combination with isoniazid. Nanomedicine (Lond.) 2014, 9, 1635–1650. [Google Scholar] [CrossRef] [PubMed]
- Halamoda-Kenzaoui, B.; Holzwarth, U.; Roebben, G.; Bogni, A.; Bremer-Hoffmann, S. Mapping of the available standards against the regulatory needs for nanomedicines. WIREs Nanomed. Nanobiotechnol. 2019, 11, e1531. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preparation Method | SP:TAβCD Molar Ratio | Nomenclature |
|---|---|---|
| Spray-drying (SD) a | 1:1 | SP1/TAβCD1 SD |
| 1:2 | SP1/TAβCD2 SD | |
| Drying with copolymer (DWC) b | 1:1 | SP1/TAβCD1 DWC |
| 1:2 | SP1/TAβCD2 DWC | |
| Physical mixture (PM) c | 1:1 | SP1/TAβCD1 PM |
| 1:2 | SP1/TAβCD2 PM |
| Sample | TAβCD | SP | ||||
|---|---|---|---|---|---|---|
| Tm [°C] | ΔHm [J g−1] | Tc [°C] | ΔHc [J g−1] | Tm [°C] | ΔHm [J g−1] | |
| Pristine TAβCD | 223 | 43 | - | - | - | - |
| Processed TAβCD | 220 | 13 | 195 | 8.4 | - | - |
| Pristine SP | - | - | - | - | 190 | - |
| Processed SP | - | - | - | - | 171 | - |
| SP1/TAβCD1 SD | 219 | 7.3 | 193 | 4.7 | - | - |
| SP1/TAβCD1 PM | 219 | 40 | 153 | 3.2 | - | - |
| SP1/TAβCD2 SD | 217 | 4.2 | 195 | 4.7 | - | - |
| SP1/TAβCD2 PM | 219 | 43 | 155 | 5.6 | - | - |
| Sample | SP Equivalent Amount used for Encapsulation [mg] | Dh [nm] a,b (± S.D.) | %Intensity b | S.D. [nm] b,c | Z-average [nm] b (± S.D.) | PdI b | %EE [%] d (± S.D.) | %DL [%] d (± S.D.) |
|---|---|---|---|---|---|---|---|---|
| Processed TAβCD e | - | 312 (25) | 100 | 66 | 278 (15) | 0.23 | - | - |
| Blank NPs f | - | 65 (3) | 100 | 28 | 58 (2) | 0.16 | - | - |
| Pristine SP NPs g | 1 | 73 (3) | 100 | 26 | 65 (1) | 0.13 | 9 (1) | 0.2 (0.1) |
| 2 | 83 (2) | 100 | 29 | 74 (1) | 0.10 | 14 (1) | 0.6 (0.1) | |
| SP1/TAβCD1 DWC NPs | 1 | 72 (7) 353 (71) | 84 16 | 13 62 | 156 (29) | 0.36 | 0 (0) | 0 (0) |
| 2 | 100 (25) 4260 (226) | 86 14 | 41 369 | 183 (47) | 0.42 | 11 (1) | 0.4 (0.1) | |
| SP1/TAβCD2 DWC NPs | 1 | 105 (5) | 100 | 30 | 109 (5) | 0.27 | 7 (1) | 0.1 (0) |
| 2 | 99 (14) 5066 (546) | 77 23 | 15 560 | 69 (1) | 0.75 | 9 (1) | 0.3 (0) | |
| SP1/TAβCD1 SD NPs | 1 | 74 (1) | 100 | 22 | 67 (1) | 0.10 | 62 (1) | 1.1 (0.1) |
| 2 | 75 (2) | 100 | 21 | 69 (2) | 0.10 | 85 (1) | 2.6 (0.1) | |
| SP1/TAβCD2 SD NPs | 1 | 72 (1) 5105 (496) | 89 11 | 14 593 | 134 (42) | 0.39 | 50 (1) | 1.5 (0.1) |
| 2 | 69 (3) 312 (35) 4071 (852) | 51 40 9 | 22 43 731 | 260 (30) | 0.69 | 53 (1) | 0.9 (0.1) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuplennik, N.; Sosnik, A. Enhanced Nanoencapsulation of Sepiapterin within PEG-PCL Nanoparticles by Complexation with Triacetyl-Beta Cyclodextrin. Molecules 2019, 24, 2715. https://doi.org/10.3390/molecules24152715
Kuplennik N, Sosnik A. Enhanced Nanoencapsulation of Sepiapterin within PEG-PCL Nanoparticles by Complexation with Triacetyl-Beta Cyclodextrin. Molecules. 2019; 24(15):2715. https://doi.org/10.3390/molecules24152715
Chicago/Turabian StyleKuplennik, Nataliya, and Alejandro Sosnik. 2019. "Enhanced Nanoencapsulation of Sepiapterin within PEG-PCL Nanoparticles by Complexation with Triacetyl-Beta Cyclodextrin" Molecules 24, no. 15: 2715. https://doi.org/10.3390/molecules24152715
APA StyleKuplennik, N., & Sosnik, A. (2019). Enhanced Nanoencapsulation of Sepiapterin within PEG-PCL Nanoparticles by Complexation with Triacetyl-Beta Cyclodextrin. Molecules, 24(15), 2715. https://doi.org/10.3390/molecules24152715