







Pharmaceutics 2026, 18(7), 826; https://doi.org/10.3390/pharmaceutics18070826 (registering DOI) - 5 Jul 2026
Abstract
Background/Objectives: Tirzepatide is a dual GIP and GLP-1 receptor agonist indicated for the treatment of type 2 diabetes and obesity. Oral delivery of tirzepatide is limited by poor gastrointestinal permeability, pH-dependent solubility, and manufacturing challenges associated with high-dose absorption enhancers. Methods: This study
[...] Read more.
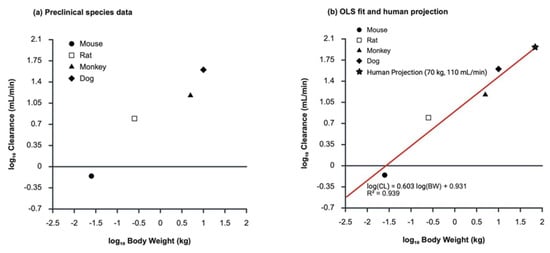
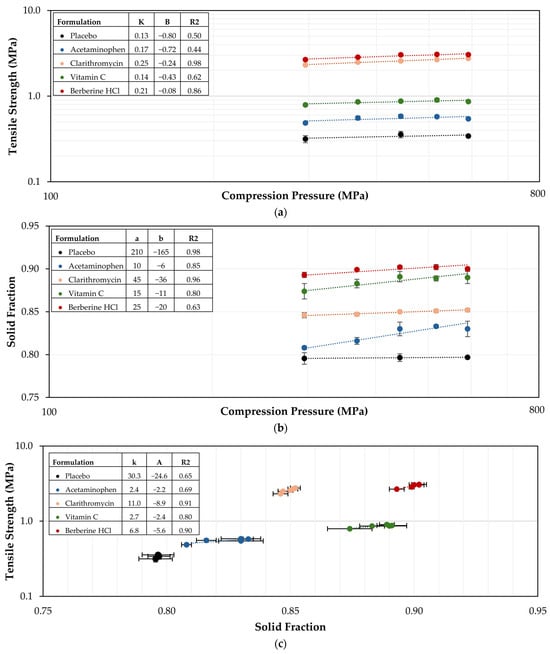
Background/Objectives: Tirzepatide is a dual GIP and GLP-1 receptor agonist indicated for the treatment of type 2 diabetes and obesity. Oral delivery of tirzepatide is limited by poor gastrointestinal permeability, pH-dependent solubility, and manufacturing challenges associated with high-dose absorption enhancers. Methods: This study developed an immediate-release oral tirzepatide tablet using a Quality by Design (QbD) approach. Sodium caprate (C10) was selected as the absorption enhancer based on acid-neutralizing capacity, Caco-2 permeability enhancement, and preliminary rat pharmacokinetic screening. Quality target product profile, critical quality attributes, preliminary hazard analysis, and failure mode and effects analysis identified binder/disintegrant ratios as critical material attributes and hammer milling conditions as critical process parameters. Face-centered central composite designs and multiple-response optimization (MRO) were applied to optimize dissolution, flowability, and tablet mechanical integrity. Results: The optimized binder/disintegrant composition produced benchmark-comparable dissolution profiles against oral semaglutide tablets in pH 1.2, 4.0, and 6.8 media, with f2 values exceeding 50 for both C10 300 mg and 500 mg formulations. The optimized process yielded tablets with low friability (0.58%) and acceptable flowability (Carr’s index, 24). In beagle dogs, the C10 300 mg formulation achieved higher systemic exposure than the C10 500 mg formulation, with a Cmax of 46.49 ± 23.79 ng/mL and AUClast of 1261.03 ± 690.44 h·ng/mL. Conclusion: These results support C10-mediated oral tirzepatide delivery and QbD-based optimization for oral peptide tablets.
Full article
(This article belongs to the Special Issue GLP-1 Receptor Agonists: Current Understanding and Emerging Therapeutic Horizons)














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}