
The Energy Status of Astrocytes Is the Achilles’ Heel of eIF2B-Leukodystrophy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Primary Astrocytes Isolation and Usage
2.3. Protein Extraction and Immunoblot Analysis
2.4. RNA Extraction and Real Time Quantitative PCR (RT-qPCR) (Detailed Version)
2.5. Quantification of PGC1α Nuclear Localization
2.6. Mitochondrial DNA (mtDNA) Quantification
2.7. Oxygen Consumption and Glycolytic Proton Efflux Rates
2.8. ATP Measurements
2.9. Assessment of Cytoplasmic ROS and Cell Death by Flow Cytometry
2.10. Wound-Scratch Assay for Migration Analysis
2.11. Statistics
2.12. Compounds Used (Stocks)
3. Results
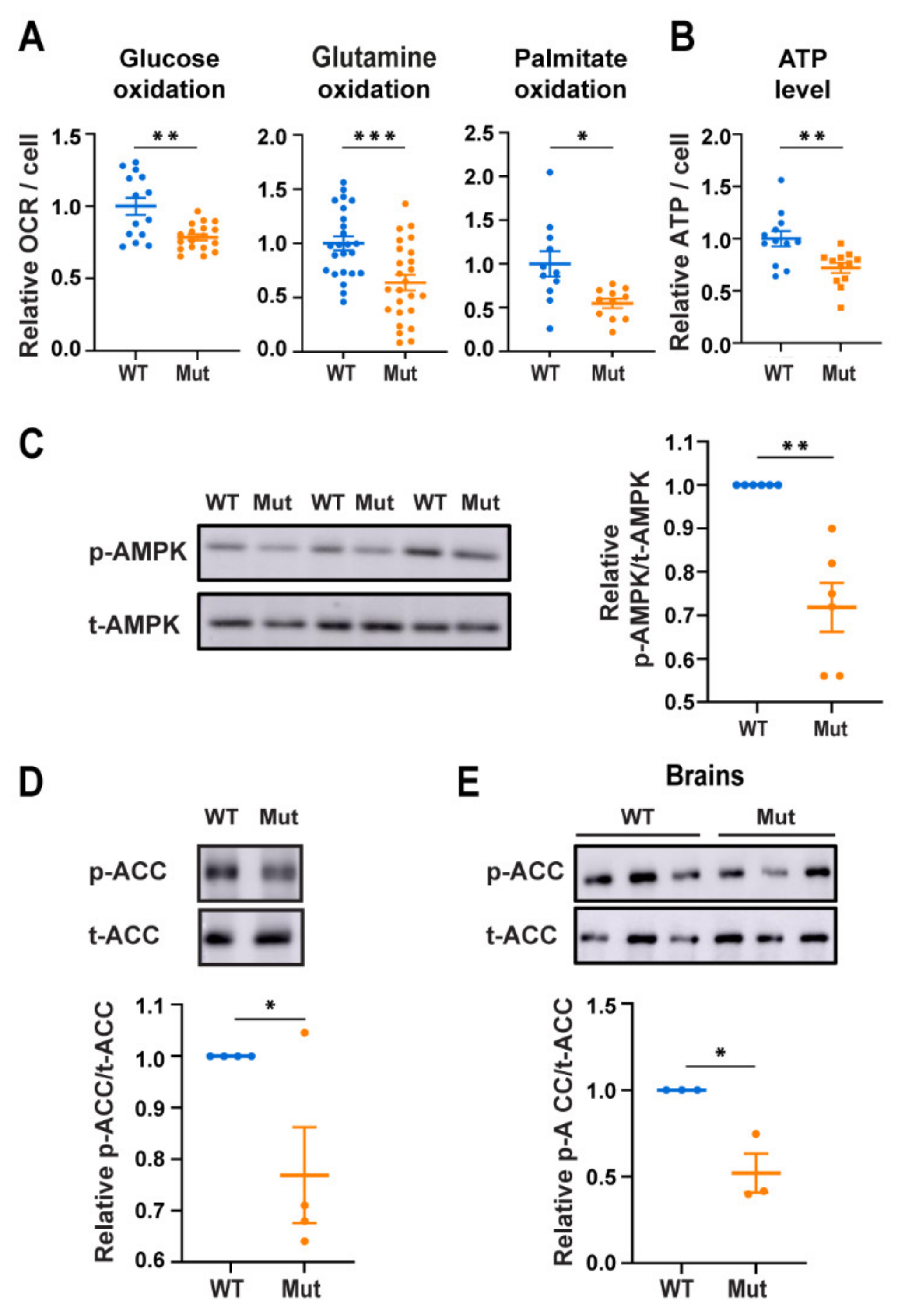
3.1. Eif2b5R132H/R132H (Mut) Astrocytes Exhibit Low AMPK Activity Despite Low Energy Status
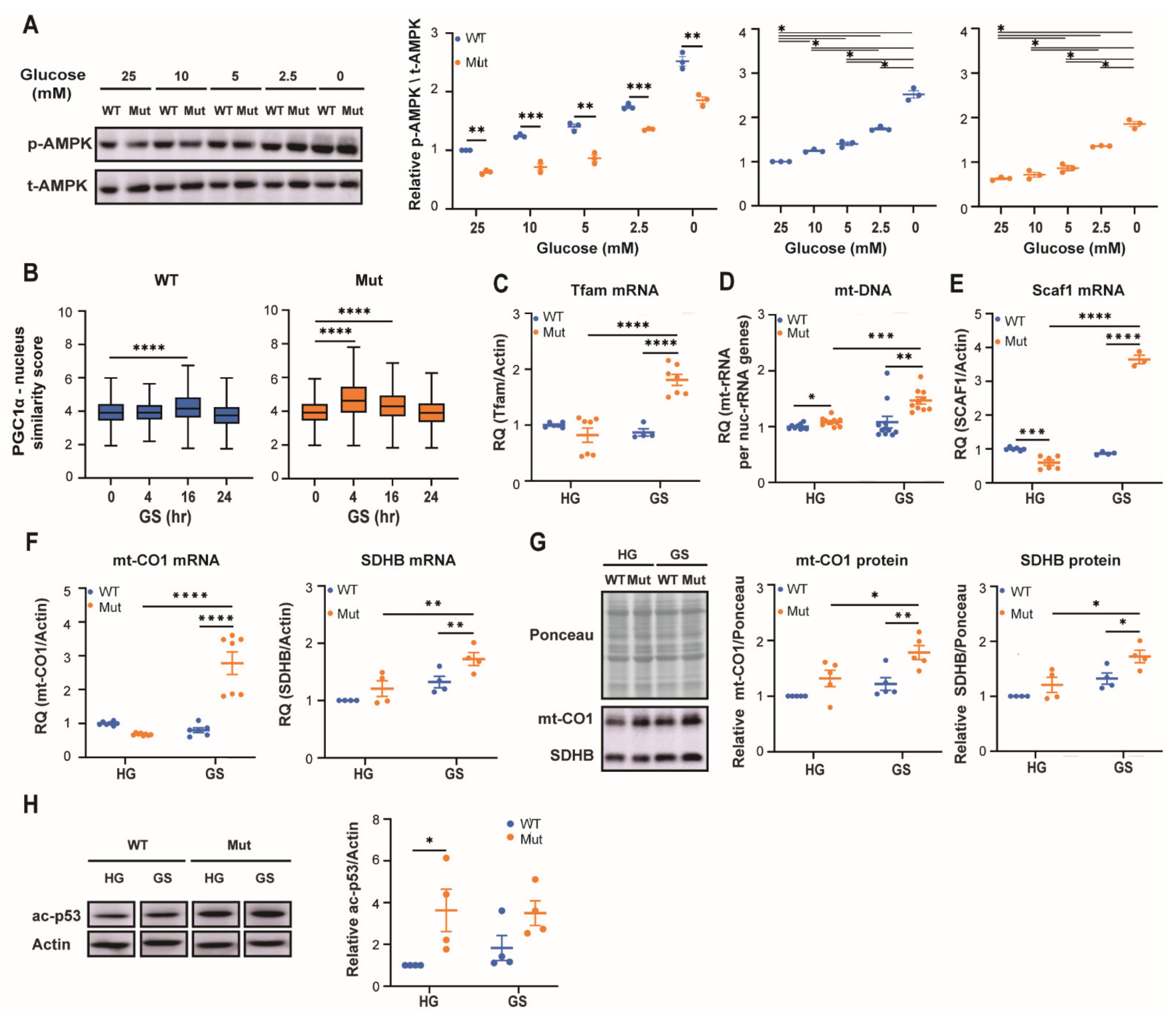
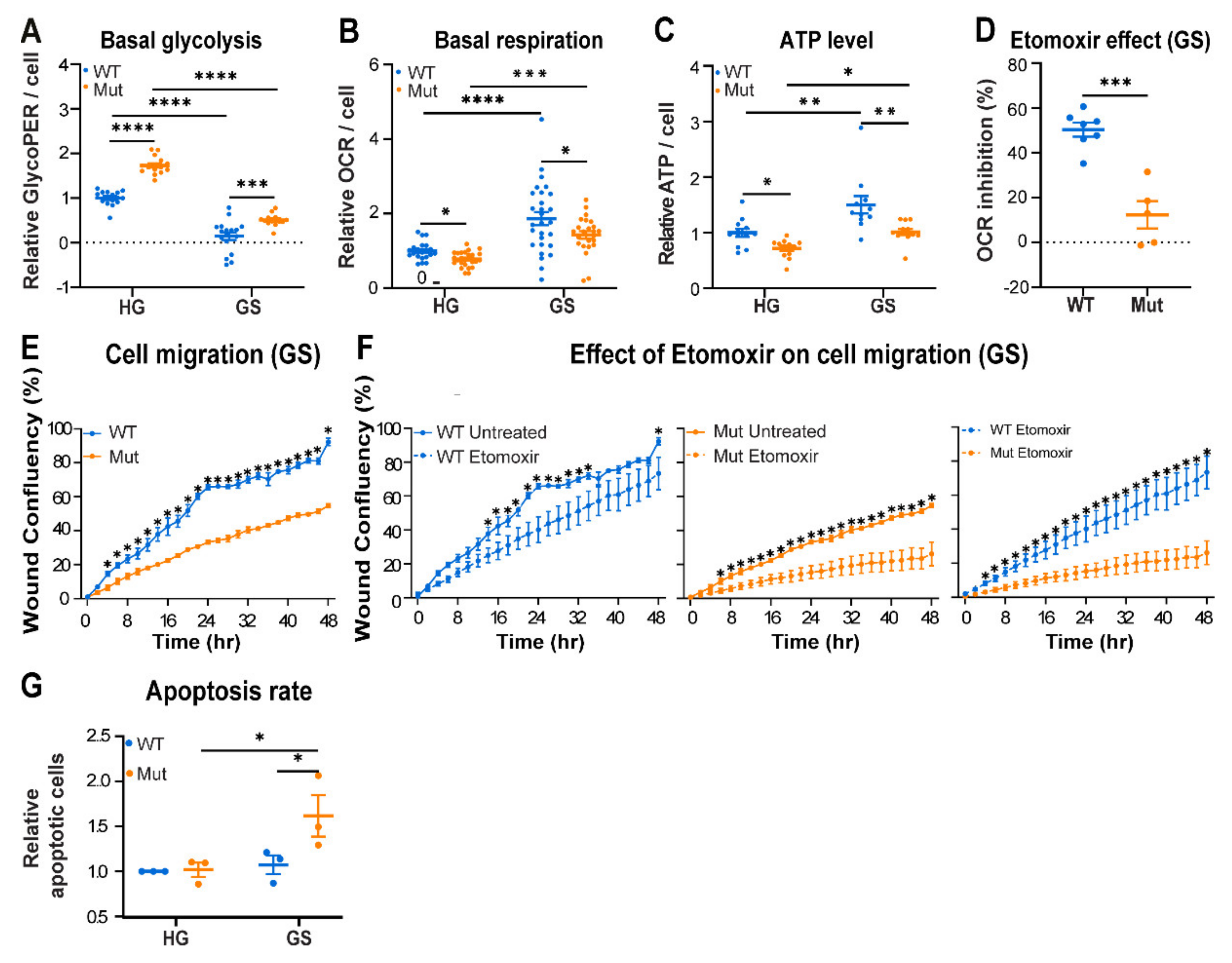
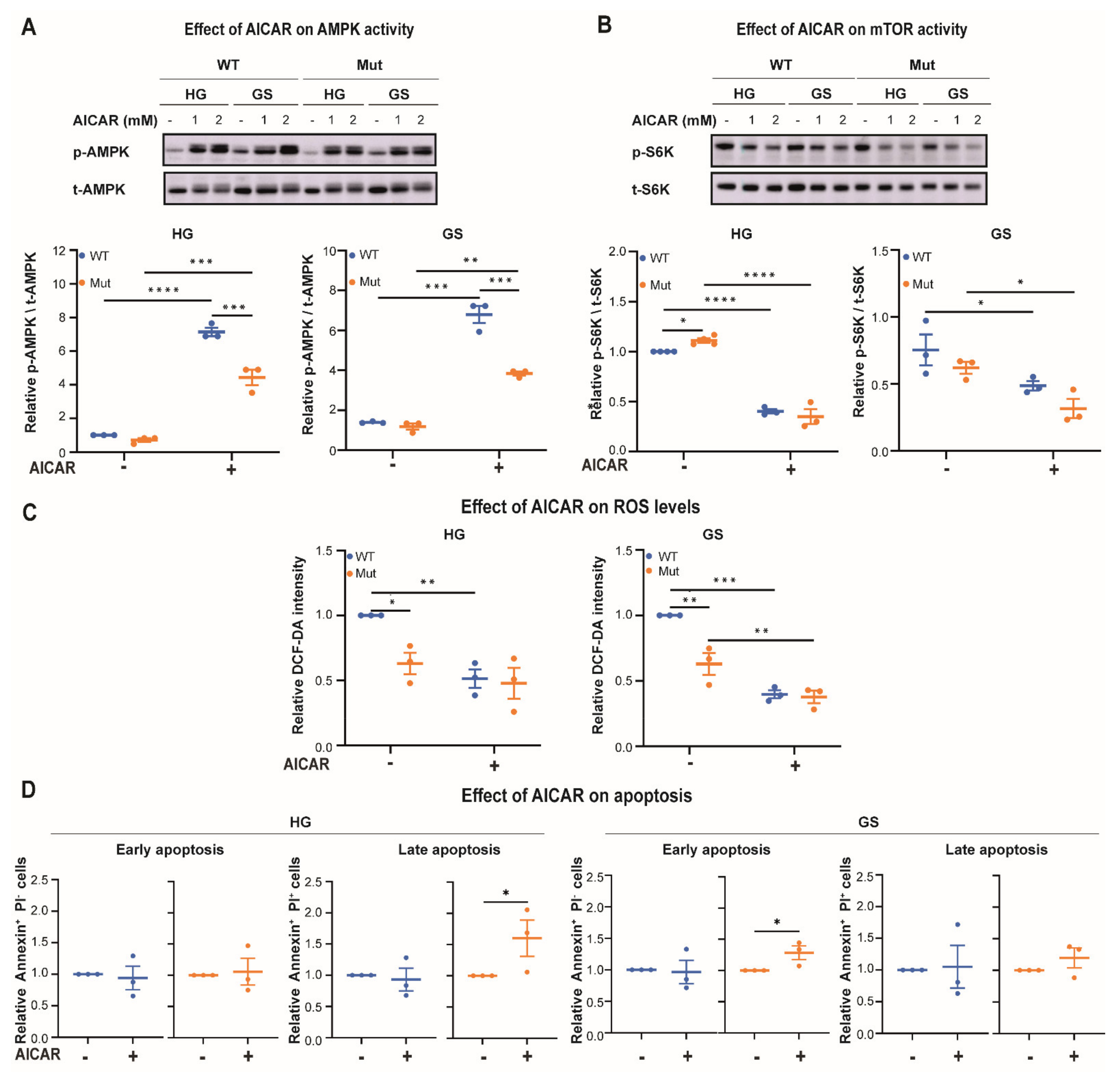
3.2. Eif2b5R132H/R132H (Mut) Astrocytes Exhibit Limited Adaptation to Energy Stress
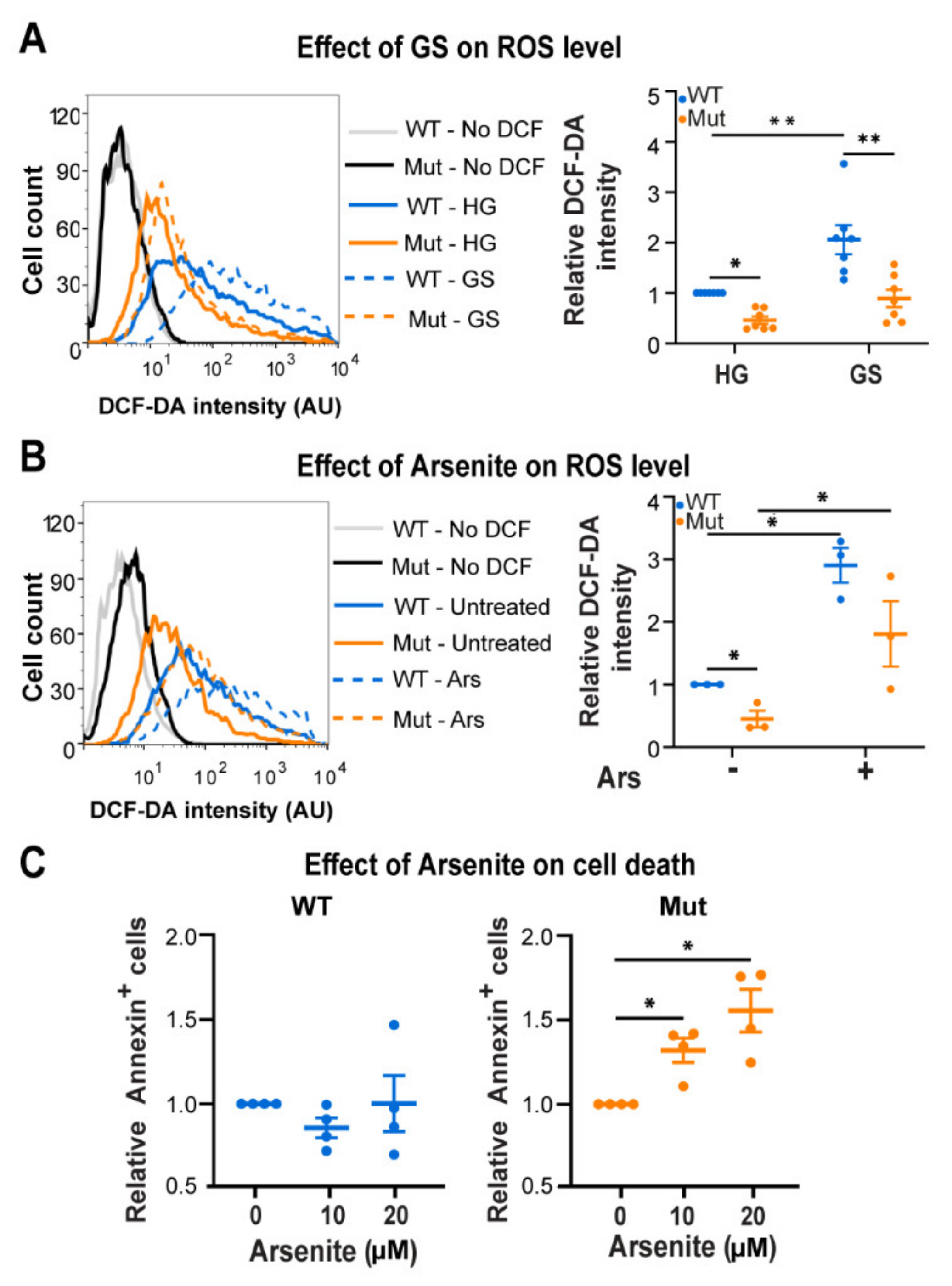
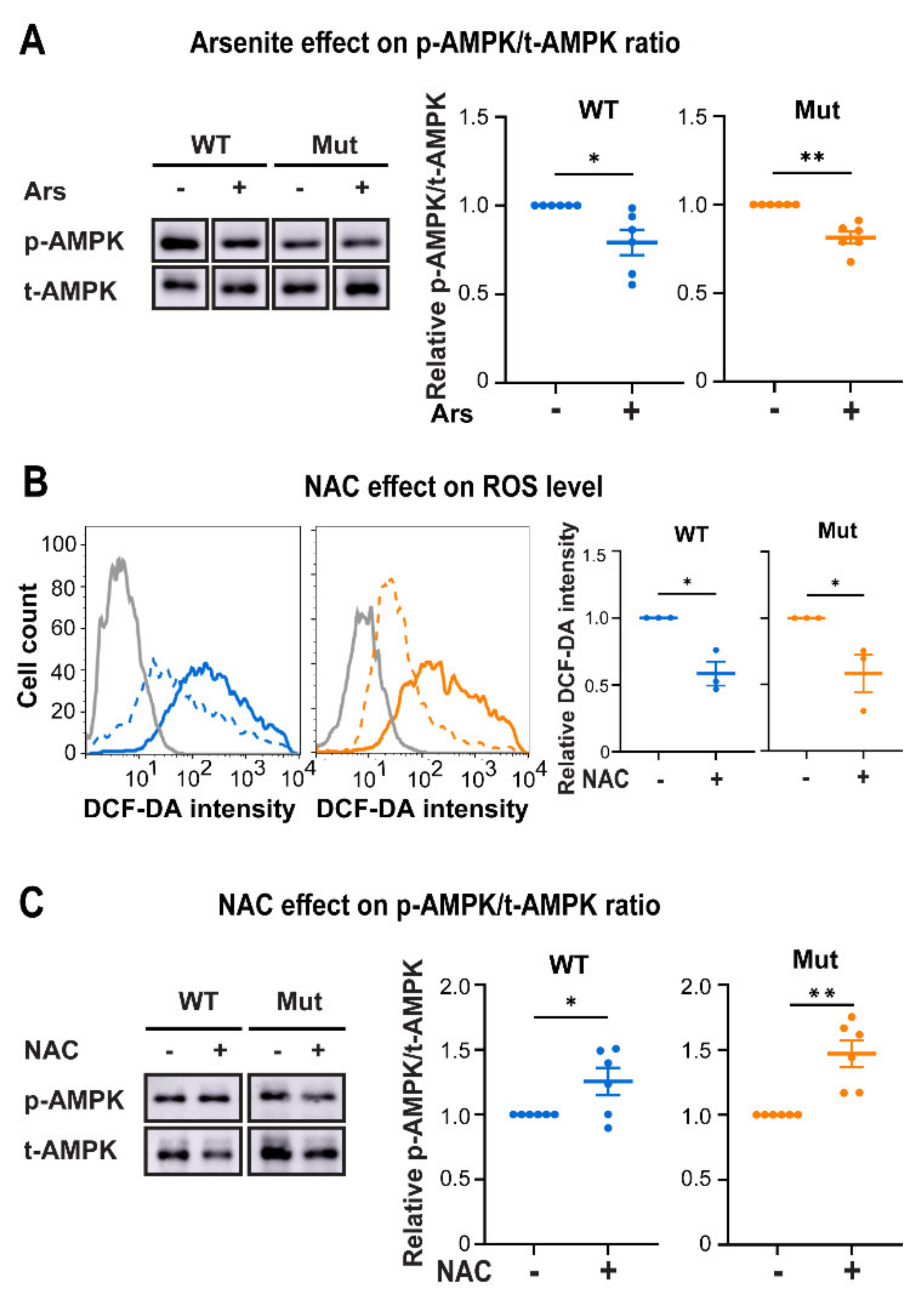
3.3. Eif2b5R132H/R132H (Mut) Astrocytes Are Hypersensitive to ROS
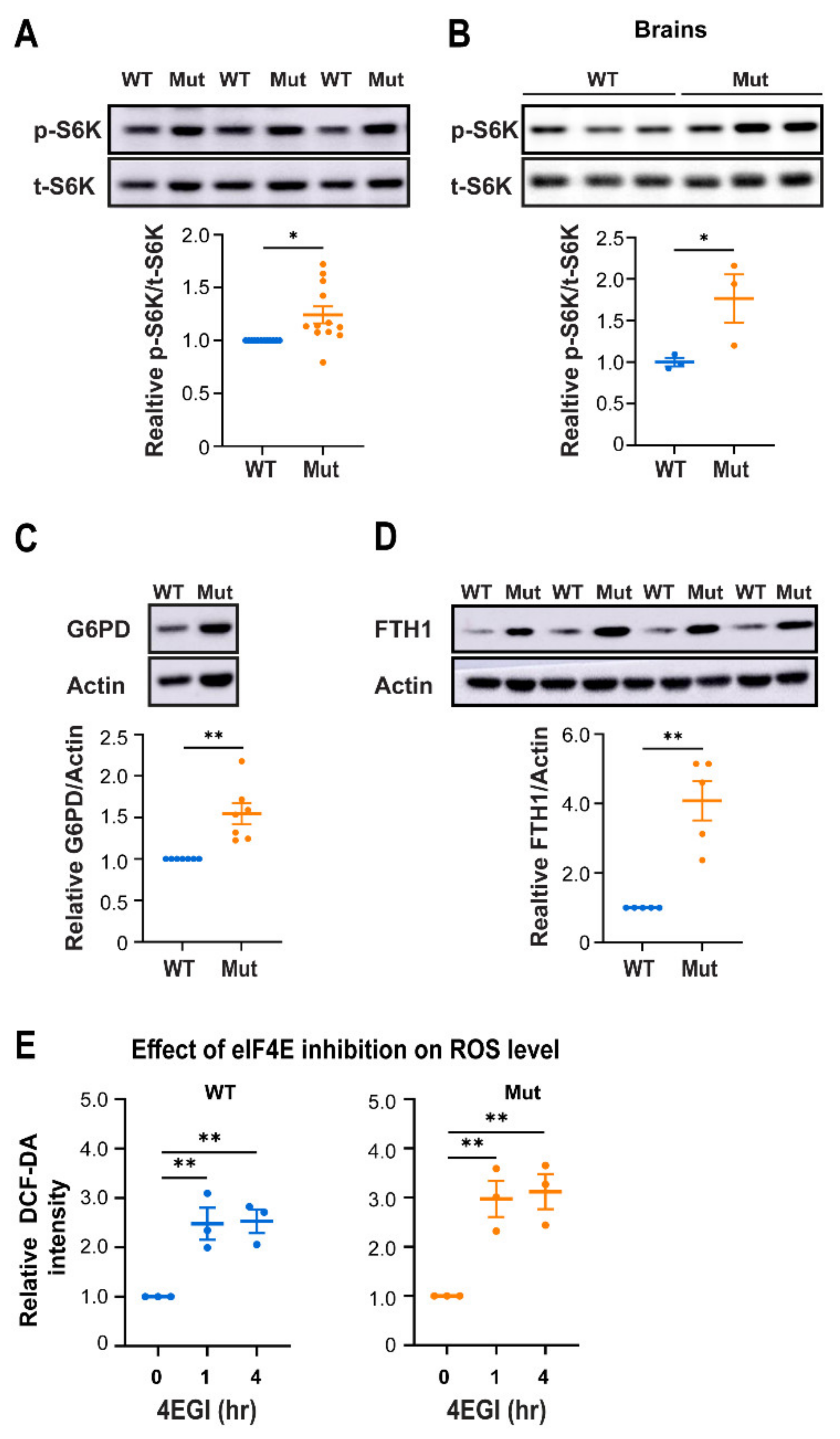
3.4. Eif2b5R132H/R132H (Mut) Astrocytes Employ Higher mTORC1 Activity for Redox Regulation
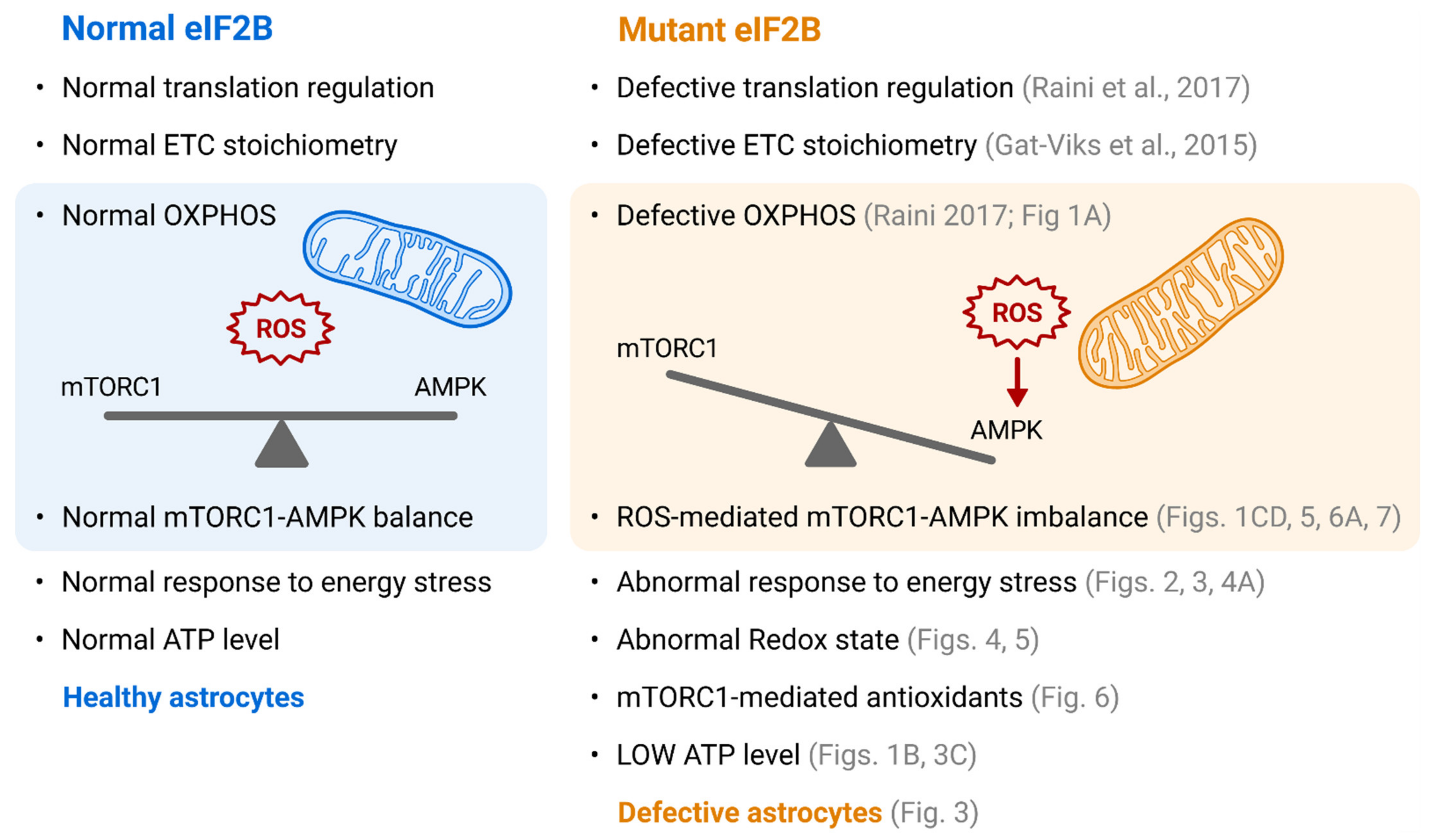
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACC | acetyl-CoA carboxylase |
| AMP | activated protein kinase–AMPK |
| Ars | Arsenite |
| DMEM-GS | DMEM without glucose |
| DMEM-HG | DMEM high glucose (25 mM) |
| ETC | electron transfer chain |
| eIF2B | eukaryotic translation initiation factor 2 (protein complex) |
| Eif2b5 | symbol of murine gene encoding the catalytic subunit of eIF2B |
| FAO | fatty acid β-oxidation |
| FTH1 | ferritin heavy chain 1 |
| GS | glucose starvation |
| mtDNA | mitochondrial DNA |
| Mut | Eif2b5R132H/R132H |
| NAC | N-acetyl-L-cystein |
| OXPHOS | oxidative phosphorylation |
| PGC1α | peroxisome proliferator-activated receptor gamma coactivator 1α |
| PPP | pentose phosphate pathway |
| SIRT1 | NAD-dependent deacetylase sirtuin-1 |
| VWMD | vanishing white matter disease |
| WT | wild-type |
References
- Tsai, J.C.; Miller-Vedam, L.E.; Anand, A.A.; Jaishankar, P.; Nguyen, H.C.; Renslo, A.R.; Frost, A.; Walter, P. Structure of the nucleotide exchange factor eIF2B reveals mechanism of memory-enhancing molecule. Science 2018, 359, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zyryanova, A.F.; Weis, F.; Faille, A.; Alard, A.A.; Crespillo-Casado, A.; Sekine, Y.; Harding, H.P.; Allen, F.; Parts, L.; Fromont, C.; et al. Binding of ISRIB reveals a regulatory site in the nucleotide exchange factor eIF2B. Science 2018, 359, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Leegwater, P.A.; Vermeulen, G.; Konst, A.A.; Naidu, S.; Mulders, J.; Visser, A.; Kersbergen, P.; Mobach, D.; Fonds, D.; van Berkel, C.G.; et al. Subunits of the translation initiation factor eIF2B are mutant in leukoencephalopathy with vanishing white matter. Nat. Genet. 2001, 29, 383–388. [Google Scholar] [CrossRef]
- Hamilton, E.M.C.; van der Lei, H.D.W.; Vermeulen, G.; Gerver, J.A.M.; Lourenco, C.M.; Naidu, S.; Mierzewska, H.; Gemke, R.; de Vet, H.C.W.; Uitdehaag, B.M.J.; et al. Natural History of Vanishing White Matter. Ann. Neurol. 2018, 84, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Elroy-Stein, O.S.R. Vanishing white matter disease. In Rosenberg’s Molecular and Genetic Basis of Neurological and Psychiatric Disease, 6th ed.; Roger, N., Rosenberg, J.M.P., Eds.; Elsevier Academic Press: Cambridge, MA, USA, 2020; Volume 2, pp. 301–317. [Google Scholar]
- Jennings, M.D.; Zhou, Y.; Mohammad-Qureshi, S.S.; Bennett, D.; Pavitt, G.D. eIF2B promotes eIF5 dissociation from eIF2*GDP to facilitate guanine nucleotide exchange for translation initiation. Genes Dev. 2013, 27, 2696–2707. [Google Scholar] [CrossRef]
- Pavitt, G.D. Regulation of translation initiation factor eIF2B at the hub of the integrated stress response. Wiley Interdiscip. Rev. RNA 2018, 9, e1491. [Google Scholar] [CrossRef]
- Pap, M.; Cooper, G.M. Role of translation initiation factor 2B in control of cell survival by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta signaling pathway. Mol. Cell Biol. 2002, 22, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368, 1–11. [Google Scholar] [CrossRef]
- Geva, M.; Cabilly, Y.; Assaf, Y.; Mindroul, N.; Marom, L.; Raini, G.; Pinchasi, D.; Elroy-Stein, O. A mouse model for eukaryotic translation initiation factor 2B-leucodystrophy reveals abnormal development of brain white matter. Brain 2010, 133 Pt 8, 2448–2461. [Google Scholar] [CrossRef]
- Marom, L.; Ulitsky, I.; Cabilly, Y.; Shamir, R.; Elroy-Stein, O. A point mutation in translation initiation factor eIF2B leads to function--and time-specific changes in brain gene expression. PLoS ONE 2011, 6, e26992. [Google Scholar] [CrossRef]
- Gat-Viks, I.; Geiger, T.; Barbi, M.; Raini, G.; Elroy-Stein, O. Proteomics-level analysis of myelin formation and regeneration in a mouse model for Vanishing White Matter disease. J. Neurochem. 2015, 134, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Raini, G.; Sharet, R.; Herrero, M.; Atzmon, A.; Shenoy, A.; Geiger, T.; Elroy-Stein, O. Mutant eIF2B leads to impaired mitochondrial oxidative phosphorylation in vanishing white matter disease. J. Neurochem. 2017, 141, 694–707. [Google Scholar] [CrossRef] [PubMed]
- Herrero, M.; Mandelboum, S.; Elroy-Stein, O. eIF2B Mutations Cause Mitochondrial Malfunction in Oligodendrocytes. Neuromol. Med. 2019, 21, 303–313. [Google Scholar] [CrossRef]
- Elroy-Stein, O. Mitochondrial malfunction in vanishing white matter disease: A disease of the cytosolic translation machinery. Neural Regen. Res. 2017, 12, 1610–1612. [Google Scholar] [CrossRef]
- Bugiani, M.; Vuong, C.; Breur, M.; van der Knaap, M.S. Vanishing white matter: A leukodystrophy due to astrocytic dysfunction. Brain Pathol. 2018, 28, 408–421. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011593. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Jager, S.; Handschin, C.; St-Pierre, J.; Spiegelman, B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc. Natl. Acad. Sci. USA 2007, 104, 12017–12022. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Chaube, B.; Malvi, P.; Singh, S.V.; Mohammad, N.; Viollet, B.; Bhat, M.K. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1alpha-mediated mitochondrial biogenesis. Cell Death Discov. 2015, 1, 15063. [Google Scholar] [CrossRef]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. AMPK Maintains Cellular Metabolic Homeostasis through Regulation of Mitochondrial Reactive Oxygen Species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef]
- Ren, Y.; Shen, H.M. Critical role of AMPK in redox regulation under glucose starvation. Redox Biol. 2019, 25, 101154. [Google Scholar] [CrossRef]
- Heiss, E.H.; Kramer, M.P.; Atanasov, A.G.; Beres, H.; Schachner, D.; Dirsch, V.M. Glycolytic switch in response to betulinic acid in non-cancer cells. PLoS ONE 2014, 9, e115683. [Google Scholar] [CrossRef]
- Anderson, R.M.; Barger, J.L.; Edwards, M.G.; Braun, K.H.; O’Connor, C.E.; Prolla, T.A.; Weindruch, R. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 2008, 7, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1alpha, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.T.; Kolesar, J.E.; Kaufman, B.A. Mitochondrial transcription factor A regulates mitochondrial transcription initiation, DNA packaging, and genome copy number. Biochim. Biophys. Acta 2012, 1819, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acin-Perez, R.; Latorre-Pellicer, A.; Colas, C.; Balsa, E.; Perales-Clemente, E.; Quiros, P.M.; Calvo, E.; Rodriguez-Hernandez, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef]
- Garcia-Poyatos, C.; Cogliati, S.; Calvo, E.; Hernansanz-Agustin, P.; Lagarrigue, S.; Magni, R.; Botos, M.; Langa, X.; Amati, F.; Vazquez, J.; et al. Scaf1 promotes respiratory supercomplexes and metabolic efficiency in zebrafish. EMBO Rep. 2020, 21, e50287. [Google Scholar] [CrossRef]
- Di, W.; Lv, J.; Jiang, S.; Lu, C.; Yang, Z.; Ma, Z.; Hu, W.; Yang, Y.; Xu, B. PGC-1: The Energetic Regulator in Cardiac Metabolism. Curr. Issues Mol. Biol. 2018, 28, 29–46. [Google Scholar] [CrossRef]
- Woo, Y.; Lee, H.J.; Jung, Y.M.; Jung, Y.J. mTOR-Mediated Antioxidant Activation in Solid Tumor Radioresistance. J. Oncol. 2019, 2019, 5956867. [Google Scholar] [CrossRef]
- Percy, M.E.; Wong, S.; Bauer, S.; Liaghati-Nasseri, N.; Perry, M.D.; Chauthaiwale, V.M.; Dhar, M.; Joshi, J.G. Iron metabolism and human ferritin heavy chain cDNA from adult brain with an elongated untranslated region: New findings and insights. Analyst 1998, 123, 41–50. [Google Scholar] [CrossRef][Green Version]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Gruning, N.M.; Kruger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef]
- Hinarejos, I.; Machuca-Arellano, C.; Sancho, P.; Espinos, C. Mitochondrial Dysfunction, Oxidative Stress and Neuroinflammation in Neurodegeneration with Brain Iron Accumulation (NBIA). Antioxidants 2020, 9, 1020. [Google Scholar] [CrossRef] [PubMed]
- Truitt, M.L.; Conn, C.S.; Shi, Z.; Pang, X.; Tokuyasu, T.; Coady, A.M.; Seo, Y.; Barna, M.; Ruggero, D. Differential Requirements for eIF4E Dose in Normal Development and Cancer. Cell 2015, 162, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Moerke, N.J.; Aktas, H.; Chen, H.; Cantel, S.; Reibarkh, M.Y.; Fahmy, A.; Gross, J.D.; Degterev, A.; Yuan, J.; Chorev, M.; et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell 2007, 128, 257–267. [Google Scholar] [CrossRef]
- Belanger, M.; Magistretti, P.J. The role of astroglia in neuroprotection. Dialogues Clin. Neurosci. 2009, 11, 281–295. [Google Scholar] [PubMed]
- Derouiche, A.; Haseleu, J.; Korf, H.W. Fine Astrocyte Processes Contain Very Small Mitochondria: Glial Oxidative Capability May Fuel Transmitter Metabolism. Neurochem. Res. 2015, 40, 2402–2413. [Google Scholar] [CrossRef]
- Lovatt, D.; Sonnewald, U.; Waagepetersen, H.S.; Schousboe, A.; He, W.; Lin, J.H.; Han, X.; Takano, T.; Wang, S.; Sim, F.J.; et al. The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J. Neurosci. 2007, 27, 12255–12266. [Google Scholar] [CrossRef]
- Hertz, L.; Peng, L.; Dienel, G.A. Energy metabolism in astrocytes: High rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 2007, 27, 219–249. [Google Scholar] [CrossRef] [PubMed]
- Falkowska, A.; Gutowska, I.; Goschorska, M.; Nowacki, P.; Chlubek, D.; Baranowska-Bosiacka, I. Energy Metabolism of the Brain, Including the Cooperation between Astrocytes and Neurons, Especially in the Context of Glycogen Metabolism. Int. J. Mol. Sci. 2015, 16, 25959–25981. [Google Scholar] [CrossRef]
- Deitmer, J.W.; Theparambil, S.M.; Ruminot, I.; Noor, S.I.; Becker, H.M. Energy Dynamics in the Brain: Contributions of Astrocytes to Metabolism and pH Homeostasis. Front Neurosci. 2019, 13, 1301. [Google Scholar] [CrossRef]
- Brown, A.M.; Rich, L.R.; Ransom, B.R. Metabolism of Glycogen in Brain White Matter. Adv. Neurobiol. 2019, 23, 187–207. [Google Scholar]
- McKenna, M.C.; Sonnewald, U.; Huang, X.; Stevenson, J.; Zielke, H.R. Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J. Neurochem. 1996, 66, 386–393. [Google Scholar] [CrossRef]
- Sonnewald, U.; Westergaard, N.; Schousboe, A. Glutamate transport and metabolism in astrocytes. Glia 1997, 21, 56–63. [Google Scholar] [CrossRef]
- Hertz, L.; Yu, A.C.; Kala, G.; Schousboe, A. Neuronal-astrocytic and cytosolic-mitochondrial metabolite trafficking during brain activation, hyperammonemia and energy deprivation. Neurochem. Int. 2000, 37, 83–102. [Google Scholar] [CrossRef]
- Hertz, L.; Hertz, E. Cataplerotic TCA cycle flux determined as glutamate-sustained oxygen consumption in primary cultures of astrocytes. Neurochem. Int. 2003, 43, 355–361. [Google Scholar] [CrossRef]
- Fiebig, C.; Keiner, S.; Ebert, B.; Schaffner, I.; Jagasia, R.; Lie, D.C.; Beckervordersandforth, R. Mitochondrial Dysfunction in Astrocytes Impairs the Generation of Reactive Astrocytes and Enhances Neuronal Cell Death in the Cortex Upon Photothrombotic Lesion. Front Mol. Neurosci. 2019, 12, 40. [Google Scholar] [CrossRef]
- Camargo, N.; Goudriaan, A.; van Deijk, A.F.; Otte, W.M.; Brouwers, J.F.; Lodder, H.; Gutmann, D.H.; Nave, K.A.; Dijkhuizen, R.M.; Mansvelder, H.D.; et al. Oligodendroglial myelination requires astrocyte-derived lipids. PLoS Biol. 2017, 15, e1002605. [Google Scholar] [CrossRef]
- Saher, G.; Stumpf, S.K. Cholesterol in myelin biogenesis and hypomyelinating disorders. Biochim. Biophys. Acta 2015, 1851, 1083–1094. [Google Scholar] [CrossRef]
- Landriscina, M.; Maddalena, F.; Laudiero, G.; Esposito, F. Adaptation to oxidative stress, chemoresistance, and cell survival. Antioxid Redox Signal 2009, 11, 2701–2716. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef]
- Moon, S.J.; Dong, W.; Stephanopoulos, G.N.; Sikes, H.D. Oxidative pentose phosphate pathway and glucose anaplerosis support maintenance of mitochondrial NADPH pool under mitochondrial oxidative stress. Bioeng. Transl. Med. 2020, 5, e10184. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Manning, B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013, 15, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Menon, S.; Manning, B.D. Common corruption of the mTOR signaling network in human tumors. Oncogene 2008, 27 (Suppl. S2), S43–S51. [Google Scholar] [CrossRef] [PubMed]
- Arai, H.; Kogure, K.; Sugioka, K.; Nakano, M. Importance of two iron-reducing systems in lipid peroxidation of rat brain: Implications for oxygen toxicity in the central nervous system. Biochem. Int. 1987, 14, 741–749. [Google Scholar]
- Samson, F.E.; Nelson, S.R. The aging brain, metals and oxygen free radicals. Cell Mol. Biol. 2000, 46, 699–707. [Google Scholar]
- Chen, N.; Dai, L.; Jiang, Y.; Wang, J.; Hao, H.; Ren, Y.; Leng, X.; Zang, L.; Wu, Y. Endoplasmic reticulum stress intolerance in EIF2B3 mutant oligodendrocytes is modulated by depressed autophagy. Brain Dev. 2016, 38, 507–515. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef]
- Schoof, M.; Boone, M.; Wang, L.; Lawrence, R.; Frost, A.; Walter, P. eIF2B conformation and assembly state regulate the integrated stress response. eLife 2021, 10, e65703. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.L.; LeBon, L.; Basso, A.M.; Kohlhaas, K.L.; Nikkel, A.L.; Robb, H.M.; Donnelly-Roberts, D.L.; Prakash, J.; Swensen, A.M.; Rubinstein, N.D.; et al. eIF2B activator prevents neurological defects caused by a chronic integrated stress response. eLife 2019, 8, e42940. [Google Scholar] [CrossRef]
- Abbink, T.E.M.; Wisse, L.E.; Jaku, E.; Thiecke, M.J.; Voltolini-Gonzalez, D.; Fritsen, H.; Bobeldijk, S.; Ter Braak, T.J.; Polder, E.; Postma, N.L.; et al. Vanishing white matter: Deregulated integrated stress response as therapy target. Ann. Clin. Transl. Neurol. 2019, 6, 1407–1422. [Google Scholar] [CrossRef]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrero, M.; Daw, M.; Atzmon, A.; Elroy-Stein, O. The Energy Status of Astrocytes Is the Achilles’ Heel of eIF2B-Leukodystrophy. Cells 2021, 10, 1858. https://doi.org/10.3390/cells10081858
Herrero M, Daw M, Atzmon A, Elroy-Stein O. The Energy Status of Astrocytes Is the Achilles’ Heel of eIF2B-Leukodystrophy. Cells. 2021; 10(8):1858. https://doi.org/10.3390/cells10081858
Chicago/Turabian StyleHerrero, Melisa, Maron Daw, Andrea Atzmon, and Orna Elroy-Stein. 2021. "The Energy Status of Astrocytes Is the Achilles’ Heel of eIF2B-Leukodystrophy" Cells 10, no. 8: 1858. https://doi.org/10.3390/cells10081858
APA StyleHerrero, M., Daw, M., Atzmon, A., & Elroy-Stein, O. (2021). The Energy Status of Astrocytes Is the Achilles’ Heel of eIF2B-Leukodystrophy. Cells, 10(8), 1858. https://doi.org/10.3390/cells10081858