



Cells 2023, 12(11), 1550; https://doi.org/10.3390/cells12111550 - 5 Jun 2023
Cited by 5 | Viewed by 2895
Abstract
►
Show Figures
Dual localization or dual targeting refers to the phenomenon by which identical, or almost identical, proteins are localized to two (or more) separate compartments of the cell. From previous work in the field, we had estimated that a third of the mitochondrial proteome
[...] Read more.
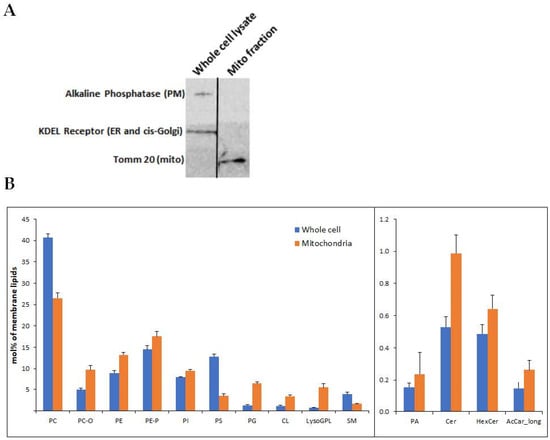
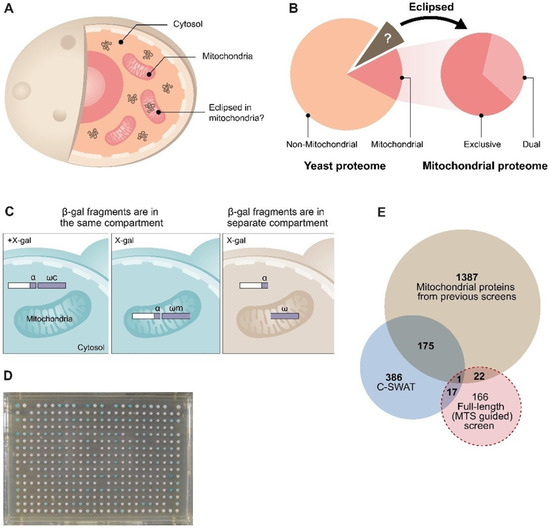
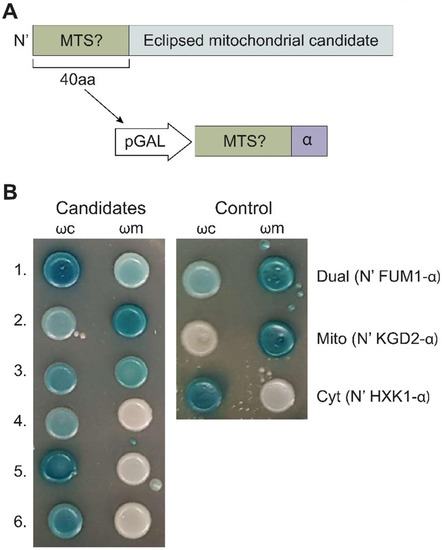
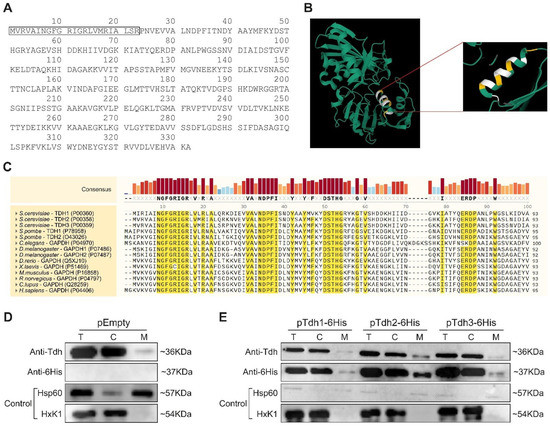
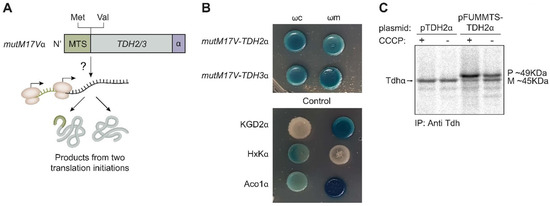
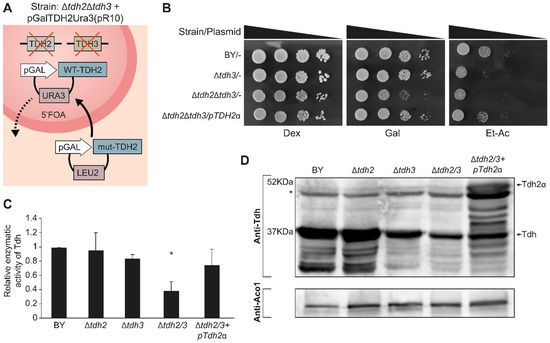
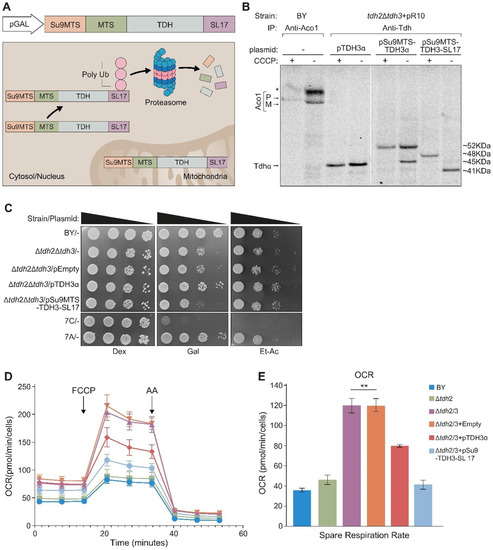
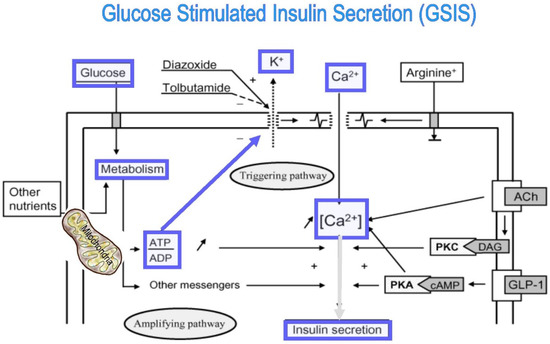
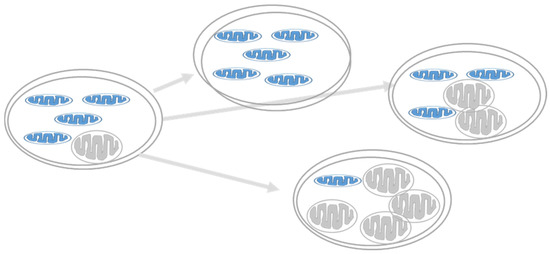
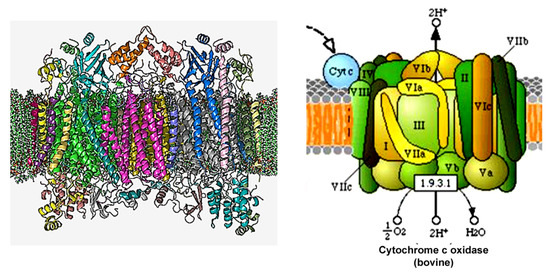
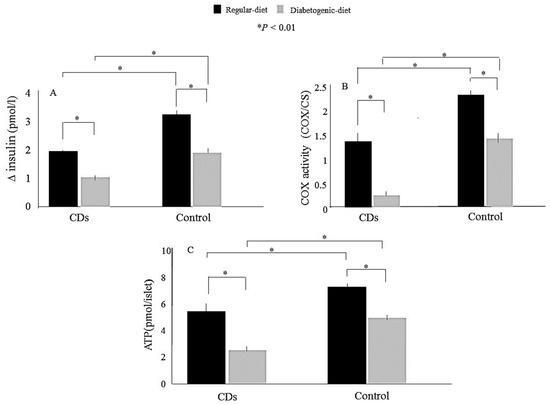
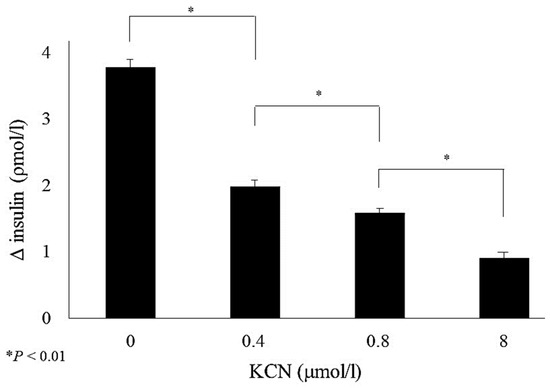
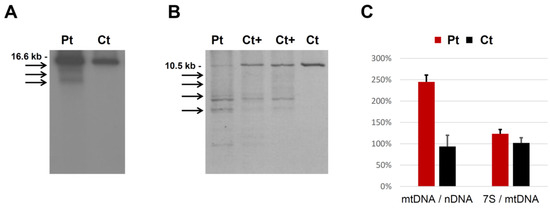
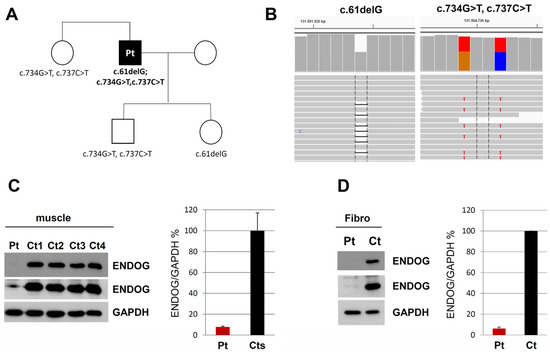
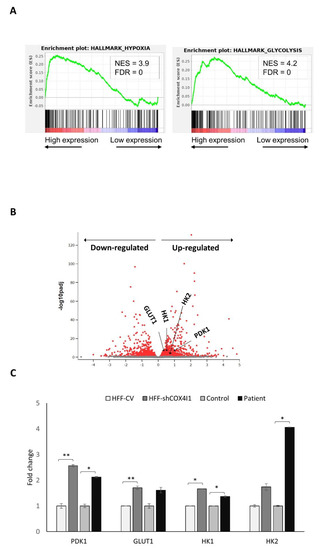
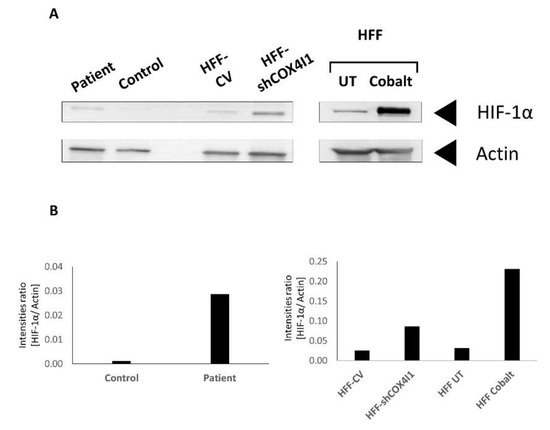
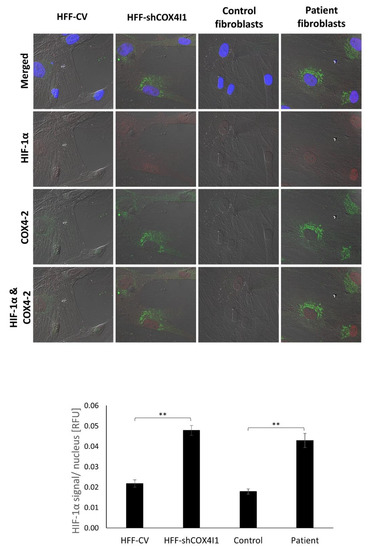
Dual localization or dual targeting refers to the phenomenon by which identical, or almost identical, proteins are localized to two (or more) separate compartments of the cell. From previous work in the field, we had estimated that a third of the mitochondrial proteome is dual-targeted to extra-mitochondrial locations and suggested that this abundant dual targeting presents an evolutionary advantage. Here, we set out to study how many additional proteins whose main activity is outside mitochondria are also localized, albeit at low levels, to mitochondria (eclipsed). To do this, we employed two complementary approaches utilizing the α-complementation assay in yeast to uncover the extent of such an eclipsed distribution: one systematic and unbiased and the other based on mitochondrial targeting signal (MTS) predictions. Using these approaches, we suggest 280 new eclipsed distributed protein candidates. Interestingly, these proteins are enriched for distinctive properties compared to their exclusively mitochondrial-targeted counterparts. We focus on one unexpected eclipsed protein family of the Triose-phosphate DeHydrogenases (TDH) and prove that, indeed, their eclipsed distribution in mitochondria is important for mitochondrial activity. Our work provides a paradigm of deliberate eclipsed mitochondrial localization, targeting and function, and should expand our understanding of mitochondrial function in health and disease.
Full article

Graphical abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}