
Client Applications and Server-Side Docker for Management of RNASeq and/or VariantSeq Workflows and Pipelines of the GPRO Suite

 , , , , , , , ,
, , , , , , , ,  , , ,
, , ,
Abstract
1. Introduction
2. Material and Methods
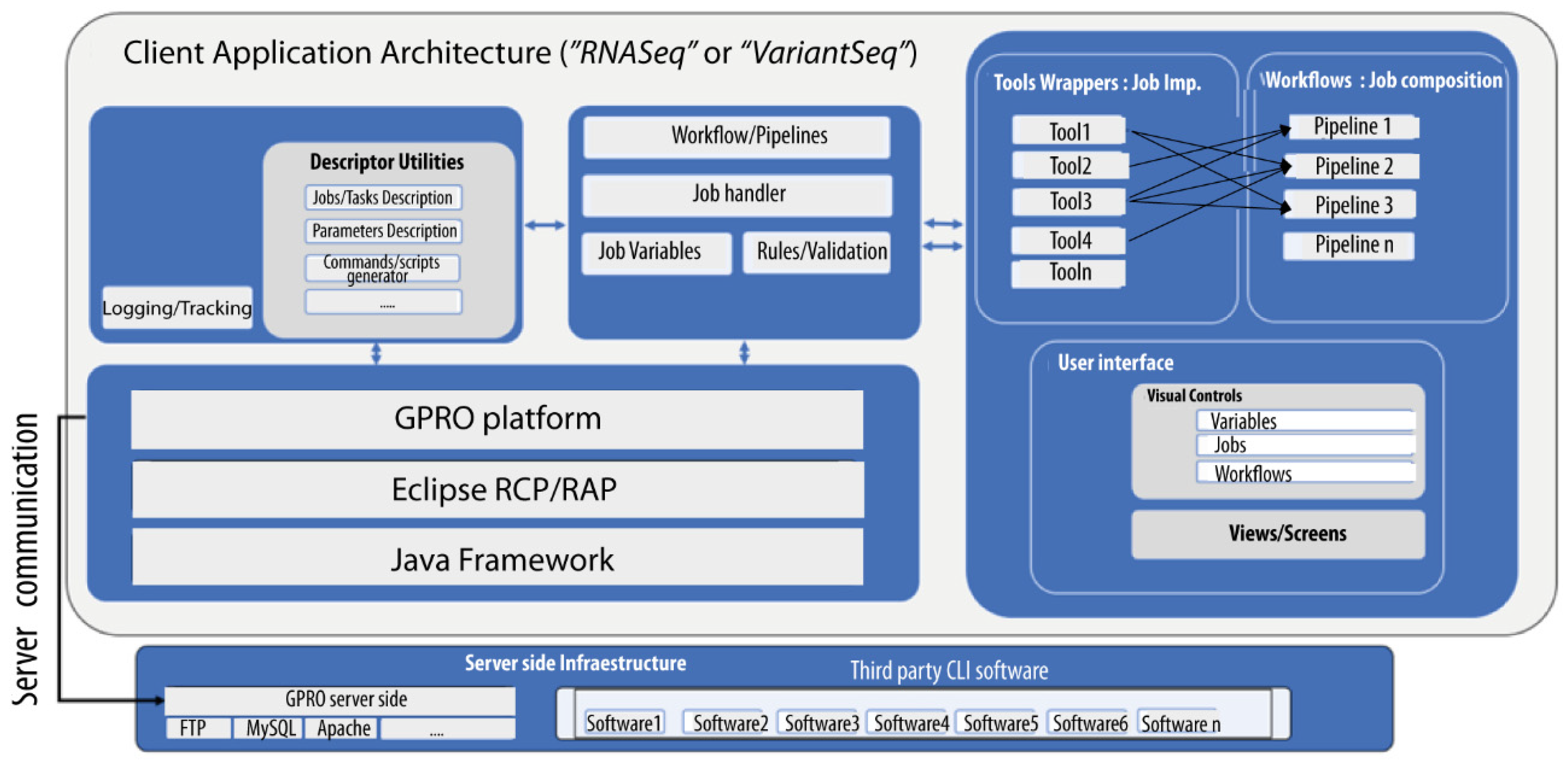
2.1. Client-Side Applications
2.2. GPRO Server-Side Platform
- Linux Operating System with at least Bash version 4;
- MySQL Server for installing databases;
- Apache HTTP server 2.2 or later;
- PHP 7 or later;
- R 3.6.0 or later and a compatible Bioconductor with R version;
- Perl 5;
- Python 2.7;
- Third-party CLI software (see Table 1 for details);
- An API for communicating between client applications and GSS.
2.3. Virtual Chatbot Assistant and Expert System
- Questions and answers database. This database identifies and stores key terms and serves as an index of answers to different questions.
- CLI tools dependency database. This database stores information on the type of input that each CLI tool receives and the output that it generates, as well as information on different parameters and customization options.
- Contextual database. This database provides a graphical representation to all pipelines/workflows and the programs implemented in each protocol.
- Key terms database. This is a database of generic questions about different protocols or programs.
- Log files database. This is a database that stores the information reported by the log files generated by the CLI software dependencies.
- Inference engine: This handles the users’ request by processing the logs and the tracking information sent by the job tracking panel of client applications, with the objective of extracting key features and errors information that can be used to query the solutions database.
- Proven facts database: This database contains the rules managed by the inference engine for recommendations of how to fix problems and errors from failed analyses.
- Administration panel: This is a website provided for administration and management of the expert system when applying rules or adjusting aspects, such as adding new task descriptors, editing databases, managing actions/recommendation templates, etc. The administration panel is only accessible by experts from our side or by users interested in contributing to the training of this tool.
- Client interface: This is the interface implemented in the pipeline jobs panel of the client applications (“RNASeq” and “VariantSeq”) to manage the interaction with the expert system engine.
- API: The API allows the interface to accept requests from the client applications and enables client applications to track and fetch the actions/recommendations proposed by the expert system.
- Online Web interface available at https://gpro.biotechvana.com/genie (accessed on 17 January 2023). This webpage includes a dialog where users can ask questions and the chatbot will respond using a graphical summarization of the different protocols of each GPRO application including “RNASeq” and “VariantSeq”.
- An interactive user interface implemented in each client application to query the chatbot directly from the application.
3. Results
3.1. General Overview
3.2. User Interface
- “FTP Browser”. This is a File Transfer Protocol (FTP) to provide users access to the GSS and to transfer files/folders from the user PC to the GSS, or vice versa.
- “Working space”. This is the framework space from which the GUIs manage the CLI tools hosted at the GSS.
- “Top Menu”. This is the main menu for each application and is located at the top of the interface. All tools and tasks are organized into different tabs as detailed below:
- “Directory”. This tab is for users to select and set the main directory for exchanging material with the GSS using the FTP browser.
- “Transcripts/Variant Protocols”. This tab provides access to the modes of computation and protocols of each application. By clicking on this tab, the user can choose between two computational modes: step-by-step or pipeline. When selecting the step-by-step mode, a “Task Menu” appears in the working space to provide access to the set of GUIs for the distinct CLI tools and/or commands implemented in the step-by-step workflow for each application. When choosing the pipeline mode, the user accesses the pipeline manager of each application.
- “Pipeline Jobs”. This tab allows the user to track the status of all jobs executed in the GSS or to obtain recommendations from the GENIE’s expert system to troubleshoot computational issues in failed analyses.
- “Preferences”. This tab allows the user to configure and activate the connection settings between the client application and the GSS.
- “Help”. This tab provides access to the user manual for each application and to the summary panel of GENIE’s chatbot.
3.3. Protocols
3.4. Usage and Tutorials
3.5. Smart Support System
- “Select in FTP Explorer”. This opens/views the output folder of the selected record.
- “View Report”. This visualizes the log file of the selected record.
- “Refresh”. This manually refreshes the history records.
- “Delete”. This deletes the selected record from the history (this only deletes the record and cached log and track information. The original files with the results are kept on the server and can only be deleted directly from the server or from the FTP Browser).
- “Restart”. This runs the analysis again with the same input data options and parameters used in the previous analysis.
- “Edit & Restart”. This runs the analysis again but allows the user to edit or modify any input data, option, or parameter from the previously used CLI tool.
- “Resolve”. This accesses the interface of the expert system, allowing the provision of recommendations on controlled actions as defined by the expert system.
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
- local_path="/path/to/local_home"
- GPRO_USER="myUserName"
- GPRO_USER_PASS="myUserNamePass"
- docker run -d -p 80:80 -p 20-22:20-22 -p 65500-65515:65500-65515 -v/path/to/local_home:/home/gpro_user biotechvana/gpro
Acknowledgments
Conflicts of Interest
References
- Consortium OPATHY; Gabaldon, T. Recent trends in molecular diagnostics of yeast infections: From PCR to NGS. FEMS Microbiol. Rev. 2019, 43, 517–547. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szczesniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Geraci, F.; Saha, I.; Bianchini, M. Editorial: RNA-Seq Analysis: Methods, Applications and Challenges. Front. Genet. 2020, 11, 220. [Google Scholar] [CrossRef] [PubMed]
- Zverinova, S.; Guryev, V. Variant calling: Considerations, practices, and developments. Hum. Mutat. 2022, 43, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C. Best practices for variant calling in clinical sequencing. Genome Med. 2020, 12, 91. [Google Scholar] [CrossRef]
- Sandmann, S.; de Graaf, A.O.; Karimi, M.; van der Reijden, B.A.; Hellstrom-Lindberg, E.; Jansen, J.H.; Dugas, M. Evaluating Variant Calling Tools for Non-Matched Next-Generation Sequencing Data. Sci. Rep. 2017, 7, 43169. [Google Scholar] [CrossRef]
- CLC OmicSoft, QIAGEN. Available online: https://digitalinsights.qiagen.com (accessed on 17 January 2023).
- Geneious, Dotmatics. Available online: http://www.geneious.com (accessed on 17 January 2023).
- Partek Genomic Suite Version 7, Partek Inc. Available online: https://www.partek.com/partek-genomics-suite (accessed on 17 January 2023).
- OmicsBox, Biobam SL. Available online: https://www.biobam.com/omicsbox (accessed on 17 January 2023).
- Okonechnikov, K.; Golosova, O.; Fursov, M.; The UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Kallio, M.A.; Tuimala, J.T.; Hupponen, T.; Klemela, P.; Gentile, M.; Scheinin, I.; Koski, M.; Kaki, J.; Korpelainen, E.I. Chipster: User-friendly analysis software for microarray and other high-throughput data. BMC Genom. 2011, 12, 507. [Google Scholar] [CrossRef]
- Golosova, O.; Henderson, R.; Vaskin, Y.; Gabrielian, A.; Grekhov, G.; Nagarajan, V.; Oler, A.J.; Quinones, M.; Hurt, D.; Fursov, M.; et al. Unipro UGENE NGS pipelines and components for variant calling, RNA-seq and ChIP-seq data analyses. PeerJ 2014, 2, e644. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R. RStudio; PBC: Boston, MA, USA, 2020; Available online: http://www.rstudio.com (accessed on 17 January 2023).
- Chang, W.; Cheng, J.; Allaire, J.; Xie, Y.; McPherson, J. Shiny: Web Application Framework for R. Available online: https://CRAN.R-project.org/package=shiny (accessed on 17 January 2023).
- Gruning, B.; Dale, R.; Sjodin, A.; Chapman, B.A.; Rowe, J.; Tomkins-Tinch, C.H.; Valieris, R.; Koster, J.; Bioconda, T. Bioconda: Sustainable and comprehensive software distribution for the life sciences. Nat. Methods 2018, 15, 475–476. [Google Scholar] [CrossRef]
- The Galaxy Community. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef] [PubMed]
- Merkel, D. Docker: Lightweight Linux containers for consistent development and deployment. Linux J. 2014, 2014, 2. [Google Scholar]
- Futami, R.; Muñoz-Pomer, L.; Viu, J.M.; Dominguez-Escriba, L.; Covelli, L.; Bernet, G.P.; Sempere, J.M.; Moya, A.; Llorens, C. GPRO: The professional tool for annotation, management and functional analysis of omic sequences and databases. Biotechvana Bioinform. 2011, SOFT3. Available online: https://www.researchgate.net/profile/Laura-Covelli-2/publication/235719764_GPRO_the_professional_tool_for_management_functional_analysis_and_annotation_of_omic_sequences_and_databases/links/0fcfd512dcbaac321b000000/GPRO-the-professional-tool-for-management-functional-analysis-and-annotation-of-omic-sequences-and-databases.pdf (accessed on 17 January 2023).
- Muñoz-Pomer, A.; Futami, R.; Covelli, L.; Dominguez-Escriba, L.; Bernet, G.P.; Sempere, J.M.; Moya, A.; Llorens, C. TIME a sequence editor for the molecular analysis of DNA and protein sequence samples. Biotechvana Bioinform. 2011, SOFT2. Available online: http://bioinformatics.biotechvana.com/article_files/34/pdf/TIME.pdf (accessed on 17 January 2023).
- Hafez, A.; Futami, R.; Arastehfar, A.; Daneshnia, F.; Miguel, A.; Roig, F.J.; Soriano, B.; Perez-Sanchez, J.; Boekhout, T.; Gabaldon, T.; et al. SeqEditor: An application for primer design and sequence analysis with or without GTF/GFF files. Bioinformatics 2020, 37, 1610–1612. [Google Scholar] [CrossRef]
- desRivieres, J.; Wiegand, I. Eclipse: A platform for integrating development tools. IBM Syst. J. 2004, 43, 371–383. [Google Scholar] [CrossRef]
- Krasner, G.E.; Pope, S.T. A cookbook approach to using MVC. JOOP 1988, 1, 26–49. [Google Scholar]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2016. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 17 January 2023).
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Hannon Lab. FASTX-Toolkit: FASTQ/a Short-Reads Pre-Processing Tools. 2016. Available online: http://hannonlab.cshl.edu/fastx_toolkit (accessed on 17 January 2023).
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.M.; Oshlack, A. Corset: Enabling differential gene expression analysis for de novo assembled transcriptomes. Genome Biol. 2014, 15, 410. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Wysoker, A.; Tibbetts, K.; Fennell, T. Picard. 2011. Available online: https://sourceforge.net/projects/picard (accessed on 17 January 2023).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Goff, L.; Trapnell, C.; Kelley, D. CummeRbund: Analysis, Exploration, Manipulation, and Visualization of Cufflinks High-Throughput Sequencing Data, R package version 2.33.0 Bioconductor; 2020; Available online: https://bioconductor.org/packages/release/bioc/html/cummeRbund.html (accessed on 17 January 2023).
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for RNA-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Bocklisch, T.; Faulkner, J.; Pawlowski, N.; Nichol, A. Rasa: Open source language understanding and dialogue management. arXiv 2017, arXiv:1712.05181. [Google Scholar] [CrossRef]
- Cer, D.; Yang, Y.; Kong, S.-y.; Hua, N.; Limtiaco, N.; John, R.S.; Constant, N.; Guajardo-Cespedes, M.; Yuan, S.; Tar, C.; et al. Universal Sentence Encoder. arXiv 2018. [Google Scholar] [CrossRef]
- Parnell, L.D.; Lindenbaum, P.; Shameer, K.; Dall’Olio, G.M.; Swan, D.C.; Jensen, L.J.; Cockell, S.J.; Pedersen, B.S.; Mangan, M.E.; Miller, C.A.; et al. BioStar: An online question & answer resource for the bioinformatics community. PLoS Comput. Biol. 2011, 7, e1002216. [Google Scholar] [CrossRef]
- Li, J.W.; Schmieder, R.; Ward, R.M.; Delenick, J.; Olivares, E.C.; Mittelman, D. SEQanswers: An open access community for collaboratively decoding genomes. Bioinformatics 2012, 28, 1272–1273. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Agarwala, R.; Bolton, E.E.; Brister, J.R.; Canese, K.; Clark, K.; Connor, R.; Fiorini, N.; Funk, K.; Hefferon, T.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2019, 47, D23–D28. [Google Scholar] [CrossRef] [PubMed]
- Raschka, S. Python Machine Learning; Packt Publishing Ltd.: Birmingham, UK, 2015. [Google Scholar]
- Gollapudi, S. Practical Machine Learning; Packt Publishing Ltd.: Birmingham, UK, 2016. [Google Scholar]
- Pérez-Sánchez, J.; Naya-Català, F.; Soriano, B.; Piazzon, M.C.; Hafez, A.; Gabaldón, T.; Llorens, C.; Sitjà-Bobadilla, A.; Calduch-Giner, J.A. Genome Sequencing and Transcriptome Analysis Reveal Recent Species-Specific Gene Duplications in the Plastic Gilthead Sea Bream (Sparus aurata). Front. Mar. Sci. 2019, 6, Article 760. [Google Scholar] [CrossRef]
- Trilla-Fuertes, L.; Ghanem, I.; Maurel, J.; G-Pastrián, L.; Mendiola, M.; Pena, C.; Lopez-Vacas, R.; Prado-Vazquez, G.; Lopez-Camacho, E.; Zapater-Moros, A.; et al. Comprehensive Characterization of the Mutational Landscape in Localized Anal Squamous Cell Carcinoma. Transl. Oncol. 2020, 13, 100778. [Google Scholar] [CrossRef] [PubMed]
- Perez-Sanchez, R.; Carnero-Moran, A.; Soriano, B.; Llorens, C.; Oleaga, A. RNA-seq analysis and gene expression dynamics in the salivary glands of the argasid tick Ornithodoros erraticus along the trophogonic cycle. Parasit Vectors 2021, 14, 170. [Google Scholar] [CrossRef]
- Oleaga, A.; Soriano, B.; Llorens, C.; Perez-Sanchez, R. Sialotranscriptomics of the argasid tick Ornithodoros moubata along the trophogonic cycle. PLoS Negl. Trop. Dis. 2021, 15, e0009105. [Google Scholar] [CrossRef]
- Llorens, C.; Soriano, B.; Trilla-Fuertes, L.; Bagan, L.; Ramos-Ruiz, R.; Gamez-Pozo, A.; Pena, C.; Bagan, J.V. Immune expression profile identification in a group of proliferative verrucous leukoplakia patients: A pre-cancer niche for oral squamous cell carcinoma development. Clin. Oral Investig. 2021, 25, 2645–2657. [Google Scholar] [CrossRef]
- Trilla-Fuertes, L.; Ghanem, I.; Gamez-Pozo, A.; Maurel, J.; G-Pastrián, L.; Mendiola, M.; Pena, C.; Lopez-Vacas, R.; Prado-Vazquez, G.; Lopez-Camacho, E.; et al. Genetic Profile and Functional Proteomics of Anal Squamous Cell Carcinoma: Proposal for a Molecular Classification. Mol. Cell. Proteom. 2020, 19, 690–700. [Google Scholar] [CrossRef]
- Trilla-Fuertes, L.; Gamez-Pozo, A.; Maurel, J.; Garcia-Carbonero, R.; Capdevila, J.; G-Pastrián, L.; Mendiola, M.; Pena, C.; Lopez-Vacas, R.; Cuatrecasas, M.; et al. Description of the genetic variants identified in a cohort of patients diagnosed with localized anal squamous cell carcinoma and treated with panitumumab. Sci. Rep. 2021, 11, 7402. [Google Scholar] [CrossRef]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587.e29. [Google Scholar] [CrossRef] [PubMed]
- Hovhannisyan, H.; Hafez, A.; Llorens, C.; Gabaldon, T. CROSSMAPPER: Estimating cross-mapping rates and optimizing experimental design in multi-species sequencing studies. Bioinformatics 2020, 36, 925–927. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CLI Third-Party Software | RNASeq | VariantSeq | |
|---|---|---|---|
| Quality Analysis and Preprocessing | FastQC v0.11.5 [24] | √ | √ |
| FastqMidCleaner 1.0.0 | √ | √ | |
| Cutadapt 1.18 [25] | √ | √ | |
| PRINSEQ-lite 0.20.4 [26] | √ | √ | |
| Trimmomatic 0.36 [27] | √ | √ | |
| FastxToolkit 0.0.13 [28] | √ | √ | |
| FastqCollapser 1.0.0 | √ | √ | |
| FastqIntersect 1.0.0 | √ | √ | |
| Mapping of Reference Genome or Transcriptome | TopHat v2.1.1 [29] | √ | √ |
| Hisat2 2.2.1 [30] | √ | √ | |
| Bowtie2 2.2.9 [31] | √ | √ | |
| BWA 0.7.15-r1140 [32] | √ | √ | |
| STAR 2.7.0f [33] | X | √ | |
| Quantification | Corset 1.06 [34] | √ | X |
| Htseq 0.12.4 [35] | √ | X | |
| Post Processing | Bed Tools v2.29.2 [36] | X | √ |
| GATK v4.1.2.0 [37,38] | X | √ | |
| Picard tools 2.19.0 [39] | X | √ | |
| SAMtools 1.8 [40] | X | √ | |
| Transcriptome Assembly | Cufflinks v2.2.1 [41] | √ | X |
| Differential Expression | DESeq 2.1.28 [42] | √ | X |
| EdgeR 3.30.3 [43] | √ | X | |
| Cuffdiff v2.2.1 [41] | √ | X | |
| CummeRbund 2.30.0 [44] | √ | X | |
| Enrichment Analysis | GOseq 1.40.0 [45] | √ | X |
| Training Sets | GATK v4.1.2.0] [37,38,46] | X | √ |
| Variant Calling | GATK) v4.1.2.0 [37,38,46] | X | √ |
| VarScan2 v2.4.3 [47] | X | √ | |
| Variant Filtering | GATK v4.1.2.0 [37,38] | X | √ |
| Annotation of Variant Effects | Variant Effect Predictor 105.0 [48] | X | √ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hafez, A.I.; Soriano, B.; Elsayed, A.A.; Futami, R.; Ceprian, R.; Ramos-Ruiz, R.; Martinez, G.; Roig, F.J.; Torres-Font, M.A.; Naya-Catala, F.; et al. Client Applications and Server-Side Docker for Management of RNASeq and/or VariantSeq Workflows and Pipelines of the GPRO Suite. Genes 2023, 14, 267. https://doi.org/10.3390/genes14020267
Hafez AI, Soriano B, Elsayed AA, Futami R, Ceprian R, Ramos-Ruiz R, Martinez G, Roig FJ, Torres-Font MA, Naya-Catala F, et al. Client Applications and Server-Side Docker for Management of RNASeq and/or VariantSeq Workflows and Pipelines of the GPRO Suite. Genes. 2023; 14(2):267. https://doi.org/10.3390/genes14020267
Chicago/Turabian StyleHafez, Ahmed Ibrahem, Beatriz Soriano, Aya Allah Elsayed, Ricardo Futami, Raquel Ceprian, Ricardo Ramos-Ruiz, Genis Martinez, Francisco Jose Roig, Miguel Angel Torres-Font, Fernando Naya-Catala, and et al. 2023. "Client Applications and Server-Side Docker for Management of RNASeq and/or VariantSeq Workflows and Pipelines of the GPRO Suite" Genes 14, no. 2: 267. https://doi.org/10.3390/genes14020267
APA StyleHafez, A. I., Soriano, B., Elsayed, A. A., Futami, R., Ceprian, R., Ramos-Ruiz, R., Martinez, G., Roig, F. J., Torres-Font, M. A., Naya-Catala, F., Calduch-Giner, J. A., Trilla-Fuertes, L., Gamez-Pozo, A., Arnau, V., Sempere-Luna, J. M., Perez-Sanchez, J., Gabaldon, T., & Llorens, C. (2023). Client Applications and Server-Side Docker for Management of RNASeq and/or VariantSeq Workflows and Pipelines of the GPRO Suite. Genes, 14(2), 267. https://doi.org/10.3390/genes14020267