
Receptor Tyrosine Kinase KIT: Mutation-Induced Conformational Shift Promotes Alternative Allosteric Pockets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
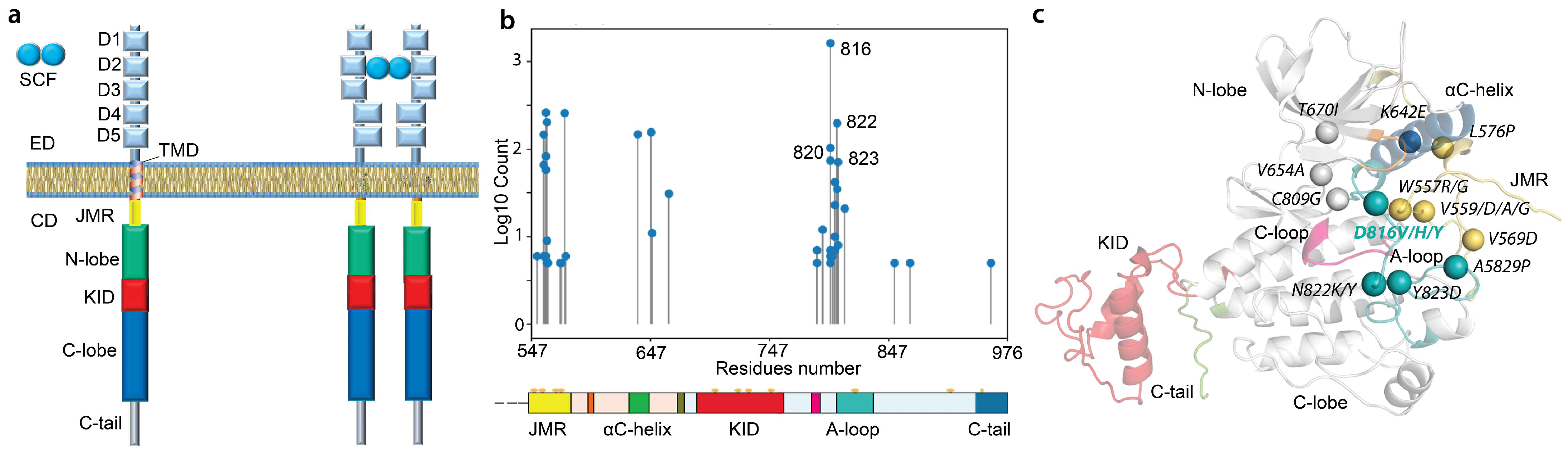
:1. Introduction
2. Results
2.1. Data Generation and Proceeding
2.2. General Characterisation of MD Trajectories
2.3. KIT Folding in Inactive (Wild Type) and Constitutively Active (Mutant) States
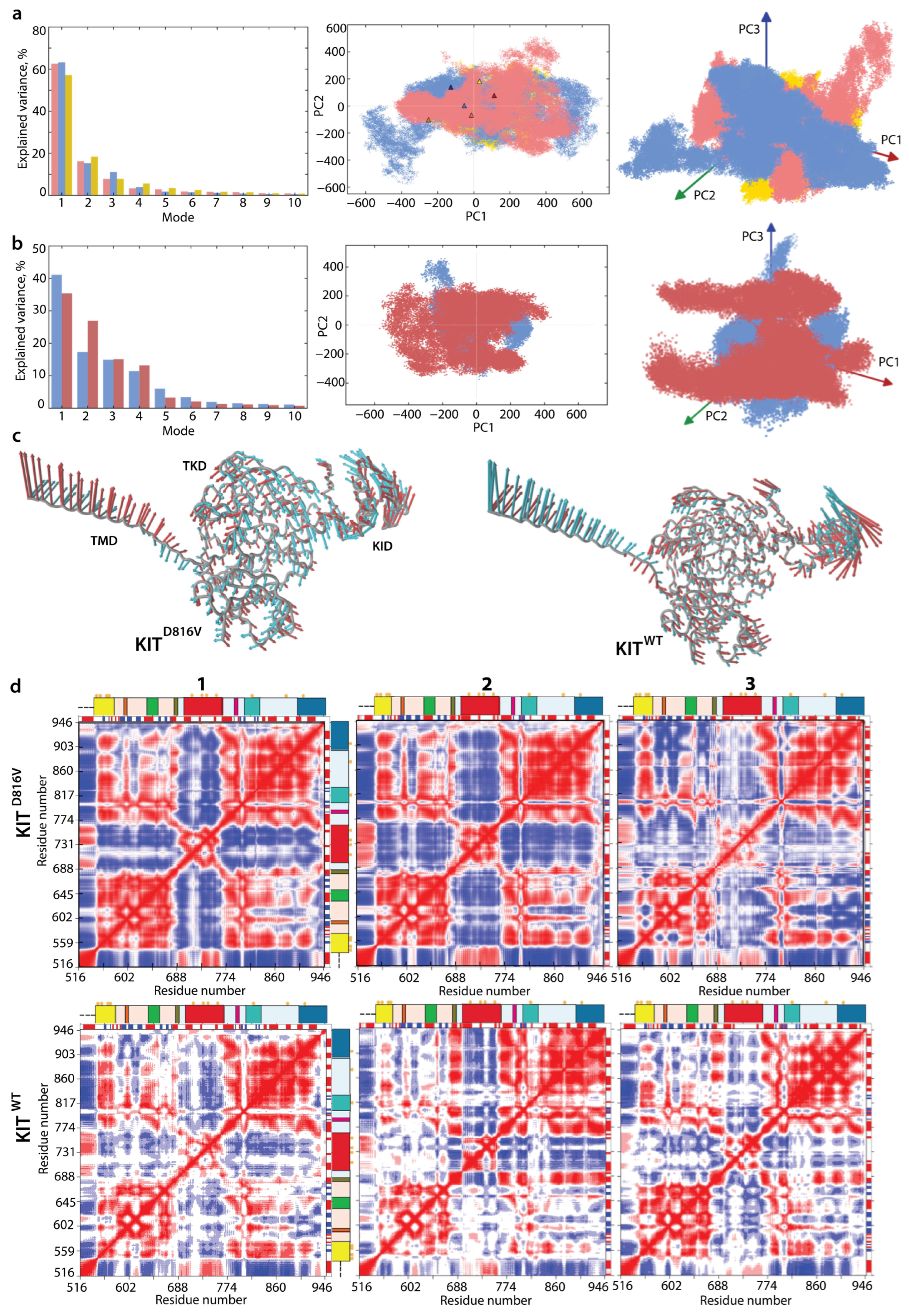
2.4. KIT Plasticity: Mutation-Induced Effects on the Conformational Space
2.5. Impact of D816V Mutation on Inter-Domain Non-Covalent Interactions Stabilising KIT
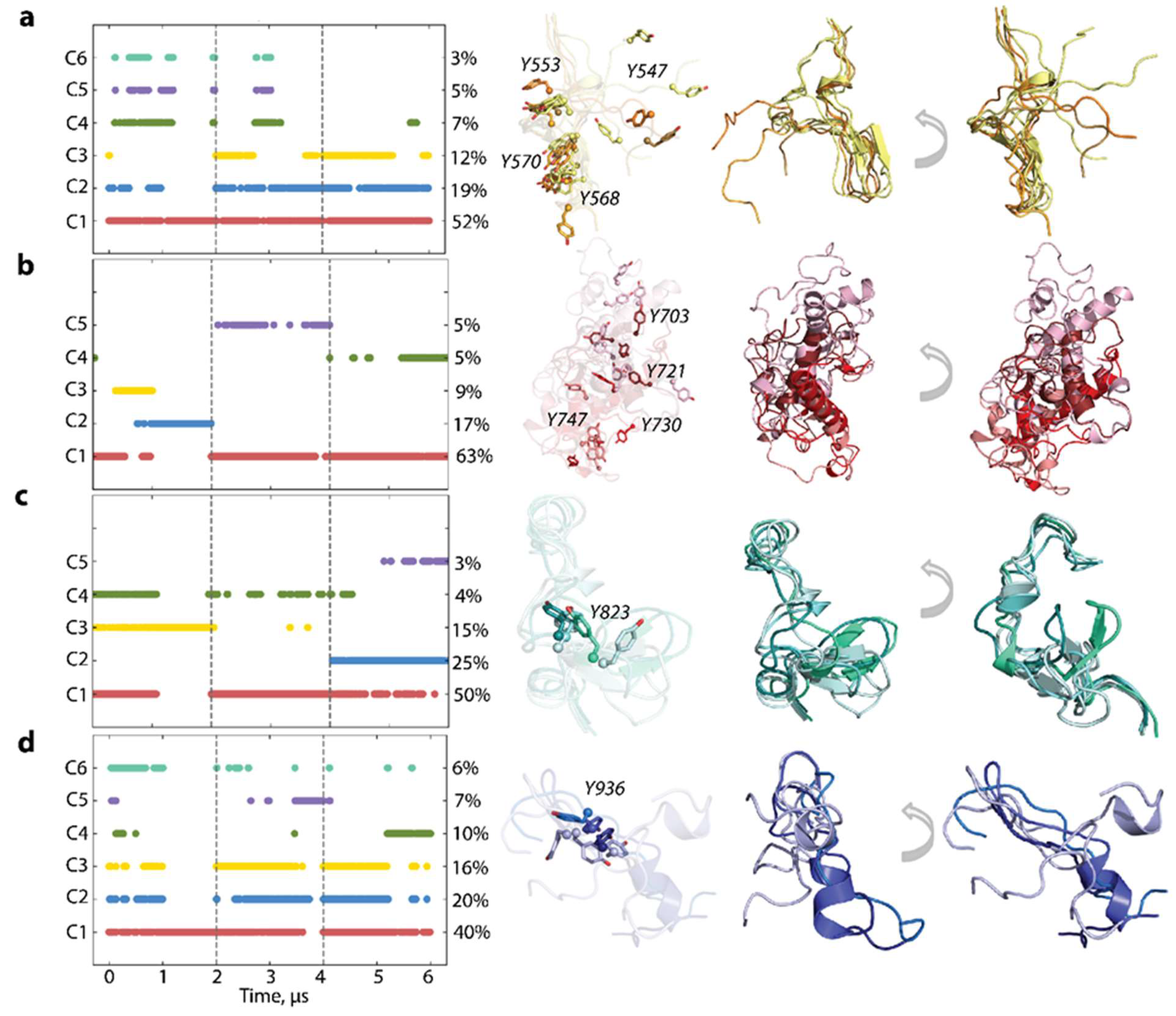
2.6. Per Domain Clustering of KITD816V Conformations
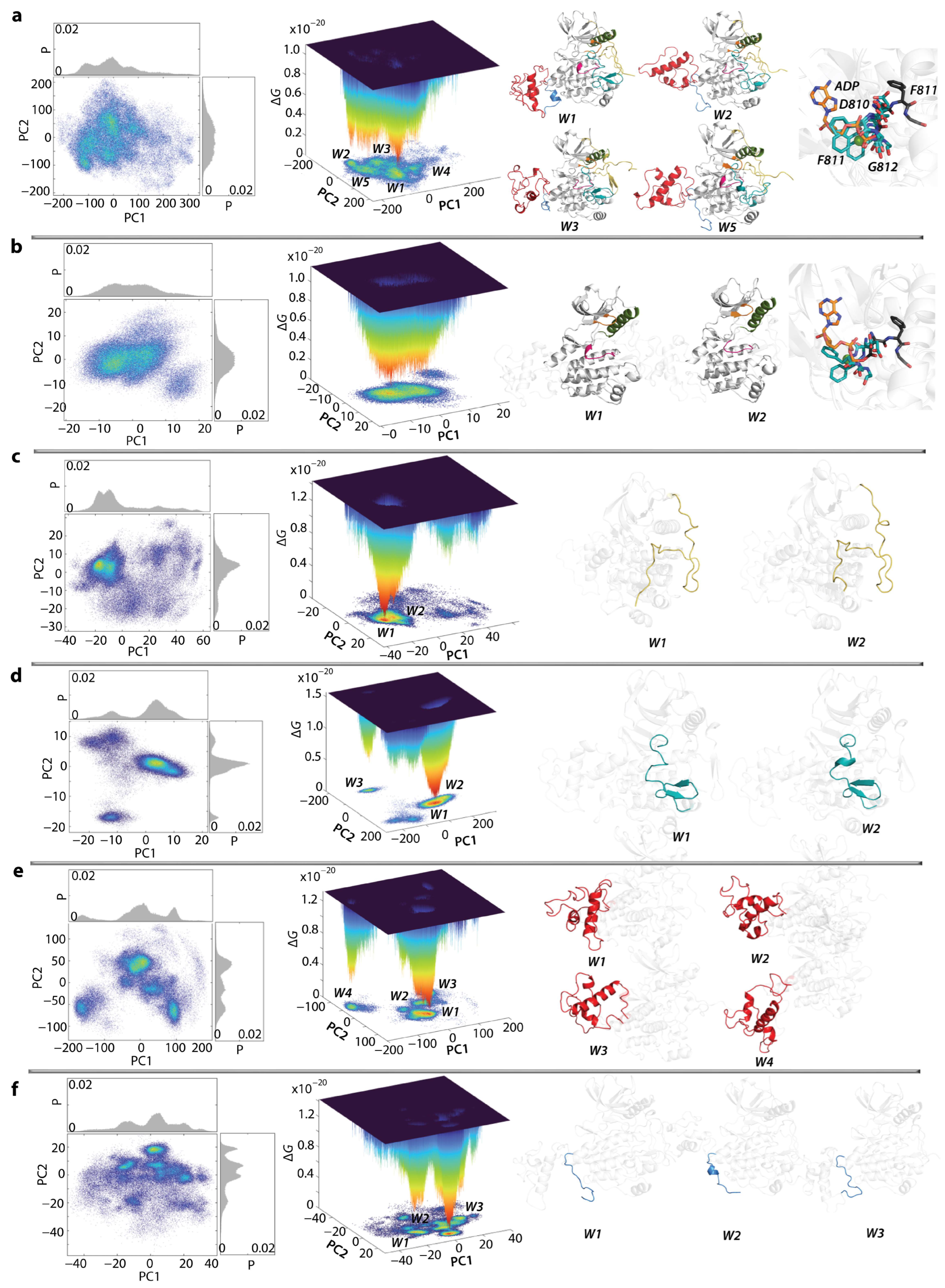
2.7. What Are We Learning from the Cumulative Free Energy Landscape of KITD816V and KITWT?
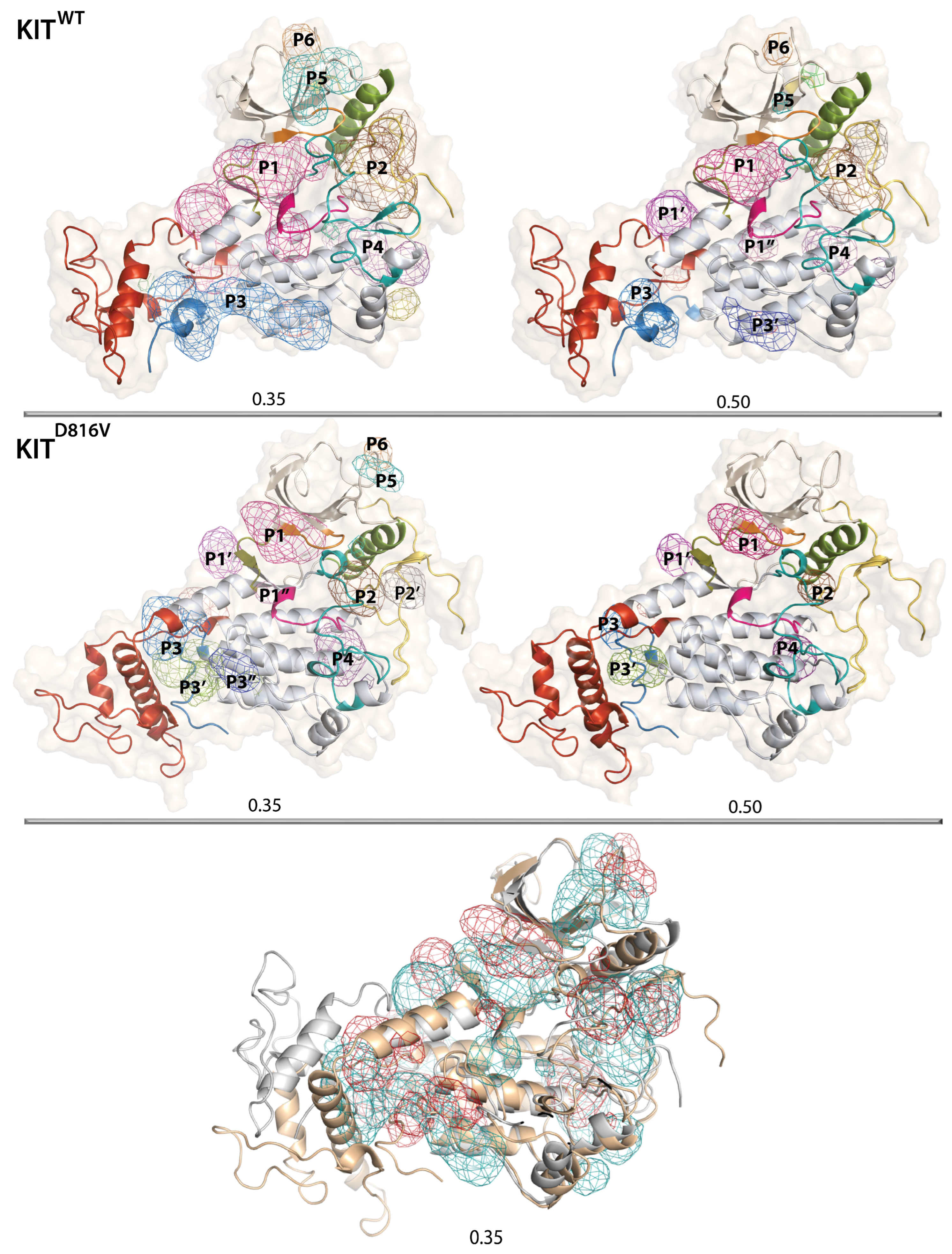
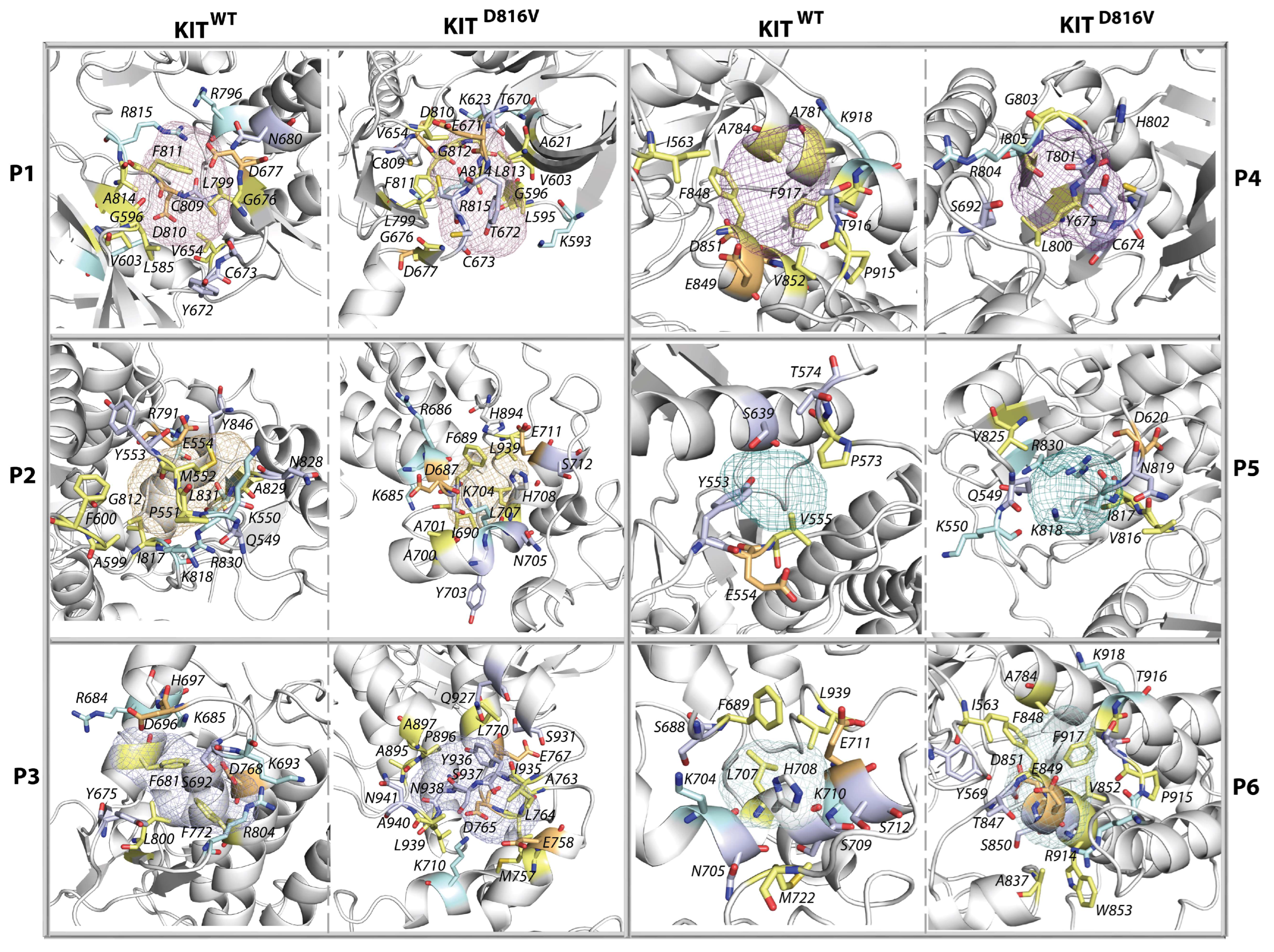
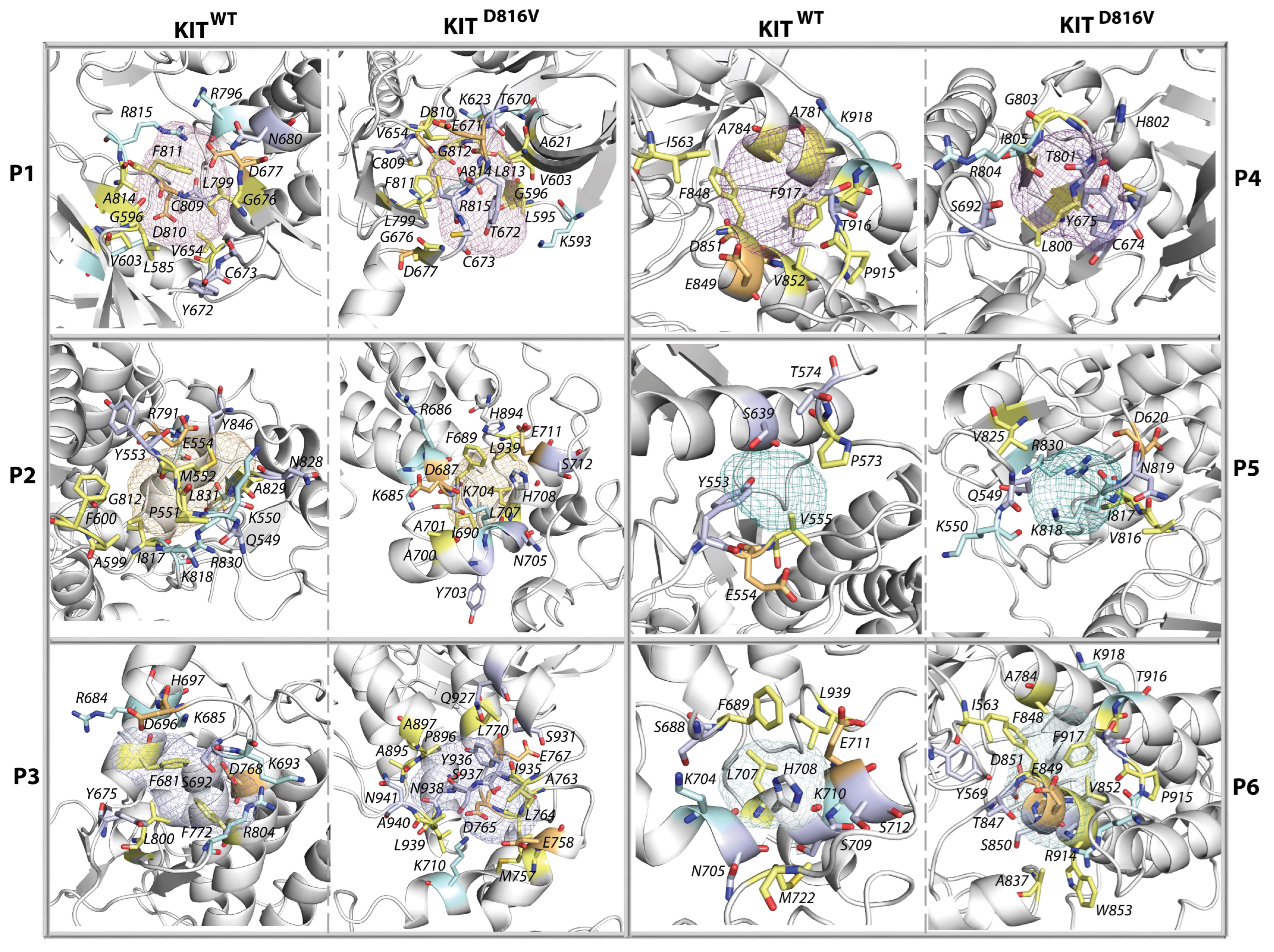
2.8. The KIT Cytoplasmic Domain Pockets Detected in the Native and Mutated Proteins
3. Discussion
4. Methods
4.1. Modelling
4.2. Molecular Dynamics Simulation
4.3. Data Analysis
- (1)
- The RMSD and RMSF values and cross-correlations were calculated for the Cα-atoms using the initial full-length model or each separated domain/region (at t = 0 µs) as a reference.
- (2)
- Secondary structural propensities for all residues were calculated using the Define Secondary Structure of Proteins (DSSP) method [98]. The secondary structure types were assigned for residues based on backbone -NH and -CO atom positions. Secondary structures were assigned every 10 and 20 ps for the individual and concatenated trajectories, respectively.
- (3)
- Difference in the probability of formation of secondary structures for each pair of residues i from KITWT and KITD816V was estimated as:
- (4)
- Clustering analysis was performed on the productive simulation time of each MD trajectory using an ensemble-based approach [55]. The algorithm extracts representative MD conformations from a trajectory by clustering the recorded snapshots according to their Cɑ-atom RMSDs. The procedure for each trajectory can be described as follows: (i) a reference structure is randomly chosen in the MD conformational ensemble, and all conformations within an arbitrary cut-off r are removed from the ensemble; this step is repeated until no conformation remains in the ensemble, providing a set of reference structures at a distance of at least r; (ii) the MD conformations are grouped into n reference clusters based on their RMSDs from each reference structure. The cut-off was varied from 3 to 5 Å. The analysis was performed every 100 ps.
- (5)
- The H-bonds between donor (D) and acceptor (A) atoms N, O, S were monitored according to the following geometrical parameters: d(D-A) ≤ 3.6 Å, ∠(DHA) ≥ 120°. Hydrophobic contacts were considered for all hydrophobic residues with side chains within a 4 Å of each other.
- (6)
- The Principal Components Analysis (PCA) modes were calculated for the backbone atoms (N, H, Cα, C, O) after least-square fitting on the average conformation calculated on the concatenated data. The eigenvectors were visualized with NMWiz module for VMD [99].
- (7)
- The relative Gibbs free energy of the canonical ensemble was computed as a function of two reaction coordinates with Equation (2) [100]:where represents the Boltzmann constant, and is the temperature. denotes the probability of states along the two reaction coordinates, which is calculated using their joint probability, and denotes the maximum probability. The population of each well was roughly estimated using a square defined with and value intervals and containing red to orange colors.
- (8)
- The pocket prediction protocol includes three steps: (i) Finding optimal criteria for pocket hunting. This step was performed by testing different isovalues ranging from 0 to 1.0 in 0.5 increments for both proteins. Two isovalues, 0.35 and 0.50, give the maximum number of pockets in KITD816V and KITWT, respectively. (ii) Pockets were identified using by Fpocket protein cavity detection algorithm, which uses Voronoi tessellation and alpha shapes [63]. (iii) Tracking the change in pocket volume along the concatenated trajectories of each protein was performed using two isovalues, 0.35 and 0.50. (iv) Pockets were ranked based on the calculated volume as well as on their local hydrophobic density.
4.4. Visualisation and Figure Preparation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Receptor tyrosine kinases: Legacy of the first two decades. Cold Spring Harb. Perspect. Biol. 2014, 6, a008912. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.H.; Lew, E.D.; Yuzawa, S.; Tome, F.; Lax, I.; Schlessinger, J. The Selectivity of Receptor Tyrosine Kinase Signaling Is Controlled by a Secondary SH2 Domain Binding Site. Cell 2009, 138, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Rönnstrand, L. Signal transduction via the stem cell factor receptor/c-Kit. Cell. Mol. Life Sci. CMLS 2004, 61, 2535–2548. [Google Scholar] [CrossRef] [PubMed]
- Weiss, F.U.; Daub, H.; Ullrich, A. Novel mechanisms of RTK signal generation. Curr. Opin. Genet. Dev. 1997, 7, 80–86. [Google Scholar] [CrossRef]
- Lennartsson, J.; Rönnstrand, L. Stem cell factor receptor/c-Kit: From basic science to clinical implications. Physiol. Rev. 2012, 92, 1619–1649. [Google Scholar] [CrossRef]
- Liang, J.; Wu, Y.-L.; Chen, B.-J.; Zhang, W.; Tanaka, Y.; Sugiyama, H. The C-kit receptor-mediated signal transduction and tumor-related diseases. Int. J. Biol. Sci. 2013, 9, 435–443. [Google Scholar] [CrossRef]
- Piao, X.H.; Paulson, R.; van der Geer, P.; Pawson, T.; Bernstein, A. Oncogenic mutation in the Kit receptor tyrosine kinase alters substrate specificity and induces degradation of the protein tyrosine phosphatase SHP-1. Proc. Natl. Acad. Sci. USA 1996, 93, 14665–14669. [Google Scholar] [CrossRef]
- Rajan, V.; Prykhozhji, S.V.; Pandey, A.; Cohen, A.M.; Rainey, J.K.; Berman, J.N. KIT D816V is dimerization-independent and activates downstream pathways frequently perturbed in mastocytosis. Br. J. Haematol. 2023, 202, 960–970. [Google Scholar] [CrossRef]
- Hirota, S.; Isozaki, K. Pathology of gastrointestinal stromal tumors. Pathol. Int. 2006, 56, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.; Cavazza, A.; Marchioni, A.; Migaldi, M.; Bavieri, M.; Facciolongo, N.; Petruzzelli, S.; Longo, L.; Tamberi, S.; Crinò, L. Kit expression in small cell carcinomas of the lung: Effects of chemotherapy. Mod. Pathol. 2003, 16, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, H.; Kanakura, Y.; Tamaki, T.; Kuriu, A.; Kitayama, H.; Ishikawa, J.; Kanayama, Y.; Yonezawa, T.; Tarui, S.; Griffin, J.D. Expression and functional role of the proto-oncogene c-kit in acute myeloblastic leukemia cells. Blood 1991, 78, 2962–2968. [Google Scholar] [CrossRef]
- Malaise, M.; Steinbach, D.; Corbacioglu, S. Clinical implications of c-Kit mutations in acute myelogenous leukemia. Curr. Hematol. Malig. Rep. 2009, 4, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Rivera, R.S.; Nagatsuka, H.; Gunduz, M.; Cengiz, B.; Gunduz, E.; Siar, C.H.; Tsujigiwa, H.; Tamamura, R.; Han, K.N.; Nagai, N. C-kit protein expression correlated with activating mutations in KIT gene in oral mucosal melanoma. Virchows Arch. Int. J. Pathol. 2008, 452, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Gilreath, J.A.; Tchertanov, L.; Deininger, M.W. Novel approaches to treating advanced systemic mastocytosis. Clin. Pharmacol. 2019, 11, 77–92. [Google Scholar] [CrossRef]
- Arock, M.; Sotlar, K.; Akin, C.; Broesby-Olsen, S.; Hoermann, G.; Escribano, L.; Kristensen, T.K.; Kluin-Nelemans, H.C.; Hermine, O.; Dubreuil, P.; et al. KIT mutation analysis in mast cell neoplasms: Recommendations of the European Competence Network on Mastocytosis. Leukemia 2015, 29, 1223–1232. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Pinchasi, D.; Zhou, M.; Lax, I.; Schlessinger, J. Kit receptor dimerization is driven by bivalent binding of stem cell factor. J. Biol. Chem. 1997, 272, 6311–6317. [Google Scholar] [CrossRef]
- Linnekin, D. Early signaling pathways activated by c-Kit in hematopoietic cells. Int. J. Biochem. Cell Biol. 1999, 31, 1053–1074. [Google Scholar] [CrossRef]
- Lennartsson, J.; Jelacic, T.; Linnekin, D.; Shivakrupa, R. Normal and oncogenic forms of the receptor tyrosine kinase kit. Stem Cells 2005, 23, 16–43. [Google Scholar] [CrossRef] [PubMed]
- Ashman, L.K.; Griffith, R. Therapeutic targeting of c-KIT in cancer. Expert. Opin. Investig. Drugs 2013, 22, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Antonescu, C.R.; Wolchok, J.D.; Chapman, P.B.; Roman, R.A.; Teitcher, J.; Panageas, K.S.; Busam, K.J.; Chmielowski, B.; Lutzky, J.; et al. KIT as a therapeutic target in metastatic melanoma. JAMA 2011, 305, 2327–2334. [Google Scholar] [CrossRef]
- Furitsu, T.; Tsujimura, T.; Tono, T.; Ikeda, H.; Kitayama, H.; Koshimizu, U.; Sugahara, H.; Butterfield, J.H.; Ashman, L.K.; Kanayama, Y.; et al. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J. Clin. Investig. 1993, 92, 1736–1744. [Google Scholar] [CrossRef] [PubMed]
- Laine, E.; Auclair, C.; Tchertanov, L. Allosteric Communication across the Native and Mutated KIT Receptor Tyrosine Kinase. PLOS Comput. Biol. 2012, 8, e1002661. [Google Scholar] [CrossRef] [PubMed]
- Laine, E.; Chauvot de Beauchêne, I.; Perahia, D.; Auclair, C.; Tchertanov, L. Mutation D816V Alters the Internal Structure and Dynamics of c-KIT Receptor Cytoplasmic Region: Implications for Dimerization and Activation Mechanisms. PLoS Comput. Biol. 2011, 7, e1002068. [Google Scholar] [CrossRef]
- Pedersen, M.; Rönnstrand, L.; Sun, J. The c-Kit/D816V mutation eliminates the differences in signal transduction and biological responses between two isoforms of c-Kit. Cell. Signal. 2009, 21, 413–418. [Google Scholar] [CrossRef]
- Chaix, A.; Lopez, S.; Voisset, E.; Gros, L.; Dubreuil, P.; De Sepulveda, P. Mechanisms of STAT protein activation by oncogenic KIT mutants in neoplastic mast cells. J. Biol. Chem. 2011, 286, 5956–5966. [Google Scholar] [CrossRef]
- Martelli, M.; Monaldi, C.; De Santis, S.; Bruno, S.; Mancini, M.; Cavo, M.; Soverini, S. Recent Advances in the Molecular Biology of Systemic Mastocytosis: Implications for Diagnosis, Prognosis, and Therapy. Int. J. Mol. Sci. 2020, 21, 3987. [Google Scholar] [CrossRef]
- Tobío, A.; Bandara, G.; Morris, D.A.; Kim, D.K.; O’Connell, M.P.; Komarow, H.D.; Carter, M.C.; Smrz, D.; Metcalfe, D.D.; Olivera, A. Oncogenic D816V-KIT signaling in mast cells causes persistent IL-6 production. Haematologica 2020, 105, 124–135. [Google Scholar] [CrossRef]
- de Toledo, M.A.S.; Fu, X.; Maié, T.; Buhl, E.M.; Götz, K.; Schmitz, S.; Kaiser, A.; Boor, P.; Braunschweig, T.; Chatain, N.; et al. KIT D816V Mast Cells Derived from Induced Pluripotent Stem Cells Recapitulate Systemic Mastocytosis Transcriptional Profile. Int. J. Mol. Sci. 2023, 24, 5275. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Pedersen, M.; Rönnstrand, L. The D816V Mutation of c-Kit Circumvents a Requirement for Src Family Kinases in c-Kit Signal Transduction. J. Biol. Chem. 2009, 284, 11039–11047. [Google Scholar] [CrossRef] [PubMed]
- Chaix, A.; Arcangeli, M.L.; Lopez, S.; Voisset, E.; Yang, Y.; Vita, M.; Letard, S.; Audebert, S.; Finetti, P.; Birnbaum, D.; et al. KIT-D816V oncogenic activity is controlled by the juxtamembrane docking site Y568-Y570. Oncogene 2014, 33, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, N.; Iqbal, N. Imatinib: A breakthrough of targeted therapy in cancer. Chemother. Res. Pract. 2014, 2014, 357027. [Google Scholar] [CrossRef]
- Yoshida, C.; Takeuchi, M.; Tsuchiyama, J.; Sadahira, Y. Successful treatment of KIT D816V-positive, imatinib-resistant systemic mastocytosis with interferon-alpha. Intern. Med. 2009, 48, 1973–1978. [Google Scholar] [CrossRef]
- Akin, C.; Brockow, K.; D’Ambrosio, C.; Kirshenbaum, A.S.; Ma, Y.; Longley, B.J.; Metcalfe, D.D. Effects of tyrosine kinase inhibitor STI571 on human mast cells bearing wild-type or mutated c-kit. Exp. Hematol. 2003, 31, 686–692. [Google Scholar] [CrossRef]
- Da Silva Figueiredo Celestino Gomes, P.; Chauvot De Beauchêne, I.; Panel, N.; Lopez, S.; De Sepulveda, P.; Geraldo Pascutti, P.; Solary, E.; Tchertanov, L. Insight on Mutation-Induced Resistance from Molecular Dynamics Simulations of the Native and Mutated CSF-1R and KIT. PLoS ONE 2016, 11, e0160165. [Google Scholar] [CrossRef]
- Shah, N.P.; Lee, F.Y.; Luo, R.; Jiang, Y.; Donker, M.; Akin, C. Dasatinib (BMS-354825) inhibits KITD816V, an imatinib-resistant activating mutation that triggers neoplastic growth in most patients with systemic mastocytosis. Blood 2006, 108, 286–291. [Google Scholar] [CrossRef]
- Patel, S.R.; Reichardt, P. An updated review of the treatment landscape for advanced gastrointestinal stromal tumors. Cancer 2021, 127, 2187–2195. [Google Scholar] [CrossRef]
- Mas, L.; Bachet, J.-B. GIST avancées: Quels traitements en 2022? Bull. Du Cancer 2022, 109, 1082–1087. [Google Scholar] [CrossRef]
- Chen, M.H.; Kerkelä, R.; Force, T. Mechanisms of cardiac dysfunction associated with tyrosine kinase inhibitor cancer therapeutics. Circulation 2008, 118, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Xu, Q.; Li, Q.; Cui, Z.; Li, W.; Zeng, F. Adverse reactions after treatment with dasatinib in chronic myeloid leukemia: Characteristics, potential mechanisms, and clinical management strategies. Front. Oncol. 2023, 13, 1113462. [Google Scholar] [CrossRef]
- Ledoux, J.; Trouvé, A.; Tchertanov, L. The Inherent Coupling of Intrinsically Disordered Regions in the Multidomain Receptor Tyrosine Kinase KIT. Int. J. Mol. Sci. 2022, 23, 1589. [Google Scholar] [CrossRef] [PubMed]
- Hensen, U.; Meyer, T.; Haas, J.; Rex, R.; Vriend, G.; Grubmüller, H. Exploring protein dynamics space: The dynasome as the missing link between protein structure and function. PLoS ONE 2012, 7, e33931. [Google Scholar] [CrossRef]
- Kufareva, I.; Ilatovskiy, A.V.; Abagyan, R. Pocketome: An encyclopedia of small-molecule binding sites in 4D. Nucleic Acids Res. 2012, 40, D535–D540. [Google Scholar] [CrossRef]
- Vidal, M. Interactome modeling. Febs Lett. 2005, 579, 1834–1838. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J.; Csermely, P. Allo-network drugs: Harnessing allostery in cellular networks. Trends Pharmacol. Sci. 2011, 32, 686–693. [Google Scholar] [CrossRef]
- Mol, C.D.; Lim, K.B.; Sridhar, V.; Zou, H.; Chien, E.Y.; Sang, B.C.; Nowakowski, J.; Kassel, D.B.; Cronin, C.N.; McRee, D.E. Structure of a c-kit product complex reveals the basis for kinase transactivation. J. Biol. Chem. 2003, 278, 31461–31464. [Google Scholar] [CrossRef]
- Mol, C.D.; Dougan, D.R.; Schneider, T.R.; Skene, R.J.; Kraus, M.L.; Scheibe, D.N.; Snell, G.P.; Zou, H.; Sang, B.C.; Wilson, K.P. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J. Biol. Chem. 2004, 279, 31655–31663. [Google Scholar] [CrossRef]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. Ser. A Math. Phys. Eng. Sci. 2016, 374, 20150202. [Google Scholar] [CrossRef]
- David, C.C.; Jacobs, D.J. Principal component analysis: A method for determining the essential dynamics of proteins. Methods Mol. Biol. 2014, 1084, 193–226. [Google Scholar] [CrossRef] [PubMed]
- Ren, Z.; Ren, P.X.; Balusu, R.; Yang, X. Transmembrane Helices Tilt, Bend, Slide, Torque, and Unwind between Functional States of Rhodopsin. Sci. Rep. 2016, 6, 34129. [Google Scholar] [CrossRef] [PubMed]
- Farrens, D.L.; Altenbach, C.; Yang, K.; Hubbell, W.L.; Khorana, H.G. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science 1996, 274, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.E.; Zoonens, M.; Engelman, D.M. Dynamic helix interactions in transmembrane signaling. Cell 2006, 127, 447–450. [Google Scholar] [CrossRef] [PubMed]
- Lyman, E.; Zuckerman, D.M. Ensemble-based convergence analysis of biomolecular trajectories. Biophys. J. 2006, 91, 164–172. [Google Scholar] [CrossRef] [PubMed]
- DiNitto, J.P.; Deshmukh, G.D.; Zhang, Y.; Jacques, S.L.; Coli, R.; Worrall, J.W.; Diehl, W.; English, J.M.; Wu, J.C. Function of activation loop tyrosine phosphorylation in the mechanism of c-Kit auto-activation and its implication in sunitinib resistance. J. Biochem. 2010, 147, 601–609. [Google Scholar] [CrossRef]
- Appadurai, R.; Nagesh, J.; Srivastava, A. High resolution ensemble description of metamorphic and intrinsically disordered proteins using an efficient hybrid parallel tempering scheme. Nat. Commun. 2021, 12, 958. [Google Scholar] [CrossRef]
- Dar, A.C.; Shokat, K.M. The evolution of protein kinase inhibitors from antagonists to agonists of cellular signaling. Annu. Rev. Biochem. 2011, 80, 769–795. [Google Scholar] [CrossRef]
- Krishnamurty, R.; Maly, D.J. Biochemical mechanisms of resistance to small-molecule protein kinase inhibitors. ACS Chem. Biol. 2010, 5, 121–138. [Google Scholar] [CrossRef]
- Wu, T.-S.; Lin, W.-H.; Tsai, H.-J.; Hsueh, C.-C.; Hsu, T.; Wang, P.-C.; Lin, H.-Y.; Peng, Y.-H.; Lu, C.-T.; Lee, L.-C.; et al. Discovery of Conformational Control Inhibitors Switching off the Activated c-KIT and Targeting a Broad Range of Clinically Relevant c-KIT Mutants. J. Med. Chem. 2019, 62, 3940–3957. [Google Scholar] [CrossRef]
- Bauer, S.; George, S.; von Mehren, M.; Heinrich, M.C. Early and Next-Generation KIT/PDGFRA Kinase Inhibitors and the Future of Treatment for Advanced Gastrointestinal Stromal Tumor. Front. Oncol. 2021, 11, 672500. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Jiang, S.; Shi, Y. Tyrosine kinase inhibitors for solid tumors in the past 20 years (2001–2020). J. Hematol. Oncol. 2020, 13, 143. [Google Scholar] [CrossRef]
- Le Guilloux, V.; Schmidtke, P.; Tuffery, P. Fpocket: An open source platform for ligand pocket detection. BMC Bioinform. 2009, 10, 168. [Google Scholar] [CrossRef] [PubMed]
- Gajiwala, K.S.; Wu, J.C.; Christensen, J.; Deshmukh, G.D.; Diehl, W.; DiNitto, J.P.; English, J.M.; Greig, M.J.; He, Y.A.; Jacques, S.L.; et al. KIT kinase mutants show unique mechanisms of drug resistance to imatinib and sunitinib in gastrointestinal stromal tumor patients. Proc. Natl. Acad. Sci. USA 2009, 106, 1542–1547. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Liang, J.; Edelsbrunner, H.; Woodward, C. Anatomy of protein pockets and cavities: Measurement of binding site geometry and implications for ligand design. Protein Sci. A Publ. Protein Soc. 1998, 7, 1884–1897. [Google Scholar] [CrossRef]
- Karplus, P.A. Hydrophobicity regained. Protein Sci. A Publ. Protein Soc. 1997, 6, 1302–1307. [Google Scholar] [CrossRef]
- Monera, O.D.; Sereda, T.J.; Zhou, N.E.; Kay, C.M.; Hodges, R.S. Relationship of sidechain hydrophobicity and alpha-helical propensity on the stability of the single-stranded amphipathic alpha-helix. J. Pept. Sci. Off. Publ. Eur. Pept. Soc. 1995, 1, 319–329. [Google Scholar] [CrossRef]
- Chauvot de Beauchêne, I.; Allain, A.; Panel, N.; Laine, E.; Trouvé, A.; Dubreuil, P.; Tchertanov, L. Hotspot mutations in KIT receptor differentially modulate its allosterically coupled conformational dynamics: Impact on activation and drug sensitivity. PLoS Comput. Biol. 2014, 10, e1003749. [Google Scholar] [CrossRef]
- Naganathan, A.N. Modulation of allosteric coupling by mutations: From protein dynamics and packing to altered native ensembles and function. Curr. Opin. Struct. Biol. 2019, 54, 1–9. [Google Scholar] [CrossRef]
- Tokuriki, N.; Tawfik, D.S. Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 2009, 19, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Varadarajan, R. Insights into protein structure, stability and function from saturation mutagenesis. Curr. Opin. Struct. Biol. 2018, 50, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Haririnia, A.; Verma, R.; Purohit, N.; Twarog, M.Z.; Deshaies, R.J.; Bolon, D.; Fushman, D. Mutations in the Hydrophobic Core of Ubiquitin Differentially Affect Its Recognition by Receptor Proteins. J. Mol. Biol. 2008, 375, 979–996. [Google Scholar] [CrossRef] [PubMed]
- Eginton, C.; Cressman, W.J.; Bachas, S.; Wade, H.; Beckett, D. Allosteric coupling via distant disorder-to-order transitions. J. Mol. Biol. 2015, 427, 1695–1704. [Google Scholar] [CrossRef]
- Changeux, J.P.; Christopoulos, A. Allosteric Modulation as a Unifying Mechanism for Receptor Function and Regulation. Cell 2016, 166, 1084–1102. [Google Scholar] [CrossRef]
- Ester, M.; Kriegel, H.P.; Sander, J.; Xiaowei, X. A density-based algorithm for discovering clusters in large spatial databases with noise. In Proceedings of the Second International Conference on Knowledge Discovery and Data Mining, Portland, OR, USA, 2–4 August 1996; Simoudis, E., Han, J., Fayyad, U., Eds.; AAAI Press: Menlo Park, CA, USA, 1996; pp. 226–231. [Google Scholar]
- Ledoux, J.; Tchertanov, L. Does Generic Cyclic Kinase Insert Domain of Receptor Tyrosine Kinase KIT Clone Its Native Homologue? Int. J. Mol. Sci. 2022, 23, 12898. [Google Scholar] [CrossRef]
- Changeux, J.P. 50 years of allosteric interactions: The twists and turns of the models. Nat. Rev. Mol. Cell Biol. 2013, 14, 819–829. [Google Scholar] [CrossRef]
- Amaro, R.E. Will the Real Cryptic Pocket Please Stand Out? Biophys. J. 2019, 116, 753–754. [Google Scholar] [CrossRef]
- Knoverek, C.R.; Amarasinghe, G.K.; Bowman, G.R. Advanced Methods for Accessing Protein Shape-Shifting Present New Therapeutic Opportunities. Trends Biochem. Sci. 2019, 44, 351–364. [Google Scholar] [CrossRef]
- Cruz, M.A.; Frederick, T.E.; Mallimadugula, U.L.; Singh, S.; Vithani, N.; Zimmerman, M.I.; Porter, J.R.; Moeder, K.E.; Amarasinghe, G.K.; Bowman, G.R. A cryptic pocket in Ebola VP35 allosterically controls RNA binding. Nat. Commun. 2022, 13, 2269. [Google Scholar] [CrossRef]
- Vajda, S.; Beglov, D.; Wakefield, A.E.; Egbert, M.; Whitty, A. Cryptic binding sites on proteins: Definition, detection, and druggability. Curr. Opin. Chem. Biol. 2018, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jérusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef] [PubMed]
- Herbert, C.; Schieborr, U.; Saxena, K.; Juraszek, J.; De Smet, F.; Alcouffe, C.; Bianciotto, M.; Saladino, G.; Sibrac, D.; Kudlinzki, D.; et al. Molecular mechanism of SSR128129E, an extracellularly acting, small-molecule, allosteric inhibitor of FGF receptor signaling. Cancer Cell 2013, 23, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Naik, R.R.; Shakya, A.K. Exploring the chemotherapeutic potential of currently used kinase inhibitors: An update. Front. Pharmacol. 2022, 13, 1064472. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.W.; Okada, M.; Sayeed, I.; Xiao, G.; Stein, D.; Jin, P.; Ye, K. Gambogic amide, a selective agonist for TrkA receptor that possesses robust neurotrophic activity, prevents neuronal cell death. Proc. Natl. Acad. Sci. USA 2007, 104, 16329–16334. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Shen, M.-Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. A Publ. Protein Soc. 2006, 15, 2507–2524. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Lomize, A.L.; Pogozheva, I.D.; Lomize, M.A.; Mosberg, H.I. Positioning of proteins in membranes: A computational approach. Protein Sci. 2006, 15, 1318–1333. [Google Scholar] [CrossRef]
- Schott-Verdugo, S.; Gohlke, H. PACKMOL-Memgen: A Simple-To-Use, Generalized Workflow for Membrane-Protein-Lipid-Bilayer System Building. J. Chem. Inf. Model. 2019, 59, 2522–2528. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.v.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Duane, S.; Kennedy, A.D.; Pendleton, B.J.; Roweth, D. Hybrid Monte Carlo. Phys. Lett. B 1987, 195, 216–222. [Google Scholar] [CrossRef]
- Andersen, H.C. Rattle: A “velocity” version of the shake algorithm for molecular dynamics calculations. J. Comput. Phys. 1983, 52, 24–34. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure—Pattern-Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [PubMed]
- Gapsys, V.; Michielssens, S.; Peters, J.H.; de Groot, B.L.; Leonov, H. Calculation of binding free energies. Methods Mol. Biol. 2015, 1215, 173–209. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bakan, A.; Dutta, A.; Mao, W.; Liu, Y.; Chennubhotla, C.; Lezon, T.R.; Bahar, I. Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics 2014, 30, 2681–2683, Epub 2014/05/23. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ledoux, J.; Botnari, M.; Tchertanov, L. Receptor Tyrosine Kinase KIT: Mutation-Induced Conformational Shift Promotes Alternative Allosteric Pockets. Kinases Phosphatases 2023, 1, 220-250. https://doi.org/10.3390/kinasesphosphatases1040014
Ledoux J, Botnari M, Tchertanov L. Receptor Tyrosine Kinase KIT: Mutation-Induced Conformational Shift Promotes Alternative Allosteric Pockets. Kinases and Phosphatases. 2023; 1(4):220-250. https://doi.org/10.3390/kinasesphosphatases1040014
Chicago/Turabian StyleLedoux, Julie, Marina Botnari, and Luba Tchertanov. 2023. "Receptor Tyrosine Kinase KIT: Mutation-Induced Conformational Shift Promotes Alternative Allosteric Pockets" Kinases and Phosphatases 1, no. 4: 220-250. https://doi.org/10.3390/kinasesphosphatases1040014
APA StyleLedoux, J., Botnari, M., & Tchertanov, L. (2023). Receptor Tyrosine Kinase KIT: Mutation-Induced Conformational Shift Promotes Alternative Allosteric Pockets. Kinases and Phosphatases, 1(4), 220-250. https://doi.org/10.3390/kinasesphosphatases1040014