- Proceeding Paper

Synthesis of Quinoxalin-2(1H)-One Derivatives via the Novel Ugi 4CR, Followed by Palladium-Catalyzed Cyclization Process
- Murali Venkata Basavanag Unnamatla,
- Rocío Gámez Montaño and
- Laurent El Kaïm
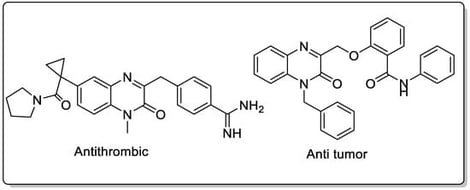
A novel synthetic methodology has been developed for the efficient preparation of Quinoxalin-2(1H)-one derivatives through a strategically designed combination of Ugi four-component reaction (4CR) followed by metal-catalyzed cross-coupling reactions. This innovative approach demonstrates exceptional versatility in generating molecular diversity by systematically modulating the reactivity profiles of the initially formed Ugi adducts, thereby enabling access to a broad library of structurally diverse quinoxalinone compounds. The synthetic protocol consistently delivered the desired final products in good to moderate yields, demonstrating the reliability and practicality of this methodology for preparative applications. Comprehensive structural characterization of all synthesized derivatives was accomplished through a combination of advanced spectroscopic techniques, including proton nuclear magnetic resonance (1H NMR), carbon-13 nuclear magnetic resonance (13C NMR), and high-resolution mass spectrometry, ensuring unambiguous confirmation of product identities and purities. The methodological framework employs 2-iodoaniline and 2-chloroacetic acid as the fixed amine and carboxylic acid components, respectively, in the multicomponent Ugi reaction, providing a consistent synthetic foundation. Subsequently, the resulting Ugi adducts undergo palladium-catalyzed cross-coupling transformations under optimized reaction conditions, facilitating the crucial cyclization and functionalization steps necessary for the formation of the target Quinoxalin-2(1H)-one derivatives. This sequential approach represents a significant advancement in heterocyclic synthesis methodology.
11 November 2025